Atripla: Package Insert / Prescribing Info
Package insert / product label
Generic name: efavirenz, emtricitabine, and tenofovir disoproxil fumarate
Dosage form: tablet, film coated
Drug class: Antiviral combinations
Medically reviewed by Drugs.com. Last updated on Jul 29, 2024.
The Atripla brand name has been discontinued in the U.S. If generic versions of this product have been approved by the FDA, there may be generic equivalents available.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
ATRIPLA® (efavirenz, emtricitabine, and tenofovir disoproxil fumarate) tablets, for oral use
Initial U.S. Approval: 2006
WARNING: POSTTREATMENT ACUTE EXACERBATION OF HEPATITIS B
See full prescribing information for complete boxed warning.
Severe acute exacerbations of hepatitis B virus (HBV) have been reported in patients coinfected with HBV and HIV-1 who have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of ATRIPLA. Closely monitor hepatic function with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue ATRIPLA. If appropriate, initiation of anti-hepatitis B therapy may be warranted. (5.1)
Recent Major Changes
Indications and Usage for Atripla
ATRIPLA is a three-drug combination of efavirenz (EFV), a non-nucleoside reverse transcriptase inhibitor, and emtricitabine (FTC) and tenofovir disoproxil fumarate (TDF), both HIV-1 nucleoside analog reverse transcriptase inhibitors, and is indicated as a complete regimen or in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 40 kg. (1)
Atripla Dosage and Administration
- Testing: Consult Full Prescribing Information for important testing recommendations prior to initiation and during treatment with ATRIPLA. (2.1)
- Recommended dosage in adults and pediatric patients weighing at least 40 kg: One tablet once daily taken orally on an empty stomach, preferably at bedtime. (2.2)
- Renal impairment: Not recommended in patients with estimated creatinine clearance below 50 mL/min. (2.3)
- Hepatic impairment: Not recommended in patients with moderate to severe hepatic impairment. (2.4)
- Dosage adjustment with rifampin coadministration: An additional 200 mg/day of efavirenz is recommended for patients weighing 50 kg or more. (2.5)
Dosage Forms and Strengths
Tablets: 600 mg of efavirenz, 200 mg of emtricitabine, and 300 mg of tenofovir disoproxil fumarate. (3)
Contraindications
Warnings and Precautions
- Rash: Discontinue if severe rash develops. (5.2, 6.1)
- Hepatotoxicity: Monitor liver function tests before and during treatment in patients with underlying hepatic disease, including hepatitis B or C coinfection, marked transaminase elevations, or who are taking medications associated with liver toxicity. Among reported cases of hepatic failure, a few occurred in patients with no pre-existing hepatic disease. (5.3, 6.2, 8.7)
- Risk of adverse reactions or loss of virologic response due to drug interactions: Consult full prescribing information prior to and during treatment for important potential drug interactions. Consider alternatives to ATRIPLA in patients taking other medications with a known risk of Torsade de Pointes or in patients at higher risk of Torsade de Pointes. (5.4)
- Serious psychiatric symptoms: Immediate medical evaluation is recommended. (5.5, 6.1)
- Nervous system symptoms (NSS): NSS are frequent, usually begin 1−2 days after initiating therapy, and resolve in 2−4 weeks. Dosing at bedtime may improve tolerability. NSS are not predictive of onset of psychiatric symptoms. (2.2, 5.6)
- New onset or worsening renal impairment: Can include acute renal failure and Fanconi syndrome. Prior to initiation and during use of ATRIPLA, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus. Avoid administering ATRIPLA with concurrent or recent use of nephrotoxic drugs. (5.7)
- Embryo fetal toxicity: Fetal harm may occur when administered to a pregnant woman during the first trimester. Avoid pregnancy while receiving ATRIPLA and for 12 weeks after discontinuation. (5.8, 8.1)
- Decreases in bone mineral density (BMD): Consider assessment of BMD in patients with a history of pathological fracture or other risk factors for osteoporosis or bone loss. (5.9)
- Convulsions: Use caution in patients with a history of seizures. (5.10)
- Lactic acidosis/severe hepatomegaly with steatosis: Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity. (5.11)
- Immune reconstitution syndrome: May necessitate further evaluation and treatment. (5.12)
- Redistribution/accumulation of body fat: Observed in patients receiving antiretroviral therapy. (5.13)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence greater than or equal to 10%) observed in an active-controlled clinical trial of EFV, FTC, and TDF are diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Consult Full Prescribing Information prior to and during treatment for important potential drug interactions. (4, 5.4, 7)
- HIV-1 protease inhibitors: Coadministration of ATRIPLA with either lopinavir/ritonavir or darunavir and ritonavir increases tenofovir concentrations. Monitor for evidence of tenofovir toxicity. Coadministration of ATRIPLA with either atazanavir or atazanavir and ritonavir is not recommended. (7.3)
Use In Specific Populations
- Pregnancy: Avoid pregnancy while receiving ATRIPLA and for 12 weeks after discontinuation. (5.8, 8.3)
- Lactation: Breastfeeding is not recommended. (8.2)
- Females and Males of Reproductive Potential: Pregnancy testing and contraception are recommended. (8.3)
- Pediatrics: The incidence of rash was higher than in adults. (5.2, 6.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2021
Full Prescribing Information
WARNING: POSTTREATMENT ACUTE EXACERBATION OF HEPATITIS B
Severe acute exacerbations of hepatitis B virus (HBV) have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF), which are components of ATRIPLA.
Closely monitor hepatic function with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue ATRIPLA. If appropriate, initiation of anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1)].
1. Indications and Usage for Atripla
ATRIPLA® is indicated as a complete regimen or in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 40 kg.
2. Atripla Dosage and Administration
2.1 Testing Prior to Initiation and During Treatment with ATRIPLA
Prior to or when initiating ATRIPLA, test patients for hepatitis B virus infection [see Warnings and Precautions (5.1)].
Prior to initiation and during use of ATRIPLA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus [see Warnings and Precautions (5.7)].
Monitor hepatic function prior to and during treatment with ATRIPLA [see Warnings and Precautions (5.3)].
Perform pregnancy testing before initiation of ATRIPLA in adolescents and adults of childbearing potential [see Warnings and Precautions (5.8), Use in Specific Populations (8.1, 8.3)].
2.2 Recommended Dosage for Adults and Pediatric Patients Weighing at Least 40 kg
ATRIPLA is a three-drug fixed-dose combination product containing 600 mg of efavirenz (EFV), 200 mg of emtricitabine (FTC), and 300 mg of tenofovir disoproxil fumarate (TDF). The recommended dosage of ATRIPLA in adults and pediatric patients weighing at least 40 kg is one tablet once daily taken orally on an empty stomach. Dosing at bedtime may improve the tolerability of nervous system symptoms [see Clinical Pharmacology (12.3)].
2.3 Not Recommended in Patients with Moderate or Severe Renal Impairment
ATRIPLA is not recommended in patients with moderate or severe renal impairment (estimated creatinine clearance below 50 mL/min) [see Warnings and Precautions (5.7), Use in Specific Populations (8.6)].
2.4 Not Recommended in Patients with Moderate to Severe Hepatic Impairment
ATRIPLA is not recommended in patients with moderate to severe hepatic impairment (Child-Pugh B or C) [see Warnings and Precautions (5.3) and Use in Specific Populations (8.7)].
2.5 Dosage Adjustment with Rifampin
If ATRIPLA is co-administered with rifampin in patients weighing 50 kg or more, take one tablet of ATRIPLA once daily followed by one additional 200 mg per day of efavirenz [see Drug Interactions (7.3) and Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
ATRIPLA tablets are pink, capsule shaped, film coated, debossed with "123" on one side, and plain faced on the other side. Each tablet contains 600 mg of efavirenz, 200 mg of emtricitabine, and 300 mg of tenofovir disoproxil fumarate (equivalent to 245 mg of tenofovir disoproxil).
4. Contraindications
- ATRIPLA is contraindicated in patients with previously demonstrated clinically significant hypersensitivity (e.g., Stevens-Johnson syndrome, erythema multiforme, or toxic skin eruptions) to efavirenz, a component of ATRIPLA [see Warnings and Precautions (5.2)].
- ATRIPLA is contraindicated to be coadministered with voriconazole or elbasvir/grazoprevir [see Drug Interactions (7.3) and Clinical Pharmacology (12.3)].
5. Warnings and Precautions
5.1 Severe Acute Exacerbation of Hepatitis B in Patients Coinfected with HIV-1 and HBV
All patients should be tested for the presence of chronic HBV before or when initiating antiretroviral therapy [see Dosage and Administration (2.1)]. Severe acute exacerbations of hepatitis B (e.g., liver decompensation and liver failure) have been reported in patients who are coinfected with HBV and HIV-1 and have discontinued FTC or TDF, two of the components of ATRIPLA. Patients who are coinfected with HIV-1 and HBV should be closely monitored, with both clinical and laboratory follow-up for at least several months after stopping treatment with ATRIPLA. If appropriate, initiation of anti-hepatitis B therapy may be warranted, especially in patients with advanced liver disease or cirrhosis, since posttreatment exacerbation of hepatitis may lead to hepatic decompensation and liver failure.
5.2 Rash
In controlled clinical trials, 26% (266/1,008) of adult subjects treated with 600 mg EFV experienced new-onset skin rash compared with 17% (111/635) of those treated in control groups. Rash associated with blistering, moist desquamation, or ulceration occurred in 0.9% (9/1,008) of subjects treated with EFV. The incidence of Grade 4 rash (e.g., erythema multiforme, Stevens-Johnson syndrome) in adult subjects treated with EFV in all trials and expanded access was 0.1%. Rashes are usually mild-to-moderate maculopapular skin eruptions that occur within the first 2 weeks of initiating therapy with EFV (median time to onset of rash in adults was 11 days) and, in most subjects continuing therapy with EFV, rash resolves within 1 month (median duration, 16 days). The discontinuation rate for rash in adult clinical trials was 1.7% (17/1,008). ATRIPLA can be reinitiated in patients interrupting therapy because of rash. ATRIPLA should be discontinued in patients developing severe rash associated with blistering, desquamation, mucosal involvement, or fever. Appropriate antihistamines and/or corticosteroids may improve the tolerability and hasten the resolution of rash. For patients who have had a life-threatening cutaneous reaction (e.g., Stevens-Johnson syndrome), alternative therapy should be considered [see Contraindications (4)].
Experience with EFV in subjects who discontinued other antiretroviral agents of the NNRTI class is limited. Nineteen subjects who discontinued nevirapine because of rash have been treated with EFV. Nine of these subjects developed mild-to-moderate rash while receiving therapy with EFV, and two of these subjects discontinued because of rash.
Rash was reported in 59 of 182 pediatric subjects (32%) treated with EFV [see Adverse Reactions (6.1)]. Two pediatric subjects experienced Grade 3 rash (confluent rash with fever, generalized rash), and four subjects had Grade 4 rash (erythema multiforme). The median time to onset of rash in pediatric subjects was 28 days (range 3−1,642 days). Prophylaxis with appropriate antihistamines before initiating therapy with ATRIPLA in pediatric patients should be considered.
5.3 Hepatotoxicity
Postmarketing cases of hepatitis, including fulminant hepatitis progressing to liver failure requiring transplantation or resulting in death, have been reported in patients treated with EFV, a component of ATRIPLA. Reports have included patients with underlying hepatic disease, including coinfection with hepatitis B or C, and patients without pre-existing hepatic disease or other identifiable risk factors [see Warnings and Precautions (5.1)].
ATRIPLA is not recommended for patients with moderate or severe hepatic impairment. Careful monitoring is recommended for patients with mild hepatic impairment receiving ATRIPLA [see Adverse Reactions (6.2) and Use in Specific Populations (8.7)].
Monitoring of liver enzymes before and during treatment is recommended for all patients [see Dosage and Administration (2.1)]. Consider discontinuing ATRIPLA in patients with persistent elevations of serum transaminases to greater than five times the upper limit of the normal range.
Discontinue ATRIPLA if elevation of serum transaminases is accompanied by clinical signs or symptoms of hepatitis or hepatic decompensation [see Adverse Reactions (6.1)].
5.4 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
The concomitant use of ATRIPLA and other drugs may result in potentially significant drug interactions [see Contraindications (4) and Drug Interactions (7.3)], some of which may lead to:
- Loss of therapeutic effect of concomitant drug or ATRIPLA and possible development of resistance.
- Possible clinically significant adverse reaction from greater exposures of ATRIPLA or concomitant drug.
QTc prolongation has been observed with the use of EFV [see Drug Interactions (7.1) and Clinical Pharmacology (12.2)]. Consider alternatives to ATRIPLA when coadministered with a drug with a known risk of Torsade de Pointes or when administered to patients at higher risk of Torsade de Pointes.
See Table 3 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during ATRIPLA therapy and review concomitant medications during ATRIPLA therapy [see Dosage and Administration (2.5), Contraindications (4), and Drug Interactions (7)].
5.5 Psychiatric Symptoms
Serious psychiatric adverse experiences have been reported in patients treated with EFV, a component of ATRIPLA. In controlled trials of 1,008 subjects treated with regimens containing EFV for a mean of 2.1 years and 635 subjects treated with control regimens for a mean of 1.5 years, the frequency (regardless of causality) of specific serious psychiatric events among subjects who received EFV or control regimens, respectively, were: severe depression (2.4%, 0.9%), suicidal ideation (0.7%, 0.3%), nonfatal suicide attempts (0.5%, 0%), aggressive behavior (0.4%, 0.5%), paranoid reactions (0.4%, 0.3%), and manic reactions (0.2%, 0.3%). When psychiatric symptoms similar to those noted above were combined and evaluated as a group in a multifactorial analysis of data from Study AI266006 (006, NCT00002410), a Phase 3 randomized, open-label trial of EFV-containing regimens versus controls in 1,266 subjects (median follow-up 180 weeks, 102 weeks, and 76 weeks for subjects treated with EFV + zidovudine + lamivudine, EFV + indinavir, and indinavir + zidovudine + lamivudine, respectively), treatment with EFV was associated with an increase in the occurrence of these selected psychiatric symptoms. Other factors associated with an increase in the occurrence of these psychiatric symptoms were history of injection drug use, psychiatric history, and receipt of psychiatric medication at trial entry; similar associations were observed in both the EFV and control treatment groups. In Study 006, onset of new serious psychiatric symptoms occurred throughout the trial for both EFV-treated and control-treated subjects. One percent of EFV-treated subjects discontinued or interrupted treatment because of one or more of these selected psychiatric symptoms. There have also been occasional postmarketing reports of death by suicide, delusions, and psychosis-like behavior, although a causal relationship to the use of EFV cannot be determined from these reports. Postmarketing cases of catatonia have also been reported and may be associated with increased EFV exposure. Patients with serious psychiatric adverse experiences should seek immediate medical evaluation to assess the possibility that the symptoms may be related to the use of EFV, and if so, to determine whether the risks of continued therapy outweigh the benefits [see Adverse Reactions (6)].
5.6 Nervous System Symptoms
Fifty-three percent (531/1,008) of subjects receiving EFV in controlled trials reported central nervous system symptoms (any grade, regardless of causality) compared to 25% (156/635) of subjects receiving control regimens. These symptoms included dizziness (28.1% of the 1,008 subjects), insomnia (16.3%), impaired concentration (8.3%), somnolence (7.0%), abnormal dreams (6.2%), and hallucinations (1.2%). Other reported symptoms were euphoria, confusion, agitation, amnesia, stupor, abnormal thinking, and depersonalization. The majority of these symptoms were mild to moderate (50.7%); symptoms were severe in 2.0% of subjects. Overall, 2.1% of subjects discontinued therapy as a result. These symptoms usually begin during the first or second day of therapy and generally resolve after the first 2–4 weeks of therapy. After 4 weeks of therapy, the prevalence of nervous system symptoms of at least moderate severity ranged from 5% to 9% in subjects treated with regimens containing EFV and from 3% to 5% in subjects treated with a control regimen. Patients should be informed that these common symptoms were likely to improve with continued therapy and were not predictive of subsequent onset of the less frequent psychiatric symptoms [see Warnings and Precautions (5.5)]. Dosing at bedtime may improve the tolerability of these nervous system symptoms [see Dosage and Administration (2.2)].
Analysis of long-term data from Study 006 showed that, beyond 24 weeks of therapy, the incidences of new-onset nervous system symptoms among EFV-treated subjects were generally similar to those in the indinavir-containing control arm.
Late-onset neurotoxicity, including ataxia and encephalopathy (impaired consciousness, confusion, psychomotor slowing, psychosis, delirium), may occur months to years after beginning EFV therapy. Some events of late-onset neurotoxicity have occurred in patients with CYP2B6 genetic polymorphisms which are associated with increased EFV levels despite standard dosing of EFV. Patients presenting with signs and symptoms of serious neurologic adverse experiences should be evaluated promptly to assess the possibility that these events may be related to EFV use, and whether discontinuation of ATRIPLA is warranted.
Patients receiving ATRIPLA should be alerted to the potential for additive central nervous system effects when ATRIPLA is used concomitantly with alcohol or psychoactive drugs.
Patients who experience central nervous system symptoms such as dizziness, impaired concentration, and/or drowsiness should avoid potentially hazardous tasks such as driving or operating machinery.
5.7 New Onset or Worsening Renal Impairment
Emtricitabine and tenofovir are principally eliminated by the kidney; however, EFV is not. Renal impairment, including cases of acute renal failure and Fanconi syndrome (renal tubular injury with severe hypophosphatemia), has been reported with the use of TDF, a component of ATRIPLA [see Adverse Reactions (6.2)].
Prior to initiation and during use of ATRIPLA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus. ATRIPLA is not recommended in patients with moderate or severe renal impairment (estimated creatinine clearance below 50 mL/min).
ATRIPLA should be avoided with concurrent or recent use of a nephrotoxic agent (e.g., high-dose or multiple non-steroidal anti-inflammatory drugs [NSAIDs]) [see Drug Interactions (7.2)]. Cases of acute renal failure after initiation of high-dose or multiple NSAIDs have been reported in HIV-infected patients with risk factors for renal dysfunction who appeared stable on TDF. Some patients required hospitalization and renal replacement therapy. Alternatives to NSAIDs should be considered, if needed, in patients at risk for renal dysfunction.
Persistent or worsening bone pain, pain in extremities, fractures, and/or muscular pain or weakness may be manifestations of proximal renal tubulopathy and should prompt an evaluation of renal function in patients at risk of renal dysfunction.
Discontinue ATRIPLA in patients who develop clinically significant decreases in renal function or evidence of Fanconi syndrome.
5.8 Embryo-Fetal Toxicity
Efavirenz may cause fetal harm when administered during the first trimester of pregnancy. Advise adults and adolescents of childbearing potential who are receiving ATRIPLA to avoid pregnancy while receiving ATRIPLA and for 12 weeks after discontinuation [see Dosage and Administration (2.1), Use in Specific Populations (8.1, 8.3)].
5.9 Bone Loss and Mineralization Defects
Bone Mineral Density
In clinical trials in HIV-1 infected adults, TDF (a component of ATRIPLA) was associated with slightly greater decreases in bone mineral density (BMD) and increases in biochemical markers of bone metabolism, suggesting increased bone turnover relative to comparators. Serum parathyroid hormone levels and 1,25 Vitamin D levels were also higher in subjects receiving TDF.
Clinical trials evaluating TDF in pediatric and adolescent subjects were conducted. Under normal circumstances, BMD increases rapidly in pediatric patients. In HIV-1 infected subjects aged 2 years to less than 18 years, bone effects were similar to those observed in adult subjects and suggest increased bone turnover. Total body BMD gain was less in the TDF-treated HIV-1 infected pediatric subjects as compared to the control groups. Similar trends were observed in chronic hepatitis-B infected adolescent subjects aged 12 years to less than 18 years. In all pediatric trials, skeletal growth (height) appeared to be unaffected.
The effects of TDF-associated changes in BMD and biochemical markers on long-term bone health and future fracture risk are unknown. Assessment of BMD should be considered for adult and pediatric patients who have a history of pathologic bone fracture or other risk factors for osteoporosis or bone loss. Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial for all patients. If bone abnormalities are suspected, then appropriate consultation should be obtained.
Mineralization Defects
Cases of osteomalacia associated with proximal renal tubulopathy, manifested as bone pain or pain in extremities and which may contribute to fractures, have been reported in association with TDF use [see Adverse Reactions (6.2)]. Arthralgias and muscle pain or weakness have also been reported in cases of proximal renal tubulopathy. Hypophosphatemia and osteomalacia secondary to proximal renal tubulopathy should be considered in patients at risk of renal dysfunction who present with persistent or worsening bone or muscle symptoms while receiving TDF-containing products [see Warnings and Precautions (5.7)].
5.10 Convulsions
Convulsions have been observed in adult and pediatric patients receiving EFV, generally in the presence of known medical history of seizures. Caution must be taken in any patient with a history of seizures.
Patients who are receiving concomitant anticonvulsant medications primarily metabolized by the liver, such as phenytoin and phenobarbital, may require periodic monitoring of plasma levels [see Drug Interactions (7.3)].
5.11 Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including TDF and FTC, components of ATRIPLA, alone or in combination with other antiretrovirals. Treatment with ATRIPLA should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
5.12 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including the components of ATRIPLA. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, Guillain-Barré syndrome, and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.13 Fat Redistribution
Redistribution/accumulation of body fat, including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance," has been observed in patients receiving antiretroviral therapy, including EFV. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in other sections of the labeling:
- Severe Acute Exacerbations of Hepatitis B in Patients Coinfected with HIV-1 and HBV [see Warnings and Precautions (5.1)].
- Rash [see Warnings and Precautions (5.2)].
- Hepatotoxicity [see Warnings and Precautions (5.3)].
- Psychiatric Symptoms [see Warnings and Precautions (5.5)].
- Nervous System Symptoms [see Warnings and Precautions (5.6)].
- New Onset or Worsening Renal Impairment [see Warnings and Precautions (5.7)].
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.8)].
- Bone Loss and Mineralization Defects [see Warnings and Precautions (5.9)].
- Convulsions [see Warnings and Precautions (5.10)].
- Lactic Acidosis/Severe Hepatomegaly with Steatosis [see Warnings and Precautions (5.11)].
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.12)].
- Fat Redistribution [see Warnings and Precautions (5.13)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials in Adult Subjects
Study 934 was an open-label active-controlled trial in which 511 antiretroviral-naïve subjects received either FTC + TDF administered in combination with EFV (N=257) or zidovudine (AZT)/lamivudine (3TC) administered in combination with EFV (N=254).
The most common adverse reactions (incidence greater than or equal to 10%, any severity) occurring in Study 934 include diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash. Adverse reactions observed in Study 934 were generally consistent with those seen in previous trials of the individual components (Table 1).
| FTC+TDF+EFV† | AZT/3TC+EFV | |
|---|---|---|
| N=257 | N=254 | |
|
||
| Fatigue | 9% | 8% |
| Depression | 9% | 7% |
| Nausea | 9% | 7% |
| Diarrhea | 9% | 5% |
| Dizziness | 8% | 7% |
| Upper respiratory tract infections | 8% | 5% |
| Sinusitis | 8% | 4% |
| Rash Event‡ | 7% | 9% |
| Headache | 6% | 5% |
| Insomnia | 5% | 7% |
| Anxiety | 5% | 4% |
| Nasopharyngitis | 5% | 3% |
| Vomiting | 2% | 5% |
In Study 073, subjects with stable, virologic suppression on antiretroviral therapy and no history of virologic failure were randomized to receive ATRIPLA or to stay on their baseline regimen. The adverse reactions observed in Study 073 were generally consistent with those seen in Study 934 and those seen with the individual components of ATRIPLA when each was administered in combination with other antiretroviral agents.
Efavirenz, Emtricitabine, or TDF
In addition to the adverse reactions in Study 934 and Study 073, the following adverse reactions were observed in clinical trials of EFV, FTC, or TDF in combination with other antiretroviral agents.
Efavirenz: The most significant adverse reactions observed in subjects treated with EFV were nervous system symptoms [see Warnings and Precautions (5.6)], psychiatric symptoms [see Warnings and Precautions (5.5)], and rash [see Warnings and Precautions (5.2)].
Selected adverse reactions of moderate-to-severe intensity observed in greater than or equal to 2% of EFV-treated subjects in two controlled clinical trials included pain, impaired concentration, abnormal dreams, somnolence, anorexia, dyspepsia, abdominal pain, nervousness, and pruritus.
Pancreatitis has also been reported, although a causal relationship with EFV has not been established. Asymptomatic increases in serum amylase levels were observed in a significantly higher number of subjects treated with EFV 600 mg than in control subjects.
Skin discoloration has been reported with higher frequency among FTC-treated subjects; it was manifested by hyperpigmentation on the palms and/or soles and was generally mild and asymptomatic. The mechanism and clinical significance are unknown.
Clinical Trials in Pediatric Subjects
Efavirenz: Assessment of adverse reactions is based on three pediatric clinical trials in 182 HIV-1 infected pediatric subjects who received EFV in combination with other antiretroviral agents for a median of 123 weeks. The type and frequency of adverse reactions in the three trials were generally similar to that of adult subjects with the exception of a higher incidence of rash, which was reported in 32% (59/182) of pediatric subjects compared to 26% of adults, and a higher frequency of Grade 3 or 4 rash reported in 3% (6/182) of pediatric subjects compared to 0.9% of adults [see Warnings and Precautions (5.2)].
Emtricitabine: In addition to the adverse reactions reported in adults, anemia and hyperpigmentation were observed in 7% and 32%, respectively, of pediatric subjects who received treatment with FTC in the larger of two open-label, uncontrolled pediatric trials (N=116).
Tenofovir DF: In a pediatric clinical trial conducted in subjects 12 to less than 18 years of age, the adverse reactions observed in pediatric subjects who received treatment with TDF (N=81) were consistent with those observed in clinical trials of TDF in adults [see Warnings and Precautions (5.9)].
Laboratory Abnormalities
Efavirenz, Emtricitabine and Tenofovir DF: Laboratory abnormalities observed in Study 934 were generally consistent with those seen in previous trials (Table 2).
| FTC+TDF+EFV* | AZT/3TC+EFV | |
|---|---|---|
| N=257 | N=254 | |
|
||
| Any ≥ Grade 3 Laboratory Abnormality | 30% | 26% |
| Fasting Cholesterol (>240 mg/dL) | 22% | 24% |
| Creatine Kinase (M: >990 U/L) (F: >845 U/L) | 9% | 7% |
| Serum Amylase (>175 U/L) | 8% | 4% |
| Alkaline Phosphatase (>550 U/L) | 1% | 0% |
| AST (M: >180 U/L) (F: >170 U/L) | 3% | 3% |
| ALT (M: >215 U/L) (F: >170 U/L) | 2% | 3% |
| Hemoglobin (<8.0 mg/dL) | 0% | 4% |
| Hyperglycemia (>250 mg/dL) | 2% | 1% |
| Hematuria (>75 RBC/HPF) | 3% | 2% |
| Glycosuria (≥3+) | <1% | 1% |
| Neutrophils (<750/mm3) | 3% | 5% |
| Fasting Triglycerides (>750 mg/dL) | 4% | 2% |
Laboratory abnormalities observed in Study 073 were generally consistent with those in Study 934.
Hepatic Events: In Study 934, 19 subjects treated with EFV, FTC, and TDF and 20 subjects treated with EFV and fixed-dose zidovudine/lamivudine were hepatitis B surface antigen or hepatitis C antibody positive. Among these coinfected subjects, one subject (1/19) in the EFV, FTC, and TDF arm had elevations in transaminases to greater than five times ULN through 144 weeks. In the fixed-dose zidovudine/lamivudine arm, two subjects (2/20) had elevations in transaminases to greater than five times ULN through 144 weeks. No HBV and/or HCV coinfected subject discontinued from the trial due to hepatobiliary disorders [see Warnings and Precautions (5.3)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of EFV, FTC, or TDF. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Efavirenz:
Cardiac Disorders
Palpitations
Ear and Labyrinth Disorders
Tinnitus, vertigo
Endocrine Disorders
Gynecomastia
Eye Disorders
Abnormal vision
Gastrointestinal Disorders
Constipation, malabsorption
General Disorders and Administration Site Conditions
Asthenia
Hepatobiliary Disorders
Hepatic enzyme increase, hepatic failure, hepatitis
Immune System Disorders
Allergic reactions
Metabolism and Nutrition Disorders
Redistribution/accumulation of body fat [see Warnings and Precautions (5.13)], hypercholesterolemia, hypertriglyceridemia
Musculoskeletal and Connective Tissue Disorders
Arthralgia, myalgia, myopathy
Nervous System Disorders
Abnormal coordination, ataxia, encephalopathy, cerebellar coordination and balance disturbances, convulsions, hypoesthesia, paresthesia, neuropathy, tremor
Psychiatric Disorders
Aggressive reactions, agitation, delusions, emotional lability, mania, neurosis, paranoia, psychosis, suicide, catatonia
Respiratory, Thoracic and Mediastinal Disorders
Dyspnea
Skin and Subcutaneous Tissue Disorders
Flushing, erythema multiforme, photoallergic dermatitis, Stevens-Johnson syndrome
Emtricitabine: No postmarketing adverse reactions have been identified for inclusion in this section.
Tenofovir DF:
Immune System Disorders
Allergic reaction, including angioedema
Metabolism and Nutrition Disorders
Lactic acidosis, hypokalemia, hypophosphatemia
Respiratory, Thoracic, and Mediastinal Disorders
Dyspnea
Gastrointestinal Disorders
Pancreatitis, increased amylase, abdominal pain
Hepatobiliary Disorders
Hepatic steatosis, hepatitis, increased liver enzymes (most commonly AST, ALT, gamma GT)
Skin and Subcutaneous Tissue Disorders
Rash
Musculoskeletal and Connective Tissue Disorders
Rhabdomyolysis, osteomalacia (manifested as bone pain and which may contribute to fractures), muscular weakness, myopathy
Renal and Urinary Disorders
Acute renal failure, renal failure, acute tubular necrosis, Fanconi syndrome, proximal renal tubulopathy, interstitial nephritis (including acute cases), nephrogenic diabetes insipidus, renal insufficiency, increased creatinine, proteinuria, polyuria
General Disorders and Administration Site Conditions
Asthenia
The following adverse reactions, listed under the body system headings above, may occur as a consequence of proximal renal tubulopathy: rhabdomyolysis, osteomalacia, hypokalemia, muscular weakness, myopathy, hypophosphatemia.
Related/similar drugs
7. Drug Interactions
7.1 Efavirenz
Efavirenz has been shown in vivo to induce CYP3A and CYP2B6. Other compounds that are substrates of CYP3A or CYP2B6 may have decreased plasma concentrations when coadministered with EFV.
Drugs that induce CYP3A activity (e.g., phenobarbital, rifampin, rifabutin) would be expected to increase the clearance of EFV, resulting in lowered plasma concentrations [see Dosage and Administration (2.2)].
There is limited information available on the potential for a pharmacodynamic interaction between EFV and drugs that prolong the QTc interval. QTc prolongation has been observed with the use of EFV [see Clinical Pharmacology (12.2)]. Consider alternatives to ATRIPLA when coadministered with a drug with a known risk of Torsade de Pointes.
7.2 Drugs Affecting Renal Function
FTC and tenofovir are primarily eliminated by the kidneys [see Clinical Pharmacology (12.3)]. Coadministration of ATRIPLA with drugs that are eliminated by active tubular secretion may increase concentrations of FTC, tenofovir, and/or the coadministered drug. Some examples include, but are not limited to, acyclovir, adefovir dipivoxil, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs [see Warnings and Precautions (5.7)]. Drugs that decrease renal function may increase concentrations of FTC and/or tenofovir.
7.3 Established and Potentially Significant Interactions
Other important drug interaction information for ATRIPLA is summarized in Table 3. The drug interactions described are based on trials conducted with either ATRIPLA, the components of ATRIPLA (EFV, FTC, or TDF) as individual agents, or are potential drug interactions [see Clinical Pharmacology (12.3)].
| Concomitant Drug Class: Drug Name | Effect | Clinical Comment |
|---|---|---|
|
||
| HIV antiviral agents | ||
| Protease inhibitor:
atazanavir | ↓ atazanavir ↑ tenofovir | Coadministration of atazanavir with ATRIPLA is not recommended. The combined effect of EFV plus TDF on atazanavir plasma concentrations is not known. There are insufficient data to support dosing recommendations for atazanavir or atazanavir/ritonavir in combination with ATRIPLA. |
| Protease inhibitor: fosamprenavir calcium | ↓ amprenavir | Fosamprenavir (unboosted): Appropriate doses of fosamprenavir and ATRIPLA with respect to safety and efficacy have not been established. Fosamprenavir/ritonavir: An additional 100 mg/day (300 mg total) of ritonavir is recommended when ATRIPLA is administered with fosamprenavir/ritonavir once daily. No change in the ritonavir dose is required when ATRIPLA is administered with fosamprenavir plus ritonavir twice daily. |
| Protease inhibitor: indinavir | ↓ indinavir | The optimal dose of indinavir, when given in combination with EFV, is not known. Increasing the indinavir dose to 1000 mg every 8 hours does not compensate for the increased indinavir metabolism due to EFV. |
| Protease inhibitor: darunavir/ritonavir | ↑ tenofovir | Monitor patients receiving ATRIPLA concomitantly with ritonavir-boosted darunavir for TDF-associated adverse reactions. Discontinue ATRIPLA in patients who develop TDF-associated adverse reactions. |
| lopinavir/ritonavir | ↓ lopinavir ↑ tenofovir | Do not use once daily administration of lopinavir/ritonavir. Dose increase of lopinavir/ritonavir is recommended for all patients when coadministered with EFV. Refer to the Full Prescribing Information for lopinavir/ritonavir for guidance on coadministration with EFV- or tenofovir-containing regimens, such as ATRIPLA. Patients should be monitored for tenofovir-associated adverse reactions. Discontinue ATRIPLA in patients who develop TDF-associated adverse reactions. |
| Protease inhibitor: ritonavir | ↑ ritonavir ↑ efavirenz | When ritonavir 500 mg every 12 hours was coadministered with EFV 600 mg once daily, the combination was associated with a higher frequency of adverse clinical experiences (e.g., dizziness, nausea, paresthesia) and laboratory abnormalities (elevated liver enzymes). Monitoring of liver enzymes is recommended when ATRIPLA is used in combination with ritonavir. |
| Protease inhibitor: saquinavir | ↓ saquinavir | Appropriate doses of the combination of EFV and saquinavir/ritonavir with respect to safety and efficacy have not been established. |
| CCR5 co-receptor antagonist: maraviroc | ↓ maraviroc | Refer to the full prescribing information for maraviroc for guidance on coadministration with ATRIPLA. |
| NRTI: didanosine | ↑ didanosine | Patients receiving ATRIPLA and didanosine should be monitored closely for didanosine-associated adverse reactions. Discontinue didanosine in patients who develop didanosine-associated adverse reactions. Higher didanosine concentrations could potentiate didanosine-associated adverse reactions, including pancreatitis, and neuropathy. Suppression of CD4+ cell counts has been observed in patients receiving TDF with didanosine 400 mg daily. In patients weighing greater than 60 kg, reduce the didanosine dose to 250 mg when it is coadministered with ATRIPLA. In patients weighing less than 60 kg, reduce the didanosine dose to 200 mg when it is coadministered with ATRIPLA. When coadministered, ATRIPLA and Videx EC may be taken under fasted conditions or with a light meal (less than 400 kcal, 20% fat). |
| NNRTI: Other NNRTIs | ↑ or ↓ efavirenz and/or NNRTI | Combining two NNRTIs has not been shown to be beneficial. ATRIPLA contains EFV and should not be coadministered with other NNRTIs. |
| Integrase strand transfer inhibitor: raltegravir | ↓ raltegravir | The clinical significance of this interaction has not been directly assessed. |
| Hepatitis C antiviral agents | ||
| boceprevir | ↓ boceprevir | Plasma trough concentrations of boceprevir were decreased when boceprevir was coadministered with EFV, which may result in loss of therapeutic effect. The combination should be avoided. |
| elbasvir/grazoprevir | ↓ elbasvir ↓ grazoprevir | Coadministration of ATRIPLA with elbasvir/grazoprevir is contraindicated [see Contraindications (4)] because it may lead to loss of virologic response to elbasvir/grazoprevir. |
| glecaprevir/pibrentasvir | ↓ glecaprevir ↓ pibrentasvir | Coadministration of ATRIPLA is not recommended because it may lead to reduced therapeutic effect of glecaprevir/pibrentasvir. |
| ledipasvir/sofosbuvir | ↑ tenofovir | Patients receiving ATRIPLA and HARVONI® (ledipasvir/sofosbuvir) concomitantly should be monitored for adverse reactions associated with TDF. |
| simeprevir | ↓ simeprevir ↔ efavirenz | Concomitant administration of simeprevir with EFV is not recommended because it may result in loss of therapeutic effect of simeprevir. |
| sofosbuvir/velpatasvir sofosbuvir/velpatasvir/voxilaprevir | ↑ tenofovir ↓ velpatasvir ↓ voxilaprevir | Coadministration of EFV-containing regimens and EPCLUSA® (sofosbuvir/velpatasvir) or VOSEVI® (sofosbuvir/velpatasvir/voxilaprevir) is not recommended. |
| Other agents | ||
| Anticoagulant: warfarin | ↑ or ↓ warfarin | Plasma concentrations and effects potentially increased or decreased by EFV. |
| Anticonvulsants: carbamazepine | ↓ carbamazepine ↓ efavirenz | There are insufficient data to make a dose recommendation for ATRIPLA. Alternative anticonvulsant treatment should be used. |
| phenytoin phenobarbital | ↓ anticonvulsant ↓ efavirenz | Potential for reduction in anticonvulsant and/or EFV plasma levels; periodic monitoring of anticonvulsant plasma levels should be conducted. |
| Antidepressants: bupropion | ↓ bupropion | The effect of EFV on bupropion exposure is thought to be due to the induction of bupropion metabolism. Increases in bupropion dosage should be guided by clinical response, but the maximum recommended dose of bupropion should not be exceeded. |
| sertraline | ↓ sertraline | Increases in sertraline dose should be guided by clinical response. |
| Antifungals: | ||
| itraconazole | ↓ itraconazole ↓ hydroxy-itraconazole | Since no dose recommendation for itraconazole can be made, alternative antifungal treatment should be considered. |
| ketoconazole | ↓ ketoconazole | Drug interaction trials with ATRIPLA and ketoconazole have not been conducted. Efavirenz has the potential to decrease plasma concentrations of ketoconazole. |
| posaconazole | ↓ posaconazole | Avoid concomitant use unless the benefit outweighs the risks. |
| voriconazole | ↓ voriconazole ↑ efavirenz | Coadministration of ATRIPLA with voriconazole is contraindicated [see Contraindications (4)] because it may lead to reduced therapeutic effect of voriconazole and increased risk of EFV-associated adverse reactions |
| Anti-infective: clarithromycin | ↓ clarithromycin ↑ 14-OH metabolite | Consider alternatives to macrolide antibiotics because of the risk of QT interval prolongation. |
| Antimycobacterial: rifabutin | ↓ rifabutin | Increase daily dose of rifabutin by 50%. Consider doubling the rifabutin dose in regimens where rifabutin is given 2 or 3 times a week. |
| rifampin | ↓ efavirenz | If ATRIPLA is coadministered with rifampin to patients weighing 50 kg or more, an additional 200 mg/day of EFV is recommended. |
| Antimalarials: artemether/lumefantrine | ↓ artemether ↓ dihydroartemisinin ↓ lumefantrine | Consider alternatives to artemether/lumefantrine because of the risk of QT interval prolongation [see Warnings and Precautions (5.4)]. |
| atovaquone/proguanil | ↓ atovaquone ↓ proguanil | Concomitant administration of atovaquone/proguanil with ATRIPLA is not recommended. |
| Calcium channel blockers: diltiazem | ↓ diltiazem ↓ desacetyl diltiazem ↓ N-monodes-methyl diltiazem | Diltiazem dose adjustments should be guided by clinical response (refer to the full prescribing information for diltiazem). No dose adjustment of ATRIPLA is necessary when administered with diltiazem. |
| Others e.g., felodipine nicardipine nifedipine verapamil | ↓ calcium channel blocker | No data are available on the potential interactions of EFV with other calcium channel blockers that are substrates of CYP3A. The potential exists for reduction in plasma concentrations of the calcium channel blocker. Dose adjustments should be guided by clinical response (refer to the full prescribing information for the calcium channel blocker). |
| HMG-CoA reductase inhibitors: atorvastatin pravastatin simvastatin | ↓ atorvastatin ↓ pravastatin ↓ simvastatin | Plasma concentrations of atorvastatin, pravastatin, and simvastatin decreased with EFV. Consult the Full Prescribing Information for the HMG-CoA reductase inhibitor for guidance on individualizing the dose. |
| Hormonal contraceptives: Oral: ethinyl estradiol/norgestimate | ↓ active metabolites of norgestimate | A reliable method of barrier contraception must be used in addition to hormonal contraceptives. Efavirenz had no effect on ethinyl estradiol concentrations, but progestin levels (norelgestromin and levonorgestrel) were markedly decreased. No effect of ethinyl estradiol/norgestimate on EFV plasma concentrations was observed. |
| Implant: etonogestrel | ↓ etonogestrel | A reliable method of barrier contraception must be used in addition to hormonal contraceptives. Decreased exposure of etonogestrel may be expected. There have been postmarketing reports of contraceptive failure with etonogestrel in EFV-exposed patients. |
| Immunosuppressants: cyclosporine, tacrolimus, sirolimus, and others metabolized by CYP3A | ↓ immuno-suppressant | Decreased exposure of the immunosuppressant may be expected due to CYP3A induction by EFV. These immunosuppressants are not anticipated to affect exposure of EFV. Dose adjustments of the immunosuppressant may be required. Close monitoring of immunosuppressant concentrations for at least 2 weeks (until stable concentrations are reached) is recommended when starting or stopping treatment with ATRIPLA. |
| Narcotic analgesic: methadone | ↓ methadone | Coadministration of EFV in HIV-1 infected individuals with a history of injection drug use resulted in signs of opiate withdrawal. Methadone dose was increased by a mean of 22% to alleviate withdrawal symptoms. Patients should be monitored for signs of withdrawal and their methadone dose increased as required to alleviate withdrawal symptoms. |
7.4 Efavirenz Assay Interference
Cannabinoid Test Interaction: Efavirenz does not bind to cannabinoid receptors. False-positive urine cannabinoid test results have been reported with some screening assays in uninfected and HIV-infected subjects receiving EFV. Confirmation of positive screening tests for cannabinoids by a more specific method is recommended.
8. Use In Specific Populations
8.1 Pregnancy
Antiretroviral Pregnancy Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in adults and adolescents exposed to ATRIPLA during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at (800) 258-4263.
Risk Summary
There are retrospective case reports of neural tube defects in infants whose mothers were exposed to EFV-containing regimens in the first trimester of pregnancy. Prospective pregnancy data from the APR are not sufficient to adequately assess this risk. Although a causal relationship has not been established between exposure to EFV in the first trimester and neural tube defects, similar malformations have been observed in studies conducted in monkeys at doses similar to the human dose (see Data). In addition, fetal and embryonic toxicities occurred in rats at a dose 10 times less than the human exposure at the recommended clinical human dose (RHD) of EFV. Because of the potential risk of neural tube defects, EFV is not recommended for use in the first trimester of pregnancy. Avoid pregnancy while receiving ATRIPLA and for 12 weeks after discontinuation. Advise pregnant patients of the potential risk to a fetus.
Available data from the APR show no increase in the overall risk of major birth defects for EFV, FTC, or TDF compared with the background rate for major birth defects of 2.7% in a U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) (see Data).
The rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in clinically recognized pregnancies in the U.S. general population is 15–20%. The background risk of major birth defects and miscarriage for the indicated population is unknown. The APR uses the MACDP as the U.S. reference population for birth defects in the general population. The MACDP evaluates mothers and infants from a limited geographic area and does not include outcomes for births that occurred at less than 20 weeks' gestation.
In animal reproduction studies, no adverse developmental effects were observed when FTC and TDF were administered separately at doses/exposures ≥60 (FTC), ≥14 (TDF) and 2.7 (tenofovir) times those at the RHD of ATRIPLA (see Data).
Data
Human Data
Efavirenz: There are retrospective postmarketing reports of findings consistent with neural tube defects, including meningomyelocele, all in infants of mothers exposed to EFV-containing regimens in the first trimester.
Based on prospective reports to the APR of 1,217 exposures to EFV-containing regimens during pregnancy resulting in live births (including over 1,023 live births exposed in the first trimester and 194 exposed in the second/third trimester), there was no increase in overall birth defects with EFV compared with the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. The prevalence of birth defects in live births was 2.3% (95% CI: 1.5% to 3.5%) with first trimester exposure to EFV-containing regimens, and 1.5% (95% CI: 0.3% to 4.5%) with the second/third trimester exposure to EFV-containing regimens. One of these prospectively reported defects with first-trimester exposure was a neural tube defect. A single case of anophthalmia with first-trimester exposure to EFV has also been prospectively reported. This case also included severe oblique facial clefts and amniotic banding, which have a known association with anophthalmia.
Emtricitabine: Based on prospective reports from the APR of 4,005 exposures to FTC-containing regimens during pregnancy resulting in live births (including 2,785 exposed in the first trimester and 1,220 exposed in the second/third trimester), there was no increase in overall major birth defects with FTC compared with the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. The prevalence of birth defects in live births was 2.4% (95% CI: 1.9% to 3.1%) with first trimester exposure to FTC-containing regimens and 2.3% (95% CI: 1.5% to 3.3%) with the second/third trimester exposure to FTC-containing regimens.
Tenofovir DF: Based on prospective reports from the APR of 5,105 exposures to TDF-containing regimens during pregnancy resulting in live births (including 3,535 exposed in the first trimester and 1,570 exposed in the second/third trimester), there was no increase in overall major birth defects with TDF compared with the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. The prevalence of birth defects in live births was 2.3% (95% CI: 1.8% to 2.9%) with first trimester exposure to TDF-containing regimens, and 2.2% (95% CI: 1.6% to 3.1%) with the second/third trimester exposure to TDF-containing regimens.
Animal Data
Efavirenz: Effects of EFV on embryo-fetal development have been studied in three nonclinical species (cynomolgus monkeys, rats, and rabbits). In monkeys, EFV 60 mg/kg/day was administered to pregnant females throughout pregnancy (gestation Days 20 through 150). The maternal systemic drug exposures (AUC) were 1.3 times the exposures at the RHD, with fetal umbilical venous drug concentrations approximately 0.7 times the maternal values. Three fetuses of 20 fetuses/infants had one or more malformations; there were no malformed fetuses or infants from placebo-treated mothers. The malformations that occurred in these three monkey fetuses included anencephaly and unilateral anophthalmia in one fetus, microphthalmia in a second, and cleft palate in the third. There was no NOAEL (no observable adverse effect level) established for this study because only one dosage was evaluated. In rats, EFV was administered either during organogenesis (gestation Days 7 to 18) or from gestation Day 7 through lactation Day 21 at 50, 100, or 200 mg/kg/day. Administration of 200 mg/kg/day in rats was associated with an increase in the incidence of early resorptions, and doses 100 mg/kg/day and greater were associated with early neonatal mortality. The AUC at the NOAEL (50 mg/kg/day) in this rat study was 0.1 times that in humans at the RHD. Drug concentrations in the milk on lactation Day 10 were approximately 8 times higher than those in maternal plasma. In pregnant rabbits, EFV was neither embryo lethal nor teratogenic when administered at doses of 25, 50, and 75 mg/kg/day over the period of organogenesis (gestation Days 6 through 18). The AUC at the NOAEL (75 mg/kg/day) in rabbits was 0.4 times that in humans at the RHD.
Emtricitabine: FTC was administered orally to pregnant mice (at 0, 250, 500, or 1,000 mg/kg/day), and rabbits (at 0, 100, 300, or 1,000 mg/kg/day) through organogenesis (on gestation days 6 through 15, and 7 through 19, respectively). No significant toxicological effects were observed in embryo-fetal toxicity studies performed with FTC in mice at exposures (AUC) approximately 60 times higher and in rabbits at approximately 120 times higher than human exposures at the RHD. In a pre/postnatal development study in mice, FTC was administered orally at doses up to 1000 mg/kg/day; no significant adverse effects directly related to drug were observed in the offspring exposed daily from before birth (in utero) through sexual maturity at daily exposures (AUC) of approximately 60 times higher than human exposures at the RHD.
Tenofovir DF: TDF was administered orally to pregnant rats (at 0, 50, 150, or 450 mg/kg/day) and rabbits (at 0, 30, 100, or 300 mg/kg/day) through organogenesis (on gestation days 7 through 17, and 6 through 18, respectively). No significant toxicological effects were observed in embryo-fetal toxicity studies performed with TDF in rats at doses up to 14 times the RHD based on body surface area comparisons and in rabbits at doses up to 19 times the RHD based on body surface area comparisons. In a pre/postnatal development study in rats, TDF was administered orally through lactation at doses up to 600 mg/kg/day; no adverse effects were observed in the offspring at tenofovir exposures of approximately 2.7 times higher than human exposures at the RHD.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-1 infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV-1.
Based on limited published data, EFV, FTC, and tenofovir have been shown to be present in human breast milk.
It is not known if the components of ATRIPLA affect milk production or have effects on the breastfed child. Because of the potential for: (1) HIV transmission (in HIV-negative infants); (2) developing viral resistance (in HIV-positive infants); and (3) adverse reactions in a breastfed infant similar to those seen in adults, instruct mothers not to breastfeed if they are receiving ATRIPLA.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Perform pregnancy testing in adults and adolescents of childbearing potential before initiation of ATRIPLA because of potential risk of neural tube defects [see Use in Specific Populations (8.1)].
Contraception
Advise adults and adolescents of childbearing potential to use effective contraception during treatment with ATRIPLA and for 12 weeks after discontinuing ATRIPLA due to the long half-life of EFV, a component of ATRIPLA. Hormonal methods that contain progesterone may have decreased effectiveness Always use barrier contraception in combination with other methods of contraception [see Drug Interactions (7.1, 7.3)].
8.4 Pediatric Use
The effectiveness and safety of ATRIPLA as a complete regimen for the treatment of HIV-1 infection was established in pediatric patients with body weight greater than or equal to 40 kg [see Dosage and Administration (2.2)]. Use of ATRIPLA in this age group is supported by adequate and well-controlled studies of ATRIPLA in adults with HIV-1 infection and data from pediatric studies of the individual components of ATRIPLA (EFV, FTC, and TDF).
ATRIPLA should only be administered to pediatric patients with a body weight greater than or equal to 40 kg. Because ATRIPLA is a fixed-dose combination tablet, the dose of ATRIPLA cannot be adjusted for patients of lower weight [see Warnings and Precautions (5.2, 5.9), Adverse Reactions (6.1), and Clinical Pharmacology (12.3)].
8.5 Geriatric Use
Clinical trials of EFV, FTC, or TDF did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for elderly patients should be cautious, keeping in mind the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Because ATRIPLA is a fixed-dose combination, and cannot be dose adjusted, it is not recommended in patients with moderate or severe renal impairment (estimated creatinine clearance below 50 mL/min) [see Dosage and Administration (2.3), Warnings and Precautions (5.7)].
8.7 Hepatic Impairment
ATRIPLA is not recommended for patients with moderate or severe hepatic impairment because there are insufficient data to determine an appropriate dose. Patients with mild hepatic impairment may be treated with ATRIPLA at the approved dose. Because of the extensive cytochrome P450-mediated metabolism of EFV and limited clinical experience in patients with hepatic impairment, caution should be exercised in administering ATRIPLA to these patients [see Dosage and Administration (2.4), Warnings and Precautions (5.3), and Clinical Pharmacology (12.3)].
10. Overdosage
If overdose occurs, the patient should be monitored for evidence of toxicity, and standard supportive treatment applied as necessary. Administration of activated charcoal may be used to aid removal of unabsorbed EFV. Hemodialysis can remove both FTC and TDF (refer to detailed information below) but is unlikely to significantly remove EFV from the blood.
Efavirenz: Some patients accidentally taking 600 mg twice daily have reported increased nervous system symptoms. One patient experienced involuntary muscle contractions.
11. Atripla Description
ATRIPLA is a fixed-dose combination tablet containing EFV, FTC, and TDF. EFV is a non-nucleoside reverse transcriptase inhibitor (NNRTI). FTC is a synthetic nucleoside analog of cytidine. TDF, which is converted in vivo to tenofovir, is an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5′-monophosphate.
ATRIPLA tablets are for oral administration. Each tablet contains 600 mg of EFV, 200 mg of FTC, and 300 mg of TDF (equivalent to 245 mg of tenofovir disoproxil) as active ingredients. The tablets include the following inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. The tablets are film coated with a coating material containing black iron oxide, polyethylene glycol, polyvinyl alcohol, red iron oxide, talc, and titanium dioxide.
Efavirenz: EFV is chemically described as (S)-6-chloro-4-(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one. Its molecular formula is C14H9ClF3NO2 and its structural formula is:
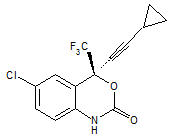
Efavirenz is a white to slightly pink crystalline powder with a molecular mass of 315.68. It is practically insoluble in water (less than 10 µg/mL).
Emtricitabine: The chemical name of FTC is 5-fluoro-1-(2R,5S)-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. FTC is the (-) enantiomer of a thio analog of cytidine, which differs from other cytidine analogs in that it has a fluorine in the 5-position.
It has a molecular formula of C8H10FN3O3S and a molecular weight of 247.24. It has the following structural formula:
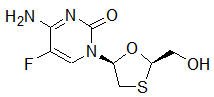
Emtricitabine is a white to off-white crystalline powder with a solubility of approximately 112 mg/mL in water at 25 °C.
Tenofovir DF: TDF is a fumaric acid salt of the bis-isopropoxycarbonyloxymethyl ester derivative of tenofovir. The chemical name of TDF is 9-[(R)-2[[bis[[(isopropoxycarbonyl)oxy]- methoxy]phosphinyl]methoxy]propyl]adenine fumarate (1:1). It has a molecular formula of C19H30N5O10P ∙ C4H4O4 and a molecular weight of 635.52. It has the following structural formula:
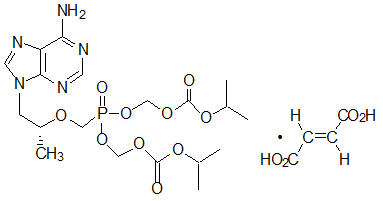
TDF is a white to off-white crystalline powder with a solubility of 13.4 mg/mL in water at 25 °C.
12. Atripla - Clinical Pharmacology
12.1 Mechanism of Action
ATRIPLA is a fixed-dose combination of antiviral drugs EFV, FTC, and TDF [see Microbiology (12.4)].
12.2 Pharmacodynamics
Cardiac Electrophysiology
Efavirenz: The effect of EFV on the QTc interval was evaluated in an open-label, positive and placebo-controlled, fixed single sequence 3-period, 3-treatment crossover QT study in 58 healthy subjects enriched for CYP2B6 polymorphisms. The mean Cmax of EFV in subjects with CYP2B6 *6/*6 genotype following the administration of 600 mg daily dose for 14 days was 2.25-fold the mean Cmax observed in subjects with CYP2B6 *1/*1 genotype. A positive relationship between EFV concentration and QTc prolongation was observed. Based on the concentration-QTc relationship, the mean QTc prolongation and its upper bound 90% confidence interval are 8.7 msec and 11.3 msec in subjects with CYP2B6*6/*6 genotype following the administration of 600 mg daily dose for 14 days [see Warnings and Precautions (5.4)].
12.3 Pharmacokinetics
ATRIPLA: One ATRIPLA tablet is bioequivalent to one Sustiva tablet (600 mg) plus one EMTRIVA® capsule (200 mg) plus one VIREAD® tablet (300 mg) following single-dose administration to fasting healthy subjects (N=45).
Efavirenz: In HIV-1 infected subjects time-to-peak plasma concentrations were approximately 3–5 hours and steady-state plasma concentrations were reached in 6–10 days. In 35 HIV-1 infected subjects receiving EFV 600 mg once daily, steady-state Cmax was 12.9 ± 3.7 µM (mean ± SD), Cmin was 5.6 ± 3.2 μM, and AUC was 184 ± 73 µM∙hr. EFV is highly bound (approximately 99.5–99.75%) to human plasma proteins, predominantly albumin. Following administration of 14C-labeled EFV, 14–34% of the dose was recovered in the urine (mostly as metabolites) and 16–61% was recovered in feces (mostly as parent drug). In vitro studies suggest CYP3A and CYP2B6 are the major isozymes responsible for EFV metabolism. EFV has been shown to induce CYP enzymes, resulting in induction of its own metabolism. EFV has a terminal half-life of 52–76 hours after single doses and 40–55 hours after multiple doses.
Emtricitabine: Following oral administration, FTC is rapidly absorbed, with peak plasma concentrations occurring at 1–2 hours postdose. Following multiple dose oral administration of FTC to 20 HIV-1 infected subjects, the steady-state plasma FTC Cmax was 1.8 ± 0.7 µg/mL (mean ± SD) and the AUC over a 24-hour dosing interval was 10.0 ± 3.1 µg∙hr/mL. The mean steady-state plasma trough concentration at 24 hours postdose was 0.09 µg/mL. The mean absolute bioavailability of FTC was 93%. Less than 4% of FTC binds to human plasma proteins in vitro, and the binding is independent of concentration over the range of 0.02–200 µg/mL. Following administration of radiolabelled FTC, approximately 86% is recovered in the urine and 13% is recovered as metabolites. The metabolites of FTC include 3′-sulfoxide diastereomers and their glucuronic acid conjugate. FTC is eliminated by a combination of glomerular filtration and active tubular secretion with a renal clearance in adults with normal renal function of 213 ± 89 mL/min (mean ± SD). Following a single oral dose, the plasma FTC half-life is approximately 10 hours.
Tenofovir DF: Following oral administration of a single 300 mg dose of TDF to HIV-1 infected subjects in the fasted state, maximum serum concentrations (Cmax) were achieved in 1.0 ± 0.4 hrs (mean ± SD) and Cmax and AUC values were 296 ± 90 ng/mL and 2287 ± 685 ng∙hr/mL, respectively. The oral bioavailability of tenofovir from TDF in fasted subjects is approximately 25%. Less than 0.7% of tenofovir binds to human plasma proteins in vitro, and the binding is independent of concentration over the range of 0.01–25 μg/mL. Approximately 70–80% of the intravenous dose of tenofovir is recovered as unchanged drug in the urine. Tenofovir is eliminated by a combination of glomerular filtration and active tubular secretion, with a renal clearance in adults with normal renal function of 243 ± 33 mL/min (mean ± SD). Following a single oral dose, the terminal elimination half-life of tenofovir is approximately 17 hours.
Effects of Food on Oral Absorption
ATRIPLA has not been evaluated in the presence of food. Administration of EFV tablets with a high-fat meal increased the mean AUC and Cmax of EFV by 28% and 79%, respectively, compared to administration in the fasted state. Compared to fasted administration, dosing of TDF and FTC in combination with either a high-fat meal or a light meal increased the mean AUC and Cmax of tenofovir by 35% and 15%, respectively, without affecting FTC exposures [see Dosage and Administration (2.2) and Patient Counseling Information (17)].
Specific Populations
Race
Efavirenz: The pharmacokinetics of EFV in HIV-1 infected subjects appear to be similar among the racial groups studied.
Pediatric Patients
Efavirenz: In an open-label trial in NRTI-experienced pediatric subjects (mean age 8 years, range 3−16 years), the pharmacokinetics of EFV in pediatric subjects were similar to the pharmacokinetics in adults who received a 600 mg daily dose of EFV. Based on mean steady-state predicted population pharmacokinetic modeling in pediatric subjects weighing >40 kg receiving the 600 mg dose of EFV, Cmax was 6.57 μg/mL, Cmin was 2.82 μg/mL, and AUC(0–24) was 254.78 μM∙hr.
Emtricitabine: The pharmacokinetics of FTC at steady state were determined in 27 HIV-1 infected pediatric subjects 13 to 17 years of age receiving a daily dose of 6 mg/kg up to a maximum dose of 240 mg oral solution or a 200-mg capsule; 26 of 27 subjects in this age group received the 200-mg capsule. Mean ± SD Cmax and AUC were 2.7 ± 0.9 μg/mL and 12.6 ± 5.4 μg∙hr/mL, respectively. Exposures achieved in pediatric subjects 12 to less than 18 years of age were similar to those achieved in adults receiving a once daily dose of 200 mg.
Tenofovir DF: Steady-state pharmacokinetics of tenofovir were evaluated in 8 HIV-1 infected pediatric subjects (12 to less than 18 years). Mean ± SD Cmax and AUCtau are 0.38 ± 0.13 μg/mL and 3.39 ± 1.22 μg∙hr/mL, respectively. Tenofovir exposure achieved in these pediatric subjects receiving oral daily doses of TDF 300 mg was similar to exposures achieved in adults receiving once-daily doses of TDF 300 mg.
Geriatric Patients
Pharmacokinetics of EFV, FTC, and tenofovir have not been fully evaluated in the elderly (65 years of age and older) [see Use in Specific Populations (8.5)].
Patients with Impaired Renal Function
Efavirenz: The pharmacokinetics of EFV have not been studied in subjects with renal insufficiency; however, less than 1% of EFV is excreted unchanged in the urine, so the impact of renal impairment on EFV elimination should be minimal.
Emtricitabine and Tenofovir DF: The pharmacokinetics of FTC and TDF are altered in subjects with renal impairment. In subjects with creatinine clearance below 50 mL/min, Cmax and AUC0–∞ of FTC and tenofovir were increased [see Warnings and Precautions (5.7)].
Patients with Hepatic Impairment
Efavirenz: A multiple-dose trial showed no significant effect on EFV pharmacokinetics in subjects with mild hepatic impairment (Child-Pugh Class A) compared with controls. There were insufficient data to determine whether moderate or severe hepatic impairment (Child-Pugh Class B or C) affects EFV pharmacokinetics [see Warnings and Precautions (5.3) and Use in Specific Populations (8.7)].
Emtricitabine: The pharmacokinetics of FTC have not been studied in subjects with hepatic impairment; however, FTC is not significantly metabolized by liver enzymes, so the impact of liver impairment should be limited.
Tenofovir DF: The pharmacokinetics of tenofovir following a 300 mg dose of TDF have been studied in non-HIV infected subjects with moderate to severe hepatic impairment. There were no substantial alterations in tenofovir pharmacokinetics in subjects with hepatic impairment compared with unimpaired subjects.
Assessment of Drug Interactions
The drug interaction trials described were conducted with either ATRIPLA or the components of ATRIPLA (EFV, FTC, or TDF) as individual agents.
Efavirenz: The steady-state pharmacokinetics of EFV and tenofovir were unaffected when EFV and TDF were administered together versus each agent dosed alone. Specific drug interaction trials have not been performed with EFV and NRTIs other than tenofovir, lamivudine, and zidovudine. Clinically significant interactions would not be expected based on NRTIs elimination pathways.
Efavirenz has been shown in vivo to cause hepatic enzyme induction, thus increasing the biotransformation of some drugs metabolized by CYP3A and CYP2B6. In vitro studies have shown that EFV inhibited CYP isozymes 2C9 and 2C19 with Ki values (8.5–17 µM) in the range of observed EFV plasma concentrations. In in vitro studies, EFV did not inhibit CYP2E1 and inhibited CYP2D6 and CYP1A2 (Ki values 82–160 µM) only at concentrations well above those achieved clinically. Coadministration of EFV with drugs primarily metabolized by CYP2C9, CYP2C19, CYP3A or CYP2B6 isozymes may result in altered plasma concentrations of the coadministered drug. Drugs which induce CYP3A and CYP2B6 activity would be expected to increase the clearance of EFV resulting in lowered plasma concentrations.
Drug interaction trials were performed with EFV and other drugs likely to be coadministered or drugs commonly used as probes for pharmacokinetic interaction. There was no clinically significant interaction observed between EFV and zidovudine, lamivudine, azithromycin, fluconazole, lorazepam, cetirizine, or paroxetine. Single doses of famotidine or an aluminum and magnesium antacid with simethicone had no effects on EFV exposures. The effects of coadministration of EFV on Cmax, AUC, and Cmin are summarized in Table 4 (effect of other drugs on EFV) and Table 5 (effect of EFV on other drugs) see [Drug Interactions (7)].
| Mean % Change of EFV Pharmacokinetic Parameters* (90% CI) | ||||||
|---|---|---|---|---|---|---|
| Coadministered Drug | Dose of Coadministered Drug (mg) | EFV Dose (mg) | N | Cmax | AUC | Cmin |
| NA = not available | ||||||
| Lopinavir/ritonavir | 400/100 mg q12h × 9 days | 600 mg qd × 9 days | 11, 12† | ↔ | ↓ 16 (↓ 38 to ↑ 15) | ↓ 16 (↓ 42 to ↑ 20) |
| Nelfinavir | 750 mg q8h × 7 days | 600 mg qd × 7 days | 10 | ↓ 12 (↓ 32 to ↑13)‡ | ↓ 12 (↓ 35 to ↑ 18)‡ | ↓ 21 (↓ 53 to ↑ 33) |
| Ritonavir | 500 mg q12h × 8 days | 600 mg qd × 10 days | 9 | ↑ 14 (↑ 4 to ↑ 26) | ↑ 21 (↑ 10 to ↑ 34) | ↑ 25 (↑ 7 to ↑ 46)‡ |
| Boceprevir | 800 mg tid × 6 days | 600 mg qd × 16 days | NA | ↑11 (↑ 2 to ↑ 20) | ↑ 20 (↑ 15 to ↑ 26) | NA |
| Rifabutin | 300 mg qd × 14 days | 600 mg qd × 14 days | 11 | ↔ | ↔ | ↓ 12 (↓ 24 to ↑ 1) |
| Rifampin | 600 mg × 7 days | 600 mg qd × 7 days | 12 | ↓ 20 (↓ 11 to ↓ 28) | ↓ 26 (↓ 15 to ↓ 36) | ↓ 32 (↓ 15 to ↓ 46) |
| Artemether/lumefantrine | Artemether 20 mg/lumefantrine 120 mg tablets (6 4-tablet doses over 3 days) | 600 mg qd × 26 days | 12 | ↔ | ↓17 | NA |
| Simvastatin | 40 mg qd × 4 days | 600 mg qd × 15 days | 14 | ↓ 12 (↓ 28 to ↑ 8) | ↔ | ↓ 12 (↓ 25 to ↑ 3) |
| Carbamazepine | 200 mg qd × 3 days, 200 mg bid × 3 days, then 400 mg qd × 15 days | 600 mg qd × 35 days | 14 | ↓ 21 (↓ 15 to ↓ 26) | ↓ 36 (↓ 32 to ↓ 40) | ↓ 47 (↓ 41 to ↓ 53) |
| Diltiazem | 240 mg × 14 days | 600 mg qd × 28 days | 12 | ↑ 16 (↑ 6 to ↑ 26) | ↑ 11 (↑ 5 to ↑ 18) | ↑ 13 (↑ 1 to ↑ 26) |
| Voriconazole | 400 mg po q12h × 1 day then 200 mg po q12h × 8 days | 400 mg qd × 9 days | NA | ↑ 38§ | ↑ 44§ | NA |
| 300 mg po q12h days 2–7 | 300 mg qd × 7 days | NA | ↓ 14¶
(↓ 7 to ↓ 21) | ↔¶ | NA | |
| 400 mg po q12h days 2–7 | 300 mg qd × 7 days | NA | ↔¶ | ↑ 17¶
(↑ 6 to ↑ 29) | NA | |
No effect on the pharmacokinetic parameters of EFV was observed with the following coadministered drugs: indinavir, saquinavir soft gelatin capsule, simeprevir, ledipasvir/sofosbuvir, sofosbuvir, clarithromycin, itraconazole, atorvastatin, pravastatin, or sertraline.
| Mean % Change of Coadministered Drug Pharmacokinetic Parameters* (90% CI) | ||||||
|---|---|---|---|---|---|---|
| Coadministered Drug | Dose of Coadministered Drug (mg) | EFV Dose (mg) | N | Cmax | AUC | Cmin |
| NA = not available | ||||||
|
||||||
| Atazanavir | 400 mg qd with a light meal d 1–20 | 600 mg qd with a light meal d 7–20 | 27 | ↓ 59 (↓ 49 to ↓ 67) | ↓ 74 (↓ 68 to ↓78) | ↓ 93 (↓ 90 to ↓ 95) |
| 400 mg qd d 1–6, then 300 mg qd d 7–20 with ritonavir 100 mg qd and a light meal | 600 mg qd 2 h after atazanavir and ritonavir d 7–20 | 13 | ↑ 14†
(↓ 17 to ↑ 58) | ↑ 39†
(↑ 2 to ↑ 88) | ↑ 48†
(↑ 24 to ↑ 76) |
|
| 300 mg qd/ritonavir 100 mg qd d 1–10 (pm), then 400 mg qd/ritonavir 100 mg qd d 11–24 (pm) (simultaneous with EFV) | 600 mg qd with a light snack d 11–24 (pm) | 14 | ↑ 17 (↑ 8 to ↑ 27) | ↔ | ↓ 42 (↓ 31 to ↓ 51) |
|
| Indinavir | 1000 mg q8h × 10 days | 600 mg qd × 10 days | 20 | |||
| After morning dose | ↔‡ | ↓ 33‡
(↓ 26 to ↓ 39) | ↓ 39‡
(↓ 24 to ↓ 51) |
|||
| After afternoon dose | ↔‡ | ↓ 37‡
(↓ 26 to ↓ 46) | ↓ 52‡
(↓ 47 to ↓ 57) |
|||
| After evening dose | ↓ 29‡
(↓ 11 to ↓ 43) | ↓ 46‡
(↓ 37 to ↓ 54) | ↓ 57‡
(↓ 50 to ↓ 63) |
|||
| Lopinavir/ritonavir | 400/100 mg q12h × 9 days | 600 mg qd × 9 days | 11, 7§ | ↔¶ | ↓ 19¶
(↓ 36 to ↑ 3) | ↓ 39¶
(↓ 3 to ↓ 62) |
| Nelfinavir | 750 mg q8h × 7 days | 600 mg qd × 7 days | 10 | ↑ 21 (↑ 10 to ↑ 33) | ↑ 20 (↑ 8 to ↑ 34) | ↔ |
| Metabolite AG-1402 | ↓ 40 (↓ 30 to ↓ 48) | ↓ 37 (↓ 25 to ↓ 48) | ↓ 43 (↓ 21 to ↓ 59) |
|||
| Ritonavir | 500 mg q12h × 8 days | 600 mg qd × 10 days | 11 | |||
| After AM dose | ↑ 24 (↑ 12 to ↑ 38) | ↑ 18 (↑ 6 to ↑ 33) | ↑ 42 (↑ 9 to ↑ 86)# |
|||
| After PM dose | ↔ | ↔ | ↑ 24 (↑ 3 to ↑ 50)# |
|||
| Saquinavir SGCÞ | 1200 mg q8h × 10 days | 600 mg qd × 10 days | 12 | ↓ 50 (↓ 28 to ↓ 66) | ↓ 62 (↓ 45 to ↓ 74) | ↓ 56 (↓ 16 to ↓ 77)# |
| Maraviroc | 100 mg bid | 600 mg qd | 12 | ↓ 51 (↓ 37 to ↓ 62) | ↓ 45 (↓ 38 to ↓ 51) | ↓ 45 (↓ 28 to ↓ 57) |
| Raltegravir | 400 mg single dose | 600 mg qd | 9 | ↓ 36 (↓ 2 to ↓ 59) | ↓ 36 (↓ 20 to ↓ 48) | ↓ 21 (↓ 51 to ↑ 28) |
| Boceprevir | 800 mg tid × 6 days | 600 mg qd × 16 days | NA | ↓ 8 (↓ 22 to ↑ 8) | ↓ 19 (↓ 11 to ↓ 25) | ↓ 44 (↓ 26 to ↓ 58) |
| Simeprevir | 150 mg qd × 14 days | 600 mg qd × 14 days | 23 | ↓ 51 (↓ 46 to ↓ 56) | ↓ 71 (↓ 67 to ↓ 74) | ↓ 91 (↓ 88 to ↓ 92) |
| Ledipasvir/sofosbuvirß | 90/400 mg qd × 14 days | 600 mg qd × 14 days | 15 | |||
| Ledipasvir | ↓ 34 (↓ 25 to ↓ 41) | ↓ 34 (↓ 25 to ↓ 41) | ↓ 34 (↓ 24 to ↓ 43) |
|||
| Sofosbuvir | ↔ | ↔ | NA | |||
| GS-331007à | ↔ | ↔ | ↔ | |||
| Sofosbuvirè | 400 mg qd single dose | 600 mg qd × 14 days | 16 | ↓ 19 (↓ 40 to ↑ 10) | ↔ | NA |
| GS-331007à | ↓ 23 (↓ 16 to ↓ 30) | ↓ 16 (↓ 24 to ↓ 8) | NA | |||
| Sofosbuvir/velpatasvirð | 400/100 mg qd × 14 days | 600 mg qd × 14 days | 14 | |||
| Sofosbuvir | ↑ 38 (↑ 14 to ↑ 67) | ↔ | NA | |||
| GS-331007à | ↓ 14 (↓ 20 to ↓ 7) | ↔ | ↔ | |||
| Velpatasvir | ↓ 47 (↓ 57 to ↓ 36) | ↓ 53 (↓ 61 to ↓ 43) | ↓ 57 (↓ 64 to ↓ 48) |
|||
| Clarithromycin | 500 mg q12h × 7 days | 400 mg qd × 7 days | 11 | ↓ 26 (↓ 15 to ↓ 35) | ↓ 39 (↓ 30 to ↓ 46) | ↓ 53 (↓ 42 to ↓ 63) |
| 14-OH metabolite | ↑ 49 (↑ 32 to ↑ 69) | ↑ 34 (↑ 18 to ↑ 53) | ↑ 26 (↑ 9 to ↑ 45) |
|||
| Itraconazole | 200 mg q12h × 28 days | 600 mg qd × 14 days | 18 | ↓ 37 (↓ 20 to ↓ 51) | ↓ 39 (↓ 21 to ↓ 53) | ↓ 44 (↓ 27 to ↓ 58) |
| Hydroxy-itraconazole | ↓ 35 (↓ 12 to ↓ 52) | ↓ 37 (↓ 14 to ↓ 55) | ↓ 43 (↓ 18 to ↓ 60) |
|||
| Posaconazole | 400 mg (oral suspension) bid × 10 and 20 days | 400 mg qd × 10 and 20 days | 11 | ↓ 45 (↓ 34 to ↓ 53) | ↓ 50 (↓ 40 to ↓ 57) | NA |
| Rifabutin | 300 mg qd × 14 days | 600 mg qd × 14 days | 9 | ↓ 32 (↓ 15 to ↓ 46) | ↓ 38 (↓ 28 to ↓ 47) | ↓ 45 (↓ 31 to ↓ 56) |
| Artemether/lumefantrine | Artemether 20 mg/lumefantrine 120 mg tablets (6 4-tablet doses over 3 days) | 600 mg qd × 26 days | 12 | |||
| Artemether | ↓ 21 | ↓ 51 | NA | |||
| dihydroartemisinin | ↓ 38 | ↓ 46 | NA | |||
| lumefantrine | ↔ | ↓ 21 | NA | |||
| Atorvastatin | 10 mg qd × 4 days | 600 mg qd × 15 days | 14 | ↓ 14 (↓ 1 to ↓ 26) | ↓ 43 (↓ 34 to ↓ 50) | ↓ 69 (↓ 49 to ↓ 81) |
| Total active (including metabolites) | ↓ 15 (↓ 2 to ↓ 26) | ↓ 32 (↓ 21 to ↓ 41) | ↓ 48 (↓ 23 to ↓ 64) |
|||
| Pravastatin | 40 mg qd × 4 days | 600 mg qd × 15 days | 13 | ↓ 32 (↓ 59 to ↑ 12) | ↓ 44 (↓ 26 to ↓ 57) | ↓ 19 (↓ 0 to ↓ 35) |
| Simvastatin | 40 mg qd × 4 days | 600 mg qd × 15 days | 14 | ↓ 72 (↓ 63 to ↓ 79) | ↓ 68 (↓ 62 to ↓ 73) | ↓ 45 (↓ 20 to ↓ 62) |
| Total active (including metabolites) | ↓ 68 (↓ 55 to ↓ 78) | ↓ 60 (↓ 52 to ↓ 68) | NAø | |||
| Carbamazepine | 200 mg qd × 3 days, 200 mg bid × 3 days, then 400 mg qd × 29 days | 600 mg qd × 14 days | 12 | ↓ 20 (↓ 15 to ↓ 24) | ↓ 27 (↓ 20 to ↓ 33) | ↓ 35 (↓ 24 to ↓ 44) |
| Epoxide metabolite | ↔ | ↔ | ↓ 13 (↓ 30 to ↑ 7) |
|||
| Diltiazem | 240 mg × 21 days | 600 mg qd × 14 days | 13 | ↓ 60 (↓ 50 to ↓ 68) | ↓ 69 (↓ 55 to ↓ 79) | ↓ 63 (↓ 44 to ↓ 75) |
| Desacetyl diltiazem | ↓ 64 (↓ 57 to ↓ 69) | ↓ 75 (↓ 59 to ↓ 84) | ↓ 62 (↓ 44 to ↓ 75) |
|||
| N-monodesmethyl diltiazem | ↓ 28 (↓ 7 to ↓ 44) | ↓ 37 (↓ 17 to ↓ 52) | ↓ 37 (↓ 17 to ↓ 52) |
|||
| Ethinyl estradiol/ norgestimate | 0.035 mg/0.25 mg × 14 days | 600 mg qd × 14 days | ||||
| Ethinyl estradiol | 21 | ↔ | ↔ | ↔ | ||
| Norelgestromin | 21 | ↓ 46 (↓ 39 to ↓ 52) | ↓ 64 (↓ 62 to ↓ 67) | ↓ 82 (↓ 79 to ↓ 85) |
||
| Levonorgestrel | 6 | ↓ 80 (↓ 77 to ↓ 83) | ↓ 83 (↓ 79 to ↓ 87) | ↓ 86 (↓ 80 to ↓ 90) |
||
| Methadone | Stable maintenance 35–100 mg daily | 600 mg qd × 14–21 days | 11 | ↓ 45 (↓ 25 to ↓ 59) | ↓ 52 (↓ 33 to ↓ 66) | NA |
| Bupropion | 150 mg single dose (sustained-release) | 600 mg qd × 14 days | 13 | ↓ 34 (↓ 21 to ↓ 47) | ↓ 55 (↓ 48 to ↓ 62) | NA |
| Hydroxybupropion | ↑ 50 (↑ 20 to ↑ 80) | ↔ | NA | |||
| Sertraline | 50 mg qd × 14 days | 600 mg qd × 14 days | 13 | ↓ 29 (↓ 15 to ↓ 40) | ↓ 39 (↓ 27 to ↓ 50) | ↓ 46 (↓ 31 to ↓ 58) |
| Voriconazole | 400 mg po q12h × 1 day then 200 mg po q12h × 8 days | 400 mg qd × 9 days | NA | ↓ 61ý | ↓ 77ý | NA |
| 300 mg po q12h days 2–7 | 300 mg qd × 7 days | NA | ↓ 36£
(↓ 21 to ↓ 49) | ↓ 55£
(↓ 45 to ↓ 62) | NA | |
| 400 mg po q12h days 2–7 | 300 mg qd × 7 days | NA | ↑ 23£
(↓ 1 to ↑ 53) | ↓ 7£
(↓ 23 to ↑ 13) | NA | |
Emtricitabine and Tenofovir DF: The steady-state pharmacokinetics of FTC and tenofovir were unaffected when FTC and TDF were administered together versus each agent dosed alone.
In vitro and clinical pharmacokinetic drug-drug interaction studies have shown that the potential for CYP mediated interactions involving FTC and tenofovir with other medicinal products is low.
TDF is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) transporters. When TDF is coadministered with an inhibitor of these transporters, an increase in absorption may be observed.
No clinically significant drug interactions have been observed between FTC and famciclovir, indinavir, sofosbuvir/velpatasvir, stavudine, TDF, and zidovudine. Similarly, no clinically significant drug interactions have been observed between TDF and abacavir, EFV, FTC, entecavir, indinavir, lamivudine, lopinavir/ritonavir, methadone, nelfinavir, oral contraceptives, ribavirin, saquinavir/ritonavir, sofosbuvir, or tacrolimus in trials conducted in healthy volunteers.
Following multiple dosing to HIV-negative subjects receiving either chronic methadone maintenance therapy, oral contraceptives, or single doses of ribavirin, steady-state tenofovir pharmacokinetics were similar to those observed in previous trials, indicating a lack of clinically significant drug interactions between these agents and TDF.
The effects of coadministered drugs on the Cmax, AUC, and Cmin of tenofovir are shown in Table 6. The effects of coadministration of TDF on Cmax, AUC, and Cmin of coadministered drugs are shown in Table 7.
| Coadministered Drug | Dose of Coadministered Drug (mg) | N | Mean % Change of Tenofovir Pharmacokinetic Parameters‡
(90% CI) |
||
|---|---|---|---|---|---|
| Cmax | AUC | Cmin | |||
| Atazanavir§ | 400 once daily × 14 days | 33 | ↑ 14 (↑ 8 to ↑ 20) | ↑ 24 (↑ 21 to ↑ 28) | ↑ 22 (↑ 15 to ↑ 30) |
| Atazanavir/ ritonavir§ | 300/100 once daily | 12 | ↑ 34 (↑ 20 to ↑ 51) | ↑ 37 (↑ 30 to ↑ 45) | ↑ 29 (↑ 21 to ↑ 36) |
| Darunavir/ ritonavir¶ | 300/100 twice daily | 12 | ↑ 24 (↑ 8 to ↑ 42) | ↑ 22 (↑ 10 to ↑ 35) | ↑ 37 (↑ 19 to ↑ 57) |
| Didanosine# | 250 or 400 once daily × 7 days | 14 | ↔ | ↔ | ↔ |
| Ledipasvir/sofosbuvir | 90/400 once daily | 15 | ↑ 79 (↑ 56 to ↑ 104) | ↑ 98 (↑ 77 to ↑ 123) | ↑ 163 (↑ 132 to ↑ 197) |
| Lopinavir/ ritonavir | 400/100 twice daily × 14 days | 24 | ↔ | ↑ 32 (↑ 25 to ↑ 38) | ↑ 51 (↑ 37 to ↑ 66) |
| Sofosbuvir | 400 once daily | 16 | ↑ 25 (↑ 8 to ↑ 45) | ↔ | ↔ |
| Sofosbuvir/velpatasvir | 400/100 once daily | 15 | ↑ 77 (↑ 53 to ↑ 104) | ↑ 81 (↑ 68 to ↑ 94) | ↑ 121 (↑ 100 to ↑ 143) |
| Tipranavir/ ritonavirÞ | 500/100 twice daily | 22 | ↓ 23 (↓ 32 to ↓ 13) | ↓ 2 (↓ 9 to ↑ 5) | ↑ 7 (↓ 2 to ↑ 17) |
| 750/200 twice daily (23 doses) | 20 | ↓ 38 (↓ 46 to ↓ 29) | ↑ 2 (↓ 6 to ↑ 10) | ↑ 14 (↑ 1 to ↑ 27) |
|
| Coadministered Drug | Dose of Coadministered Drug (mg) | N | Mean % Change of Coadministered Drug Pharmacokinetic Parameters‡
(90% CI) |
||
|---|---|---|---|---|---|
| Cmax | AUC | Cmin | |||
|
|||||
| Atazanavir§ | 400 once daily × 14 days | 34 | ↓ 21 (↓ 27 to ↓ 14) | ↓ 25 (↓ 30 to ↓ 19) | ↓ 40 (↓ 48 to ↓ 32) |
| Atazanavir/ritonavir 300/100 once daily × 42 days | 10 | ↓ 28 (↓ 50 to ↑ 5) | ↓ 25¶
(↓ 42 to ↓ 3) | ↓ 23¶
(↓ 46 to ↑ 10) |
|
| Darunavir# | Darunavir/ritonavir 300/100 once daily | 12 | ↑ 16 (↓ 6 to ↑ 42) | ↑ 21 (↓ 5 to ↑ 54) | ↑ 24 (↓ 10 to ↑ 69) |
| DidanosineÞ | 250 once, simultaneously with TDF and a light mealß | 33 | ↓ 20à
(↓ 32 to ↓ 7) | ↔à | NA |
| Lopinavir | Lopinavir/ritonavir 400/100 twice daily × 14 days | 24 | ↔ | ↔ | ↔ |
| Ritonavir | Lopinavir/ritonavir 400/100 twice daily × 14 days | 24 | ↔ | ↔ | ↔ |
| Tipranavirè | Tipranavir/ritonavir 500/100 twice daily | 22 | ↓ 17 (↓ 26 to ↓ 6) | ↓ 18 (↓ 25 to ↓ 9) | ↓ 21 (↓ 30 to ↓ 10) |
| Tipranavir/ritonavir 750/200 twice daily (23 doses) | 20 | ↓ 11 (↓ 16 to ↓ 4) | ↓ 9 (↓ 15 to ↓ 3) | ↓ 12 (↓ 22 to 0) |
|
12.4 Microbiology
Mechanism of Action
Efavirenz: EFV is a non-nucleoside reverse transcriptase (RT) inhibitor of HIV-1. Efavirenz activity is mediated predominantly by noncompetitive inhibition of HIV-1 reverse transcriptase. HIV-2 RT and human cellular DNA polymerases α, β, γ, and δ are not inhibited by EFV.
Emtricitabine: Emtricitabine, a synthetic nucleoside analog of cytidine, is phosphorylated by cellular enzymes to form FTC 5'-triphosphate. Emtricitabine 5'-triphosphate inhibits the activity of the HIV-1 RT by competing with the natural substrate deoxycytidine 5'-triphosphate and by being incorporated into nascent viral DNA which results in chain termination. Emtricitabine 5'-triphosphate is a weak inhibitor of mammalian DNA polymerases α, β, ε, and mitochondrial DNA polymerase γ.
Tenofovir DF: TDF is an acyclic nucleoside phosphonate diester analog of adenosine monophosphate. TDF requires initial diester hydrolysis for conversion to tenofovir and subsequent phosphorylations by cellular enzymes to form tenofovir diphosphate. Tenofovir diphosphate inhibits the activity of HIV-1 RT by competing with the natural substrate deoxyadenosine 5'-triphosphate and, after incorporation into DNA, by DNA chain termination. Tenofovir diphosphate is a weak inhibitor of mammalian DNA polymerases α, β, and mitochondrial DNA polymerase γ.
Antiviral Activity
Efavirenz, Emtricitabine, and Tenofovir DF: In combination studies evaluating the antiviral activity in cell culture of FTC and EFV together, EFV and tenofovir together, and FTC and tenofovir together, additive to synergistic antiviral effects were observed.
Efavirenz: The concentration of EFV inhibiting replication of wild-type laboratory adapted strains and clinical isolates in cell culture by 90–95% (EC90–95) ranged from 1.7–25 nM in lymphoblastoid cell lines, peripheral blood mononuclear cells, and macrophage/monocyte cultures. Efavirenz demonstrated additive antiviral activity against HIV-1 in cell culture when combined with non-nucleoside reverse transcriptase inhibitors (NNRTIs) (delavirdine and nevirapine), nucleoside reverse transcriptase inhibitors (NRTIs) (abacavir, didanosine, lamivudine, stavudine, zalcitabine, and zidovudine), protease inhibitors (PIs) (amprenavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir), and the fusion inhibitor enfuvirtide. Efavirenz demonstrated additive to antagonistic antiviral activity in cell culture with atazanavir. Efavirenz demonstrated antiviral activity against clade B and most non-clade B isolates (subtypes A, AE, AG, C, D, F, G, J, and N), but had reduced antiviral activity against group O viruses. Efavirenz is not active against HIV-2.
Emtricitabine: The antiviral activity in cell culture of FTC against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, the MAGI-CCR5 cell line, and peripheral blood mononuclear cells. The 50% effective concentration (EC50) values for FTC were in the range of 0.0013–0.64 µM (0.0003–0.158 µg/mL). In drug combination studies of FTC with NRTIs (abacavir, lamivudine, stavudine, zalcitabine, and zidovudine), NNRTIs (delavirdine, EFV, and nevirapine), and PIs (amprenavir, nelfinavir, ritonavir, and saquinavir), additive to synergistic effects were observed. Emtricitabine displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, and G (EC50 values ranged from 0.007–0.075 µM) and showed strain-specific activity against HIV-2 (EC50 values ranged from 0.007–1.5 µM).
Tenofovir DF: The antiviral activity in cell culture of tenofovir against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, primary monocyte/macrophage cells and peripheral blood lymphocytes. The EC50 values for tenofovir were in the range of 0.04–8.5 µM. In drug combination studies of tenofovir with NRTIs (abacavir, didanosine, lamivudine, stavudine, zalcitabine, and zidovudine), NNRTIs (delavirdine, EFV, and nevirapine), and PIs (amprenavir, indinavir, nelfinavir, ritonavir, and saquinavir), additive to synergistic effects were observed. Tenofovir displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, G, and O (EC50 values ranged from 0.5–2.2 µM) and showed strain-specific activity against HIV-2 (EC50 values ranged from 1.6–5.5 µM).
Resistance
EFV, FTC, and TDF: HIV-1 isolates with reduced susceptibility to the combination of FTC and tenofovir have been selected in cell culture and in clinical trials. Genotypic analysis of these isolates identified the M184V/I and/or K65R amino acid substitutions in the viral RT. In addition, a K70E substitution in HIV-1 reverse transcriptase has been selected by tenofovir and results in reduced susceptibility to tenofovir.
In a clinical trial of treatment-naïve subjects [Study 934, see Clinical Studies (14)] resistance analysis was performed on HIV-1 isolates from all confirmed virologic failure subjects with greater than 400 copies/mL of HIV-1 RNA at Week 144 or early discontinuations. Genotypic resistance to EFV, predominantly the K103N substitution, was the most common form of resistance that developed. Resistance to EFV occurred in 13/19 analyzed subjects in the FTC + TDF group and in 21/29 analyzed subjects in the zidovudine/lamivudine fixed-dose combination group. The M184V amino acid substitution, associated with resistance to FTC and lamivudine, was observed in 2/19 analyzed subject isolates in the FTC + TDF group and in 10/29 analyzed subject isolates in the zidovudine/lamivudine group. Through 144 weeks of Study 934, no subjects developed a detectable K65R substitution in their HIV-1 as analyzed through standard genotypic analysis.
In a clinical trial of treatment-naïve subjects, isolates from 8/47 (17%) analyzed subjects receiving TDF developed the K65R substitution through 144 weeks of therapy; 7 of these occurred in the first 48 weeks of treatment and one at Week 96. In treatment experienced subjects, 14/304 (5%) of TDF treated subjects with virologic failure through Week 96 showed greater than 1.4-fold (median 2.7) reduced susceptibility to tenofovir. Genotypic analysis of the resistant isolates showed a substitution in the HIV-1 RT gene resulting in the K65R amino acid substitution.
Efavirenz: Clinical isolates with reduced susceptibility in cell culture to EFV have been obtained. The most frequently observed amino acid substitution in clinical trials with EFV is K103N (54%). One or more RT substitutions at amino acid positions 98, 100, 101, 103, 106, 108, 188, 190, 225, 227, and 230 were observed in subjects failing treatment with EFV in combination with other antiretrovirals. Other resistance substitutions observed to emerge commonly included L100I (7%), K101E/Q/R (14%), V108I (11%), G190S/T/A (7%), P225H (18%), and M230I/L (11%).
HIV-1 isolates with reduced susceptibility to EFV (greater than 380-fold increase in EC90 value) emerged rapidly under selection in cell culture. Genotypic characterization of these viruses identified substitutions resulting in single amino acid substitutions L100I or V179D, double substitutions L100I/V108I, and triple substitutions L100I/V179D/Y181C in RT.
Emtricitabine: Emtricitabine-resistant isolates of HIV-1 have been selected in cell culture and in clinical trials. Genotypic analysis of these isolates showed that the reduced susceptibility to FTC was associated with a substitution in the HIV-1 RT gene at codon 184 which resulted in an amino acid substitution of methionine by valine or isoleucine (M184V/I).
Cross Resistance
Efavirenz, Emtricitabine, and Tenofovir DF: Cross resistance has been recognized among NNRTIs. Cross resistance has also been recognized among certain NRTIs. The M184V/I and/or K65R substitutions selected in cell culture by the combination of FTC and tenofovir are also observed in some HIV-1 isolates from subjects failing treatment with tenofovir in combination with either lamivudine or FTC, and either abacavir or didanosine. Therefore, cross resistance among these drugs may occur in patients whose virus harbors either or both of these amino acid substitutions.
Efavirenz: Clinical isolates previously characterized as EFV resistant were also phenotypically resistant in cell culture to delavirdine and nevirapine compared to baseline. Delavirdine- and/or nevirapine-resistant clinical viral isolates with NNRTI resistance-associated substitutions (A98G, L100I, K101E/P, K103N/S, V106A, Y181X, Y188X, G190X, P225H, F227L, or M230L) showed reduced susceptibility to EFV in cell culture. Greater than 90% of NRTI-resistant isolates tested in cell culture retained susceptibility to EFV.
Emtricitabine: Emtricitabine-resistant isolates (M184V/I) were cross resistant to lamivudine but retained susceptibility in cell culture to didanosine, stavudine, tenofovir, zidovudine, and NNRTIs (delavirdine, EFV, and nevirapine). HIV-1 isolates containing the K65R substitution, selected in vivo by abacavir, didanosine, and tenofovir, demonstrated reduced susceptibility to inhibition by FTC. Viruses harboring substitutions conferring reduced susceptibility to stavudine and zidovudine (M41L, D67N, K70R, L210W, T215Y/F, and K219Q/E) or didanosine (L74V) remained sensitive to FTC.
Tenofovir DF: Cross resistance has been observed among NRTIs. The K65R substitution in HIV-1 RT selected by tenofovir is also selected in some HIV-1 infected patients treated with abacavir, or didanosine. HIV-1 isolates with the K65R substitution also showed reduced susceptibility to FTC and lamivudine. Therefore, cross resistance among these drugs may occur in patients whose virus harbors the K65R substitution. The K70E substitution selected clinically by TDF results in reduced susceptibility to abacavir, didanosine, FTC, and lamivudine. HIV-1 isolates from subjects (N=20) whose HIV-1 expressed a mean of 3 zidovudine-associated RT amino acid substitutions (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E/N) showed a 3.1-fold decrease in the susceptibility to tenofovir. Subjects whose virus expressed an L74V substitution without zidovudine resistance associated substitutions (N=8) had reduced response to TDF. Limited data are available for patients whose virus expressed a Y115F substitution (N=3), Q151M substitution (N=2), or T69 insertion (N=4), all of whom had a reduced response.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Efavirenz: Long-term carcinogenicity studies in mice and rats were carried out with EFV. Mice were dosed with 0, 25, 75, 150, or 300 mg/kg/day for 2 years. Incidences of hepatocellular adenomas and carcinomas and pulmonary alveolar/bronchiolar adenomas were increased above background in females. No increases in tumor incidence above background were seen in males. In studies in which rats were administered EFV at doses of 0, 25, 50, or 100 mg/kg/day for 2 years, no increases in tumor incidence above background were observed. The systemic exposure (based on AUCs) in mice was approximately 1.7-fold that in humans receiving the 600-mg/day dose. The exposure in rats was lower than that in humans. The mechanism of the carcinogenic potential is unknown. However, in genetic toxicology assays, EFV showed no evidence of mutagenic or clastogenic activity in a battery of in vitro and in vivo studies. These included bacterial mutation assays in S. typhimurium and E. coli, mammalian mutation assays in Chinese hamster ovary cells, chromosome aberration assays in human peripheral blood lymphocytes or Chinese hamster ovary cells, and an in vivo mouse bone marrow micronucleus assay. Given the lack of genotoxic activity of EFV, the relevance to humans of neoplasms in EFV-treated mice is not known.
Efavirenz did not impair mating or fertility of male or female rats, and did not affect sperm of treated male rats. The reproductive performance of offspring born to female rats given EFV was not affected. Because of the rapid clearance of EFV in rats, systemic drug exposures achieved in these studies were equivalent to or below those achieved in humans given therapeutic doses of EFV.
Emtricitabine: In long-term carcinogenicity studies of FTC, no drug-related increases in tumor incidence were found in mice at doses up to 750 mg/kg/day (26 times the human systemic exposure at the therapeutic dose of 200 mg/day) or in rats at doses up to 600 mg/day (31 times the human systemic exposure at the therapeutic dose).
Emtricitabine was not genotoxic in the reverse mutation bacterial test (Ames test), or the mouse lymphoma or mouse micronucleus assays.
Emtricitabine did not affect fertility in male rats at approximately 140-fold or in male and female mice at approximately 60-fold higher exposures (AUC) than in humans given the recommended 200 mg daily dose. Fertility was normal in the offspring of mice exposed daily from before birth (in utero) through sexual maturity at daily exposures (AUC) of approximately 60-fold higher than human exposures at the recommended 200 mg daily dose.
Tenofovir DF: Long-term oral carcinogenicity studies of TDF in mice and rats were carried out at exposures up to approximately 16 times (mice) and 5 times (rats) those observed in humans at the therapeutic dose for HIV-1 infection. At the high dose in female mice, liver adenomas were increased at exposures 16 times that in humans. In rats, the study was negative for carcinogenic findings at exposures up to 5 times that observed in humans at the therapeutic dose.
TDF was mutagenic in the in vitro mouse lymphoma assay and negative in an in vitro bacterial mutagenicity test (Ames test). In an in vivo mouse micronucleus assay, TDF was negative when administered to male mice.
There were no effects on fertility, mating performance, or early embryonic development when TDF was administered to male rats at a dose equivalent to 10 times the human dose based on body surface area comparisons for 28 days prior to mating and to female rats for 15 days prior to mating through Day 7 of gestation. There was, however, an alteration of the estrous cycle in female rats.
13.2 Animal Toxicology and/or Pharmacology
Efavirenz: Nonsustained convulsions were observed in 6 of 20 monkeys receiving EFV at doses yielding plasma AUC values 4- to 13-fold greater than those in humans given the recommended dose.
Tenofovir DF: Tenofovir and TDF administered in toxicology studies to rats, dogs, and monkeys at exposures (based on AUCs) greater than or equal to 6-fold those observed in humans caused bone toxicity. In monkeys the bone toxicity was diagnosed as osteomalacia. Osteomalacia observed in monkeys appeared to be reversible upon dose reduction or discontinuation of tenofovir. In rats and dogs, the bone toxicity manifested as reduced bone mineral density. The mechanism(s) underlying bone toxicity is unknown.
Evidence of renal toxicity was noted in 4 animal species administered tenofovir and TDF. Increases in serum creatinine, BUN, glycosuria, proteinuria, phosphaturia and/or calciuria and decreases in serum phosphate were observed to varying degrees in these animals. These toxicities were noted at exposures (based on AUCs) 2- to 20-times higher than those observed in humans. The relationship of the renal abnormalities, particularly the phosphaturia, to the bone toxicity is not known.
14. Clinical Studies
Clinical Study 934 (NCT00112047) supports the use of ATRIPLA tablets in antiretroviral treatment-naïve HIV-1 infected patients.
Clinical Study 073 (NCT00365612) provides clinical experience in subjects with stable, virologic suppression and no history of virologic failure who switched from their current regimen to ATRIPLA.
In antiretroviral treatment-experienced patients, the use of ATRIPLA tablets may be considered for patients with HIV-1 strains that are expected to be susceptible to the components of ATRIPLA as assessed by treatment history or by genotypic or phenotypic testing [see Microbiology (12.4)].
Study 934: Data through 144 weeks are reported for Study 934, a randomized, open-label, active-controlled multicenter trial comparing FTC + TDF administered in combination with EFV versus zidovudine/lamivudine fixed-dose combination administered in combination with EFV in 511 antiretroviral-naïve subjects. From Weeks 96 to 144 of the trial, subjects received FTC/TDF fixed-dose combination with EFV in place of FTC + TDF with EFV. Subjects had a mean age of 38 years (range 18–80); 86% were male, 59% were Caucasian, and 23% were Black. The mean baseline CD4+ cell count was 245 cells/mm3 (range 2–1191), and median baseline plasma HIV-1 RNA was 5.01 log10 copies/mL (range 3.56–6.54). Subjects were stratified by baseline CD4+ cell count (< or ≥200 cells/mm3), and 41% had CD4+ cell counts <200 cells/mm3. Fifty-one percent (51%) of subjects had baseline viral loads >100,000 copies/mL. Treatment outcomes through 48 and 144 weeks for those subjects who did not have EFV resistance at baseline (N=487) are presented in Table 8.
| Outcomes | At Week 48 | At Week 144 | ||
|---|---|---|---|---|
| FTC+TDF+EFV (N=244) | AZT/3TC+EFV (N=243) | FTC+TDF+EFV (N=227)* | AZT/3TC+EFV (N=229)* |
|
|
||||
| Responder† | 84% | 73% | 71% | 58% |
| Virologic failure‡ | 2% | 4% | 3% | 6% |
| Rebound | 1% | 3% | 2% | 5% |
| Never suppressed | 0% | 0% | 0% | 0% |
| Change in antiretroviral regimen | 1% | 1% | 1% | 1% |
| Death | <1% | 1% | 1% | 1% |
| Discontinued due to adverse event | 4% | 9% | 5% | 12% |
| Discontinued for other reasons§ | 10% | 14% | 20% | 22% |
Through Week 48, 84% and 73% of subjects in the FTC + TDF group and the zidovudine/lamivudine group, respectively, achieved and maintained HIV-1 RNA <400 copies/mL (71% and 58% through Week 144). The difference in the proportion of subjects who achieved and maintained HIV-1 RNA <400 copies/mL through 48 weeks largely results from the higher number of discontinuations due to adverse events and other reasons in the zidovudine/lamivudine group in this open-label trial. In addition, 80% and 70% of subjects in the FTC + TDF group and the zidovudine/lamivudine group, respectively, achieved and maintained HIV-1 RNA <50 copies/mL through Week 48 (64% and 56% through Week 144). The mean increase from baseline in CD4+ cell count was 190 cells/mm3 in the FTC + TDF group and 158 cells/mm3 in the zidovudine/lamivudine group at Week 48 (312 and 271 cells/mm3 at Week 144).
Through 48 weeks, 7 subjects in the FTC + TDF group and 5 subjects in the zidovudine/lamivudine group experienced a new CDC Class C event (10 and 6 subjects through 144 weeks).
Study 073: Study 073 was a 48-week open-label, randomized clinical trial in subjects with stable virologic suppression on combination antiretroviral therapy consisting of at least two NRTIs administered in combination with a protease inhibitor (with or without ritonavir) or a NNRTI.
To be enrolled, subjects were to have HIV-1 RNA <200 copies/mL for at least 12 weeks on their current regimen prior to trial entry with no known HIV-1 substitutions conferring resistance to the components of ATRIPLA and no history of virologic failure.
The trial compared the efficacy of switching to ATRIPLA or staying on the baseline antiretroviral regimen (SBR). Subjects were randomized in a 2:1 ratio to switch to ATRIPLA (N=203) or stay on SBR (N=97). Subjects had a mean age of 43 years (range 22–73 years); 88% were male, 68% were white, 29% were Black or African-American, and 3% were of other races. At baseline, median CD4+ cell count was 516 cells/mm3, and 96% had HIV-1 RNA <50 copies/mL. The median time since onset of antiretroviral therapy was 3 years, and 88% of subjects were receiving their first antiretroviral regimen at trial enrollment.
At Week 48, 89% and 87% of subjects who switched to ATRIPLA maintained HIV RNA <200 copies/mL and <50 copies/mL, respectively, compared to 88% and 85% who remained on SBR; this difference was not statistically significant. No changes in CD4+ cell counts from baseline to Week 48 were observed in either treatment arm.
16. How is Atripla supplied
ATRIPLA tablets are pink, capsule shaped, film coated, debossed with "123" on one side and plain faced on the other side. Each bottle contains 30 tablets (NDC 15584-0101-1) and silica gel desiccant, and is closed with a child-resistant closure.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Severe Acute Exacerbation of Hepatitis B in Patients Coinfected with HIV-1 and HBV
Inform patients that severe acute exacerbations of hepatitis B have been reported in patients who are coinfected with HBV and HIV-1 and have discontinued FTC or TDF, and may occur with discontinuation of ATRIPLA. Advise patients not to discontinue ATRIPLA without first informing their healthcare provider. All patients should be tested for HBV infection before or when starting ATRIPLA and those who are infected with HBV need close medical follow-up for several months after stopping ATRIPLA to monitor for exacerbations of hepatitis [see Warnings and Precautions (5.1)].
Rash
Inform patients that a common side effect is rash, and that rashes usually go away without any change in treatment. However, since rash may be serious, advise patients to contact their physician promptly if rash occurs [see Warnings and Precautions (5.2)].
Hepatotoxicity
Inform patients to watch for early warning signs of liver inflammation or failure, such as fatigue, weakness, lack of appetite, nausea and vomiting, as well as later signs such as jaundice, confusion, abdominal swelling, and discolored feces, and to consult their health care professional without delay if such symptoms occur [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
Drug Interactions
Advise patients that ATRIPLA may interact with many drugs; therefore, advise patients to report to their healthcare provider the use of any other medication, including other drugs for treatment of hepatitis C virus [see Warnings and Precautions (5.4) and Drug Interactions (7)].
Psychiatric Symptoms
- Inform patients that serious psychiatric symptoms including severe depression, suicide attempts, aggressive behavior, delusions, paranoia, psychosis-like symptoms, and catatonia have been reported in patients receiving EFV, a component of ATRIPLA [see Warnings and Precautions (5.5)].
- Advise patients to seek immediate medical evaluation if they experience severe psychiatric adverse experiences.
- Advise patients to inform their physician of any history of mental illness or substance abuse.
Nervous System Symptoms
- Inform patients that central nervous system symptoms (NSS) including dizziness, insomnia, impaired concentration, drowsiness, and abnormal dreams, are commonly reported during the first weeks of therapy with EFV, a component of ATRIPLA. Dosing at bedtime may improve the tolerability of these symptoms, which are likely to improve with continued therapy.
- Alert patients to the potential for additive effects when ATRIPLA is used concomitantly with alcohol or psychoactive drugs.
- Instruct patients that if they experience NSS to avoid potentially hazardous tasks such as driving or operating machinery [see Warnings and Precautions (5.6) and Dosage and Administration (2.2)].
- Inform patients that there is a risk of developing late-onset neurotoxicity, including ataxia and encephalopathy, which may occur months to years after beginning therapy with EFV, a component of ATRIPLA [see Warnings and Precautions (5.6)].
New Onset or Worsening Renal Impairment
Inform patients that renal impairment, including cases of acute renal failure and Fanconi syndrome, has been reported. Advise patients to avoid using ATRIPLA with concurrent or recent use of a nephrotoxic agent (e.g., high-dose or multiple NSAIDs) [see Warnings and Precautions (5.7)].
Embryo-Fetal Toxicity
Apprise patients of the potential harm to the fetus if ATRIPLA is used during the first trimester of pregnancy, or if the patient becomes pregnant while taking this drug. Instruct adults and adolescents of childbearing potential receiving ATRIPLA to avoid pregnancy and to notify their healthcare provider if they become pregnant or plan to become pregnant while taking ATRIPLA [see Warnings and Precautions (5.8)]. A reliable form of barrier contraception must always be used in combination with other methods of contraception, including oral or other hormonal contraception. Because of the long half-life of EFV, recommend use of adequate contraceptive measures for 12 weeks after discontinuation of ATRIPLA [see Use in Specific Populations (8.1, 8.3)].
Bone Loss and Mineralization Defects
Inform patients that decreases in bone mineral density have been observed with the use of TDF, a component of ATRIPLA. Advise patients that bone mineral density monitoring may be performed in patients who have a history of pathologic bone fracture or other risk factors for osteoporosis or bone loss [see Warnings and Precautions (5.9)].
Convulsions
Inform patients that convulsions have been reported with the use of EFV, a component of ATRIPLA. Patients who are receiving concomitant anticonvulsant medications primarily metabolized by the liver may require periodic monitoring of plasma levels [see Warnings and Precautions (5.10) and Drug Interactions (7.3)].
Lactic Acidosis and Severe Hepatomegaly
Inform patients that lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported. Treatment with ATRIPLA should be suspended in any patient who develops clinical symptoms suggestive of lactic acidosis or pronounced hepatotoxicity [see Warnings and Precautions (5.11)].
Immune Reconstitution Syndrome
Inform patients that in some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started. It is believed that these symptoms are due to an improvement in the body's immune response, enabling the body to fight infections that may have been present with no obvious symptoms. Advise patients to inform their healthcare provider immediately of any symptoms of infection [see Warnings and Precautions (5.12)].
Fat Redistribution
Inform patients that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy, including ATRIPLA and that the cause and long-term health effects of these conditions are not known [see Warnings and Precautions (5.13)].
Dosing Instructions
Advise patients to take ATRIPLA orally on an empty stomach and that it is important to take ATRIPLA on a regular dosing schedule to avoid missing doses. Advise patients that dosing at bedtime may improve the tolerability of nervous system symptoms [see Dosage and Administration (2.1)].
Pregnancy Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in patients exposed to ATRIPLA during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Instruct patients not to breastfeed because HIV-1 can be passed to the baby in the breast milk [see Use in Specific Populations (8.2)].
Manufactured and distributed by:
Gilead Sciences, Inc.
Foster City, CA 94404
ATRIPLA, EMTRIVA, EPCLUSA, HARVONI, SOVALDI, VIREAD, and VOSEVI are trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners.
© 2019 Gilead Sciences, LLC. All rights reserved.
21937-GS-020
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: October/2019 | |||
| Patient Information
ATRIPLA® (uh TRIP luh) (efavirenz, emtricitabine, and tenofovir disoproxil fumarate) tablets |
||||
| What is the most important information I should know about ATRIPLA? ATRIPLA can cause serious side effects, including:
|
||||
| What is ATRIPLA?
ATRIPLA is a prescription medicine that contains efavirenz, emtricitabine, and tenofovir disoproxil fumarate combined in 1 tablet. ATRIPLA is used alone as a complete regimen, or in combination with other anti-HIV-1 medicines to treat people with HIV-1 infection who weigh at least 88 lbs (40 kg). It is not known if ATRIPLA is safe and effective for use in children with HIV-1 infection who weigh less than 88 lbs (40 kg). |
||||
| Who should not take ATRIPLA? Do not take ATRIPLA if you:
|
||||
Before taking ATRIPLA, tell your healthcare provider about all of your medical conditions, including if you:
Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine. ATRIPLA and some medicines may interact with each other causing serious side effects. You can ask your healthcare provider or pharmacist for a list of medicines that interact with ATRIPLA. Do not start a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take ATRIPLA with other medicines. |
||||
How should I take ATRIPLA?
|
||||
What should I avoid while taking ATRIPLA?
|
||||
| What are the possible side effects of ATRIPLA? ATRIPLA may cause serious side effects, including:
|
||||
|
|
|||
| If you have dizziness, trouble concentrating or sleepiness, do not drive a car, use machinery, or do anything that needs you to be alert. | ||||
|
||||
|
|
|||
|
||||
|
|
|||
| These are not all the possible side effects of ATRIPLA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||||
How should I store ATRIPLA?
|
||||
| General information about the safe and effective use of ATRIPLA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ATRIPLA for a condition for which it was not prescribed. Do not give ATRIPLA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ATRIPLA that is written for health professionals. |
||||
| What are the ingredients of ATRIPLA? Active Ingredients: efavirenz, emtricitabine, and tenofovir disoproxil fumarate Inactive Ingredients: croscarmellose sodium, hydroxypropyl cellulose, magnesium sterate, microcrystalline cellulose, and sodium lauryl sulfate. The film coating contains black iron oxide, polyethylene glycol, polyvinyl alcohol, red iron oxide, talc, and titanium dioxide. Manufactured and distributed by: Gilead Sciences, Inc. Foster City, CA 94404 For more information go to www.ATRIPLA.com or call 1-800-445-3235. ATRIPLA, EMTRIVA, TRUVADA, and VIREAD are trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners. © 2019 Gilead Sciences, LLC. All rights reserved. 21937-GS-020 |
||||
PRINCIPAL DISPLAY PANEL - 30 Tablet Bottle Label
NDC 15584-0101-1
ATRIPLA®
(efavirenz 600 mg/
emtricitabine 200 mg/tenofovir
disoproxil fumarate 300 mg)
tablets
30 tablets
Note to pharmacist: Do not cover ALERT box with pharmacy label.
ALERT: Find out about medicines that
should NOT be taken with ATRIPLA
Rx only

| ATRIPLA
efavirenz, emtricitabine, and tenofovir disoproxil fumarate tablet, film coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| Labeler - Gilead Sciences, LLC (780297904) |
Frequently asked questions
More about Atripla (efavirenz / emtricitabine / tenofovir disoproxil)
- Check interactions
- Compare alternatives
- Reviews (105)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- FDA approval history
- Drug class: antiviral combinations
