Krystexxa: Package Insert / Prescribing Info
Package insert / product label
Generic name: pegloticase
Dosage form: injection, solution
Drug class: Antihyperuricemic agents
J Code (medical billing code): J2507 (1 mg, injection)
Medically reviewed by Drugs.com. Last updated on Apr 29, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
KRYSTEXXA® (pegloticase) injection, for intravenous use
Initial U.S. Approval: 2010
WARNING: ANAPHYLAXIS and INFUSION REACTIONS, G6PD DEFICIENCY ASSOCIATED HEMOLYSIS and METHEMOGLOBINEMIA
See full prescribing information for complete boxed warning.
- Anaphylaxis and infusion reactions have been reported to occur during and after administration of KRYSTEXXA. (5.1, 5.2)
- Anaphylaxis may occur with any infusion, including a first infusion, and generally manifest within 2 hours of the infusion. However, delayed hypersensitivity reactions have also been reported. (5.1)
- KRYSTEXXA should be administered in healthcare settings and by healthcare providers prepared to manage anaphylaxis and infusion reactions. (5.1, 5.2)
- Pre-medicate with antihistamines and corticosteroids and closely monitor for anaphylaxis for an appropriate period of time after administration of KRYSTEXXA. (5.1, 5.2)
- Monitor serum uric acid levels prior to each infusion and discontinue treatment if levels increase to above 6 mg/dL, particularly when 2 consecutive levels above 6 mg/dL are observed. (5.2)
- Screen patients at risk for G6PD deficiency prior to starting KRYSTEXXA. Hemolysis and methemoglobinemia have been reported with KRYSTEXXA in patients with G6PD deficiency. KRYSTEXXA is contraindicated in patients with G6PD deficiency. (4, 5.3)
Recent Major Changes
Indications and Usage for Krystexxa
Krystexxa Dosage and Administration
- Recommended Dosage: The recommended dosage is KRYSTEXXA 8 mg every two weeks given as an intravenous infusion, co-administered with weekly methotrexate 15 mg orally. KRYSTEXXA alone may be used in patients for whom methotrexate is contraindicated or not clinically appropriate. (2.2)
- Methotrexate with folic acid or folinic acid supplementation should be initiated at least 4 weeks prior to initiating, and throughout treatment with KRYSTEXXA. (2.2)
- Discontinue oral urate-lowering agents before starting KRYSTEXXA. (2.1)
- Monitor serum uric acid levels before each infusion. (2.1)
- Pre-medicate patients with antihistamines and corticosteroids. (2.1, 5.1, 5.2)
- KRYSTEXXA must be diluted prior to use. (2.1)
- Do not administer as an intravenous push or bolus. The KRYSTEXXA admixture should only be administered by intravenous infusion over no less than 120 minutes via gravity feed, syringe-type pump, or infusion pump. (2.1)
Dosage Forms and Strengths
- Injection: pegloticase containing 8 mg/mL of uricase protein in single-dose vials. (3)
Contraindications
Warnings and Precautions
- Anaphylaxis: Anaphylaxis may occur with any KRYSTEXXA infusion. Pre-medicate and monitor patients. (5.1)
- Infusion Reactions: Infusion reactions occurred in patients treated with KRYSTEXXA. Pre-medicate and monitor patients. (5.2)
- G6PD Deficiency Associated Hemolysis and Methemoglobinemia: Screen patients at risk for G6PD deficiency. Do not administer KRYSTEXXA to patients with G6PD deficiency. (5.3)
- Gout Flares: Gout flare prophylaxis is recommended for at least the first 6 months of KRYSTEXXA therapy. (5.4)
- Congestive Heart Failure: Congestive heart failure exacerbation may occur. Monitor patients closely following infusion. (5.5)
Adverse Reactions/Side Effects
- Co-administration with methotrexate: The most common adverse reactions (≥5% of patients) are gout flares, arthralgia, COVID-19, nausea and fatigue. (6.1)
- KRYSTEXXA alone: The most common adverse reactions (≥5% of patients) are gout flares, infusion reactions, nausea, contusion or ecchymosis, nasopharyngitis, constipation, chest pain, anaphylaxis and vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Inc. at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2025
Full Prescribing Information
WARNING: ANAPHYLAXIS and INFUSION REACTIONS, G6PD DEFICIENCY ASSOCIATED HEMOLYSIS and METHEMOGLOBINEMIA
- Anaphylaxis and infusion reactions have been reported to occur during and after administration of KRYSTEXXA. (5.1, 5.2)
- Anaphylaxis may occur with any infusion, and generally manifests within 2 hours of the infusion. However, delayed hypersensitivity reactions have also been reported. (5.1)
- KRYSTEXXA should be administered in healthcare settings and by healthcare providers prepared to manage anaphylaxis and infusion reactions. (5.1, 5.2)
- Premedicate with antihistamines and corticosteroids and closely monitor for anaphylaxis for an appropriate period of time after administration of KRYSTEXXA. (5.1, 5.2)
- Monitor serum uric acid levels prior to each infusion and discontinue treatment if levels increase to above 6 mg/dL, particularly when 2 consecutive levels above 6 mg/dL are observed. (5.2)
- Screen patients at risk for G6PD deficiency prior to starting KRYSTEXXA. Hemolysis and methemoglobinemia have been reported with KRYSTEXXA in patients with G6PD deficiency. KRYSTEXXA is contraindicated in patients with G6PD deficiency. (4, 5.3)
1. Indications and Usage for Krystexxa
KRYSTEXXA® (pegloticase) is indicated, for the treatment of chronic gout in adult patients refractory to conventional therapy.
Gout refractory to conventional therapy occurs in patients who have failed to normalize serum uric acid and whose signs and symptoms are inadequately controlled with xanthine oxidase inhibitors at the maximum medically appropriate dose or for whom these drugs are contraindicated.
2. Krystexxa Dosage and Administration
2.1 Important Administration Instructions
Precautions Prior to Treatment
It is recommended that before starting KRYSTEXXA patients discontinue oral urate-lowering medications and not institute therapy with oral urate-lowering agents while patients are on KRYSTEXXA therapy.
Monitoring Therapy: The risk of infusion reactions, including anaphylaxis, is higher in patients who have lost therapeutic response. Monitor serum uric acid levels prior to each infusion and discontinue treatment if levels increase to above 6 mg/dL, particularly when 2 consecutive levels above 6 mg/dL are observed [see Warnings and Precautions (5.1, 5.2)].
Infusion Reaction Precautions
KRYSTEXXA should be administered in a healthcare setting by healthcare providers prepared to manage infusion reactions, including anaphylaxis. Observe patients for an appropriate period of time after administration [see Warnings and Precautions (5.1, 5.2)].
Patients should receive pre-infusion medications (e.g., antihistamines, corticosteroids), to minimize the risk of infusion reactions, including anaphylaxis.
If an infusion reaction occurs during the administration of KRYSTEXXA, the infusion may be slowed, or stopped and restarted at a slower rate, at the discretion of the physician. Since infusion reactions can occur after completion of infusion, observation of patients for approximately an hour post-infusion should be considered [see Warnings and Precautions (5.2), Adverse Reactions (6.1)].
2.2 Recommended Dosage and Administration
The recommended dosage is KRYSTEXXA 8 mg given as an intravenous infusion every two weeks, co-administered with weekly oral methotrexate 15 mg and folic acid or folinic acid supplementation. KRYSTEXXA alone may be used in patients for whom methotrexate is contraindicated or not clinically appropriate.
If co-administering with methotrexate, start weekly methotrexate and folic acid or folinic acid supplementation at least 4 weeks prior to initiating, and throughout treatment with KRYSTEXXA. [see Clinical Studies (14)]. Refer to the Full Prescribing Information for methotrexate.
The optimal treatment duration with KRYSTEXXA has not been established.
2.3 Preparation Instructions
Visually inspect KRYSTEXXA for particulate matter and discoloration before administration, whenever solution and container permit. Do not use vials if either is present.
Use appropriate aseptic technique. Withdraw 1 mL of KRYSTEXXA from the vial into a sterile syringe. Discard any unused portion of product remaining in the vial. Inject into a single 250 mL bag of 0.9% Sodium Chloride Injection, USP or 0.45% Sodium Chloride Injection, USP for intravenous infusion. Do not mix or dilute with other drugs.
Invert the infusion bag containing the dilute KRYSTEXXA solution a number of times to ensure thorough mixing. Do not shake.
KRYSTEXXA diluted in infusion bags is stable for 4 hours at 2ºC to 8ºC (36ºF to 46ºF) and at room temperature (20ºC to 25ºC, 68ºF to 77ºF). However, it is recommended that diluted solutions be stored under refrigeration, not frozen, protected from light, and used within 4 hours of dilution [see How Supplied/Storage and Handling (16)].
Before administration, allow the diluted solution of KRYSTEXXA to reach room temperature. KRYSTEXXA in a vial or in an intravenous infusion fluid should never be subjected to artificial heating (e.g., hot water, microwave).
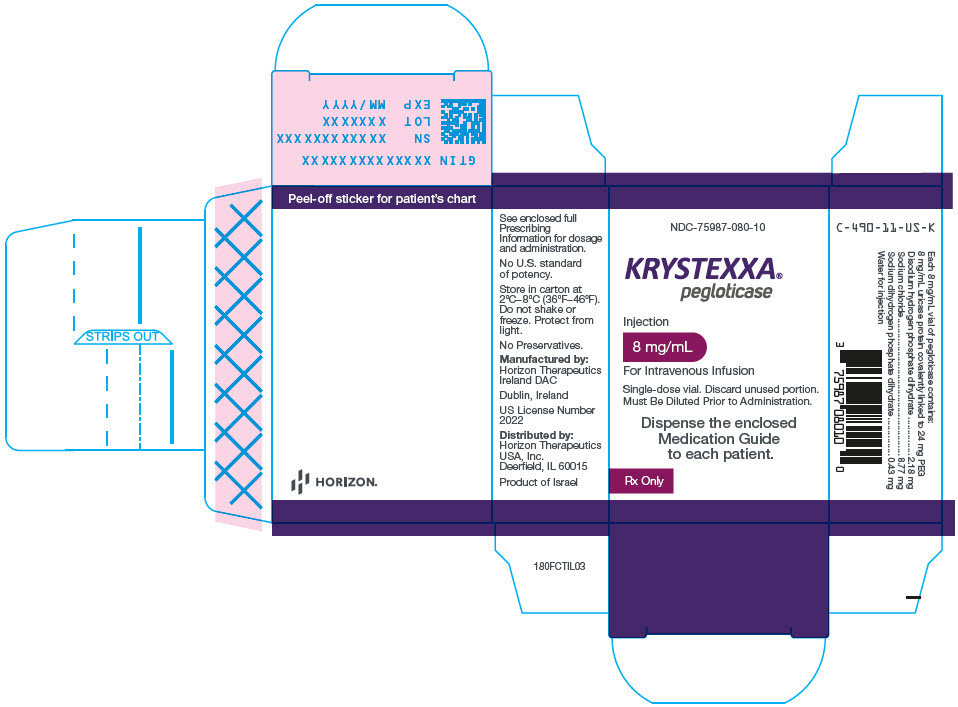
3. Dosage Forms and Strengths
Injection: 8 mg/mL as a clear and colorless solution for dilution in a single-dose vial. KRYSTEXXA concentrations are expressed as concentrations of uricase protein. Each mL of pegloticase contains 8 mg of uricase protein conjugated to monomethoxypoly (ethylene glycol) [mPEG].
4. Contraindications
KRYSTEXXA is contraindicated in:
- Patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency [see Warnings and Precautions (5.3)]
- Patients with history of serious hypersensitivity reactions, including anaphylaxis, to KRYSTEXXA or any of its components
5. Warnings and Precautions
5.1 Anaphylaxis
In a 52-week controlled trial, which evaluated KRYSTEXXA co-administered with methotrexate compared to KRYSTEXXA alone, patients were pre-treated with standardized infusion reaction prophylaxis and were discontinued from treatment with KRYSTEXXA if serum uric acid levels increased to above 6 mg/dL at 2 consecutive visits after the initiation of KRYSTEXXA therapy to reduce the risk of anaphylaxis. One patient randomized to the group treated with KRYSTEXXA co-administered with methotrexate (1%) experienced anaphylaxis during the first infusion and no patients experienced anaphylaxis in the group treated with KRYSTEXXA alone [see Adverse Reactions (6.1), Clinical Studies (14)].
During pre-marketing clinical trials with KRYSTEXXA alone, KRYSTEXXA was not discontinued following 2 consecutive serum uric acid levels above 6 mg/dL. Anaphylaxis was reported with a frequency of 6.5% (8/123) of patients treated with KRYSTEXXA every 2 weeks and 4.8% (6/126) for the every 4-week dosing regimen. There were no cases of anaphylaxis in patients receiving placebo. Anaphylaxis generally occurred within 2 hours after treatment.
Diagnostic criteria of anaphylaxis were skin or mucosal tissue involvement, and, either airway compromise, and/or reduced blood pressure with or without associated symptoms, and a temporal relationship to KRYSTEXXA or placebo injection with no other identifiable cause. Manifestations included wheezing, peri-oral or lingual edema, or hemodynamic instability, with or without rash or urticaria, nausea or vomiting. Cases occurred in patients being pre-treated with one or more doses of an oral antihistamine, an intravenous corticosteroid and/or acetaminophen. This pre-treatment may have blunted or obscured symptoms or signs of anaphylaxis and therefore the reported frequency may be an underestimate.
KRYSTEXXA should be administered in a healthcare setting by healthcare providers prepared to manage anaphylaxis. Patients should be pre-treated with antihistamines and corticosteroids. Anaphylaxis may occur with any infusion, including a first infusion, and generally manifests within 2 hours of the infusion. However, delayed type hypersensitivity reactions have also been reported. Patients should be closely monitored for an appropriate period of time for anaphylaxis after administration of KRYSTEXXA. Patients should be informed of the symptoms and signs of anaphylaxis and instructed to seek immediate medical care should anaphylaxis occur after discharge from the healthcare setting.
The risk of anaphylaxis is higher in patients whose uric acid level increases to above 6 mg/dL, particularly when 2 consecutive levels above 6 mg/dL are observed. Monitor serum uric acid levels prior to infusions and discontinue treatment if levels increase to above 6 mg/dL. Because of the possibility that concomitant use of oral urate-lowering therapy and KRYSTEXXA may potentially blunt the rise of serum uric acid levels, it is recommended that before starting KRYSTEXXA patients discontinue oral urate-lowering medications and not institute therapy with oral urate-lowering agents while taking KRYSTEXXA.
5.2 Infusion Reactions
In a 52-week, controlled trial which evaluated KRYSTEXXA co-administered with methotrexate compared to KRYSTEXXA alone [see Adverse Reactions (6.1), Clinical Studies (14)], patients were pre-treated with standardized infusion reaction prophylaxis and were discontinued from treatment with KRYSTEXXA if serum uric acid levels increased to above 6 mg/dL at 2 consecutive visits after the initiation of KRYSTEXXA therapy to reduce the risk of infusion reactions. Infusion reactions were reported in 4% of patients in the KRYSTEXXA co-administered with methotrexate group compared to 31% of patients treated with KRYSTEXXA alone experienced infusion reactions [see Adverse Reactions (6.1), Clinical Studies (14). In both treatment groups, the majority of infusion reactions occurred at the first or second KRYSTEXXA infusion and during the time of infusion. Manifestations of these infusion reactions were similar to that observed in the pre-marketing trials.
During pre-marketing 24-week controlled clinical trials with KRYSTEXXA alone, KRYSTEXXA was not discontinued following 2 consecutive serum uric acid levels above 6 mg/dL. Infusion reactions were reported in 26% of patients treated with KRYSTEXXA 8 mg every 2 weeks, and 41% of patients treated with KRYSTEXXA 8 mg every 4 weeks, compared to 5% of patients treated with placebo. These infusion reactions occurred in patients being pre-treated with an oral antihistamine, intravenous corticosteroid and/or acetaminophen. This pre-treatment may have blunted or obscured symptoms or signs of infusion reactions and therefore the reported frequency may be an underestimate.
Manifestations of these reactions included urticaria (frequency of 10.6%), dyspnea (frequency of 7.1%), chest discomfort (frequency of 9.5%), chest pain (frequency of 9.5%), erythema (frequency of 9.5%), and pruritus (frequency of 9.5%). These manifestations overlap with the symptoms of anaphylaxis, but in a given patient did not occur together to satisfy the clinical criteria for diagnosing anaphylaxis. Infusion reactions are thought to result from release of various mediators, such as cytokines. Infusion reactions occurred at any time during a course of treatment with approximately 3% occurring with the first infusion, and approximately 91% occurred during the time of infusion.
KRYSTEXXA should be administered in a healthcare setting by healthcare providers prepared to manage infusion reactions. Patients should be pre-treated with antihistamines and corticosteroids. KRYSTEXXA should be infused slowly over no less than 120 minutes. In the event of an infusion reaction, the infusion should be slowed, or stopped and restarted at a slower rate.
The risk of infusion reaction is higher in patients whose uric acid level increases to above 6 mg/dL, particularly when 2 consecutive levels above 6 mg/dL are observed. Monitor serum uric acid levels prior to infusions and discontinue treatment if levels increase to above 6 mg/dL. Because of the possibility that concomitant use of oral urate-lowering therapy and KRYSTEXXA may potentially blunt the rise of serum uric acid levels, it is recommended that before starting KRYSTEXXA patients discontinue oral urate-lowering medications and not institute therapy with oral urate-lowering agents while taking KRYSTEXXA.
5.3 G6PD Deficiency Associated Hemolysis and Methemoglobinemia
Life threatening hemolytic reactions and methemoglobinemia have been reported with KRYSTEXXA in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Because of the risk of hemolysis and methemoglobinemia, do not administer KRYSTEXXA to patients with G6PD deficiency [see Contraindications (4)]. Screen patients at risk for G6PD deficiency prior to starting KRYSTEXXA. For example, patients of African, Mediterranean (including Southern European and Middle Eastern), and Southern Asian ancestry are at increased risk for G6PD deficiency.
5.4 Gout Flares
In a 52-week, randomized, double-blind trial which evaluated KRYSTEXXA co-administered with methotrexate compared to KRYSTEXXA alone, patients were administered gout flare prophylaxis similar to that in the pre-marketing, placebo-controlled trials. In this trial, the percentages of patients with any flare for the first 3 months were 66% and 69% for the group treated with KRYSTEXXA co-administered with methotrexate and the group treated with KRYSTEXXA alone, respectively. In the group treated with KRYSTEXXA co-administered with methotrexate, the percentages of patients with any flare for the subsequent 3 month increments of treatment were 27% during Month 6, 8% during Month 9 and 9% during Month 12. In the group treated with KRYSTEXXA alone, the percentages of patients with any flare were 14% during Month 6, 9% during Month 9 and 21% during Month 12.
During pre-marketing, 24-week controlled clinical trials with KRYSTEXXA alone, the frequencies of gout flares were high in all treatment groups, but more so with KRYSTEXXA treatment during the first 3 months of treatment, and decreased in the subsequent 3 months of treatment. The percentages of patients with any flare for the first 3 months were 74%, 81%, and 51%, for KRYSTEXXA 8 mg every 2 weeks, KRYSTEXXA 8 mg every 4 weeks, and placebo, respectively. The percentages of patients with any flare for the subsequent 3 months were 41%, 57%, and 67%, for KRYSTEXXA 8 mg every 2 weeks, KRYSTEXXA 8 mg every 4 weeks, and placebo, respectively. Patients received gout flare prophylaxis with colchicine and/or nonsteroidal anti-inflammatory drugs (NSAIDs) starting at least one week before receiving KRYSTEXXA.
Gout flares may occur after initiation of KRYSTEXXA. An increase in gout flares is frequently observed upon initiation of anti-hyperuricemic therapy, due to changing serum uric acid levels resulting in mobilization of urate from tissue deposits. Gout flare prophylaxis with a non-steroidal anti-inflammatory drug (NSAID) or colchicine is recommended starting at least 1 week before initiation of KRYSTEXXA therapy and lasting at least 6 months, unless medically contraindicated or not tolerated. KRYSTEXXA does not need to be discontinued because of a gout flare. The gout flare should be managed concurrently as appropriate for the individual patient [see Dosage and Administration (2)].
5.5 Congestive Heart Failure
KRYSTEXXA has not been formally studied in patients with congestive heart failure, but some patients in the pre-marketing, 24-week controlled clinical trials experienced exacerbation of congestive heart failure. Two cases of congestive heart failure exacerbation occurred during the trials in patients receiving treatment with KRYSTEXXA 8 mg every 2 weeks. No cases were reported in placebo-treated patients. Four subjects had exacerbations of pre-existing congestive heart failure while receiving KRYSTEXXA 8 mg every 2 weeks during the open-label extension study.
Exercise caution when using KRYSTEXXA in patients who have congestive heart failure and monitor patients closely following infusion.
5.6 Re-treatment with KRYSTEXXA
No controlled trial data are available on the safety and efficacy of re-treatment with KRYSTEXXA after stopping treatment for longer than 4 weeks. Due to the immunogenicity of KRYSTEXXA, patients receiving re-treatment may be at increased risk of anaphylaxis and infusion reactions. Therefore, patients receiving re-treatment after a drug-free interval should be monitored carefully [see Adverse Reactions (6.2)].
6. Adverse Reactions/Side Effects
The following serious adverse reactions are discussed in greater detail in other sections of the label:
- Anaphylaxis [see Warnings and Precautions (5.1)]
- Infusion Reactions [see Warnings and Precautions (5.2)]
- G6PD Deficiency Associated Hemolysis and Methemoglobinemia [see Warnings and Precautions (5.3)]
- Gout Flares [see Warnings and Precautions (5.4)]
- Congestive Heart Failure [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying and controlled conditions, adverse reaction rates observed in clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug, and may not predict the rates observed in a broader patient population in clinical practice.
Co-administration with Methotrexate
A 52-week, randomized, double-blind trial was conducted in adult patients with chronic gout refractory to conventional therapy to evaluate administration of KRYSTEXXA 8 mg every 2 weeks co-administered with weekly administration of oral methotrexate 15 mg, compared to KRYSTEXXA alone. In this trial, patients who were able to tolerate two weeks on methotrexate 15 mg were then randomized to receive four additional weeks on either methotrexate 15 mg or matching placebo prior to initiating KRYSTEXXA therapy. A total of 152 subjects were randomized, and of these, 145 subjects completed the 4-week methotrexate run-in period and received KRYSTEXXA (96 subjects received KRYSTEXXA co-administered with methotrexate and 49 received KRYSTEXXA plus placebo) during the treatment period. All patients received pre-treatment with an oral antihistamine, intravenous corticosteroid and acetaminophen. These patients were between the ages of 24 and 83 years (average 55 years); 135 patients were male and 17 and were female; 105 patients were White/Caucasian, 22 were Black/African American, 14 were Asian, 5 were Native Hawaiian/Other Pacific Islander and 5 identified as Other; 28 were Hispanic or Latino. Common co-morbid conditions among the enrolled patients included hypertension (63%), osteoarthritis (25%), hyperlipidemia (24%), gastroesophageal reflux disease (22%), obesity (20%), type 2 diabetes (18%) and depression (16%). Patients with an eGFR <40 mL/min/1.73 m2 were excluded from this trial.
The most commonly reported adverse reaction during the methotrexate pre-treatment periods was gout flare. The most commonly reported adverse reactions that occurred in ≥ 5% in either treatment group during the KRYSTEXXA co-administered with methotrexate or KRYSTEXXA alone period are provided in Table 1.
| Adverse Reaction | KRYSTEXXA with Methotrexate (N=96) n (%) | KRYSTEXXA Alone (N=49) n (%) |
|---|---|---|
|
||
| Gout flare | 64 (67%) | 35 (71%) |
| Arthralgia | 13 (14%) | 5 (10%) |
| COVID-19 | 9 (9%) | 3 (6%) |
| Nausea | 5 (5%) | 6 (12%) |
| Fatigue | 5 (5%) | 2 (4%) |
| Infusion reaction | 4 (4%)* | 15 (31%) |
| Pain in extremity | 1 (1%) | 3 (6%) |
| Hypertension | 1 (1%) | 3 (6%) |
| Vomiting | 0 | 4 (8%) |
KRYSTEXXA ALONE
The data described below reflect exposure to KRYSTEXXA in patients with chronic gout refractory to conventional therapy in two replicate randomized, placebo-controlled, double-blind 24-week clinical trials: 85 patients were treated with KRYSTEXXA 8 mg every 2 weeks; 84 patients were treated with KRYSTEXXA 8 mg every 4 weeks; and 43 patients were treated with placebo. These patients were between the ages of 23 and 89 years (average 55 years); 173 patients were male and 39 were female; and 143 patients were White/Caucasian, 27 were Black/African American, 24 were Hispanic/Latino and 18 were all other ethnicities. Common co-morbid conditions among the enrolled patients included hypertension (72%), dyslipidemia (49%), chronic kidney disease (28%), diabetes (24%), coronary artery disease (18%), arrhythmia (16%), and cardiac failure/left ventricular dysfunction (12%).
During the pre-marketing placebo-controlled clinical trials, the most commonly reported adverse reactions that occurred in greater than or equal to 5% of patients treated with KRYSTEXXA 8 mg every 2 weeks are provided in Table 2.
| Adverse Reaction | KRYSTEXXA 8 mg every 2 weeks | Placebo |
|---|---|---|
| (N=85) n* (%) | (N=43) n (%) |
|
|
||
| Gout flare | 65 (77%) | 35 (81%) |
| Infusion reaction | 22 (26%) | 2 (5%) |
| Nausea | 10 (12%) | 1 (2%) |
| Contusion† or Ecchymosis† | 9 (11%) | 2 (5%) |
| Nasopharyngitis | 6 (7%) | 1 (2%) |
| Constipation | 5 (6%) | 2 (5%) |
| Chest Pain | 5 (6%) | 1 (2%) |
| Anaphylaxis | 4 (5%) | 0 (0%) |
| Vomiting | 4 (5%) | 1 (2%) |
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The observed incidence of antibody positivity in an assay is highly dependent on several factors including assay sensitivity and specificity and assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, the comparison of the incidence of antibodies to pegloticase with the incidence of antibodies to other products may be misleading.
In a 52-week, randomized, double-blind trial which evaluated KRYSTEXXA co-administered with methotrexate compared to KRYSTEXXA alone, approximately 26% of patients had pre-existing antibodies to pegloticase. Patients with an increase in titer from baseline or who were negative at baseline and developed an anti-pegloticase response at one or more post dose time points was 30% and 51%, for the KRYSTEXXA co-administered with methotrexate and KRYSTEXXA alone treatment groups, respectively. Patients with higher antibody titers were more likely to have faster clearance and lower efficacy.
During pre-marketing 24-week controlled clinical trials with KRYSTEXXA alone, anti-pegloticase antibodies developed in 92% of patients treated with KRYSTEXXA every 2 weeks, and 28% for placebo. Anti-PEG antibodies were also detected in 42% of patients treated with KRYSTEXXA. High anti-pegloticase antibody titer was associated with a failure to maintain pegloticase-induced normalization of uric acid. The impact of anti-PEG antibodies on patients' responses to other PEG-containing therapeutics is unknown.
There was a higher incidence of infusion reactions in patients with high anti-pegloticase antibody titer: 53% (16 of 30) in the KRYSTEXXA every 2 weeks group compared to 6% in patients who had undetectable or low antibody titers.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of KRYSTEXXA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship.
General disorders and administration site conditions: asthenia, malaise, peripheral swelling
Related/similar drugs
7. Drug Interactions
7.1 Methotrexate
KRYSTEXXA 8 mg every 2 weeks has been studied in patients with chronic gout refractory to conventional therapy taking concomitant oral methotrexate 15 mg weekly [see Clinical Studies (14)]. Co-administration of methotrexate with KRYSTEXXA may increase pegloticase concentration compared to KRYSTEXXA alone [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies of KRYSTEXXA in pregnant women.
Based on animal reproduction studies, no structural abnormalities were observed when pegloticase was administered by subcutaneous injection to pregnant rats and rabbits during the period of organogenesis at doses up to 50 and 75 times, respectively, the maximum recommended human dose (MRHD). Decreases in mean fetal and pup body weights were observed at approximately 50 and 75 times the MRHD, respectively [see Data].
All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the US general population, the estimated background risk of major birth defects and miscarriage in clinical recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In 2 separate embryo-fetal developmental studies, pregnant rats and rabbits received pegloticase during the period of organogenesis at doses up to approximately 50 and 75 times the maximum recommended human dose (MRHD), respectively (on a mg/m2 basis at maternal doses up to 40 and 30 mg/kg twice weekly, in rats and rabbits, respectively). No evidence of structural abnormalities was observed in rats or rabbits. However, decreases in mean fetal and pup body weights were observed at approximately 50 and 75 times the MRHD in rats and rabbits, respectively (on a mg/m2 basis at maternal doses up to 40 and 30 mg/kg every other day, in rats and rabbits, respectively). No effects on mean fetal body weights were observed at approximately 10 and 25 times the MRHD in rats and rabbits, respectively (on a mg/m2 basis at maternal doses up to 10 mg/kg twice weekly in both species).
8.4 Pediatric Use
The safety and effectiveness of KRYSTEXXA in pediatric patients less than 18 years of age have not been established.
8.5 Geriatric Use
Of the total number of patients treated with KRYSTEXXA 8 mg every 2 weeks in the controlled studies, 34% (29 of 85) were 65 years of age and older and 12% (10 of 85) were 75 years of age and older. No overall differences in safety or effectiveness were observed between older and younger patients, but greater sensitivity of some older individuals cannot be ruled out. No dose adjustment is needed for patients 65 years of age and older.
8.6 Renal Impairment
No dose adjustment is required for patients with renal impairment. In a 52-week, randomized, double-blind trial which evaluated KRYSTEXXA co-administered with methotrexate compared to KRYSTEXXA alone, 85% of patients had chronic kidney disease based on estimated glomerular filtration rate (eGFR) of ≥ 40 to < 90 mL/min/1.73m2 at baseline. In the pre-marketing 24-week controlled clinical trials with KRYSTEXXA alone, a total of 32% (27 of 85) of patients treated with KRYSTEXXA 8 mg every 2 weeks had a creatinine clearance of ≤62.5 mL/min. No overall differences in efficacy were observed.
10. Overdosage
No reports of overdosage with KRYSTEXXA have been reported. The maximum dose that has been administered as a single intravenous dose is 12 mg as uricase protein.
Patients suspected of receiving an overdose should be monitored, and general supportive measures should be initiated as no specific antidote has been identified.
11. Krystexxa Description
KRYSTEXXA (pegloticase) is a uric acid specific enzyme which is a PEGylated product that consists of recombinant modified mammalian urate oxidase (uricase) produced by a genetically modified strain of Escherichia coli. Uricase is covalently conjugated to monomethoxypoly (ethylene glycol) [mPEG] (10 kDa molecular weight). The cDNA coding for uricase is based on mammalian sequences. Each uricase subunit has a molecular weight of approximately 34 kDa per subunit. The average molecular weight of pegloticase (tetrameric enzyme conjugated to mPEG) is approximately 540 kDa.
KRYSTEXXA (pegloticase) injection is a sterile, clear and colorless solution in a single-dose vial intended for intravenous infusion after dilution.
KRYSTEXXA (pegloticase) concentrations are expressed as concentrations of uricase protein. Each mL of KRYSTEXXA contains 8 mg of uricase protein (conjugated to 24 mg of 10 kDa mPEG), 2.18 mg Disodium Hydrogen Phosphate Dihydrate (Na2HPO4∙2H2O), 8.77 mg Sodium Chloride (NaCl), 0.43 mg Sodium Dihydrogen Phosphate Dihydrate (NaH2PO4∙2H2O), and Water for Injection to deliver 8 mg of pegloticase (as uricase protein).
12. Krystexxa - Clinical Pharmacology
12.1 Mechanism of Action
KRYSTEXXA is a uric acid specific enzyme which is a recombinant uricase and achieves its therapeutic effect by catalyzing the oxidation of uric acid to allantoin, thereby lowering serum uric acid. Allantoin is an inert and water-soluble purine metabolite; it is readily eliminated, primarily by renal excretion.
12.2 Pharmacodynamics
Approximately 24 hours following the first dose of KRYSTEXXA, mean plasma uric acid levels for subjects in the KRYSTEXXA groups were 0.7 mg/dL for the KRYSTEXXA 8 mg every 2 weeks group. In comparison, the mean plasma uric acid level for the placebo group was 8.2 mg/dL.
In a single-dose, dose-ranging trial, following 1-hour intravenous infusions of 0.5, 1, 2, 4, 8 or 12 mg of pegloticase in 24 patients with symptomatic gout (n=4 subjects/dose group), plasma uric acid decreased with increasing pegloticase dose or concentrations. The duration of suppression of plasma uric acid appeared to be positively associated with pegloticase dose. Sustained decrease in plasma uric acid below the solubility concentration of 6 mg/dL for more than 300 hours was observed with doses of 8 mg and 12 mg.
12.3 Pharmacokinetics
Pegloticase levels were determined in serum based on measurements of uricase enzyme activity.
Following single intravenous infusions of 0.5 mg to 12 mg pegloticase in 23 patients with symptomatic gout, maximum serum concentrations of pegloticase increased in proportion to the dose administered.
The population pharmacokinetic analysis showed that age, sex, weight, and creatinine clearance did not influence the pharmacokinetics of pegloticase. Significant covariates impacting pegloticase pharmacokinetics were body surface area and anti-pegloticase antibodies.
Special Populations
Drug Interactions
Co-administration of methotrexate with KRYSTEXXA decreased anti-pegloticase antibody incidence rate and titers compared to KRYSTEXXA alone, therefore increased pegloticase exposure levels. In patients with chronic gout refractory to conventional therapy receiving KRYSTEXXA 8 mg every 2 weeks co-administered with weekly administration of oral methotrexate 15 mg, higher steady-state peak and trough concentrations (2.65 µg/ml and 1.13 µg/ml, respectively) of pegloticase were observed compared to when KRYSTEXXA is given alone (steady-state peak and trough concentrations at 2.13 µg/ml and 0.59 µg/ml, respectively).
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to evaluate the carcinogenic potential of pegloticase.
The genotoxic potential of pegloticase has not been evaluated.
There was no evidence of impairment on fertility at pegloticase doses up to 40 mg/kg (approximately 50 times the MRHD on mg/m2 basis) every other day in rats.
13.2 Animal Toxicology and/or Pharmacology
Pegloticase at similar to and higher than the maximum recommended human dose (MRHD) on a plasma AUC basis [at intravenous (IV) doses of ≥ 0.4 mg/kg in dogs] caused cytoplasmic vacuoles in multiple organs, and edema and histiocyte infiltration in the aortic outflow tract in dogs. Organs with cytoplasmic vacuoles included the spleen, adrenal gland, liver, heart, duodenum, and jejunum. Vacuoles in the spleen, adrenal glands, and heart persisted after a 1-year recovery period at pegloticase doses (≥ 1.5 mg/kg in dogs) approximately 5 times the MRHD, but were absent at a dose similar to the MRHD. Vacuoles in the liver, duodenum, and jejunum persisted after a 3-month recovery period at a pegloticase dose (10 mg/kg in dogs) approximately 30 times the MRHD, but were absent at doses (≤ 1.5 mg/kg) approximately 5 times and similar to the MRHD. The edema and histiocyte infiltration in the aortic outflow tract was absent after recovery periods of 6 and 12 months, respectively.
Vacuoles in the spleen, liver, duodenum, and jejunum were within macrophages and most likely represented phagocytic removal of pegloticase from the circulation. However, the vacuolated cells in the heart and adrenal gland did not stain as macrophages. In the aortic outflow tract of the heart, vacuoles were in the cytoplasm of endothelial cells in the intimal lining of the aorta. In the adrenal gland, vacuoles were located within cortical cells in the zona reticularis and zona fasciculata. The clinical significance of these findings and the functional consequences are unknown.
14. Clinical Studies
Co-administration with Methotrexate
A 52-week, randomized, double-blind trial was conducted in adult patients with chronic gout refractory to conventional therapy, to evaluate administration of KRYSTEXXA 8 mg every 2 weeks co-administered with weekly administration of methotrexate 15 mg, compared to KRYSTEXXA alone: Trial 1 (NCT03994731). In this trial, patients were naïve to KRYSTEXXA therapy. Patients who were able to tolerate two weeks on oral methotrexate 15 mg were then randomized to receive four additional weeks on either methotrexate 15 mg or matching placebo prior to initiating KRYSTEXXA therapy in a 2:1 ratio. Patients were pre-treated with a standardized infusion reaction prophylaxis regimen consisting of an oral fexofenadine, acetaminophen and intravenous methylprednisolone prior to each KRYSTEXXA infusion. Methotrexate or placebo was continued weekly throughout the KRYSTEXXA treatment period with daily oral folic acid in order to evaluate the immunomodulatory effect of methotrexate to attenuate development of anti-drug antibodies.
Entry criteria for patients to be eligible for this trial were: baseline serum uric acid ≥7 mg/dL and inability to maintain serum uric acid <6 mg/dL on other urate-lowering therapy, intolerable side effects associated with current urate-lowering therapy, and/or presence of clinically evident tophaceous deposits.
The primary endpoint was the proportion of Month 6 responders, where a responder was defined as achieving and maintaining serum uric acid less than 6 mg/dL for at least 80% of the time during Month 6. The proportion of Month 12 responders was a key secondary endpoint. A significantly greater proportion of patients receiving KRYSTEXXA co-administered with methotrexate compared to KRYSTEXXA alone achieved both the primary and secondary endpoints (see Table 3).
| Treatment Group | ||
|---|---|---|
| Trial 1 | KRYSTEXXA with Methotrexate (N=100) | KRYSTEXXA Alone (N=52) |
|
||
| Number, % of Patients Who Met Response Criteria During Month 6 | 71 (71%) | 20 (39%) |
| Difference* (95% Confidence Interval) | 32% (16%, 48%) | |
| P-Value | <0.0001 | |
| Number, % of Patients Who Met Response Criteria During Month 12 | 60 (60%) | 16 (31%) |
| Difference* (95% Confidence Interval) | 29% (13%, 45%) | |
| P-Value | 0.0003 | |
The effect of KRYSTEXXA co-administered with methotrexate and KRYSTEXXA alone on tophi was assessed using standardized digital photography, image analysis and Central Readers blinded to treatment assignment. Approximately 53.3% (81/152) of randomized patients had tophi at baseline (Week -6) that were confirmed by digital photography. Of those, 54% (28/52) in the KRYSTEXXA co-administered with methotrexate group and 31% (9/29) in the KRYSTEXXA alone group achieved a complete response at Month 12 (defined as 100% resolution of at least one target tophus, no new tophi appear and no single tophus showing progression). The difference between the two treatment groups was statistically significant (22.8%, 95% CI: 1.2%, 44.4%).
KRYSTEXXA Alone
The efficacy of KRYSTEXXA was studied in adult patients with chronic gout refractory to conventional therapy in two replicate, multicenter, randomized, double-blind, placebo-controlled studies of six months duration: Trial 2 and Trial 3. Patients were randomized to receive KRYSTEXXA 8 mg every 2 weeks or every 4 weeks or placebo in a 2:2:1 ratio.
Studies were stratified for the presence of tophi. Seventy-one percent (71%) of patients had baseline tophi. All patients were prophylaxed with an oral antihistamine, intravenous corticosteroid and acetaminophen. Patients also received prophylaxis for gout flares with non-steroidal anti-inflammatory drugs (NSAIDs) or colchicine, or both, beginning at least one week before KRYSTEXXA treatment unless medically contraindicated or not tolerated. Patients who completed the randomized clinical trials were eligible to enroll in a 2-year open label extension study.
Entry criteria for patients to be eligible for the trials were: baseline serum uric acid of at least 8 mg/dL; had symptomatic gout with at least 3 gout flares in the previous 18 months or at least 1 gout tophus or gouty arthritis; and had a self-reported medical contraindication to allopurinol or medical history of failure to normalize uric acid (to less than 6 mg/dL) with at least 3 months of allopurinol treatment at the maximum medically appropriate dose.
The mean age of study subjects was 55 years (23-89); 82% were male, mean body mass index (BMI) was 33 kg/m2, mean duration of gout was 15 years, and mean baseline serum uric acid was 10 mg/dL.
To assess the efficacy of KRYSTEXXA in lowering uric acid, the primary endpoint in both trials was the proportion of patients who achieved plasma uric acid (PUA) less than 6 mg/dL for at least 80% of the time during Month 3 and Month 6. As shown in Table 4, a greater proportion of patients treated with KRYSTEXXA every 2 weeks achieved urate lowering to below 6 mg/dL than patients receiving placebo. Although the 4 week regimen also demonstrated efficacy for the primary endpoint, this regimen was associated with increased frequency of anaphylaxis and infusion reactions and less efficacy with respect to tophi.
| Treatment Group | N | Number (%) of Subjects Who Met Response Criteria | 95% Confidence Interval* | P-Value† |
|---|---|---|---|---|
| Note: Based on post-hoc analyses of the clinical trial data, if KRYSTEXXA had been stopped when a patient's uric acid level rose to greater than 6 mg/dL on a single occasion, the incidence of infusion reactions would have been reduced by approximately 67%, but the success rates for the primary efficacy endpoint would have been reduced by approximately 20%. If KRYSTEXXA had been stopped after 2 consecutive uric acid levels greater than 6 mg/dL, the incidence of infusion reactions would have been half, and there would have been little change in the efficacy outcome. | ||||
| Trial 2 | ||||
| KRYSTEXXA 8 mg every 2 weeks | 43 | 20 (47%) | [32%, 61%] | <0.001 |
| KRYSTEXXA 8 mg every 4 weeks | 41 | 8 (20%) | [7%, 32%] | 0.044 |
| Placebo | 20 | 0 (0%) | ||
| Trial 3 | ||||
| KRYSTEXXA 8 mg every 2 weeks | 42 | 16 (38%) | [23%, 53%] | <0.001 |
| KRYSTEXXA 8 mg every 4 weeks | 43 | 21 (49%) | [34%, 64%] | <0.001 |
| Placebo | 23 | 0 (0%) | ||
The effect of treatment on tophi was a secondary efficacy endpoint and was assessed using standardized digital photography, image analysis, and a Central Reader blinded to treatment assignment. Approximately 70% of patients had tophi at baseline. A pooled analysis of data from Trial 2 and Trial 3 (NCT00325195) was performed as pre-specified in the protocols. At Month 6, the percentage of patients who achieved a complete response (defined as 100% resolution of at least one target tophus, no new tophi appear and no single tophus showing progression) was 45%, 26%, and 8%, with KRYSTEXXA 8 mg every 2 weeks, KRYSTEXXA 8 mg every 4 weeks, and placebo, respectively. The difference between KRYSTEXXA and placebo was statistically significant for the every 2 week dosing regimen, but not for the every 4 week dosing regimen.
16. How is Krystexxa supplied
How Supplied
KRYSTEXXA (pegloticase) injection is supplied as a sterile, clear and colorless solution containing 8 mg of uricase protein in 1 mL in phosphate buffered saline intended for intravenous infusion after dilution. KRYSTEXXA is supplied in a single-dose glass vial with a coated (latex-free) rubber injection stopper.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Anaphylaxis and Infusion Reactions
- Anaphylaxis and infusion reactions can occur at any infusion while on therapy. Counsel patients on the importance of adhering to any prescribed medications, to help prevent or lessen the severity of these reactions.
- Educate patients on the signs and symptoms of anaphylaxis, including wheezing, peri-oral or lingual edema, hemodynamic instability, and rash or urticaria, nausea or vomiting.
- Educate patients on the most common signs and symptoms of an infusion reaction, including urticaria (skin rash), erythema (redness of the skin), dyspnea (difficulty breathing), flushing, chest discomfort, chest pain, and rash.
- Advise patients to seek medical care immediately if they experience any symptoms of an allergic reaction during or at any time after the infusion of KRYSTEXXA [see Warnings and Precautions (5.1, 5.2), Adverse Reactions (6.1)].
- Advise patients to discontinue any oral urate-lowering agents before starting on KRYSTEXXA and not to take any oral urate-lowering agents while on KRYSTEXXA.
Glucose-6-phosphate dehydrogenase (G6PD) Deficiency
Inform patients not to take KRYSTEXXA if they have a condition known as G6PD deficiency. Explain to patients that G6PD deficiency is more frequently found in individuals of African, Mediterranean, or Southern Asian ancestry and that they may be tested to determine if they have G6PD deficiency, unless already known [see Warnings and Precautions (5.3), Contraindications (4)].
Gout Flares
Explain to patients that gout flares may initially increase when starting treatment with KRYSTEXXA, and that medications to help reduce flares may need to be taken regularly for the first few months after KRYSTEXXA is started [see Warnings and Precautions (5.4), Adverse Reactions (6.1)]. Advise patients that they should not stop KRYSTEXXA therapy if they have a flare.
AMGEN
Manufactured by:
Horizon Therapeutics Ireland DAC
Dublin, Ireland
US License Number 2022
Patent: https://pat.amgen.com/krystexxa/
©2025 Horizon Therapeutics Ireland DAC. All rights reserved.
1XXXXXX-v5
Medication Guide
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 01/2025 |
| Medication Guide KRYSTEXXA® (kris-TEX-a) (pegloticase) injection, for intravenous use |
|
| Read this Medication Guide before you start receiving KRYSTEXXA and before each treatment. There may be new information. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment. Talk to your doctor if you have any questions about your treatment with KRYSTEXXA. | |
| What is the most important information I should know about KRYSTEXXA?
Serious allergic reactions may happen in some people who receive KRYSTEXXA. These allergic reactions can be life threatening and usually happen within 2 hours of the infusion. KRYSTEXXA should be given to you by a doctor or nurse in a healthcare setting where serious allergic reactions can be treated. Your doctor or nurse should watch you for any signs of a serious allergic reaction during and after your treatment with KRYSTEXXA. Tell your doctor or nurse right away if you have any of these symptoms during or after your treatment with KRYSTEXXA:
|
|
What is KRYSTEXXA?
|
|
| Who should not receive KRYSTEXXA?
Do not receive KRYSTEXXA if you:
|
|
| What should I tell my doctor before receiving treatment with KRYSTEXXA?
Before you receive KRYSTEXXA, tell your doctor about all of your medical conditions, including if you:
Know the medicines you take. Keep a list of your medicines and show them to your doctor and pharmacist when you get a new medicine. |
|
How will I receive KRYSTEXXA?
|
|
| What are the possible side effects of KRYSTEXXA?
KRYSTEXXA may cause serious side effects. See "What is the most important information I should know about KRYSTEXXA?" The most common side effects of KRYSTEXXA when given together with methotrexate include:
These are not all of the side effects of KRYSTEXXA. For more information, ask your doctor or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Horizon at 1-866-479-6742. |
|
| General information about the safe and effective use of KRYSTEXXA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. This Medication Guide summarizes the most important information about KRYSTEXXA. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about KRYSTEXXA that is written for health professionals. For more information, go to www.KRYSTEXXA.com or call 1-866-479-6742. |
|
| What are the ingredients in KRYSTEXXA?
Active ingredient: pegloticase Inactive ingredients: disodium hydrogen phosphate dihydrate, sodium chloride, sodium dihydrogen phosphate dihydrate, and water for injection AMGEN Manufactured by: Horizon Therapeutics Ireland DAC Dublin, Ireland US License Number 2022 ©2025 Horizon Therapeutics Ireland DAC. All rights reserved |
|
| KRYSTEXXA
pegloticase injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Horizon Therapeutics USA, Inc. (033470838) |
| Registrant - Amgen, Inc (039976196) |
Biological Products Related to Krystexxa
Find detailed information on biosimilars for this medication.
More about Krystexxa (pegloticase)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (18)
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: antihyperuricemic agents
- Breastfeeding
- En español