Thiola Tablets: Package Insert / Prescribing Info
Package insert / product label
Generic name: tiopronin
Dosage form: tablet, sugar coated
Drug class: Miscellaneous genitourinary tract agents
Medically reviewed by Drugs.com. Last updated on Jan 14, 2024.
On This Page
Thiola Tablets Description
THIOLA ® (Tiopronin) is a reducing and complexing thiol compound. Tiopronin is N-(2-Mercaptopropionyl) glycine and has the following structure:
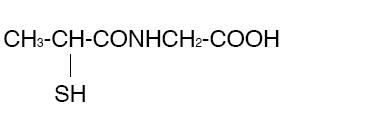
Tiopronin has the empirical formula C5H9NO3S and a molecular weight of 163.20. In this drug product tiopronin exists as a dl racemic mixture.
Tiopronin is a white crystalline powder which is freely soluble in water.
THIOLA ® tablets are white, sugar coated tablets, each containing 100 mg. of Tiopronin and are taken orally.
Inactive ingredients: Calcium carbonate, carnauba wax, ethyl cellulose, Eudragit E 100, hydroxy-propyl cellulose, lactose, magnesium stearate, povidone, sugar, talc, titanium dioxide.
Thiola Tablets - Clinical Pharmacology
THIOLA ® is an active reducing agent which undergoes thiol-disulfide exchange with cystine to form a mixed disulfide of Thiola-cysteine.
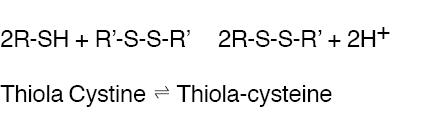
From this reaction, a water-soluble mixed disulfide is formed and the amount of sparingly soluble cystine is reduced. When THIOLA ® is given orally, up to 48% of dose appears in urine during the first 4 hours and up to 78% by 72 hours. Thus, in patients with cystinuria, sufficient amount of THIOLA ® or its active metabolites could appear in urine to react with cystine, lowering cystine excretion.
The decrement in urinary cystine produced by THIOLA ® is generally proportional to the dose. A reduction in urinary cystine of 250-350 mg/day at a THIOLA ® dosage of 1 g/day, and a decline of approximately 500 mg/day at a dosage of 2 g/day, might be expected. THIOLA ® causes a sustained reduction in cystine excretion without apparent loss of effectiveness. THIOLA ® has a rapid onset and offset of action, showing a fall in cystine excretion on the first day of administration and a rise on the first day of drug withdrawal.
Indications and Usage for Thiola Tablets
THIOLA ® is indicated for the prevention of cystine (kidney) stone formation in patients with severe homozygous cystinuria with urinary cystine greater than 500 mg/day, who are resistant to treatment with conservative measures of high fluid intake, alkali and diet modification, or who have adverse reactions to d-penicillamine.
Cystine stones typically occur in approximately 10,000 persons in the United States who are homozygous for cystinuria. These persons excrete abnormal amounts of cystine in urine of over 250 mg/g creatinine, as well as excessive amounts of other dibasic amino acids (lysine, arginine and ornithine). In addition, they show varying intestinal transport defects for these same amino acids. The stone formation is the result of poor aqueous solubility of cystine.
Since there are no known inhibitors of the crystallization of cystine, the stone formation is determined primarily by the urinary supersaturation of cystine. Thus, cystine stones could theoretically form whenever urinary cystine concentration exceeds the solubility limit. Cystine solubility in urine is pH-dependent, and ranges from 170-300 mg/liter at pH 5, 190-400 mg/liter at pH 7 and 220-500 mg/liter at pH 7.5.
The goal of therapy is to reduce urinary cystine concentration below its solubility limit. It may be accomplished by dietary means aimed at reducing cystine synthesis and by a high fluid intake in order to increase urine volume and thereby lower cystine concentration.
Unfortunately, the above conservative measures alone may be ineffective in controlling cystine stone formation in some homozygous patients with severe cystinuria (urinary cystine exceeding 500 mg/day). In such patients, d-penicillamine has been used as an additional therapy. Like THIOLA ™, dpenicillamine undergoes thiol-disulfide exchange with cystine, thereby lowering the amount of sparingly soluble cystine in urine.
However, d-penicillamine treatment is frequently accompanied by adverse reactions, such as dermatologic complications, hypersensitivity reactions, hematologic abnormalities and renal disturbances. THIOLA ® may have a particular therapeutic role in such patients.
Contraindications
The use of THIOLA ® during pregnancy is contraindicated, except in those with severe cystinuria where the anticipated benefit of inhibited stone formation clearly outweighs possible hazards of treatment (see PRECAUTIONS).
THIOLA ® should not be begun again in patients with a prior history of developing agranulocytosis, aplastic anemia or thrombocytopenia on this medication.
Mothers maintained on THIOLA ® treatment should not nurse their infants.
Warnings
Despite apparent lower toxicity of THIOLA ™, THIOLA ® may potentially cause all the serious adverse reactions reported for d-penicillamine. Thus, although no death has been reported to result directly from THIOLA ® treatment, a fatal outcome from THIOLA ® is possible, as has been reported with d-penicillamine therapy from such complications as aplastic anemia, agranulocytosis, thrombocytopenia, Goodpasture’s syndrome or myasthenia gravis.
Leukopenia of the granulocytic series may develop without eosinophilia. Thrombocytopenia may be immunologic in origin or occur on an idiosyncratic basis. The reduction in peripheral blood white count to less than 3500/cubic mm or in platelet count to below 100,000 cubic mm mandates cessation of therapy. Patients should be instructed to report promptly the occurrence of any symptom or sign of these hematological abnormalities, such as fever, sore throat, chills, bleeding or easy bruisability.
Proteinuria, sometimes sufficiently severe to cause nephrotic syndrome, may develop from membranous glomerulopathy. A close observation of affected patients is mandatory.
The following complications, though rare, have been reported during d-penicillamine therapy and could occur during THIOLA ® treatment. When there are abnormal urinary findings associated with hemoptysis and pulmonary infiltrates suggestive of Goodpasture’s syndrome, THIOLA ® treatment should be stopped. Appearance of myasthenic syndrome or myasthenia gravis requires cessation of treatment. When pemphigus-type reactions develop, THIOLA ® therapy should be stopped. Steroid treatment may be necessary.
Precautions
Patients should be advised of the potential development of complications and to report promptly the occurrence of any symptom or sign of them.
To help monitor potential complications, the following tests are recommended: peripheral blood counts, direct platelet count, hemoglobin, serum albumin, liver function tests, 24-hour urinary protein and routine urinalysis at 3- 6 month intervals during treatment. In order to assess effect on stone disease, urinary cystine analysis should be monitored frequently during the first 6 months when the optimum dose schedule is being determined, and at 6-month intervals thereafter. Abdominal roentogenogram (KUB) is advised on a yearly basis to monitor the size and appearance/disappearance of stone(s).
- CARCINOGENESIS, MUTAGENESIS, IMPAIRMENT OF FERTILITY: Long-term carcino-genicity studies in animals have not been performed. High doses of THIOLA
® in experimental animals have been shown to interfere with maintenance of pregnancy and viability of the fetus.
- USE IN PREGNANCY: Pregnancy category C. D-penicillamine has been shown to cause skeletal defects and cleft palates in the fetus when given to pregnant rats at 10 times the dose recommended for human use. A similar teratogenicity might be expected for THIOLA
® although no such findings could be related to the drug in studies in mice and rats at doses up to 10 times the highest recommended human dose.
There are no adequate and well-controlled studies in pregnant women. THIOLA ® should be used during pregnancy only if the potential benefit justifies potential risk to the fetus.
- NURSING MOTHERS: Because THIOLA
® may be excreted in milk and because of the potential serious adverse reactions of nursing infants from THIOLA
®, mothers taking THIOLA
® should not nurse their infants.
- PEDIATRIC USE: Safety and effectiveness below the age of 9 have not been established.
Adverse Reactions/Side Effects
Some patients may develop drug fever, usually during the first month of therapy. THIOLA ® treatment should be discontinued until the fever subsides. It may be reinstated at a small dose, with a gradual increase in dosage until the desired level is achieved.
A generalized rash (erythematous, maculopapular or morbilliform) accompanied by pruritis may develop during the first few months of treatment. It may be controlled by antihistamine therapy, typically recedes when THIOLA ® treatment is discontinued, and seldom recurs when THIOLA ® treatment is restarted at a lower dosage. Less commonly, rash may appear late in the course of treatment (of more than 6 months). Located usually in the trunk, the late rash is associated with intense pruritis, recedes slowly after discontinuing treatment, and usually recurs upon resumption of treatment.
A drug reaction simulating lupus erythematous, manifested by fever, arthralgia and lymphadenopathy may develop. It may be associated with a positive antinuclear antibody test, but not necessarily with nephropathy. It may require discontinuance of THIOLA ® treatment.
A reduction in taste perception may develop. It is believed to be the result of chelation of trace metals by THIOLA ™. Hypogeusia is often self-limiting.
Unlike during d-penicillamine therapy, vitamin B6 deficiency is uncommonly associated with THIOLA ® treatment.
Some patients may complain of wrinkling and friability of skin. This complication usually occurs after long-term treatment, and is believed to result from the effect of THIOLA ® on collagen.
A multiclinic trial involving 66 cystinuric patients in the United States indicated that THIOLA ® is associated with fewer or less severe adverse reactions than d-penicillamine. Among those who had to stop taking d-penicillamine due to toxicity, 64.7% could take THIOLA ® . In those without prior history of d-penicillamine treatment, only 5.9% developed reactions of sufficient severity to require THIOLA ® withdrawal. A review of available literature supports the findings from this trial.
Despite this apparent reduced toxicity to THIOLA
® relative to d-penicillamine, THIOLA
® treatment may potentially be associated with all the adverse reactions reported with d-penicillamine. They include:
Gastrointestinal side-effects (nausea, emesis, diarrhea or softstools, anorexia, abdominal pain, bloating or flatus) in about 1 in 6 patients;
Impairment in taste and smell in about 1 in 25 patients;
Dermatologic complications (pharyngitis, oral ulcers, rash, ecchymosis, prurites, uritcaria, warts, skin wrinkling, pemphigus, elastosis perforans serpiginosa) in about 1 in 6 patients;
Hypersensitivity reactions (laryngeal edema, dyspnea, respiratory distress, fever, chills, arthralgia, weakness, fatigue, myalgia, adenopathy) in about 1 in 25 patients;
Hematologic abnormalities (increased bleeding, anemia, leukopenia, thrombocytopenia, eosinophilia) in about 1 in 25 patients;
Renal complications (proteinuria, nephrotic syndrome, hematuria) in about 1 in 20 patients;
Pulmonary manifestations (bronchiolitis, hemoptysis, pulmonary infiltrates, dyspnea) in about 1 in 50 patients;
Neurologic complications (myasthenic syndrome) in about 1 in 50 patients.
These reactions are more likely to develop during THIOLA ® therapy among patients who had previously shown toxicity to d-penicillamine.
In patients who had previously manifested adverse reactions to d-penicillamine, adverse reactions to THIOLA ® are more likely to occur than in patients who took THIOLA ® for the first time. A close supervision with a careful monitoring of potential side effects is mandatory during THIOLA ® treatment. Patients should be told to report promptly any symptoms suggesting toxicity. The treatment with THIOLA ® should be stopped if severe toxicity develops.
Jaundice and abnormal liver function tests have been reported during THIOLA ® therapy for non-cystinuric conditions. A direct cause and effect relationship, based upon these foreign reports, has not been established. Although such complications were not encountered in the small multi-center trials in the United States, patients should be carefully monitored and if any abnormalities are noted, the drug should be discontinued and the patient treated by appropriate measures.
Related/similar drugs
Thiola Tablets Dosage and Administration
It is recommended that a conservative treatment program should be attempted first. At least 3 liters of fluid (10-10 oz. glassfuls) should be provided, including two glasses with each meal and at bedtime. The patients should be expected to awake at night to urinate; they should drink two more glasses of fluids before returning to bed. Additional fluids should be consumed if there is excessive sweating or intestinal fluid loss. A minimum urine output of 2 liters/day on a consistent basis should be sought. A modest amount of alkali should be provided in order to maintain urinary pH at a high normal range (6.5-7.0). Potassium alkali are advantageous over sodium alkali, because they do not cause hypercalciuria and are less likely to cause the complication of calcium stones.
Excessive alkali therapy is not advisable. When urinary pH increases above 7.0 with alkali therapy, the complication of calcium phosphate nephrolithiasis may ensue because of the enhanced urinary supersaturation of hydroxyapatite in an alkaline environment.
In patients who continue to form cystine stones on the above conservative program, THIOLA ® may be added to the treatment program. THIOLA ® may also be substituted for d-penicillamine in patients who have developed toxicity to the latter drug. In both situations, the conservative treatment program should be continued.
The dose of THIOLA ® should not be arbitrary but should be based on that amount required to reduce urinary cystine concentration to below its solubility limit (generally <250 mg/liter). The extent of the decline in cystine excretion is generally dependent on the THIOLA ® dosage.
THIOLA ® may be begun at a dosage of 800 mg/day in adult patients with cystine stones. In a multiclinic trial, average dose of THIOLA ® was about 1000 mg/day. However, some patients require a smaller dose. In children, initial dosage may be based on 15 mg/kg/day. Urinary cystine should be measured at 1 month after THIOLA ® treatment, and every 3 months thereafter. THIOLA ® dosage should be readjusted depending on the urinary cystine value. Whenever possible, THIOLA ® should be given in divided doses 3 times/day at least one hour before or 2 hours after meals.
In patients who had shown severe toxicity to d-penicillamine, THIOLA ® might be begun at a lower dosage.
How is Thiola Tablets supplied
THIOLA ® (NDC 0178-0900-01), is available for oral administration as 100 mg. round, white, sugar coated tablets in bottles of 100 tablets each. Each tablet is imprinted in red with “M” on one side and blank on the other side. Store at 25°C (77°F); excursions permitted to 15- 30°C (59-86°F) [see USP Controlled Room Temperature].
| THIOLA
tiopronin tablet, sugar coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Mission Pharmacal Company (008117095) |
| Registrant - Mission Pharmacal Company (927726893) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Mission Pharmacal Company | 927726893 | manufacture(0178-0900) | |
More about Thiola (tiopronin)
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- Drug class: miscellaneous genitourinary tract agents
- En español
