Rethymic: Package Insert / Prescribing Info
Package insert / product label
Generic name: allogenic thymocyte-depleted thymus tissue-agdc
Dosage form: implant
Drug class: Other immunostimulants
Medically reviewed by Drugs.com. Last updated on Jan 8, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
RETHYMIC (Allogeneic processed thymus tissue–agdc)
For surgical implantation
Initial U.S. Approval: 2021
Indications and Usage for Rethymic
RETHYMIC is indicated for immune reconstitution in pediatric patients with congenital athymia. (1)
Limitations of Use:
- RETHYMIC is not indicated for the treatment of patients with severe combined immunodeficiency (SCID).
Rethymic Dosage and Administration
RETHYMIC is administered by a surgical procedure. The recommended dose range is 5,000 to 22,000 mm2 of RETHYMIC/m2 recipient body surface area (BSA). (2) Immunosuppressive therapy is recommended for patients receiving RETHYMIC based on disease phenotype and PHA levels. (14)
Dosage Forms and Strengths
RETHYMIC consists of yellow to brown slices of processed tissue with varying thickness and shape. The dosage is determined by the surface area of the RETHYMIC slices and recipient BSA. (3)
Contraindications
None. (4)
Warnings and Precautions
- Immune reconstitution sufficient to protect from infection is unlikely to develop prior to 6 to 12 months after treatment with RETHYMIC. Given the immunocompromised condition of athymic patients, infection control measures should be followed until the development of thymic function can be established. ( 5.1)
- Monitor and treat patients at risk for the development of graft versus host disease (GVHD). ( 5.2)
- Monitor for the development of autoimmune disorders, including complete blood counts with differential, liver enzymes, serum creatinine, urinalysis, and thyroid function. ( 5.3)
- Pre-existing renal impairment is a risk factor for death. ( 5.4)
- Pre-existing cytomegalovirus infection may result in death prior to the development of thymic function. ( 5.5)
- Monitor for the development of lymphoproliferative disorder (blood cancer). ( 5.6)
- Transmission of infectious diseases may occur because RETHYMIC is derived from human tissue. ( 5.7)
- Immunizations should not be administered in patients who have received RETHYMIC until immune-function criteria have been met. ( 5.8)
- Patients should be tested for anti-HLA antibodies prior to treatment. ( 5.9)
Adverse Reactions/Side Effects
The most common (>10%) adverse events related to RETHYMIC included: hypertension (high blood pressure, 19%), cytokine release syndrome (18%), rash (15%), hypomagnesemia (low magnesium, 16%), renal impairment / failure (decrease of kidney function, 12%), thrombocytopenia (low platelets, 12%), and graft versus host disease, (10%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Sumitomo Pharma America at 833-369-9868 or FDA at 1-800-FDA-1088 or https://www.fda.gov/safety/medwatch-fda-safety-information-and-adverse-event-reporting-program.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2023
Full Prescribing Information
1. Indications and Usage for Rethymic
RETHYMIC® is indicated for immune reconstitution in pediatric patients with congenital athymia.
2. Rethymic Dosage and Administration
2.1 Dosage
RETHYMIC is administered by a surgical procedure. The dosage is determined by the total surface area of the RETHYMIC slices and recipient body surface area (BSA). A RETHYMIC slice is defined as the contents on a single filter membrane; the RETHYMIC slices are variable in size and shape. The recommended dose range is 5,000 to 22,000 mm2 of RETHYMIC surface area/m2 recipient BSA. The manufacturer calculates the dose in advance for the specific patient; the amount of product provided is adjusted at the manufacturing facility to ensure the maximum dose for the patient cannot be exceeded. Up to 42 cultured RETHYMIC slices will be provided for each patient. At the time of surgery, the manufacturing personnel communicate to the surgical team the portion of the product that represents the minimum dose. Patients with evidence of maternal engraftment or an elevated response to phytohemagglutinin (PHA) should receive RETHYMIC with immunosuppressive medications (Table 2).
2.2 Administration Instructions
Surgical implantation of RETHYMIC should be done by a qualified surgical team in a single surgical session at a qualified hospital. RETHYMIC should be implanted in the quadriceps muscle in accordance with the instructions provided below. Implantation of RETHYMIC into the quadriceps requires a healthy bed of muscle tissue.
Preparation for the Implantation Procedure:
- Operating room culture dishes (sterile 100 mm tissue culture dishes) and saline for injection are supplied by the operating room; a sufficient supply of operating room culture dishes and saline must be provided by the hospital for use in the implantation procedure.
- The product is delivered to the operating room by manufacturing personnel. The recommended dose is determined based on the patient's BSA. The manufacturer calculates the dose in advance for the specific patient. Manufacturing personnel and the operating room staff confirm that the lot delivered is for the intended recipient.
- Manufacturing personnel communicate to the surgical team the minimum number of RETHYMIC slices to be implanted to achieve the minimum dose. The product expiration date and time for the entire lot is labeled on each polystyrene dish (drug product dish).
- Always handle RETHYMIC slices aseptically. Do not use if there is evidence of contamination.
- Outside the sterile field, manufacturing personnel unpack RETHYMIC from the shipping box. One drug product dish at a time is removed from the drug product box and shipping box. Manufacturing personnel inspect the drug product box and each drug product dish for signs of contamination, damage, spills, or leakage. If damage to the drug product dishes, leaks, spillage or evidence of contamination is noted, manufacturing personnel will notify the surgical team that the lot cannot be implanted.
- When the surgical team is ready, manufacturing personnel and surgical staff begin the transfer of drug product to the sterile operative field. Manufacturing personnel carry one drug product dish, which contains up to 4 RETHYMIC slices on up to 2 surgical sponges, with each RETHYMIC slice on a filter membrane, to the surgical staff near the sterile field. Manufacturing personnel open the drug product dish to expose the RETHYMIC slices.
- The surgical staff team member uses a pair of forceps to remove individual RETHYMIC slices with their filter membranes from the drug product dish (Figure 1). The surgical team member places each RETHYMIC slice with its filter membrane into a sterile 100 mm tissue culture dish ("operating room culture dish") containing approximately 2 mL preservative-free saline that resides in the sterile field on the instrument table. This is repeated to transfer all RETHYMIC slices from the first drug product dish into a sterile operating room culture dish. After the first set of RETHYMIC slices has been prepared for surgical implantation and provided to the surgeon, another drug product dish with RETHYMIC slices is passed to the surgical staff member for removal from their filter membranes as described above.
- Using 2 pairs of sterile forceps, the surgical staff team member should use one pair of forceps to hold the filter in place while using the other forceps to scrape and loosen the RETHYMIC slice from the filter membrane (Figure 1). Then, while using one pair of forceps to hold the filter in place, the surgical staff team member uses the other forceps to lift the RETHYMIC slice away from the filter membrane by pulling the tissue up. The surgical staff team member places each RETHYMIC slice separately into the saline-containing operating room culture dish in the sterile field on top of its original filter membrane. The RETHYMIC slice will change from a flatter slice to a condensed, lumpy shape at this stage of the procedure. The surgeon then implants the first set of RETHYMIC slices. The surgical team staff member should process the next set of up to 4 RETHYMIC slices from the next drug product dish into a second operating room culture dish in the same manner while the surgeon continues implanting the first set of up to 4 slices. When the surgeon finishes implanting the first set of RETHYMIC slices, the surgical staff team member transfers and prepares the next operating room culture dish on the surgical field. Continue this cycle until all the desired tissue is transferred during the implantation procedure.
Figure 1: Preparing for the Implantation Procedure
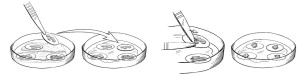
Figure 1: Within the sterile field, forceps are used to move individual RETHYMIC slices with their filter membranes from the drug product dish to the operating room culture dish (left images). A pair of forceps is used to gently scrape and lift the RETHYMIC slice off the filter membrane in the operating room culture dish in preparation for easy removal prior to implantation (right images).
Implantation of RETHYMIC:
- After induction of general anesthesia, a cranial-caudal skin incision (typically ~5 cm in length; Figure 2) should be made over the anterior thigh compartment. The size of the incision and the use of one or both legs for the implantation procedure is determined by the size of the patient, his/her muscle mass, and the amount of tissue to be implanted. If all or nearly all of the tissue can be implanted in one leg, then only one leg should be used.
Figure 2: Surgical Incision and Opening of Fascia
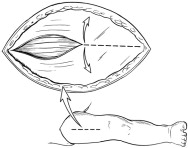
- Open fascia to expose the anterior compartment muscles (Figure 2).
- Create a pocket in between the muscle fibers using a tonsil clamp or similar instrument. Each pocket should be made along the natural furrows throughout the quadriceps muscle group.
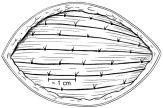
- Individual RETHYMIC slices should be implanted approximately 1 cm in depth and approximately 1 cm apart into the pockets between the muscle fibers in the quadriceps muscle (Figure 3).
Figure 3: Implant Individual RETHYMIC Slices
- A large or thick RETHYMIC slice may be cut in half, at the surgeon's discretion, to ensure the slice is surrounded by muscle tissue once implanted. Implant as many RETHYMIC slices as possible within the recommended dose range of 5,000 to 22,000 mm2 of processed thymus tissue/m2 recipient BSA. During the procedure, the surgeon uses their judgment to balance the benefit of implanting additional RETHYMIC slices against the risk(s) that may be associated with implantation into limited muscle mass, number of implantations sites and other patient considerations.
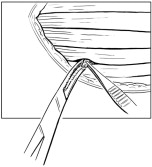
- Once each RETHYMIC slice has been implanted, it should be fully covered by muscle tissue. Then a single absorbable suture should be used to close the pocket where the RETHYMIC slice was implanted (Figure 4).
Figure 4: Close the Site of Implantation
- Once the intended dose has been implanted, confirm hemostasis. Close the skin incision with 2 layers of absorbable sutures and apply a standard dressing, such as wound closure strips or skin glue. Leave the fascia open to allow room for muscle compartment swelling. An occlusive dressing may be used to prevent contamination.
3. Dosage Forms and Strengths
RETHYMIC consists of yellow to brown slices of processed thymus tissue with varying thickness and shape. Each drug product dish contains up to 4 RETHYMIC slices that adhere to circular filter membranes on top of surgical sponges in 5 mL of medium. The RETHYMIC slices are variable in size and shape; a RETHYMIC slice is defined as the contents of a single filter membrane. The dosage is based on the total surface area of the RETHYMIC slices, and the amount administered is calculated based on recipient BSA. The surgeon should implant as many RETHYMIC slices as possible within the recommended dose range of 5,000 to 22,000 mm2 of RETHYMIC/m2 recipient BSA. The manufacturer calculates the dose in advance for the specific patient; the amount of product provided is adjusted at the manufacturing facility to ensure the maximum dose for the patient cannot be exceeded. Up to 42 RETHYMIC slices will be provided for each patient. At the time of surgery, the manufacturing personnel will inform the surgical team of the portion of the product that represents the minimum dose.
5. Warnings and Precautions
5.1 Infection Control and Immunoprophylaxis
Immune reconstitution sufficient to protect from infection is unlikely to develop prior to 6-12 months after treatment with RETHYMIC. Given the immunocompromised condition of athymic patients, follow infection control measures until the development of thymic function is established as measured through flow cytometry. This should include counseling patients and their caregivers on good handwashing practices and minimizing exposure to visitors. Monitor patients closely for signs of infection, including fever. If a fever develops, assess the patient by blood and other cultures and treat with antimicrobials as clinically indicated.
Patients should be maintained on immunoglobulin replacement therapy until all of the following criteria are met:
- No longer on immunosuppression (at least 10% of CD3+ T cells are naïve in phenotype).
- At least 9 months post-treatment.
- Phytohemagglutinin (PHA) response within normal limits.
- Normal serum IgA is also desirable but not required.
Two months after stopping immunoglobulin replacement therapy, the IgG trough level should be checked.
- If the IgG trough level is in the normal range for age, the patient can remain off of immunoglobulin replacement.
- If the IgG trough level is lower than the normal range for age, immunoglobulin replacement therapy should be restarted and continued for a year before being retested using the above guidelines.
Prior to and after treatment with RETHYMIC, patients should be maintained on Pneumocystis jiroveci pneumonia prophylaxis until all of the following criteria are met:
- No longer on immunosuppression (at least 10% of CD3+ T cells are naïve in phenotype).
- At least 9 months post-treatment.
- PHA response within normal limits.
- CD4+ T cell count > 200 cells/mm3.
5.2 Graft versus Host Disease
In clinical studies with RETHYMIC, GVHD occurred in 11 (10%) RETHYMIC-treated patients of whom 6 (55%) died. RETHYMIC may cause or exacerbate pre-existing GVHD. Seven patients (7%) experienced autologous GVHD, 3 patients (3%) experienced GVHD due to maternal cells and 1 patient (1%) experienced GVHD due to cells from a prior hematopoietic cell transplant (HCT). Risk factors for GVHD include atypical complete DiGeorge anomaly phenotype, prior HCT and maternal engraftment. GVHD may manifest as fever, rash, lymphadenopathy, elevated bilirubin and liver enzymes, enteritis, and/or diarrhea. Patients with elevated baseline T cell proliferative response to PHA > 5,000 cpm or > 20-fold over background should receive immunosuppressive therapies to decrease the risk of GVHD (Table 2 and Table 3). Development of GVHD symptoms should be closely monitored and promptly treated.
5.3 Autoimmune Disorders
Thirty-seven patients (35%) in the RETHYMIC clinical program experienced autoimmune-related adverse reactions. These events included: thrombocytopenia (including idiopathic thrombocytopenic purpura) in 13 patients (12%), neutropenia in 9 patients (9%), proteinuria in 7 patients (7%), hemolytic anemia in 7 patients (7%), alopecia in 4 patients (4%), hypothyroidism in 2 patients (2%), autoimmune hepatitis in 2 patients (2%), and autoimmune arthritis (juvenile idiopathic and psoriatic arthritis) in 2 patients (2%). One patient (1%) each experienced transverse myelitis, albinism, hyperthyroidism, and ovarian failure. The onset of autoimmune related events ranged from the three days before the surgical implantation procedure until 16 years post-treatment. Most events occurred within the first year after treatment.
Monitor complete blood counts with differential weekly for the first 2 months post-treatment and then monthly through 12 months post-treatment. Liver enzymes including aspartate aminotransferase and alanine aminotransferase, serum creatinine levels, and urinalysis should be performed monthly for 3 months and then every 3 months through 12 months post-treatment. Thyroid function studies should be performed prior to treatment and then at 6 months and 12 months post-treatment. After 12 months, testing should be performed annually.
5.4 Renal Impairment
Ten patients with renal impairment (elevated serum creatinine at baseline) were treated in studies with RETHYMIC. Five of these patients died within 1 year and a sixth patient died 3 years after treatment with RETHYMIC. Renal impairment at baseline is considered a risk factor for death.
5.5 Cytomegalovirus Infection
In clinical studies with RETHYMIC, 4 out of 4 patients with preexisting CMV infection prior to treatment with RETHYMIC died. The benefits/risks of treatment should be considered prior to treating patients with pre-existing CMV infection.
5.6 Malignancy
Because of the underlying immune deficiency, patients who receive RETHYMIC may be at risk of developing post-treatment lymphoproliferative disorder (blood cancer). The infant tissue donor is screened for Epstein-Barr virus (EBV) and cytomegalovirus (CMV), but patients should be tested for EBV and CMV using PCR prior to and 3 months following treatment with RETHYMIC, or after any exposure to or suspected infection with CMV or EBV.
5.7 Transmission of Serious Infections and Transmissible Infectious Diseases
Transmission of infectious disease may occur because RETHYMIC is derived from human tissue. Disease may be caused by known or unknown infectious agents. Donors are screened for increased risk of infection with human immunodeficiency virus (HIV), human T-cell lymphotropic virus (HTLV), hepatitis B virus (HBV), hepatitis C virus (HCV), Treponema pallidum, Trypanosoma cruzi, West Nile virus (WNV), transmissible spongiform encephalopathy (TSE) agents, vaccinia and Zika virus. Donors are also screened for clinical evidence of sepsis, and communicable disease risks associated with xenotransplantation. Blood samples (from the infant tissue donor or the birth mother, as applicable) are tested for HIV types 1, 2, and O, HTLV types I and II, HBV, HCV, T. pallidum, WNV, and T. cruzi. Blood from the infant tissue donor is also tested for Toxoplasma gondii, Epstein-Barr virus (EBV) and CMV. RETHYMIC is tested for sterility, endotoxin, and mycoplasma. These measures do not eliminate the risk of transmitting these or other infectious diseases and disease agents.
Testing of maternal and infant donor blood is also performed for evidence of donor infection due to cytomegalovirus (CMV).
Product manufacturing includes porcine- and bovine-derived reagents. While all animal-derived reagents are tested for animal viruses, bacteria, fungi, and mycoplasma before use, these measures do not eliminate the risk of transmitting these or other transmissible infectious diseases and disease agents.
Final sterility and mycoplasma test results are not available at the time of use, but manufacturing personnel will communicate any positive results from sterility testing to the physician. Report the occurrence of transmitted infection to Sumitomo Pharma America at 833-369-9868.
5.8 Vaccine Administration
Immunizations should not be administered in patients who have received RETHYMIC until immune-function criteria have been met.
Inactivated vaccines:
Inactivated vaccines may be administered once all of the following criteria are met:
- Immunosuppressive therapies have been discontinued.
- Immunoglobulin (IgG) replacement therapy has been discontinued.
- The total CD4+ T cell count is > 200 cells/mm3 and there are more CD4+ T cells than CD8+ T cells (CD4+ > CD8+).
It is recommended that no more than 2 inactivated vaccines be given per month.
Live Vaccines:
Live virus vaccines should not be administered until patients have met the criteria for inactivated vaccines and received vaccinations with inactivated agents (e.g., tetanus toxoid). No additional vaccines (live or inactivated), except the inactivated influenza vaccine, should be given within 6 months after vaccination with a measles-containing vaccine or within 2 months after the varicella vaccine. Consider verifying response to vaccination with appropriate testing, in particular varicella and measles.
5.9 Anti-HLA Antibodies
All patients should be screened for anti-HLA antibodies prior to receiving RETHYMIC. Patients testing positive for anti-HLA antibodies should receive RETHYMIC from a donor who does not express those HLA alleles.
5.10 HLA Typing
HLA matching is required in patients who have received a prior hematopoietic cell transplantation (HCT) or a solid organ transplant. Patients who have received a prior HCT are at increased risk of developing GVHD after RETHYMIC if the HCT donor did not fully match the recipient. To minimize this risk, HLA matching of RETHYMIC to recipient alleles that were not expressed in the HCT donor is recommended.
6. Adverse Reactions/Side Effects
The most common adverse reactions (incidence in at least 10% of patients) reported following administration of RETHYMIC were hypertension (high blood pressure), cytokine release syndrome, rash, hypomagnesemia (low magnesium), renal impairment / failure (decrease of kidney function), thrombocytopenia (low platelets), and graft versus host disease.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described in this section are derived from 10 prospective, single-center, open-label studies, and include 105 patients who were treated with RETHYMIC in these studies and who had at least one year of follow-up. Table 1 lists the adverse reactions occurring in 105 patients who were treated with RETHYMIC in these studies.
| System Organ Class Preferred Term | RETHYMIC (N=105) n (%) |
|---|---|
|
|
| Number of Patients with Adverse Reactions* | 80 (76) |
| Hypertension (high blood pressure) | 20 (19) |
| Cytokine release syndrome† | 19 (18) |
| Hypomagnesemia (low magnesium) | 17 (16) |
| Rash‡ | 16 (15) |
| Renal impairment / failure§ (decrease of kidney function) | 13 (12) |
| Thrombocytopenia¶ (low platelets) | 13 (12) |
| Graft versus host disease# | 11 (10) |
| Hemolytic anemiaÞ (low red bloods cells) | 9 (9) |
| Neutropenia (low white blood cells) | 9 (9) |
| Respiratory distressß (difficulty breathing) | 8 (8) |
| Proteinuria (protein in urine) | 7 (7) |
| Pyrexia (fever) | 6 (6) |
| Acidosisà | 6 (6) |
| Diarrheaè | 5 (5) |
| Seizureð | 5 (5) |
Of the 105 patients, 29 patients died after receiving RETHYMIC, including 23 deaths in the first year (<365 days) after treatment with RETHYMIC. Causes of death in the first year included 13 deaths due to infection or complications due to infection, 5 deaths due to respiratory failure / hypoxia, 3 deaths due to hemorrhage-related events, and 2 deaths due to cardiorespiratory arrest. Of the 6 patients who died more than 1 year after treatment with RETHYMIC, the deaths were considered unrelated to study treatment: 2 died due to respiratory failure and 1 died due to each of the following: cardiopulmonary arrest, intracranial hemorrhage, infection, and unknown cause.
Related/similar drugs
7. Drug Interactions
No drug interaction studies have been conducted with RETHYMIC. If possible, prolonged use of immunosuppressive therapies, including high-dose corticosteroids, should be avoided.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no clinical data with RETHYMIC in pregnant women. No animal reproductive and developmental toxicity studies have been conducted with RETHYMIC. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of cellular components of RETHYMIC in human milk, the effect breastfeeding may have on RETHYMIC, the effect of being breastfed from a mother who received RETHYMIC as a child, or the effects of RETHYMIC on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for RETHYMIC and potential adverse effects on the breastfed infant from RETHYMIC.
8.3 Females and Males of Reproductive Potential
No nonclinical or clinical studies were performed to evaluate the effects of RETHYMIC on fertility.
8.4 Pediatric Use
The efficacy and safety of RETHYMIC have been established in pediatric patients with congenital athymia. The efficacy of RETHYMIC has been established in 95 pediatric patients (median age 9 months [range: 33 days to 3 years], including 65 patients age <1 year, 24 patients age 1 to <2 years, and 6 patients age 2 to <3 years at time of treatment) who were treated with RETHYMIC and included in the analysis of efficacy [see Clinical Studies (14)]. The safety of RETHYMIC has been established in 105 pediatric patients (median age 9 months [range: 33 days to 16.9 years] at time of treatment) with congenital athymia who were evaluated for safety following RETHYMIC administration. The safety population included 65 patients age <1 year, 27 patients age 1 to <2 years, 9 patients age 2 to <3 years, 1 patient age 3 to <6 years, and 3 patients age 13 to 17 years at time of treatment. Within the safety population, survival was similar across age groups. Adverse reactions were reported at similar frequencies across the age groups and were generally of similar types and severities.
8.6 Renal Impairment
In the clinical studies with RETHYMIC, 10 of 105 patients had impaired renal function at baseline based on elevated screening creatinine [see Warnings and Precautions (5.4)]. Baseline renal function should be considered when selecting immunosuppressants. Ensure appropriate involvement of a nephrologist in care of patients with renal impairment.
10. Overdosage
The maximum recommended dose is 22,000 mm2 of RETHYMIC/m2 recipient body surface area (BSA). Standard clinical care is recommended for patients receiving a dose > 22,000 mm2 of RETHYMIC/m2 recipient BSA. The product, as provided, has been adjusted at the manufacturing facility to not exceed the maximum dose based on the patient body surface area.
During clinical development one patient received a dose higher (23,755 mm2/m2) than the maximum recommended dose. This patient developed enteritis. A biopsy showed T cell, B cell, and neutrophil infiltration of the gut which resolved after treatment with immunosuppression, 5 months after treatment with RETHYMIC. The enteritis may have been related to the high dose of RETHYMIC.
11. Rethymic Description
RETHYMIC consists of yellow to brown slices of allogeneic processed thymus tissue for administration by surgical implantation. Three to 11 drug product containers, with a total of 10 to 42 RETHYMIC slices, are provided for each patient. Each drug product container provides up to 4 RETHYMIC slices of variable size. The total dose, based on the number of slices administered to the patient, is 5,000 to 22,000 mm2 of RETHYMIC/m2 recipient BSA.
Thymus tissue is obtained from donors less than or equal to 9 months of age undergoing cardiac surgery. This thymus tissue is aseptically processed and cultured for 12 to 21 days to produce RETHYMIC slices. Each product lot is manufactured from a single unrelated donor and one product lot treats a single patient. The manufacturing process preserves the thymic epithelial cells and tissue structure and depletes most of the donor thymocytes from the tissue. These RETHYMIC slices are then surgically implanted into patients with congenital athymia.
The product manufacture uses reagents derived from animal materials. The surgical sponge used during culturing is porcine-derived. Fetal bovine serum is a component in the culture medium used to culture the thymus slices and RETHYMIC is formulated in media that is supplemented with fetal bovine serum. Therefore, bovine- and porcine-derived proteins will be present in RETHYMIC. These animal-derived reagents are tested for animal viruses, retroviruses, bacteria, fungi, yeast, and mycoplasma before use.
12. Rethymic - Clinical Pharmacology
12.1 Mechanism of Action
RETHYMIC is intended to reconstitute immunity in patients who are athymic. The proposed mechanism of action involves the migration of recipient T cell progenitors from the bone marrow to the implanted RETHYMIC slices, where they develop into naïve immunocompetent recipient T cells. Evidence of thymic function can be observed with the development of naïve T cells in the peripheral blood; this is unlikely to be observed prior to 6-12 months after treatment with RETHYMIC.
14. Clinical Studies
The efficacy of RETHYMIC was evaluated in 10 prospective, single-center, open-label studies that enrolled a total of 105 patients, including 95 patients in the primary efficacy analysis. The demographics and baseline characteristics of the patients enrolled in the clinical studies were similar across studies. Across the efficacy population, 59% were male; 70% were White, 22% were Black, 4% were Asian/Pacific Islander; 2% were American Indian/Alaskan Native; and 2% were multi-race. The median (range) age at the time of treatment was 9 months (1-36). The diagnosis of congenital athymia was based on flow cytometry documenting fewer than 50 naïve T cells/mm3 (CD45RA+, CD62L+) in the peripheral blood or less than 5% of total T cells being naïve in phenotype in 91/95 patients (range 0-98 naïve T cells/mm3). In addition to congenital athymia, patients also had complete DiGeorge syndrome (cDGS; also referred to as complete DiGeorge anomaly (cDGA)) if they also met at least one of the following criteria: congenital heart defect, hypoparathyroidism (or hypocalcemia requiring calcium replacement), 22q11 hemizygosity, 10p13 hemizygosity, CHARGE (coloboma, heart defect, choanal atresia, growth and development retardation, genital hypoplasia, ear defects including deafness) syndrome, or CHD7 mutation. Across the efficacy population, 93 patients (98%) were diagnosed with cDGS, and the most common DiGeorge gene mutations or syndromic associations were Chromosome 22q11.2 deletion (36 patients; 38%) and CHARGE syndrome (23 patients; 24%). There were 35 patients with missing or no identified genetic mutations. Two (2%) patients had FOXN1 deficiency, and 1 patient (1%) had a TBX variant. There were 50 (53%) patients with typical cDGS; these patients had congenital athymia with the absence of a T cell-related rash. There were 42 (44%) patients diagnosed with atypical cDGS; these patients may have had a rash, lymphadenopathy, or oligoclonal T cells. Patients who did not have congenital athymia (e.g. SCID) and patients with prior transplants, including thymus and HCT, were excluded from the efficacy analysis population. The baseline demographics and disease characteristics were similar in the safety population.
Patients with heart surgery anticipated within 4 weeks prior to, or 3 months after, the planned RETHYMIC treatment date, patients with human immunodeficiency virus (HIV) infection, and patients who were not considered good surgical candidates were excluded from study participation.
Patients in the efficacy population received RETHYMIC in a single surgical procedure at a dose of 4,900 to 24,000 mm2 of RETHYMIC / recipient BSA in m2. Patients were assigned to receive immunosuppressive therapy prior to and/or after treatment according to their disease phenotype and pre-RETHYMIC PHA response. Table 2 summarizes the criteria used to administer immunosuppression. Table 3 summarizes the specific immunosuppressant dosing used in RETHYMIC clinical studies. No patients were retreated with RETHYMIC.
| Complete DiGeorge Anomaly Phenotype | Phytohemagglutinin (PHA) Response* | Immunosuppression Used During Clinical Studies with RETHYMIC |
|---|---|---|
| Abbreviations: ATG-R: anti-thymocyte globulin [rabbit] (Thymoglobulin); cpm: counts per minute; MMF: mycophenylate mofetil; PHA: phytohemagglutinin | ||
|
||
| Typical | < 5,000 cpm or < 20-fold response to PHA over background | None |
| Typical | ≥ 5,000 cpm and < 50,000 cpm or Evidence of maternal engraftment |
|
| Typical | ≥ 50,000 cpm |
|
| Atypical | < 40,000 cpm on immunosuppression or < 75,000 cpm when not on immunosuppression |
|
| Atypical | ≥ 40,000 cpm on immunosuppression or ≥ 75,000 cpm when not on immunosuppression or Evidence of maternal engraftment | |
| Immunosuppressant | Dose of Immunosuppressant |
|---|---|
| Abbreviations: ATG-R: anti-thymocyte globulin [rabbit] (Thymoglobulin); IV: intravenous; MMF: mycophenylate mofetil; PO: oral | |
|
|
| ATG-R |
|
| Methylprednisolone*,† |
|
| Cyclosporine‡,§,¶ |
|
| Basiliximab |
|
| MMF |
|
| Alemtuzumab# |
|
The Kaplan-Meier estimated survival rates were 77% (95% CI [0.670, 0.841]) at 1 year and 76% (95% CI [0.658, 0.832]) at 2 years. For patients who were alive at 1 year after treatment with RETHYMIC, the survival rate was 94% at a median follow-up of 10.7 years.
Without treatment, congenital athymia is fatal in childhood. In a natural history population observed from 1991 through 2017, 49 patients diagnosed with congenital athymia received supportive care only. The 2-year survival rate was 6%, with all patients dying by 3 years of age. This population included 33 (67%) males. The most common cause of death was infection in 26 (53%) patients. Other common causes (≥10%) included support withdrawn in 7 (14%) patients, respiratory arrest in 5 (10%) patients, and cardiac arrest in 5 (10%) patients.
The Kaplan-Meier estimated survival rates for the RETHYMIC clinical trial population and the natural history population are shown in Figure 5. Four patients with >50 naïve T cells/mm3 (CD45RA+, CD62L+) at time of RETHYMIC administration have been treated; 2 (50%) were alive with follow-up less than 2 years.
Figure 5: Kaplan-Meier Survival by Year (RETHYMIC Efficacy Analysis Population and Natural History Population)
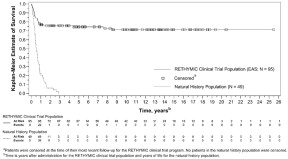
RETHYMIC significantly reduced the number of infections over time. In the first year after treatment with RETHYMIC, the number of patients with an infection event onset 6 to ≤ 12 months after treatment decreased by 38% (from 63 to 39) relative to the number of patients with an infection event onset in the first 6 months post-treatment. A two-year analysis showed a decrease in both the number of patients with an infection event and the mean number of infection events per patient, with an onset in the first 12 months post-treatment as compared to 12 to ≤ 24 months after treatment. There was a mean difference of 2.9 events (p<0.001) per patient.
Naïve CD4+ and CD8+ T cells reconstituted over the first year, with a durable increase through Year 2. Median (minimum, maximum) naïve CD4+ T cells/mm3 increased from a baseline of 1 (0, 38) to values of 42 (0, 653), 212 (1, 751), and 275 (33, 858) at 6, 12, and 24 months after treatment with RETHYMIC, respectively. Median naïve CD8+ T cells/mm3 increased from a baseline of 0 (0, 46) to values of 9 (0, 163), 58 (0, 304), and 86 (6, 275) at 6, 12, and 24 months after treatment with RETHYMIC, respectively. This was accompanied by functional improvements based on T cell proliferative responses to PHA.
16. How is Rethymic supplied
How Supplied
- RETHYMIC, NDC 72359-001-01, contains a single-dose unit, supplied ready for use as slices of processed thymus tissue, in sterile, polystyrene dishes (drug product dishes). Each drug product dish contains up to 4 RETHYMIC slices, adhered to circular filter membranes on top of surgical sponges in 5 mL of medium containing fetal bovine serum.
- Up to 42 RETHYMIC slices are supplied in a single-dose unit according to the dosage calculated in advance by the manufacturer for the specific patient. The dosage is determined by the total surface area of the RETHYMIC slices and recipient body surface area (BSA). The recommended dose range is 5,000 to 22,000 mm2 of RETHYMIC surface area/m2 recipient BSA. At the time of surgery, the manufacturing personnel communicate to the surgical team the portion of the product that represents the minimum dose.
- All drug product dishes are supplied in a polycarbonate container in an insulated shipping box.
Storage and Handling
- Use RETHYMIC prior to the time and date of expiration printed on the polycarbonate container.
- Store RETHYMIC at room temperature in the polycarbonate container in the insulated shipping box until ready for use. Do not refrigerate, freeze, agitate, or sterilize RETHYMIC.
- In the operating room, manufacturing personnel inspect the drug product containers as they are removed from the shipping box. If damage to the drug product dishes, leaks, spillage or evidence of contamination is noted, manufacturing personnel will notify the surgical team that the lot cannot be implanted.
- Match the patient's identity with the patient identifiers on the patient label on the polycarbonate container. Do not remove the drug product containers from the polycarbonate container if the information on the patient label does not match the intended patient.
- Manufacturing personnel record which RETHYMIC slices are used during the surgery. If any RETHYMIC slices are not administered to the patient, manufacturing personnel return this tissue to the manufacturing facility and dispose of this tissue as biohazardous waste in accordance with local requirements. Manufacturing personnel calculate the total dose that was administered to the patient.
17. Patient Counseling Information
Advise patients and/or their caregivers that:
- Immune reconstitution sufficient to protect from infection usually develops between 6-12 months after treatment with RETHYMIC, but for some patients elevated naïve T cell numbers are not observed until 2 years after treatment. Strict infection control measures should be observed until the healthcare provider confirms that immune function has been reconstituted through the evaluation of blood using flow cytometry and the criteria for the discontinuation of immunoglobulin replacement therapy and Pneumocystis jiroveci pneumonia prophylaxis have been met. Patients and caregivers should follow good handwashing practices, minimize contact with others, and immediately report signs and symptoms of infection to their healthcare provider [see Warnings and Precautions (5.1)].
- Congenital athymia alters the immune response to vaccines. Instruct patients and/or their caregivers to notify their healthcare professional to evaluate the immune status of RETHYMIC recipients prior to receiving vaccinations [see Warnings and Precautions (5.8)].
- Immunosuppression should be administered in patients with elevated T cell response, maternal engraftment, or oligoclonal T cell expansion and autoreactive T cells manifested by rash, lymphadenopathy and/or diarrhea. Inform patients and/or their caregivers on risks associated with short-term and long-term use of immunosuppression and refer them to review the risks of the specific immunosuppressants prescribed with their physician.
- Congenital athymia is associated with a wide spectrum of genetic anomalies. Instruct patients and/or their caregiver to consult with a clinical geneticist prior to receiving RETHYMIC.
Advise patients and/or their caregivers of the following risks:
- Graft versus Host Disease [see Warnings and Precautions (5.2)]
- Autoimmune Disorders (patient's immune (defense) system mistakenly attacks patient's body) [see Warnings and Precautions (5.3)]
- Renal Impairment (decrease of kidney function) [see Warnings and Precautions (5.4)]
- Cytomegalovirus Infection [see Warnings and Precautions (5.5)]
- Malignancy (Cancer) [see Warnings and Precautions (5.6)]
- Transmission of Serious Infections and Transmissible Infectious Diseases [see Warnings and Precautions (5.7)]
PRINCIPAL DISPLAY PANEL - 22,000 mm Container Label
NDC 72359–001–02
allogeneic processed thymus tissue–agdc
RETHYMIC
Dosage for entire lot is 5000 – 22,000 mm^2
RETHYMIC /m^2 recipient body surface area.
Handle aseptically. Do not agitate or sterilize.
Formulated in media that is supplemented with fetal
bovine serum. Preservative Free. Store at room
temperature. Do not freeze or refrigerate.
Lot # GMP–423
Container 1 of 11
Expiration: 13JUN2023
Contains up to 4 slices of RETHYMIC
adhered to filter membranes on top of
surgical sponges in 5 mL of media.
For intended recipient only. Rx Only.
For administration by surgical implantation.
See package insert for full prescribing
information and instructions for administration.
Manufactured for Sumitomo Pharma America, Inc.
Marlborough, MA 01752
Telephone: 1–833–369–9868
Lic. #: 2368

PRINCIPAL DISPLAY PANEL - 22,000 mm Container Box Label
NDC 72359–001–01
allogeneic processed thymus tissue–agdc
RETHYMIC
Dosage for entire lot is 5000 – 22,000 mm^2
RETHYMIC /m^2 recipient body surface area.
This entire lot contains 22,000 mm^2 RETHYMIC.
Formulated in media that is supplemented with fetal
bovine serum. Preservative free.
Handle aseptically. Do not agitate or sterilize.
Store at room temperature. Do not freeze or refrigerate.
Lot # GMP–423
Expiration: 13JUN2023
Manufactured for Sumitomo Pharma America, Inc.
Marlborough, MA 01752
Telephone: 1–833–369–9868
Lic. #: 2368

| RETHYMIC
allogenic thymocyte-depleted thymus tissue-agdc implant |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Sumitomo Pharma America, Inc. (131661746) |
| Registrant - Sumitomo Pharma Switzerland GmbH (480146015) |
More about Rethymic (allogeneic processed thymus tissue)
- Check interactions
- Compare alternatives
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: other immunostimulants