Oxlumo Injection: Package Insert / Prescribing Info
Package insert / product label
Generic name: lumasiran
Dosage form: injection, solution
Drug class: Miscellaneous metabolic agents
J Code (medical billing code): J0224 (0.5 mg, injection)
Medically reviewed by Drugs.com. Last updated on Apr 15, 2025.
On This Page
Highlights of Prescribing Information
OXLUMO (lumasiran) injection, for subcutaneous use
Initial U.S. Approval: 2020
Indications and Usage for Oxlumo Injection
OXLUMO is a HAO1-directed small interfering ribonucleic acid (siRNA) indicated for the treatment of primary hyperoxaluria type 1 (PH1) to lower urinary and plasma oxalate levels in pediatric and adult patients. (1)
Oxlumo Injection Dosage and Administration
- The recommended dose of OXLUMO by subcutaneous injection is based on body weight. (2.1)
| Body Weight | Loading Dose | Maintenance Dose |
|---|---|---|
| less than 10 kg | 6 mg/kg once monthly for 3 doses | 3 mg/kg once monthly, beginning 1 month after the last loading dose |
| 10 kg to less than 20 kg | 6 mg/kg once monthly for 3 doses | 6 mg/kg once every 3 months (quarterly), beginning 1 month after the last loading dose |
| 20 kg and above | 3 mg/kg once monthly for 3 doses | 3 mg/kg once every 3 months (quarterly), beginning 1 month after the last loading dose |
- See Full Prescribing Information for important preparation and administration instructions. (2.2)
Dosage Forms and Strengths
- Injection: 94.5 mg/0.5 mL in a single-dose vial. (3)
Contraindications
- None. (4)
Adverse Reactions/Side Effects
The most common adverse reaction (reported in ≥20% of patients) is injection site reactions. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Alnylam Pharmaceuticals at 1-877-256-9526 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Revised: 4/2025
Full Prescribing Information
1. Indications and Usage for Oxlumo Injection
OXLUMO is indicated for the treatment of primary hyperoxaluria type 1 (PH1) to lower urinary and plasma oxalate levels in pediatric and adult patients [see Clinical Pharmacology (12.1), Clinical Studies (14.1, 14.2, 14.3)].
2. Oxlumo Injection Dosage and Administration
2.1 Recommended Dosage
The recommended dosing regimen of OXLUMO consists of loading doses (monthly for 3 doses) followed by maintenance doses (beginning 1 month after the last loading dose) administered subcutaneously as shown in Table 1.
Dosing is based on actual body weight.
| Body Weight | Loading Dose | Maintenance Dose |
|---|---|---|
| Less than 10 kg | 6 mg/kg once monthly for 3 doses | 3 mg/kg once monthly, beginning 1 month after the last loading dose |
| 10 kg to less than 20 kg | 6 mg/kg once monthly for 3 doses | 6 mg/kg once every 3 months (quarterly), beginning 1 month after the last loading dose |
| 20 kg and above | 3 mg/kg once monthly for 3 doses | 3 mg/kg once every 3 months (quarterly), beginning 1 month after the last loading dose |
2.2 Administration Instructions
OXLUMO is intended for subcutaneous use and should be administered by a healthcare professional.
Visually inspect the drug product solution. Do not use if it contains particulate matter or if it is cloudy or discolored. OXLUMO is a sterile, preservative-free, clear, colorless-to-yellow solution. It is supplied in a single-dose vial, as a ready-to-use solution that does not require additional reconstitution or dilution prior to administration.
- Use aseptic technique.
- Divide injection volumes greater than 1.5 mL equally into multiple syringes.
- For volumes less than 0.3 mL, a sterile 0.3-mL syringe is recommended. If using a 0.3 mL (30 unit) insulin syringe, 1-unit markings indicate 0.01 mL.
- Administer subcutaneous injection into the abdomen, thigh, or the side or back of the upper arms. Rotate injection sites. Do not inject into scar tissue or areas that are reddened, inflamed, or swollen.
- If injecting into the abdomen, avoid the area around the navel.
- If more than one injection is needed for a single dose of OXLUMO, the injection sites should be at least 2 cm apart.
- Discard unused portion of the drug.
3. Dosage Forms and Strengths
Injection: 94.5 mg/0.5 mL clear, colorless-to-yellow solution in a single-dose vial.
6. Adverse Reactions/Side Effects
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of OXLUMO has been evaluated in a placebo-controlled trial and two single-arm clinical trials. Across these trials, 98 patients with PH1 have been treated with OXLUMO, including 71 pediatric patients and 15 patients on hemodialysis. Overall, 92 patients were treated for at least 6 months, 78 patients for at least 12 months, and 29 patients for at least 24 months.
In the randomized, placebo-controlled, double-blind study ILLUMINATE-A in pediatric and adult patients with PH1 aged 6 to 61 years, 26 patients received OXLUMO, and 13 patients received placebo. Of these, 25 patients received ≥5 months of treatment.
In two single-arm studies in patients with PH1, ILLUMINATE-B (patients <6 years of age) and ILLUMINATE-C (pediatric and adult patients with moderately or severely reduced GFR [eGFR ≤45 mL/min/1.73 m2 or pediatric patients <12 months of age with serum creatinine above the upper limit of normal for age] and patients with kidney failure on hemodialysis), the OXLUMO safety profile was similar to that seen in ILLUMINATE-A [see Clinical Studies (14)].
In placebo-controlled and open-label clinical studies the most common adverse reaction reported was injection site reaction. Injection site reactions included erythema, swelling, pain, hematoma, pruritus, and discoloration. These symptoms were generally mild and resolved within one day of the injection and did not lead to discontinuation of treatment.
| Adverse Reaction | OXLUMO N=26 N (%) | Placebo N=13 N (%) |
|---|---|---|
|
||
| Injection site reaction | 10 (38) | 0 (0) |
| Abdominal pain* | 4 (15) | 1 (8) |
6.2 Postmarketing Experience
The following additional adverse reaction has been reported during post-approval use. Because these events are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency.
Immune system disorder: Hypersensitivity
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no available data with the use of OXLUMO in pregnant women to evaluate a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes.
No adverse effects on pregnancy or embryo-fetal development related to OXLUMO were observed in rats at 45 times and in rabbits at 90 times the maximum recommended human dose in women (see Data).
The estimated background risk of major birth defects and miscarriage in the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in pregnant rats, lumasiran was administered subcutaneously at doses of 3, 10, and 30 mg/kg/day during organogenesis (gestational days 6-17). Administration of lumasiran resulted in no effects on embryo-fetal survival or fetal body weights and no lumasiran-related fetal malformations were observed. The 30 mg/kg/day dose in rats is 45 times the maximum recommended human dose (MRHD) for women of 3 mg/kg/month normalized to 0.1 mg/kg/day, based on body surface area. In an embryo-fetal development study in female rabbits, lumasiran was administered subcutaneously at doses of 3, 10, and 30 mg/kg/day during organogenesis (gestational days 7-19). There were decreases in maternal food consumption and decreases in maternal body weight gains at doses ≥3 mg/kg/day. There were no lumasiran-related fetal findings identified at doses up to 30 mg/kg/day (90 times the normalized MRHD based on body surface area).
In a postnatal development study, lumasiran administered subcutaneously to pregnant female rats on gestational days 7, 13, 19 and on lactation days 6, 12, and 18 through weaning at doses up to 50 mg/kg did not produce maternal toxicity or developmental effects in the offspring.
8.2 Lactation
Risk Summary
There are no data on the presence of OXLUMO in human milk, the effects on the breastfed child, or the effects of the drug on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for OXLUMO and any potential adverse effects on the breastfed child from OXLUMO or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of OXLUMO have been established in pediatric patients aged birth and older. Use of OXLUMO in these age groups is supported by evidence from an adequate and well controlled study of OXLUMO in pediatric patients 6 years or older and adults with PH1 (ILLUMINATE-A), a single-arm clinical study in pediatric patients less than 6 years of age with PH1 (ILLUMINATE-B), and a single-arm clinical study in pediatric and adult patients with PH1 who had advanced chronic kidney disease including patients on hemodialysis (ILLUMINATE-C) [see Adverse Reactions (6.1), Clinical Studies (14)].
8.5 Geriatric Use
Clinical studies of OXLUMO did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients.
8.6 Hepatic Impairment
No dose adjustment is recommended for patients with mild [total bilirubin > upper limit of normal (ULN) to 1.5 × ULN or AST > ULN] or moderate hepatic impairment (total bilirubin > 1.5 to 3 × ULN with any AST). OXLUMO has not been studied in patients with severe hepatic impairment (total bilirubin > 3 × ULN with any AST) [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dose adjustment is necessary in patients with renal impairment including patients with kidney failure treated with hemodialysis [see Clinical Pharmacology (12.3)]. OXLUMO has not been studied in patients on peritoneal dialysis.
11. Oxlumo Injection Description
OXLUMO injection contains lumasiran, a HAO1-directed double-stranded small interfering ribonucleic acid (siRNA), covalently linked to a ligand containing N-acetylgalactosamine (GalNAc).
The structural formula of lumasiran sodium is presented below:
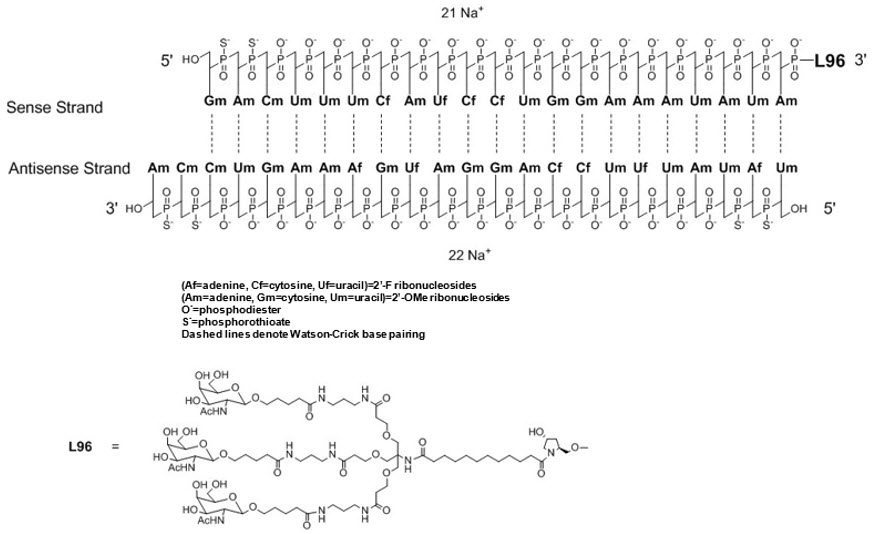
The molecular formula of lumasiran sodium is C530H669F10N173O320P43S6Na43 and the molecular weight is 17,286 Da.
OXLUMO is supplied as a sterile, preservative-free, clear, colorless-to-yellow solution for subcutaneous administration containing the equivalent of 94.5 mg of lumasiran (provided as lumasiran sodium) in 0.5 mL of water for injection and sodium hydroxide and/or phosphoric acid to adjust the pH to ~ 7.0.
12. Oxlumo Injection - Clinical Pharmacology
12.1 Mechanism of Action
Lumasiran reduces levels of glycolate oxidase (GO) enzyme by targeting the hydroxyacid oxidase 1 (HAO1) messenger ribonucleic acid (mRNA) in hepatocytes through RNA interference. Decreased GO enzyme levels reduce the amount of available glyoxylate, a substrate for oxalate production. As the GO enzyme is upstream of the deficient alanine: glyoxylate aminotransferase (AGT) enzyme that causes PH1, the mechanism of action of lumasiran is independent of the underlying AGXT gene mutation. OXLUMO is not expected to be effective in primary hyperoxaluria type 2 (PH2) or type 3 (PH3) because its mechanism of action does not affect the metabolic pathways causing hyperoxaluria in PH2 and PH3.
12.2 Pharmacodynamics
The pharmacodynamic effects of OXLUMO have been evaluated in adult and pediatric patients with PH1 across a range of doses and dosing frequency. Dose-dependent reductions in urinary oxalate levels were observed, resulting in the selection of the recommended body weight-based loading and maintenance dosing regimens. With the recommended dosing regimens, onset of effect was observed within two weeks after the first dose and maximal reductions in urinary oxalate were observed by Month 2 and persisted with continued use of OXLUMO maintenance dosage [see Figures 1 and 2 in Clinical Studies (14.1, 14.2)].
12.3 Pharmacokinetics
The pharmacokinetic (PK) properties of OXLUMO were evaluated following administration of single and multiple dosages in patients with PH1 as summarized in Table 3.
| Lumasiran | ||
|---|---|---|
|
||
| General Information | ||
| Steady-State Exposure | Cmax [Median (Range)] | 462 (38.5 to 1500) ng/mL |
| AUC0-last [Median (Range)] | 6810 (2890 to 10700) ng∙h/mL | |
| Dose Proportionality |
|
|
| Accumulation |
|
|
| Absorption | ||
| Tmax [Median (Range)] | 4 (0.5 to 12) hours | |
| Distribution* | ||
| Estimated Vd/F | 4.9 L | |
| Protein Binding | 85% | |
| Elimination | ||
| Apparent Half-Life [Mean (%CV)] | 5.2 (47%) hours | |
| Estimated CL/F | 26.5 L/hour | |
| Metabolism | ||
| Primary Pathway | Lumasiran is metabolized by endo- and exonucleases to oligonucleotides of shorter lengths. | |
| Excretion | ||
| Primary Pathway | Less than 26% of the administered dose of lumasiran is excreted unchanged into the urine within 24 hours with the rest excreted as inactive metabolite. | |
Specific Populations
No clinically significant differences in the pharmacokinetics or pharmacodynamics of lumasiran were observed based on age (4 months to <65 years old), sex, race/ethnicity, renal impairment, use of hemodialysis, or mild to moderate hepatic impairment (total bilirubin ≤ ULN and AST > ULN; or total bilirubin ≤ 3× ULN). The effect of severe hepatic impairment on the pharmacokinetics of lumasiran is unknown.
Body Weight
In children <20 kg, lumasiran Cmax was twice as high due to the higher 6 mg/kg dose and faster absorption rate. At the approved recommended dosage, lumasiran AUC was similar across the 6.2 kg to 110 kg body weight range [see Dosage and Administration (2.1)].
12.6 Immunogenicity
The observed incidence of anti-drug antibody (ADA, including neutralizing antibody) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of OXLUMO or of other siRNA products.
Across all clinical studies in the lumasiran development program, including patients with PH1 and healthy volunteers dosed with OXLUMO, 7 of 120 (6%) lumasiran-treated individuals with mean follow-up duration of 8.9 months, tested positive for ADA, as early as from Day 29.
No clinically significant differences in the safety, pharmacokinetic, or pharmacodynamic profiles of lumasiran were observed in patients who tested positive for ADA.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies were conducted in Tg-rasH2 mice and Sprague Dawley rats.
Lumasiran was not carcinogenic in transgenic Tg-rasH2 mice following monthly subcutaneous administration of lumasiran for 26 weeks at doses of 150, 500, or 1500 mg/kg.
In a 2-year carcinogenicity study, lumasiran was not carcinogenic up to the highest dose tested. Sprague Dawley rats were administered subcutaneous doses of 20, 55, or 110 mg/kg lumasiran once every 4 weeks (3, 9, or 18 times the normalized maintenance MRHD, based on body surface area).
Lumasiran was not genotoxic in an in vitro bacterial reverse mutation (Ames) assay, in the in vitro chromosomal aberration assay in cultured human peripheral blood lymphocytes, or the in vivo micronucleus assay in rats.
Administration of lumasiran by weekly subcutaneous doses of 0, 5, 15, and 50 mg/kg in male and female rats prior to and during mating and continuing in females once on Day 6 of presumed gestation resulted in no adverse effects upon the male or female fertility endpoints evaluated.
14. Clinical Studies
14.1 ILLUMINATE-A
ILLUMINATE-A was a randomized, double-blind trial comparing lumasiran and placebo in 39 patients 6 years of age and older with PH1 and an eGFR ≥30 mL/min/1.73 m2 (ILLUMINATE-A; NCT03681184). Patients received 3 loading doses of 3 mg/kg OXLUMO (N=26) or placebo (N=13) administered once monthly, followed by quarterly maintenance doses of 3 mg/kg OXLUMO or placebo [see Dosage and Administration (2.1)]. After six months, all patients received OXLUMO.
The median age of patients at first dose was 15 years (range 6 to 61 years), 67% were male, and 77% were White. At baseline, the median 24-hour urinary oxalate excretion corrected for body surface area (BSA) was 1.7 mmol/24 h/1.73 m2, the median plasma oxalate level was 13.1 µmol/L, 33% of patients had eGFR ≥90 mL/min/1.73 m2, 49% had eGFR of 60 to <90 mL/min/1.73 m2, and 18% had eGFR 30 to <60 mL/min/1.73 m2, 56% were on pyridoxine, and 85% reported a history of symptomatic kidney stone events.
The primary endpoint was the percent reduction from baseline in 24-hour urinary oxalate excretion corrected for BSA averaged over Months 3 through 6. The LS mean percent change from baseline in 24-hour urinary oxalate in the OXLUMO group was -65% (95% CI: -71, -59) compared with -12% (95% CI: -20, -4) in the placebo group, resulting in a between-group LS mean difference of 53% (95% CI: 45, 62; p<0.0001) [Figure 1].
| Abbreviation: CI ꞊ Confidence Interval. Results are plotted as mean (95% CI) of percent change from baseline. |
| Figure 1. ILLUMINATE-A: Percent Change from Baseline in 24-hour Urinary Oxalate by Month |
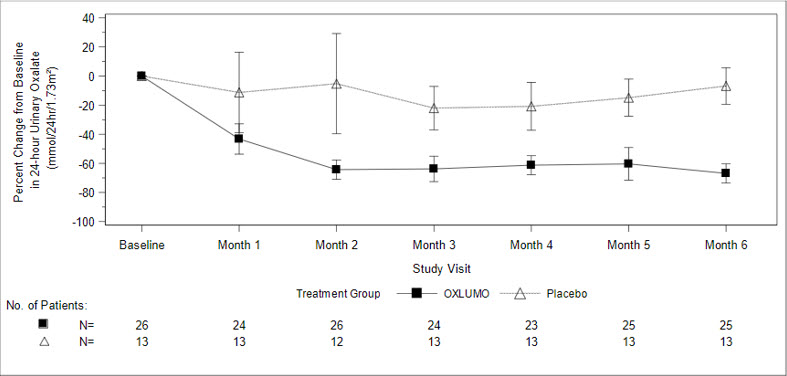 |
By Month 6, 52% (95% CI: 31, 72) of patients treated with OXLUMO achieved a normal 24-hour urinary oxalate corrected for BSA (≤0.514 mmol/24 hr/1.73 m2) compared to 0% (95% CI: 0, 25) placebo-treated patients (p=0.001). Reduced urinary oxalate levels were maintained through Month 24 in patients treated with OXLUMO.
14.2 ILLUMINATE-B
ILLUMINATE-B was a single-arm study in 18 patients <6 years of age with PH1 and an eGFR >45 mL/min/1.73 m2 for patients ≥12 months of age or a normal serum creatinine for patients <12 months of age (ILLUMINATE-B; NCT03905694). Dosing was based on body weight [see Dosage and Administration (2.1)].
The median age of patients at first dose was 51 months (range 4 to 74 months), 56% were female, and 88% were White. Three patients were less than 10 kg, 12 were 10 kg to <20 kg, and 3 were ≥20 kg. The median spot urinary oxalate: creatinine ratio at baseline was 0.47 mmol/mmol.
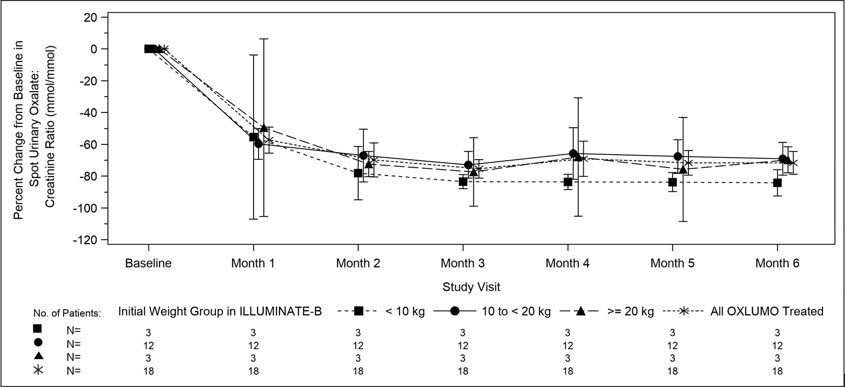
The primary endpoint was the percent reduction from baseline in spot urinary oxalate: creatinine ratio averaged over Months 3 through 6. Patients treated with OXLUMO achieved a reduction in spot urinary oxalate: creatinine ratio from baseline of 72% (95% CI: 66, 78) (Figure 2). The reduction in urinary oxalate excretion was maintained with continued OXLUMO treatment through Month 12.
| Abbreviation: CI ꞊ Confidence Interval. Results are plotted as mean (95% CI) of percent change from baseline. |
| Figure 2. ILLUMINATE-B: Percent Change from Baseline in Spot Urinary Oxalate: Creatinine Ratio by Month |
|
|
14.3 ILLUMINATE-C
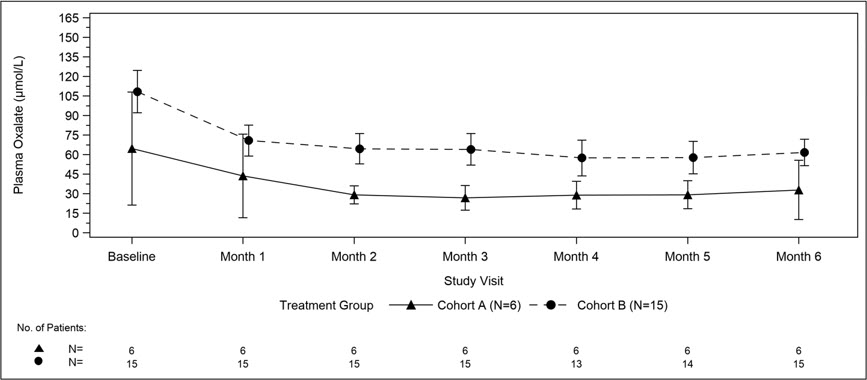
A total of 21 patients were enrolled and treated with OXLUMO in a multi-center, single-arm study in patients with PH1 and an eGFR ≤45 mL/min/1.73 m2 in patients 12 months of age and older or an elevated serum creatinine for age in patients less than 12 months of age, including patients on hemodialysis. ILLUMINATE-C included 2 cohorts. Cohort A included 6 patients who did not require dialysis at the time of study enrollment. Cohort B included 15 patients who were on a stable regimen of hemodialysis; the hemodialysis regimen was to remain stable in these patients for the first 6 months of the study. Patients received the recommended dosing regimen of OXLUMO based on body weight [see Dosage and Administration (2.1)]. Patients requiring peritoneal dialysis were excluded.
The median age of patients at first dose was 9 years (range 0 to 59 years), 57% were male, and 76% were White. For Cohort A, the median plasma oxalate level was 58 µmol/L. For Cohort B, the median pre-dialysis plasma oxalate level was 104 µmol/L.
The primary endpoint was the percent change in plasma oxalate from baseline to Month 6 (average from Month 3 to Month 6) for Cohort A (N=6) and the percent change in pre-dialysis plasma oxalate from baseline to Month 6 (average from Month 3 to Month 6) for Cohort B (N=15). The percent change from baseline to Month 6 in plasma oxalate levels in Cohort A was an LS mean difference of -33% (95% CI: -82, 15) and in Cohort B was -42% (95% CI: -51, -34).
Mean plasma oxalate decreased from 65 µmol/L (95% CI: 21, 108) at baseline to 33 µmol/L (95% CI: 10, 56) at Month 6 in Cohort A, and from 108 µmol/L (95% CI: 92, 125) at baseline to 62 µmol/L (95% CI: 51, 72) at Month 6 in Cohort B. The time course for changes in plasma oxalate is shown in Figure 3.
| Abbreviation: CI ꞊ Confidence Interval. Results are plotted as mean (95% CI) of actual values. For Cohort A, the baseline is defined as the mean of all plasma oxalate samples collected prior to the first dose of lumasiran; for Cohort B, the baseline is defined as the last four pre-dialysis plasma oxalate samples collected prior to the first dose of lumasiran. In Cohort B, only pre-dialysis samples are utilized. |
| Figure 3. ILLUMINATE-C: Plasma Oxalate Levels (µmol/L) during the Primary Analysis Period by Month |
 |
| OXLUMO
lumasiran injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Alnylam Pharmaceuticals, Inc. (115524410) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Vetter Pharma-Fertigung GmbH & Co. KG | 344217323 | MANUFACTURE(71336-1002) , ANALYSIS(71336-1002) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Ajinomoto Althea, Inc. | 023050730 | MANUFACTURE(71336-1002) | |
More about Oxlumo (lumasiran)
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: miscellaneous metabolic agents
- En español