Cabergoline: Package Insert / Prescribing Info
Package insert / product label
Dosage form: tablet
Drug class: Prolactin inhibitors
J Code (medical billing code): J8515 (0.25 mg, oral)
Medically reviewed by Drugs.com. Last updated on Jul 16, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
CABERGOLINE tablets, for oral use
Initial U.S. Approval: 1996
Recent Major Changes
Indications and Usage for Cabergoline
CABERGOLINE is an ergot derivative indicated for the treatment of hyperprolactinemic disorders, either idiopathic or due to pituitary adenomas in adults. (1)
Limitations of Use
Avoid use of CABERGOLINE for the inhibition or suppression of postpartum physiologic lactation because of the risk of serious adverse reactions. (5.4)
Cabergoline Dosage and Administration
- •
- Before initiating CABERGOLINE evaluate for valvular heart disease, including with an echocardiogram. If valvular disease is detected, do not administer CABERGOLINE. (2.1)
- •
- Recommended starting dosage of CABERGOLINE is 0.25 mg orally twice weekly. (2.2)
- •
- Titrate CABERGOLINE to achieve normal serum prolactin levels by increasing CABERGOLINE by 0.25 mg orally twice weekly at intervals of no less than 4 weeks. (2.2)
- •
- Maximum recommended dosage is 1 mg orally, twice weekly. (2.2)
Dosage Forms and Strengths
Tablets: 0.5 mg, white, with functional score. (3)
Contraindications
Warnings and Precautions
- •
- Cardiac Valvulopathy and Pericardial Fibrosis: Before initiating CABERGOLINE, perform a cardiovascular evaluation, including echocardiogram, to evaluate for valvular disease. During CABERGOLINE treatment, monitor for the development of valvulopathy with a cardiac echocardiogram at intervals of 6 to 12 months or as clinically indicated and monitor for chest pain and signs and symptoms of heart failure (if heart failure occurs, exclude valvular fibrosis and pericarditis). Consider additional clinical and diagnostic monitoring at baseline and as necessary during CABERGOLINE treatment. Use CABERGOLINE in patients treated with other drugs associated with valvulopathy only if the potential benefit of CABERGOLINE outweighs the risk. Discontinue CABERGOLINE if the patient has a new diagnosis of valvular regurgitation, valvular restriction, valve leaflet thickening, or pericarditis. (5.1)
- •
- Pleural, Pulmonary and Retroperitoneal Fibrosis: During CABERGOLINE treatment monitor for signs and symptoms of progressive fibrosis, (e.g., pleuro-pulmonary disease, renal impairment, ureteral/abdominal vascular obstruction). Consider clinical and diagnostic monitoring for pleural, pulmonary, and retroperitoneal fibrosis at baseline and as necessary during CABERGOLINE treatment. If pleural, pericardial, retroperitoneal, or pulmonary fibrosis occur, discontinue CABERGOLINE. (5.2)
- •
- Orthostatic Hypotension: Check blood pressure at baseline and during treatment with CABERGOLINE and monitor for orthostatic hypotension. (5.3)
- •
- Risks with Use of CABERGOLINE for Postpartum Lactation Inhibition or Suppression: Avoid use of CABERGOLINE for the inhibition or suppression of physiologic lactation. Use of bromocriptine, another dopamine agonist for this unapproved use has been associated with cases of hypertension, stroke, myocardial infarction, seizures, and death. (5.4)
- •
- Impulse Control Disorders and Compulsive Behaviors: Specifically ask patients about the development of new or increased gambling urges, sexual urges, uncontrolled spending, or other urges while being treated with CABERGOLINE. Consider dosage reduction or stopping CABERGOLINE if a patient develops such urges while taking CABERGOLINE. (5.5)
Adverse Reactions/Side Effects
The most common adverse reactions (incidence >10%) are nausea, headache, and dizziness. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Greenstone LLC at 1-877-446-3679 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
CABERGOLINE, a dopamine receptor agonist, is not recommended for concomitant use with D2-antagonists, such as phenothiazines, butyrophenones, thioxanthenes, or metoclopramide. (7)
Use In Specific Populations
Pregnancy: If conception occurs during CABERGOLINE therapy, discontinue CABERGOLINE if the risks to the mother or fetus outweigh the benefits to the mother. (8.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2025
Full Prescribing Information
1. Indications and Usage for Cabergoline
CABERGOLINE is an ergot derivative indicated for the treatment of hyperprolactinemic disorders, either idiopathic or due to pituitary adenomas in adults.
Limitations of Use
Avoid use of CABERGOLINE for the inhibition or suppression of postpartum physiologic lactation because of the risk of serious adverse reactions [see Warnings and Precautions (5.4)].
2. Cabergoline Dosage and Administration
2.1 Recommended Evaluation Before Initiating CABERGOLINE
Before initiating CABERGOLINE evaluate for valvular heart disease, including with an echocardiogram. If valvular disease is detected, do not administer CABERGOLINE [see Contraindications (4) and Warnings and Precautions (5.1)].
2.2 Recommended Dosage
The recommended starting dosage of CABERGOLINE is 0.25 mg orally twice weekly. Titrate CABERGOLINE to achieve normal serum prolactin levels by increasing CABERGOLINE by 0.25 mg orally twice weekly at intervals of no less than 4 weeks. The maximum recommended dosage is 1 mg orally, twice weekly [see Warnings and Precautions (5.2)]. Administer CABERGOLINE with or without food [see Clinical Pharmacology (12.3)].
If CABERGOLINE is discontinued, monitor the serum prolactin level periodically to determine whether CABERGOLINE should be reinstituted.
3. Dosage Forms and Strengths
Tablets: 0.5 mg, white, with functional score, oblong scored on one side with the letter P and the letter U on either side of the breakline, engraved with the number 700 on the opposite side.
4. Contraindications
CABERGOLINE is contraindicated in patients with:
- •
- Uncontrolled hypertension.
- •
- Known hypersensitivity to ergot derivatives.
- •
- History of cardiac valvular disorders, as suggested by anatomical evidence of valvulopathy of any valve, determined by pre-treatment evaluation including echocardiographic demonstration of valve leaflet thickening, valve restriction, or mixed valve restriction-stenosis, or history of pericardial fibrosis [see Warnings and Precautions (5.1)].
- •
- History of pleural, pulmonary, or retroperitoneal fibrotic disorders [see Warnings and Precautions (5.2)].
5. Warnings and Precautions
5.1 Cardiac Valvulopathy and Pericardial Fibrosis
Before initiating CABERGOLINE, perform a cardiovascular evaluation, including with an echocardiogram, to evaluate for valvular disease. CABERGOLINE is contraindicated in the presence of valvular disease or pericardial fibrosis [see Contraindications (4)].
Cases of valvular and pericardial fibrosis have often manifested as heart failure. Following CABERGOLINE treatment initiation, monitor for the development of valvulopathy with a cardiac echocardiogram at intervals of 6 to 12 months or as clinically indicated with new onset edema, cardiac murmur, dyspnea, or heart failure. During CABERGOLINE treatment, monitor for chest pain and signs and symptoms of heart failure and if heart failure occurs, valvular fibrosis and pericarditis should be excluded. Consider clinical and diagnostic monitoring such as erythrocyte sedimentation rate, serum creatinine measurements, chest-x- ray, and other investigations and cardiac imaging at baseline and as necessary while patients are treated with during CABERGOLINE treatment. Use CABERGOLINE in patients treated with other drugs associated with valvulopathy only if the potential benefit of CABERGOLINE outweighs the risk.
Discontinue CABERGOLINE if the patient has a new diagnosis of valvular regurgitation, valvular restriction, valve leaflet thickening, or pericarditis.
Postmarketing cases of cardiac valvulopathy have been reported in patients who received CABERGOLINE. These cases have generally occurred during administration of high doses of CABERGOLINE (>2 mg/day) for the treatment of Parkinson’s disease (PD) (CABERGOLINE is not approved for the treatment of PD). Cases of cardiac valvulopathy have also been reported in patients who received lower dosages of CABERGOLINE for the treatment of hyperprolactinemic disorders. In a 12-year, multi-country retrospective cohort study, the use of CABERGOLINE for PD was associated with an increased risk of cardiac valvular regurgitation (CVR). Compared to non-ergot-derived dopamine agonists and levodopa, CVR with CABERGOLINE use had an incidence rate per 10,000 person years of 68 (95% CI: 37, 115) versus 10 (95% CI: 5, 19) for non-ergot dopamine agonists and 11 (95% CI: 7, 17) for levodopa.
5.2 Pleural, Pulmonary and Retroperitoneal Fibrosis
CABERGOLINE is contraindicated in patients with a history of pleural, pulmonary, or retroperitoneal fibrosis. During CABERGOLINE treatment monitor for signs and symptoms of progressive fibrosis, including:
- •
- Pleuro-pulmonary disease (e.g., dyspnea, shortness of breath, persistent cough, chest pain).
- •
- Renal impairment or ureteral/abdominal vascular obstruction (e.g., pain in the loin/flank, lower limb edema, abdominal masses or tenderness that may indicate retroperitoneal fibrosis).
Consider clinical and diagnostic monitoring for pleural, pulmonary, and retroperitoneal fibrosis such as with erythrocyte sedimentation rate, serum creatinine measurements, chest-x-ray, and other investigations at baseline and as necessary during CABERGOLINE treatment. If pleural, pericardial, retroperitoneal, or pulmonary fibrosis occur, discontinue CABERGOLINE.
Postmarketing cases of pleural, pulmonary, and retroperitoneal fibrosis have been reported following CABERGOLINE administration. Some reports were in patients previously treated with other ergotinic dopamine agonists. CABERGOLINE-treated patients who developed a pleural effusion or pulmonary fibrosis and subsequently discontinued CABERGOLINE had improvement of their pulmonary symptoms.
5.3 Orthostatic Hypotension
Check blood pressure at baseline and during treatment with CABERGOLINE and monitor for orthostatic hypotension. Warn patients about the risk of orthostatic hypotension and precautions to take when rising from a supine or sitting position. Instruct patients to report dizziness or lightheadedness with changes in position to their healthcare provider.
CABERGOLINE can cause orthostatic hypotension [see Adverse Reactions (6.1)]. In a 4-week, placebo-controlled trial in patients with hyperprolactinemic disorders, the percentage of CABERGOLINE-treated patients and placebo-treated patients who developed orthostatic hypotension was 4% and 0%, respectively [see Adverse Reactions (6.1)]. The risk of orthostatic hypotension is greater in CABERGOLINE-treated patients when taking concomitant drugs that lower blood pressure.
5.4 Risks with Use of CABERGOLINE for Postpartum Lactation Inhibition or Suppression
Avoid use of CABERGOLINE for the inhibition or suppression of postpartum physiologic lactation because of the risk of serious adverse reactions. Use of bromocriptine, another dopamine agonist for this unapproved use has been associated with cases of hypertension, stroke, myocardial infarction seizures, and death.
5.5 Impulse Control Disorders and Compulsive Behaviors
Because patients may not recognize impulse control and compulsive behaviors as abnormal, it is important for health care providers to specifically ask patients about the development of new or increased gambling urges, sexual urges, uncontrolled spending, or other urges while being treated with CABERGOLINE. Consider dosage reduction or stopping CABERGOLINE if a patient develops such urges while taking CABERGOLINE.
Patients can experience intense urges to gamble or to spend money, increased sexual urges, binge eating, and/or other intense urges, and the inability to control these urges while taking one or more drugs that increase central dopaminergic tone, including CABERGOLINE. In some cases, these urges were reported to have stopped when the dosage was reduced, or the drug was stopped.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- •
- Cardiac Valvulopathy and Pericardial Fibrosis [see Warnings and Precautions (5.1)].
- •
- Pleural, Retroperitoneal, and Pulmonary Fibrosis [see Warnings and Precautions (5.2)].
- •
- Orthostatic Hypotension [see Warnings and Precautions (5.3)].
- •
- Impulse Control Disorders and Compulsive Behaviors [see Warnings and Precautions (5.5)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of CABERGOLINE has been evaluated in more than 900 patients with hyperprolactinemic disorders. In a 4-week, double-blind, placebo-controlled trial (CABERGOLINE vs. placebo) [see Clinical Studies (14)], the incidence of the most common adverse reactions during the placebo-controlled trial in patients with hyperprolactinemic disorders is presented in Table 1.
|
||
|
CABERGOLINE (n=168) |
Placebo (n=20) |
|
|
Nausea |
45 (27%) |
4 (20%) |
|
Headache |
43 (26%) |
5 (25%) |
|
Dizziness |
25 (15%) |
1 (5%) |
|
Constipation |
16 (10%) |
0% |
|
Fatigue |
12 (7%) |
0% |
|
Postural hypotension |
6 (4%) |
0% |
|
Dyspepsia |
4 (2%) |
0% |
|
Vomiting |
4 (2%) |
0% |
|
Nervousness |
4 (2%) |
0% |
|
Vertigo |
2 (1%) |
0% |
|
Paresthesia |
2 (1%) |
0% |
|
Breast pain |
2 (1%) |
0% |
|
Dysmenorrhea |
2 (1%) |
0% |
|
Abnormal vision |
2 (1%) |
0% |
In the 8-week, double-blind period of the comparative trial with bromocriptine, 2% (4/221) of CABERGOLINE-treated patients (0.5 mg twice weekly) discontinued treatment because of an adverse event and 6% (14/231) of bromocriptine-treated patients (at a dose of 2.5 mg twice daily) discontinued treatment because of an adverse event. The most common reasons for CABERGOLINE discontinuation were headache, nausea, and vomiting (3, 2, and 2 patients, respectively). The incidence of the most common adverse events during the double-blind period of the comparative trial with bromocriptine is presented in Table 2.
|
||
|
CABERGOLINE (n=221) |
Bromocriptine (n=231) |
|
|
Nausea |
63 (29%) |
100 (43%) |
|
Headache |
58 (26%) |
62 (27%) |
|
Dizziness |
38 (17%) |
42 (18%) |
|
Constipation |
15 (7%) |
21 (9%) |
|
Asthenia |
13 (6%) |
15 (6%) |
|
Abdominal pain |
12 (5%) |
19 (8%) |
|
Dyspepsia |
11 (5%) |
16 (7%) |
|
Fatigue |
10 (5%) |
18 (8%) |
|
Vertigo |
9 (4%) |
10 (4%) |
|
Vomiting |
9 (4%) |
16 (7%) |
|
Depression |
7 (3%) |
5 (2%) |
|
Hot flashes |
6 (3%) |
3 (1%) |
|
Breast pain |
5 (2%) |
8 (3%) |
|
Dry mouth |
5 (2%) |
2 (1%) |
|
Paresthesia |
5 (2%) |
6 (3%) |
|
Somnolence |
5 (2%) |
5 (2%) |
|
Diarrhea |
4 (2%) |
7 (3%) |
|
Flatulence |
4 (2%) |
3 (1%) |
|
Pain |
4 (2%) |
6 (3%) |
|
Acne |
3 (1%) |
0% |
|
Anorexia |
3 (1%) |
3 (1%) |
|
Anxiety |
3 (1%) |
3 (1%) |
|
Hypotension |
3 (1%) |
4 (2%) |
|
Insomnia |
3 (1%) |
2 (1%) |
|
Syncope |
3 (1%) |
3 (1%) |
|
Abnormal vision |
2 (1%) |
2 (1%) |
|
Arthralgia |
2 (1%) |
0% |
|
Dependent edema |
2 (1%) |
1 (<1%) |
|
Dysmenorrhea |
2 (1%) |
1 (<1%) |
|
Impaired concentration |
2 (1%) |
1 (<1%) |
|
Influenza-like symptoms |
2 (1%) |
0% |
|
Malaise |
2 (1%) |
0% |
|
Nervousness |
2 (1%) |
5 (2%) |
|
Palpitation |
2 (1%) |
5 (2%) |
|
Periorbital edema |
2 (1%) |
2 (1%) |
|
Peripheral edema |
2 (1%) |
1 (<1%) |
|
Pruritus |
2 (1%) |
1 (<1%) |
|
Rhinitis |
2 (1%) |
9 (4%) |
|
Throat irritation |
2 (1%) |
0% |
|
Toothache |
2 (1%) |
0% |
Events that were reported at an incidence of <1% in the clinical studies follow:
- •
- Body As a Whole: facial edema, influenza-like symptoms, malaise
- •
- Cardiovascular System: hypotension, syncope, palpitations
- •
- Digestive System: dry mouth, flatulence, diarrhea, anorexia
- •
- Metabolic and Nutritional System: weight loss, weight gain
- •
- Nervous System: somnolence, nervousness, paresthesia, insomnia, anxiety
- •
- Respiratory System: nasal stuffiness, epistaxis
- •
- Skin and Appendages: acne, pruritus
- •
- Special Senses: abnormal vision
- •
- Urogenital System: dysmenorrhea, increased libido
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of CABERGOLINE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- •
- Psychiatric: impulse control disorders and compulsive behaviors including, hypersexuality, increased libido, pathological gambling; psychotic disorder, aggression
- •
- Skin and subcutaneous: alopecia
Related/similar drugs
7. Drug Interactions
CABERGOLINE, a dopamine receptor agonist, is not recommended for concomitant use with D2-antagonists, such as phenothiazines, butyrophenones, thioxanthenes, or metoclopramide.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
If conception occurs during CABERGOLINE therapy, discontinue CABERGOLINE if the risks to the mother or fetus outweigh the benefits to the mother. There are risks to the mother associated with the use of CABERGOLINE (see Clinical Considerations).
The estimated background risk of major birth defects and miscarriage in patients with hyperprolactinemic disorders, either idiopathic or due to pituitary adenomas is unknown. All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Maternal Adverse Reactions: In general, avoid use of dopamine agonists, including CABERGOLINE, during pregnancy and the postpartum period. The risks of CABERGOLINE use increase in pregnant females with pregnancy-induced hypertension, preeclampsia, and eclampsia.
Data
Human Data: Published case reports have not reported a clear association with CABERGOLINE and major birth defects, miscarriage, or adverse fetal outcomes when CABERGOLINE was used during early pregnancy. However, these case reports cannot definitely establish the absence of CABERGOLINE-associated risk.
Animal Data: Embryo-fetal development studies have been performed with cabergoline administered by oral gavage in mice, rats, and rabbits:
- •
- There were no teratogenic effects in the presence of maternal toxicity in mice given cabergoline at doses up to 8 mg/kg/day (approximately 55 times the maximum recommended human dose based on body surface area) during the period of organogenesis.
- •
- A dose of 0.012 mg/kg/day (approximately 0.14 times the maximum recommended human dose) administered during the period of organogenesis in rats caused an increase in post-implantation loss. This finding is likely due to the role of prolactin in implantation in rats and is not thought to be relevant to humans.
- •
- At doses of 0.5 mg/kg/day (approximately 19 times the maximum recommended human dose) administered during the period of organogenesis in rabbits, cabergoline caused maternal toxicity characterized by a loss of body weight and decreased food consumption. Doses of 4 mg/kg/day (approximately 150 times the maximum recommended human dose) administered during the period of organogenesis in the rabbit caused an increased occurrence of various malformations. However, in another study in rabbits, no treatment-related malformations or embryofetal toxicity were observed at doses up to 8 mg/kg/day (approximately 300 times the maximum recommended human dose).
8.2 Lactation
Risk Summary
CABERGOLINE is not recommended in postpartum women who are breastfeeding or who are planning to breastfeed. Avoid use of CABERGOLINE for the inhibition or suppression of physiologic lactation [see Indications and Usage (1) and Warnings and Precautions (5.4)].
8.4 Pediatric Use
Safety and effectiveness of CABERGOLINE in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of CABERGOLINE did not include sufficient numbers of patients 65 years of age and older to determine whether they respond differently from younger adult patients.
8.6 Hepatic Impairment
The use of CABERGOLINE in patients with severe hepatic impairment (HI) (Child-Pugh C) is not recommended. When using CABERGOLINE in patients with moderate HI (Child-Pugh B) increase monitoring of CABERGOLINE-associated adverse reactions. The recommendations for use of CABERGOLINE in patients with mild HI (Child Pugh A) is the same as those with normal hepatic function.
Patients with moderate or severe HI had increased cabergoline exposure [see Clinical Pharmacology (12.3)], which may increase the risk of CABERGOLINE-associated adverse reactions.
10. Overdosage
Take measures to support blood pressure, if necessary. Consider contacting the Poison Help line (1-800-222-1222) or a medical toxicologist for additional overdose management recommendations.
11. Cabergoline Description
Cabergoline is an ergot derivative and dopamine receptor agonist. The chemical name for cabergoline is 1-[(6-allylergolin-8ß-yl)-carbonyl]-1-[3-(dimethylamino) propyl]-3-ethylurea. Its empirical formula is C26H37N5O2, and its molecular weight is 451.62. The structural formula is as follows
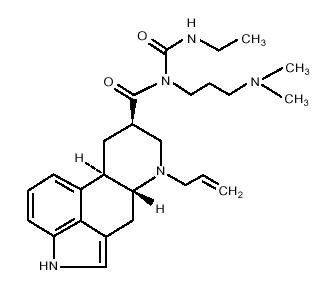
Cabergoline is a white powder soluble in ethyl alcohol, chloroform, and N, N-dimethylformamide (DMF); slightly soluble in 0.1N hydrochloric acid; very slightly soluble in n-hexane; and insoluble in water.
CABERGOLINE tablets, for oral administration, contain 0.5 mg of cabergoline and the inactive ingredients of leucine, USP, and lactose, NF.
12. Cabergoline - Clinical Pharmacology
12.1 Mechanism of Action
Cabergoline is an ergot derivative and dopamine receptor agonist with a high affinity for D2 receptors. Results of in vitro studies demonstrate that cabergoline exerts a direct inhibitory effect on the secretion of prolactin by rat pituitary lactotrophs. Cabergoline decreased serum prolactin levels in reserpinized rats. Receptor-binding studies indicate that cabergoline has low affinity for dopamine D1, α1- and α2-adrenergic, and 5-HT1- and 5-HT2-serotonin receptors.
12.2 Pharmacodynamics
The exposure-response relationship and time course of pharmacodynamic response for the safety and effectiveness of CABERGOLINE have not been fully characterized.
12.3 Pharmacokinetics
Absorption
The time to reach maximum cabergoline plasma concentration was 2 to 3 hours after single oral doses of 0.5 mg to 1.5 mg (1.5 times the maximum recommended dose) of CABERGOLINE in healthy subjects. Following dosing of CABERGOLINE between 0.5 mg to 7 mg (7 times the maximum recommended dose), cabergoline plasma levels appeared to be dose-proportional. The absolute bioavailability of cabergoline is unknown. A significant fraction of the administered dose undergoes a first-pass effect.
Effect of Food: High-fat food did not alter the pharmacokinetics of cabergoline [see Dosage and Administration (2.2)].
Distribution
Protein binding of cabergoline was 40% to 42%.
Elimination
The elimination half-life of cabergoline estimated from urinary data of 12 healthy subjects ranged between 63 to 69 hours.
Metabolism: Cabergoline is extensively metabolized, predominately via hydrolysis of the acylurea bond or the urea moiety. Hydrolysis of the acylurea or urea moiety abolishes the prolactin-lowering effect of cabergoline, and major metabolites identified thus far do not contribute to the therapeutic effect.
Excretion: After oral dosing of radioactive cabergoline to 5 healthy volunteers, approximately 22% and 60% of the dose was excreted within 20 days in the urine and feces, respectively. Less than 4% of the dose was excreted unchanged in the urine. Nonrenal and renal clearances for cabergoline are about 3.2 L/min and 0.08 L/min, respectively. Urinary excretion in hyperprolactinemic patients was similar.
Specific Populations
Patients with Hepatic Impairment:
In a pharmacokinetic hepatic impairment (HI) study [see Use in Specific Populations (8.6)]:
- •
- In 4 CABERGOLINE-treated patients with mild HI (Child-Pugh A), no effect on mean area under the cabergoline plasma concentration-time curve (AUC) was observed.
- •
- In 4 CABERGOLINE-treated patients with moderate HI (Child-Pugh B) there was a 1.5-fold increase in mean cabergoline AUC.
- •
- In 4 CABERGOLINE-treated patients with severe HI (Child-Pugh C) there was a 5.6-fold increase in the mean cabergoline AUC.
Male and Female Patients: Males aged 20 to 34 years were shown to have had higher Cmax than females aged 20 to 27 years) while males aged 66 to 75 years had lower Cmax compared to females aged 66 to 74 years. The clinical significance of the findings is unknown.
Patients with Renal Impairment: The pharmacokinetics of cabergoline were not altered in 12 patients with moderate-to-severe renal impairment as assessed by creatinine clearance.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies were conducted in mice and rats with cabergoline given by gavage at doses up to 0.98 mg/kg/day and 0.32 mg/kg/day, respectively. These doses are 7 times and 4 times the maximum recommended human dose calculated on a body surface area basis using total mg/m2/week in rodents and mg/m2/week for a 50 kg human.
There was a slight increase in the incidence of cervical and uterine leiomyomas and uterine leiomyosarcomas in mice. In rats, there was a slight increase in malignant tumors of the cervix and uterus and interstitial cell adenomas. The occurrence of tumors in female rodents may be related to the prolonged suppression of prolactin secretion because prolactin is needed in rodents for the maintenance of the corpus luteum. In the absence of prolactin, the estrogen/progesterone ratio is increased, thereby increasing the risk for uterine tumors. In male rodents, the decrease in serum prolactin levels was associated with an increase in serum luteinizing hormone, which is thought to be a compensatory effect to maintain testicular steroid synthesis. Since these hormonal mechanisms are thought to be species-specific, the relevance of these tumors to humans is not known.
Mutagenesis
The mutagenic potential of cabergoline was evaluated and found to be negative in a battery of in vitro tests. These tests included the bacterial mutation (Ames) test with Salmonella typhimurium, the gene mutation assay with Schizosaccharomyces pombe P1 and V79 Chinese hamster cells, DNA damage and repair in Saccharomyces cerevisiae D4, and chromosomal aberrations in human lymphocytes. Cabergoline was also negative in the bone marrow micronucleus test in the mouse.
Impairment of Fertility
In female rats, a daily cabergoline dose of 0.003 mg/kg for 2 weeks prior to mating and throughout the mating period inhibited conception. This dose represents approximately 0.04 times the maximum recommended human dose calculated on a body surface area basis using total mg/m2/week in rats and mg/m2/week for a 50 kg human. This finding is likely due to the role of prolactin in implantation in rats and is not thought to be relevant to humans.
14. Clinical Studies
The prolactin-lowering efficacy of CABERGOLINE was demonstrated in 647 females with hyperprolactinemic disorders (including 55% microprolactinomas, 7% macroprolactinomas, 3% empty sella syndromes, and 3% idiopathic) in two randomized, double-blind, studies: one 4-week placebo-controlled dose-response study with an 12-month open-label extension (Study 1) and one 8-week active comparator study comparing CABERGOLINE and bromocriptine with 16-week open extension (Study 2).
Study 1: 4-Week Placebo-Controlled Dose-Response Study
Study 1 enrolled 188 non-pregnant, hyperprolactinemic females (these patients had a prolactin level >20 ng/ml (the upper normal reference limit)) with the following hyperprolactinemic etiologies: macroprolactinoma (60%, n=113), idiopathic (36%, n=67) and another etiology (4%, n=8). Patients had a mean age of 32 years (range, 16-46 years of age), 99% were White (n=186), 0.5% were Asian (n=1) and 0.5% were another race (n=1). Patients were randomized one of the following five oral treatments given twice weekly for four weeks (for the CABERGOLINE groups, the first week the CABERGOLINE dosage was lower to reduce the risk of hypotensive reactions):
- •
- Group 1: placebo twice weekly (n=20),
- •
- Group 2: 0.0625 mg of CABERGOLINE twice weekly for the first week followed by 0.125 mg twice weekly for the next three weeks (n=42)-this dosage is not recommended because this dosage was not effective and results from this group are not presented below [see Dosage and Administration (2.2)],
- •
- Group 3: 0.25 mg of CABERGOLINE twice weekly for the first week followed 0.5 mg twice weekly for the next three weeks (n=42),
- •
- Group 4: 0.375 mg of CABERGOLINE twice weekly for the first week followed 0.75 mg twice weekly for the next three weeks (n=42), and
- •
- Group 5: 0.5 mg of CABERGOLINE twice weekly for the first week followed 1 mg twice weekly for the next three weeks 0.75 mg, (n=42).
In Study 1, the endpoint was the percentage of patients who achieved a normal serum prolactin level (<20 ng/dL) at the end of the 4-week treatment period. At 4-weeks, 0%, 76%, 74% and 95% of patients in the placebo group (group 1), group 3, group 4, and group 5, achieved normal serum prolactin levels, respectively (p <0.0001 across all the CABERGOLINE groups vs. placebo).
Study 2: 8-Week Active Comparator Study
Study 2 was an 8-week, randomized, double-blind active-control study that compared CABERGOLINE (n=223) with bromocriptine (n=236) for the treatment of hyperprolactinemic amenorrhea. In this study, patients had a mean age of 31 years (range 16 – 46 years of age). Patients were randomized to oral CABERGOLINE 0.5 mg twice weekly or oral bromocriptine 2.5 mg twice daily for eight weeks (there was an attrition rate respectively of 46% and 43%, in the CABERGOLINE and bromocriptine groups, respectively). Endpoints included percentage of patients who achieved the following at 8 weeks:
- •
- A normal serum prolactin level (<20 ng/dL)
- •
- Restoration of menses
- •
- Disappearance of galactorrhea
In Study 2, at 8 weeks, CABERGOLINE-treated and bromocriptine-treated patients had a normal serum prolactin level (77% and 59%, respectively), restoration of menses (77% and 70% respectively), and disappearance of galactorrhea, (73% and 56%, respectively).
The durability of the efficacy of CABERGOLINE beyond 24 months of therapy has not been established.
16. How is Cabergoline supplied
CABERGOLINE tablets are white, scored, oblong, with functional score on one side with the letter P and the letter U on either side of the breakline, engraved with the number 700 on the opposite side and they are available in the following configuration: 0.5 mg strength, 8-count bottle, and NDC number 59762-1005-1.
Store at controlled room temperature 20°C to 25°C (68°F to 77°F) [see USP]. Store the tablets in the original container.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Fibrotic Conditions
There is a risk of cardiac valvulopathy, and pericardial, pleural, pulmonary, and retroperitoneal fibrosis with CABERGOLINE treatment. Advise patients to notify their healthcare provider if they develop shortness of breath, chest pain, persistent cough, difficulty with breathing when lying down, or swelling in their extremities [see Warnings and Precautions (5.1)].
Orthostatic Hypotension
Warn patients about the risk of orthostatic hypotension and instruct patients to rise slowly from a supine or sitting position. Advise patients to notify their healthcare provider if they develop dizziness or lightheadedness [see Warnings and Precautions (5.3)].
Impulse Control Disorders and Compulsive Behaviors
Patients should be alerted to the possibility that patients may experience intense urges to spend money uncontrollably, intense urges to gamble, increased sexual urges, and other intense urges and the inability to control these urges while taking CABERGOLINE. Advise patients to inform their health care provider if they develop new or increased uncontrolled spending, gambling urges, sexual urges, or other urges while being treated with CABERGOLINE [see Warnings and Precautions (5.5)].
Pregnancy
Advise patients to notify their health care provider if they suspect they are pregnant, become pregnant, or intend to become pregnant during therapy. A pregnancy test should be done if there is any suspicion of pregnancy and continuation of CABERGOLINE treatment should be discussed with their health care provider [see Use in Specific Populations (8.1)].

This product’s labeling may have been updated. For the most recent Prescribing Information, please visit www.Greenstonellc.com.
LAB-0304-9.0



| CABERGOLINE
cabergoline tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Mylan Pharmaceuticals Inc. (059295980) |
| Registrant - Pfizer Inc (113480771) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Italia S.r.l. | 458521908 | ANALYSIS(59762-1005) , LABEL(59762-1005) , MANUFACTURE(59762-1005) , PACK(59762-1005) | |
More about cabergoline
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (118)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: prolactin inhibitors
- Breastfeeding
- En español
