Soltamox: Package Insert / Prescribing Info
Package insert / product label
Generic name: tamoxifex citrate
Dosage form: oral liquid
Drug classes: Hormones / antineoplastics, Selective estrogen receptor modulators
Medically reviewed by Drugs.com. Last updated on Nov 25, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
SOLTAMOX® (tamoxifen citrate) oral solution
Initial U.S. Approval: 1977
WARNING: UTERINE MALIGNANCIES and THROMBOEMBOLIC EVENTS
See full prescribing information for complete boxed warning.
- Serious, life-threatening, and fatal events from use of tamoxifen include uterine malignancies, stroke, and pulmonary embolism. (5.1, 5.2)
- Discuss risks and benefits of tamoxifen with women at high risk for breast cancer and women with ductal carcinoma in situ (DCIS) when considering tamoxifen use to reduce the risk of developing breast cancer. (5.1, 5.2)
- For most patients already diagnosed with breast cancer, the benefits of tamoxifen outweigh its risks. (5.1, 5.2)
Indications and Usage for Soltamox
SOLTAMOX is an estrogen agonist/antagonist indicated:
- For treatment of adult patients with estrogen receptor-positive metastatic breast cancer (1.1)
- For adjuvant treatment of adult patients with early stage estrogen receptor- positive breast cancer (1.2)
- To reduce risk of invasive breast cancer following breast surgery and radiation in adult women with ductal carcinoma in situ (DCIS) (1.3)
- To reduce the incidence of breast cancer in adult women at high risk (1.4)
Soltamox Dosage and Administration
Dosage Forms and Strengths
- Oral solution: Each 10 mL of solution contains 20 mg tamoxifen, equivalent to 30.4 mg tamoxifen citrate. (3)
Contraindications
- Known hypersensitivity to tamoxifen or any other SOLTAMOX ingredient (4)
- In patients who require concomitant warfarin therapy or have a history of deep vein thrombosis or pulmonary embolus, if the indication for treatment is either reduction of breast cancer incidence in high-risk patients or risk reduction of invasive breast cancer after treatment of DCIS (4)
Warnings and Precautions
- Uterine malignancies: Promptly evaluate abnormal vaginal bleeding in a woman with current or past tamoxifen use. (5.1)
- Thromboembolic events: Risk increases with coadministered chemotherapy. For treatment of breast cancer, consider risks and benefits in patients with a history of thromboembolic events. (5.2)
- Embryo-fetal toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.3, 8.1, 8.3)
- Effects on the liver: Liver cancer and liver abnormalities, some fatal, have occurred. Perform periodic liver function testing. (5.4, 5.9)
Adverse Reactions/Side Effects
Most common adverse reactions: hot flashes, mood disturbances, vaginal discharge, vaginal bleeding, nausea, and fluid retention (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Mayne Pharma at 1-844-825-8500 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Anastrozole and letrozole: Should not be used in combination with tamoxifen. (7.1)
- Warfarin: Do not use in patients taking tamoxifen for DCIS and for reduction in breast cancer incidence in women at high risk. (4) Closely monitor coagulation indices for increased anticoagulant effect when used with tamoxifen for metastatic breast cancer or as adjuvant therapy. (7.2)
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2020
Full Prescribing Information
WARNING: UTERINE MALIGNANCIES and THROMBOEMBOLIC EVENTS
Serious and life-threatening events from the use of SOLTAMOX include uterine malignancies, stroke, and pulmonary embolism [see Warnings and Precautions (5.1, 5.2)]. Fatal cases of each type of event have occurred.
Incidence rates per 1000 women-years for these events were estimated from the National Surgical Adjuvant Breast and Bowel Project (NSABP) P-1 trial in women at high risk for breast cancer [see Clinical Studies (14.4)]:
- Endometrial adenocarcinoma: 2.20 for tamoxifen vs. 0.71 for placebo
- Uterine sarcoma: 0.17 for tamoxifen vs. 0.04 for placebo
- Stroke: 1.43 for tamoxifen vs. 1.00 for placebo.
- Pulmonary embolism: 0.75 for tamoxifen versus 0.25 for placebo.
Discuss the potential benefits of tamoxifen versus the potential risks of these serious events with women at high risk for breast cancer and women with ductal carcinoma in situ (DCIS) considering tamoxifen to reduce the risk of developing breast cancer [see Warnings and Precautions (5)]. For most patients already diagnosed with breast cancer, the benefits of tamoxifen outweigh its risks.
1. Indications and Usage for Soltamox
1.1 Metastatic Breast Cancer
SOLTAMOX is indicated for the treatment of adult patients with estrogen receptor-positive metastatic breast cancer.
1.2 Adjuvant Treatment of Breast Cancer
SOLTAMOX is indicated:
- for the adjuvant treatment of adult patients with early stage estrogen receptor-positive breast cancer
- to reduce the occurrence of contralateral breast cancer in adult patients when used as adjuvant therapy for the treatment of breast cancer.
1.3 Ductal Carcinoma in Situ
In adult women with DCIS, following breast surgery and radiation, SOLTAMOX is indicated to reduce the risk of invasive breast cancer [see Boxed Warning and Clinical Studies (14.3)].
1.4 Reduction in Breast Cancer Incidence in Women at High Risk
SOLTAMOX is indicated to reduce the incidence of breast cancer in adult women at high risk for breast cancer. [see Boxed Warning and Clinical Studies (14.4)].
2. Soltamox Dosage and Administration
Metastatic Breast Cancer
For patients with breast cancer, the recommended daily dose of SOLTAMOX is 20 to 40 mg. Doses greater than 20 mg per day should be given in divided doses (morning and evening).
Adjuvant Treatment of Breast Cancer
For use in the adjuvant setting, the recommended dose of SOLTAMOX is 20 mg daily for 5-10 years [see Clinical Studies (14.2)]. Doses greater than 20 mg daily yield no additional clinical benefit.
3. Dosage Forms and Strengths
SOLTAMOX oral solution: Each 10 mL of solution contains 20 mg tamoxifen, equivalent to 30.4 mg tamoxifen citrate.
4. Contraindications
- SOLTAMOX is contraindicated in patients with known hypersensitivity (e.g., angioedema, serious skin reactions) to tamoxifen or any other SOLTAMOX ingredient [see Adverse Reactions (6.2)].
- SOLTAMOX is contraindicated in patients who require concomitant warfarin therapy or have a history of deep vein thrombosis or pulmonary embolus if the indication for treatment is either reduction of breast cancer incidence in high-risk patients or risk reduction of invasive breast cancer after treatment of DCIS [see Warnings and Precautions (5.2) and Drug Interactions (7.2)].
5. Warnings and Precautions
5.1 Endometrial Cancer, Uterine Sarcoma, and Other Effects on the Uterus
Endometrial Cancer and Uterine Sarcoma
An increased incidence of uterine malignancies (endometrial adenocarcinoma and uterine sarcoma), including fatal cases, has been reported with tamoxifen treatment. The underlying mechanism is unknown, but may be related to the estrogen-like effect of tamoxifen. Most uterine malignancies seen with tamoxifen are classified as adenocarcinoma of the endometrium; however, uterine sarcomas, including malignant mixed mullerian tumors (MMMT), have also been reported. Uterine sarcoma was generally associated with a higher FIGO stage (III/IV) at diagnosis, poor prognosis, and short survival. Uterine sarcoma has been reported to occur more frequently among long-term users (≥2 years) of tamoxifen than non-users.
Promptly evaluate any patient receiving or who has previously received tamoxifen who reports abnormal vaginal bleeding. Patients receiving or who have previously received tamoxifen should have annual gynecological examinations. Advise patients to promptly inform a healthcare provider if they experience any abnormal gynecological symptoms (e.g., menstrual irregularities, abnormal vaginal bleeding, changes in vaginal discharge, or pelvic pain or pressure). There are no data to suggest that routine endometrial sampling in asymptomatic women taking tamoxifen is beneficial.
In a review of long-term data (median length of total follow-up was 6.9 years, including blinded follow-up) on 8,306 women with an intact uterus at randomization in the NSABP P-1 risk reduction trial, the incidence of both adenocarcinomas and uterine sarcomas was increased in women taking tamoxifen.
- During blinded follow-up, there were 36 cases of FIGO Stage I endometrial adenocarcinoma (22 were FIGO Stage IA, 13 IB, and 1 IC) in women receiving tamoxifen and 15 cases in women receiving placebo [14 were FIGO Stage I (9 IA and 5 IB), and 1 case was FIGO Stage IV].
- During total follow-up, endometrial adenocarcinoma was reported in 53 women randomized to tamoxifen (30 cases of FIGO Stage IA, 20 were Stage IB, 1 was Stage IC, and 2 were Stage IIIC), and 17 women randomized to placebo (9 cases were FIGO Stage IA, 6 were Stage IB, 1 was Stage IIIC, and 1 was Stage IVB) (incidence per 1,000 women-years of 2.20 and 0.71, respectively).
- Uterine sarcomas were reported in 4 women randomized to tamoxifen (1 was FIGO IA, 1 was FIGO IB, 1 was FIGO IIA, and 1 was FIGO IIIC) and 1 patient randomized to placebo (FIGO IA); incidence per 1,000 women-years of 0.17 and 0.04, respectively. Of the patients randomized to tamoxifen, the FIGO IA and IB cases were a MMMT and sarcoma, respectively; the FIGO II was a MMMT; and the FIGO III was a sarcoma; and the 1 patient randomized to placebo had a MMMT.
- A similar increased incidence in endometrial adenocarcinoma and uterine sarcoma was observed among women receiving tamoxifen in 5 other NSABP clinical trials.
In the NSABP P-1 trial, endometrial sampling did not alter the endometrial cancer detection rate compared to women who did not undergo endometrial sampling (0.6% with sampling, 0.5% without sampling) for women who had not undergone hysterectomy.
Non-Malignant Effects on the Uterus
An increased incidence of endometrial changes including hyperplasia and polyps has been reported with tamoxifen treatment. The incidence and pattern of this increase suggest that the underlying mechanism is related to the partial estrogenic effect of tamoxifen. There have been reports of endometriosis and uterine fibroids in women receiving tamoxifen. Ovarian cysts have also been observed in premenopausal patients with advanced breast cancer who have been treated with tamoxifen. Tamoxifen has been reported to cause menstrual irregularity or amenorrhea.
5.2 Thromboembolic Events
There is an increased incidence of thromboembolic events, including deep vein thrombosis and pulmonary embolism, during tamoxifen therapy. When tamoxifen is coadministered with chemotherapy, there is a further increase in the risk of thromboembolic events.
For treatment of breast cancer, carefully consider the risks and benefits of tamoxifen in women with a history of thromboembolic events. For women with DCIS and for the reduction in breast cancer incidence in women at high risk, tamoxifen is contraindicated in women who require concomitant warfarin-type anticoagulant therapy or in women with a history of deep vein thrombosis or pulmonary embolus. Advise patients to seek medical attention immediately if signs or symptoms of a thromboembolic event occur.
Data from the NSABP P-1 trial in women at high risk for breast cancer show that participants receiving tamoxifen without a history of pulmonary emboli (PE) had a statistically significant increase in pulmonary emboli (events: 18 tamoxifen, 6 placebo; incidence rate per 1,000 women-years: 0.75 tamoxifen versus 0.25 placebo; RR = 3.01, 95% CI: 1.15 to 9.27) [see Clinical Studies (14.4)]. Three of the pulmonary emboli, all in the tamoxifen arm, were fatal. Eighty-seven percent of the cases of pulmonary embolism occurred in women at least 50 years of age at randomization. Among women receiving tamoxifen, PE events occurred between 2 and 60 months (average = 27 months) from the start of treatment.
In this same population, a non-statistically significant increase in deep vein thrombosis (DVT) was seen in the tamoxifen group (30 tamoxifen, 19 placebo; relative risk (RR) = 1.59, 95% CI: 0.86 to 2.98). The same increase in relative risk was seen in women ≤49 and in women ≥50, although fewer events occurred in younger women. Women with thromboembolic events were at risk for a second related event (7 out of 25 women on placebo, 5 out of 48 women on tamoxifen) and were at risk for thromboembolic event and treatment related complications (0/25 on placebo, 4/48 on tamoxifen). Among women receiving tamoxifen, DVT events occurred between 2 and 57 months (average = 19 months) from the start of treatment.
In a small substudy (N = 81) of the NSABP-1 trial, there appeared to be no benefit to screening women for Factor V Leiden and Prothrombin mutations G20210A as a means to identify those who may not be appropriate candidates for tamoxifen therapy.
In the NSABP P-1 trial, there was an increase in stroke among women randomized to tamoxifen compared to women randomized to placebo (events: 24 placebo; 34 tamoxifen; incidence rate per 1,000 women-years: 1.43 tamoxifen versus 1.00 placebo; RR = 1.42, 95% CI: 0.82 to 2.51). Six of the 24 strokes in the placebo group were considered hemorrhagic in origin and 10 of the 34 strokes in the tamoxifen group were categorized as hemorrhagic. Seventeen of the 34 strokes in the tamoxifen group were considered occlusive and 7 were considered to be of unknown etiology. Fourteen of the 24 strokes on the placebo arm were reported to be occlusive and 4 of unknown etiology. Three strokes in the placebo group and 4 strokes in the tamoxifen group were fatal. Eighty-eight percent of the strokes occurred in women at least 50 years of age at the time of randomization. Among women receiving tamoxifen, the stroke events occurred between 1 and 63 months (average = 30 months) from the start of treatment.
5.3 Embryo-Fetal Toxicity
Tamoxifen can cause fetal harm when administered to a pregnant woman. There are postmarketing reports of vaginal bleeding, spontaneous abortions, birth defects, and fetal deaths in pregnant women taking tamoxifen. In a primate model, administration of tamoxifen at doses 2 times the maximum recommended human dose resulted in spontaneous abortion. In rat and rabbit studies, doses of tamoxifen less than or equal to human doses resulted in increased embryotoxicity, abortions, and altered learning behaviors in the offspring. Additionally, rodent models showed reproductive tract changes often associated with diethylstilbestrol (DES) in offspring of both sexes.
Advise pregnant women of the potential risks to a fetus, including the potential long-term risk of a DES-like syndrome. Advise females of reproductive potential to use effective non-hormonal contraception during treatment with tamoxifen and for 2 months following the last dose [see Use in Specific Populations (8.1, 8.3)].
5.4 Liver Cancer and Other Effects on the Liver
Liver Cancer
In the Swedish Breast Cancer Cooperative Group trial using adjuvant tamoxifen 40 mg per day (two times the recommended dosage) for 2 to 5 years, 3 cases of liver cancer were reported in the tamoxifen-treated group versus 1 case in the observation group [see Clinical Studies (14.2)]. One case of liver cancer was reported in NSABP P-1 (women at high risk for breast cancer) in a participant randomized to tamoxifen [see Clinical Studies (14.4)].
Hepatocellular carcinoma has been observed in animals receiving tamoxifen [see Nonclinical Toxicology (13.1)].
Non-Malignant Effects on the Liver
Tamoxifen has been associated with changes in liver enzyme levels, and on rare occasions, a spectrum of more severe liver abnormalities including fatty liver, cholestasis, hepatitis and hepatic necrosis. Some of these serious cases included fatalities. In most reported cases, the relationship to tamoxifen is uncertain. However, some positive rechallenges and dechallenges have been reported. Monitor liver function periodically.
In the NSABP P-1 trial, grade 3 to 4 changes in liver function (SGOT, SGPT, bilirubin, alkaline phosphatase) were observed (10 on placebo and 6 on tamoxifen). Serum lipids were not systematically collected.
5.5 Other Cancers
A number of second primary tumors occurring at sites other than the endometrium have been reported following the treatment of breast cancer with tamoxifen in clinical trials. Data from the NSABP B-14 (adjuvant breast cancer study in women with axillary node-negative breast cancer) and P-1 studies show no increase in other (non-uterine) cancers among patients receiving tamoxifen [see Clinical Studies (14.2, 14.4)]. Whether an increased risk for other (non-uterine) cancers is associated with tamoxifen is still unknown.
5.6 Hypercalcemia in Patients with Metastatic Breast Cancer
As with other additive hormonal therapy (estrogens and androgens), hypercalcemia has been reported in some breast cancer patients with bone metastases within a few weeks of starting treatment with tamoxifen. If hypercalcemia occurs, treat as appropriate; if the hypercalcemia is severe, discontinue tamoxifen.
5.7 Hematologic Effects
Decreases in platelet counts, usually to 50,000-100,000/mm3, infrequently lower, have been reported in patients taking tamoxifen for breast cancer. In the NSABP P-1 trial, 6 women on tamoxifen and 2 on placebo experienced grade 3 to 4 decreases in platelet counts (≤50,000/mm3). In patients with significant thrombocytopenia, hemorrhagic episodes have occurred, but it is uncertain if these episodes were due to tamoxifen therapy.
Leukopenia has been observed, sometimes in association with anemia and/or thrombocytopenia. There have been reports of neutropenia and pancytopenia, including some severe cases, in patients receiving tamoxifen. Perform periodic complete blood counts, including platelet counts.
5.8 Effects on the Eye
Ocular disturbances, including corneal changes, decrement in color vision perception, retinal vein thrombosis, and retinopathy have been reported in patients receiving tamoxifen. An increased incidence of cataracts and the need for cataract surgery have also been reported. Patients should be advised to seek medical attention if they experience any visual disturbance.
In the NSABP P-1 trial, an increased risk of borderline significance of developing cataracts among those women without cataracts at baseline (540 tamoxifen; 483 placebo; RR = 1.13, 95% CI: 1.00 to 1.28) was observed. Among these same women, tamoxifen was associated with an increased risk of having cataract surgery (101 tamoxifen; 63 placebo; RR = 1.62, 95% CI: 1.18 to 2.22) [see Clinical Studies (14.4)]. Among all women on the trial (with or without cataracts at baseline), tamoxifen was associated with an increased risk of having cataract surgery (201 tamoxifen; 129 placebo; RR = 1.58, 95% CI: 1.26 to 1.97).
6. Adverse Reactions/Side Effects
The following serious adverse reactions are discussed below and elsewhere in the labeling:
- Uterine malignancies [see Boxed Warning, Warnings and Precautions (5.1), and Clinical Studies (14.4)]
- Thromboembolic events [see Boxed Warning, Warnings and Precautions (5.2), and Clinical Studies (14.4)]
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.3), and Use in Specific Populations (8.1, 8.3)]
- Liver cancer [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Patients with Metastatic Breast Cancer
In patients treated with tamoxifen for metastatic breast cancer, the most frequent adverse reaction was hot flashes. Other adverse reactions which were seen less commonly are hypercalcemia, peripheral edema, distaste for food, pruritus vulvae, depression, dizziness, lightheadedness, headache, hair thinning and/or partial hair loss, and vaginal dryness.
Increased bone, tumor pain and local disease flare have occurred. Patients with soft tissue disease may have sudden increases in the size of preexisting lesions, sometimes associated with marked erythema within and surrounding the lesions and/or the development of new lesions. When they occurred, the bone pain or disease flares were seen shortly after starting tamoxifen and generally subsided rapidly.
Premenopausal Women with Metastatic Breast Cancer
Table 1 summarizes the incidence of adverse reactions reported at a frequency of 2% or greater from clinical trials that compared tamoxifen therapy to ovarian ablation in premenopausal patients with metastatic breast cancer.
| % of Women | ||
|---|---|---|
| Tamoxifen N=104 | Ovarian Ablation N=100 | |
| Adverse Reactions* | ||
|
||
| Flush | 33 | 46 |
| Amenorrhea | 16 | 69 |
| Altered menses | 13 | 5 |
| Oligomenorrhea | 9 | 1 |
| Bone pain | 6 | 6 |
| Menstrual disorder | 6 | 4 |
| Nausea | 5 | 4 |
| Cough/coughing | 4 | 1 |
| Edema | 4 | 1 |
| Fatigue | 4 | 1 |
| Musculoskeletal pain | 3 | 0 |
| Pain | 3 | 4 |
| Ovarian cyst(s) | 3 | 2 |
| Depression | 2 | 2 |
| Abdominal cramps | 1 | 2 |
| Anorexia | 1 | 2 |
Adverse Reactions in Adjuvant Breast Cancer
In the NSABP B-14 study, women with axillary node-negative breast cancer were randomized to 5 years of tamoxifen 20 mg per day or placebo following primary surgery [see Clinical Studies (14.2)]. Table 2 presents the most common adverse reactions (mean follow-up of approximately 6.9 years) that were more common on tamoxifen than placebo.
| % of Women | ||
|---|---|---|
| Tamoxifen N=1,422 | Placebo N=1,437 | |
| Hot flashes | 64 | 48 |
| Fluid retention | 32 | 30 |
| Vaginal discharge | 30 | 15 |
| Nausea | 26 | 24 |
| Irregular menses | 25 | 19 |
| Weight loss (>5%) | 23 | 18 |
| Skin changes | 19 | 15 |
| Increased SGOT | 5 | 3 |
| Increased bilirubin | 2 | 1 |
| Increased creatinine | 2 | 1 |
| Thrombocytopenia* | 2 | 1 |
| Thrombotic events† | ||
| Deep vein thrombosis | 0.8 | 0.2 |
| Pulmonary embolism | 0.5 | 0.2 |
| Superficial phlebitis | 0.4 | 0 |
In the Eastern Cooperative Oncology Group (ECOG) adjuvant breast cancer trial [see Clinical Studies (14.2)], tamoxifen or placebo was administered for 2 years to women following mastectomy. When compared to placebo, tamoxifen showed a higher incidence of hot flashes (19% vs. 8% for placebo). The incidence of all other adverse reactions was similar in the two treatment groups with the exception of thrombocytopenia, where the incidence for tamoxifen was 10% vs. 3% for placebo.
In other adjuvant studies [the Toronto study and Tamoxifen Adjuvant Trial Organization (NATO)], women received either tamoxifen or no therapy [see Clinical Studies (14.2)]. In the Toronto study, hot flashes were observed in 29% of patients for tamoxifen vs. 1% in the untreated group. In the NATO trial, hot flashes and vaginal bleeding were reported in 2.8% and 2.0% of women, respectively, for tamoxifen vs. 0.2% for each in the untreated group.
Anastrozole Adjuvant Trial (ATAC: Arimidex, Tamoxifen, Alone or in Combination) – Study of Anastrozole Compared to Tamoxifen for Adjuvant Treatment of Early Breast Cancer
At a median follow-up of 33 months, the combination of anastrozole and tamoxifen did not demonstrate an efficacy benefit when compared to tamoxifen monotherapy in all patients as well as in the hormone receptor- positive subpopulation. The combination treatment arm was discontinued from the trial. The median duration of adjuvant treatment for safety evaluation was 59.8 months and 59.6 months for patients receiving anastrozole 1 mg and tamoxifen 20 mg monotherapy, respectively.
Adverse reactions occurring with an incidence of at least 5% in either single-drug treatment group during treatment or within 14 days of the end of treatment are presented in Table 3.
| % of Women | ||
|---|---|---|
| Body system and adverse reactions by COSTART-preferred term* | Tamoxifen N=3,094 | Anastrozole N=3,092 |
| Body as a whole | ||
| Asthenia | 18 | 19 |
| Pain | 16 | 17 |
| Back pain | 10 | 10 |
| Accidental injury | 10 | 10 |
| Abdominal pain | 9 | 9 |
| Infection | 9 | 9 |
| Headache | 8 | 10 |
| Flu syndrome | 6 | 6 |
| Cyst | 5 | 5 |
| Chest pain | 5 | 7 |
| Neoplasm | 5 | 5 |
| Cardiovascular | ||
| Vasodilatation | 41 | 36 |
| Hypertension | 11 | 13 |
| Digestive | ||
| Nausea | 11 | 11 |
| Constipation | 8 | 8 |
| Diarrhea | 7 | 9 |
| Dyspepsia | 6 | 7 |
| Gastrointestinal disorder | 5 | 7 |
| Hemic and lymphatic | ||
| Lymphedema | 11 | 10 |
| Anemia | 5 | 4 |
| Metabolic and nutritional | ||
| Peripheral edema | 11 | 10 |
| Weight gain | 9 | 9 |
| Hypercholesterolemia | 3 | 9 |
| Musculoskeletal | ||
| Arthritis | 14 | 17 |
| Arthralgia | 11 | 15 |
| Osteoporosis | 7 | 11 |
| Fracture | 7 | 10 |
| Bone pain | 6 | 7 |
| Joint disorder | 5 | 6 |
| Myalgia | 5 | 6 |
| Arthrosis | 5 | 7 |
| Nervous system | ||
| Depression | 12 | 13 |
| Insomnia | 9 | 10 |
| Dizziness | 8 | 8 |
| Anxiety | 6 | 6 |
| Paresthesia | 5 | 7 |
| Respiratory | ||
| Pharyngitis | 14 | 14 |
| Cough increased | 9 | 8 |
| Dyspnea | 8 | 8 |
| Sinusitis | 5 | 6 |
| Bronchitis | 5 | 5 |
| Skin and appendages | ||
| Rash | 13 | 11 |
| Sweating | 6 | 5 |
| Special senses | ||
| Cataract specified | 7 | 6 |
| Urogenital | ||
| Urinary tract infection | 10 | 8 |
| Leukorrhea | 9 | 3 |
| Vaginal hemorrhage† | 6 | 4 |
| Breast pain | 6 | 8 |
| Vaginitis | 5 | 4 |
| Vulvovaginitis | 5 | 6 |
| Breast neoplasm | 5 | 5 |
Certain adverse reactions and combinations of adverse reactions were prospectively specified for analysis in the ATAC trial, based on the known pharmacologic properties and safety profiles of the two drugs (Table 4).
| % of Women | Odds Ratio † | 95% CI | ||
|---|---|---|---|---|
| Tamoxifen N=3,094 | Anastrozole N=3,092 |
|||
|
||||
| Hot flashes | 41 | 36 | 0.80 | 0.73 to 0.89 |
| Musculoskeletal events ‡ | 29 | 36 | 1.32 | 1.19 to 1.47 |
| Mood disturbances | 18 | 19 | 1.10 | 0.97 to 1.25 |
| Fatigue/asthenia | 18 | 19 | 1.07 | 0.94 to 1.22 |
| Vaginal discharge | 13 | 4 | 0.24 | 0.19 to 0.30 |
| Nausea and vomiting | 12 | 13 | 1.03 | 0.88 to 1.19 |
| Vaginal bleeding | 10 | 5 | 0.50 | 0.41 to 0.61 |
| Cataracts | 7 | 6 | 0.85 | 0.69 to 1.04 |
| All fractures | 7 | 10 | 1.57 | 1.30 to 1.88 |
| Fractures of spine, hip, or wrist | 3 | 4 | 1.48 | 1.13 to 1.95 |
| Wrist/Colles' fractures | 2 | 2 | ||
| Hip fractures | 1 | 1 | ||
| Spine fractures | 1 | 1 | ||
| Venous thromboembolic events | 5 | 3 | 0.61 | 0.47 to 0.80 |
| Deep venous thromboembolic events | 2 | 2 | 0.64 | 0.45 to 0.93 |
| Ischemic cerebrovascular events | 3 | 2 | 0.70 | 0.50 to 0.97 |
| Ischemic cardiovascular disease | 3 | 4 | 1.23 | 0.95 to 1.60 |
| Endometrial cancer § | 0.6 | 0.2 | 0.31 | 0.10 to 0.94 |
Adverse Reactions in Ductal Carcinoma in Situ
The types and frequency of adverse reactions in the NSABP B-24 trial in women with DCIS were consistent with those observed in the other adjuvant trials conducted with tamoxifen [see Clinical Studies (14.3)].
Adverse Reactions in Women at High Risk for Breast Cancer
In the NSABP P-1 trial, there was an increase in five serious adverse reactions in the tamoxifen group [see Warnings and Precautions (5.1, 5.2, and 5.8) and Clinical Studies (14.4)]:
- endometrial cancer (33 cases in the tamoxifen group vs. 14 in the placebo group);
- pulmonary embolism (18 cases in the tamoxifen group vs. 6 in the placebo group);
- deep vein thrombosis (30 cases in the tamoxifen group vs. 19 in the placebo group);
- stroke (34 cases in the tamoxifen group vs. 24 in the placebo group);
- cataract formation (540 cases in the tamoxifen group vs. 483 in the placebo group), and
- cataract surgery (101 cases in the tamoxifen group vs. 63 in the placebo group).
Table 5 presents the adverse reactions observed in NSABP P-1 by treatment arm. Only adverse reactions more common on tamoxifen than placebo are shown.
| % of Women | ||
|---|---|---|
| Tamoxifen N=6,681 | Placebo N=6,707 | |
| Self-Reported Symptoms | N=6,441 * | N=6,469 * |
| Hot flashes | 80 | 68 |
| Vaginal discharge | 55 | 35 |
| Vaginal bleeding | 23 | 22 |
| Laboratory Abnormalities | N=6,520 † | N=6,535 † |
| Platelets decreased | 0.7 | 0.3 |
| Adverse Reactions | N=6,492 ‡ | N=6,484 ‡ |
| Mood changes | 11.6 | 10.8 |
| Infection/sepsis | 6.0 | 5.1 |
| Constipation | 4.4 | 3.2 |
| Skin changes | 5.6 | 4.7 |
| Alopecia | 5.2 | 4.4 |
| Allergy | 2.5 | 2.1 |
In the NSABP P-1 trial, 15.0% and 9.7% of participants receiving tamoxifen and placebo therapy, respectively, withdrew from the trial for medical reasons including the following: hot flashes (3.1% vs. 1.5%, respectively) and vaginal discharge (0.5% vs. 0.1% respectively).
Severe hot flashes occurred in 28% of women on placebo and 45% of women on tamoxifen. Vaginal discharge was severe in 4.5% on placebo and 12.3% on tamoxifen.
SOLTAMOX-related Adverse Reactions
In a single-dose pharmacokinetic study in healthy perimenopausal and postmenopausal women, throat irritation was reported by 3 of 60 evaluable subjects (5%) in the SOLTAMOX treatment group while none of the subjects in the tamoxifen citrate tablet group reported this event. All cases were mild, occurred within an hour after dosing, and resolved within 24 hours.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of tamoxifen. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Skin and Subcutaneous Disorders: Erythema multiforme, Stevens-Johnson syndrome, bullous pemphigoid
Respiratory, Thoracic, Mediastinal Disorders: Interstitial pneumonitis
Immune System Disorders: Hypersensitivity reactions including angioedema; in some of these cases, the time to onset was more than one year.
Gastrointestinal Disorders: Elevation of serum triglyceride levels, in some cases with pancreatitis
Related/similar drugs
7. Drug Interactions
7.1 Aromatase Inhibitors
Anastrozole
The combination of anastrozole and tamoxifen did not demonstrate any benefit when compared to tamoxifen alone and should be avoided in all patients [see Clinical Studies (14.2)]. In the ATAC trial, coadministration of anastrozole and tamoxifen in breast cancer patients reduced the anastrozole plasma concentration by 27% compared to that achieved with anastrozole alone. The tamoxifen concentration was not altered [see Clinical Pharmacology (12.3)].
Letrozole
The concomitant use of letrozole with tamoxifen is not recommended because the efficacy of the combination in the adjuvant treatment of breast cancer has not been established. Tamoxifen reduced the plasma concentration of letrozole by 38% when these drugs were co-administered [see Clinical Pharmacology (12.3)].
7.2. Warfarin
A marked increase in anticoagulant effect may occur when tamoxifen is used in combination with warfarin. Closely monitor coagulation indices in patients who are taking tamoxifen for either the treatment of metastatic breast cancer or as adjuvant therapy who require concomitant use of warfarin [see Contraindications (4)].
7.3 Inducers of CYP3A4
Strong CYP3A4 inducers should not be used with tamoxifen. Strong CYP3A4 inducers (e.g., rifampin) reduce tamoxifen AUC and Cmax [see Clinical Pharmacology (12.3)].
7.4 Strong Inhibitors of CYP2D6
The impact on the efficacy of tamoxifen with co-administration of strong CYP2D6 inhibitors (e.g., paroxetine) is not well established. Some studies have shown that the efficacy of tamoxifen may be reduced when the drugs are co-administered as a result of reduced levels of potent active metabolites of tamoxifen. However, other studies have failed to demonstrate such an effect [see Clinical Pharmacology (12.3)].
7.5 Drug-Laboratory Test Interactions
There are postmarketing reports of T4 elevations in postmenopausal patients taking tamoxifen that may be explained by increases in thyroid-binding globulin. These elevations were not accompanied by clinical hyperthyroidism.
Variations in the karyopyknotic index on vaginal smears and various degrees of estrogen effect on Pap smears have been seen in postmenopausal patients taking tamoxifen.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
SOLTAMOX can cause fetal harm when administered to a pregnant woman. There are no adequate and well- controlled studies of tamoxifen in pregnant women. There are limited postmarketing reports of vaginal bleeding, spontaneous abortions, birth defects, and fetal deaths in pregnant women taking tamoxifen. In a primate model administration of tamoxifen at doses 2 times higher than the maximum recommended human dose resulted in spontaneous abortion. In rat and rabbit studies, doses of tamoxifen less than or equal to human doses resulted in increased embryotoxicity, abortions, and altered learning behaviors. Additionally, rodent models showed reproductive tract changes often associated with DES in offspring of both sexes [see Data]. Advise pregnant women of the potential risk to a fetus, including the potential long-term risk of a DES-like syndrome.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In embryofetal development studies in rats using doses significantly below those used in humans (0.01 to 0.03 times the recommended human dose on a mg/m2 basis), a lower incidence of embryo implantation and a higher incidence of fetal death or delayed in utero growth occurred, along with slower learning behavior in some rat pups when compared to historical controls. When pregnant rabbits received tamoxifen during organogenesis, abortion and premature delivery occurred at doses of greater than 0.125 mg/kg/day (about 0.05 times the daily maximum recommended human dose on a mg/m2 basis). Although reversible nonteratogenic skeletal variations occurred in rat reproductive studies at doses less than or equal to the human dose, there were no teratogenic changes in either rats or rabbits. In pregnant marmosets, administration of 10 mg/kg/day tamoxifen (about 2 times the daily maximum recommended human dose on a mg/m2 basis) during organogenesis or in the last half of pregnancy terminated pregnancy in some animals. In pregnancies that continued, there were no malformations or deformations in offspring.
In rodent models of fetal reproductive tract development, tamoxifen (at doses 0.002 to 2.4 times the daily maximum recommended human dose on a mg/m2 basis) caused changes in both sexes that are similar to those caused by estradiol, ethynylestradiol and diethylstilbestrol. Although the clinical relevance of these changes is unknown, some of these changes, especially vaginal adenosis, are similar to those seen in young women who were exposed to diethylstilbestrol in utero and who have a 1 in 1,000 risk of developing clear-cell adenocarcinoma of the vagina or cervix. To date, in utero exposure to tamoxifen has not been shown to cause vaginal adenosis, or clear-cell adenocarcinoma of the vagina or cervix, in young women; however, only a small number of young women have been exposed to tamoxifen in utero, and a smaller number have been followed long enough (to age 15 to 20) to determine whether vaginal or cervical neoplasia could occur as a result of this exposure.
8.2 Lactation
Risk Summary
There is no information regarding the presence of tamoxifen or its metabolites in human milk or its effects on the breastfed infant. Additional information from juvenile animal studies suggests a potential for serious long- term adverse effects for breastfed infants exposed to tamoxifen through breast milk [see Use in Specific Populations (8.4)]. Because of the potential for serious adverse reactions in breastfed infants from tamoxifen, advise a woman not to breastfeed during treatment with SOLTAMOX and for 3 months following the last dose.
Tamoxifen has been reported to inhibit lactation. Two placebo-controlled studies in over 150 women have shown that tamoxifen significantly inhibits early postpartum milk production. In both studies tamoxifen was administered within 24 hours of delivery for between 5 and 18 days. The effect of tamoxifen on established milk production is unknown.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
A negative pregnancy test should be confirmed in females of reproductive potential before initiation of SOLTAMOX therapy.
Contraception
Females
SOLTAMOX can cause fetal harm. Advise females of reproductive potential to use effective nonhormonal contraception during treatment with SOLTAMOX and for 2 months following the last dose [see Use in Specific Populations (8.1)].
Infertility
Females
Based on animal studies, SOLTAMOX may impair embryo implantation in females of reproductive potential, however, may not reliably cause infertility. Advise women that SOLTAMOX does not always cause infertility, even in the presence of menstrual irregularity [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of SOLTAMOX in pediatric patients have not been established.
Tamoxifen Use in McCune-Albright Syndrome
The safety and effectiveness of SOLTAMOX for the treatment of girls with McCune-Albright syndrome, including long-term effects, have not been established.
A single, uncontrolled multicenter trial of tamoxifen 20 mg once a day was conducted in a heterogeneous group of girls with McCune-Albright syndrome and precocious puberty manifested by physical signs of pubertal development, episodes of vaginal bleeding, and/or advanced bone age (bone age of at least 12 months beyond chronological age). Twenty-eight female pediatric patients, aged 2 to 10 years, were treated for up to 12 months. Effect of treatment on frequency of vaginal bleeding, bone age advancement, and linear growth rate was assessed relative to prestudy baseline. Tamoxifen treatment was associated with a 50% reduction in frequency of vaginal bleeding episodes by patient or family report (mean annualized frequency of 3.56 episodes at baseline and 1.73 episodes on-treatment). Among the patients who reported vaginal bleeding during the prestudy period, 62% (13 out of 21 patients) reported no bleeding for a 6-month period and 33% (7 out of 21 patients) reported no vaginal bleeding for the duration of the trial. Not all patients improved on treatment and a few patients not reporting vaginal bleeding in the 6 months prior to enrollment reported menses on treatment.
Tamoxifen therapy was associated with a reduction in mean rate of increase of bone age (defined as the ratio of bone age change and chronological age); individual responses with regard to this endpoint were highly heterogeneous. Linear growth rate (height velocity) was reduced during the course of tamoxifen treatment in a majority of patients (mean change of 1.68 cm/year relative to baseline; change from 7.47 cm/year at baseline to 5.79 cm/year on study). This change was not uniformly seen across all stages of bone maturity; all recorded response failures occurred in patients with bone ages less than 7 years at screening.
Mean uterine volume increased after 6 months of treatment and doubled at the end of the one-year study. A causal relationship to tamoxifen has not been established; however, as an increase in the incidence of endometrial adenocarcinoma and uterine sarcoma has been noted in adults treated with tamoxifen, continued monitoring of McCune-Albright patients treated with tamoxifen for long-term uterine effects is recommended.
Pharmacokinetics in Pediatric Patients
The pharmacokinetics of tamoxifen and N-desmethyl tamoxifen were characterized using a population pharmacokinetic analysis with sparse samples per patient obtained from 27 female pediatric patients aged 2 to 10 years enrolled in a study of tamoxifen in treating McCune-Albright syndrome. Clearance (CL/F), adjusted for body weight, in female pediatric patients was approximately 2.3-fold higher than in adult female breast cancer patients. In the youngest cohort of female pediatric patients (2 to 6 year olds), CL/F was 2.6-fold higher; in the oldest cohort (7 to 10.9 year olds), CL/F was approximately 1.9-fold higher. Exposure to N-desmethyl tamoxifen was comparable between the pediatric and adult patients.
In pediatric patients, an average steady-state peak plasma concentration (Css, max) and AUC were 187 ng/mL and 4,110 ng hr/mL, respectively, and Css, max occurred approximately 8 hours after dosing.
Animal Data
Direct neonatal exposure of tamoxifen to mice and rats produced: 1) reproductive tract lesions in female rodents (similar to those seen in humans after intrauterine exposure to DES) and 2) functional defects of the reproductive tract in male rodents such as testicular atrophy and arrest of spermatogenesis.
8.5 Geriatric Use
Ductal Carcinoma in Situ
In the NSABP B-24 trial, the percentage of women at least 65 years of age was 23%. Women at least 70 years of age accounted for 10% of participants. A total of 14 and 12 invasive breast cancers were seen among participants 65 and older in the placebo and tamoxifen groups, respectively. This subset is too small to reach any conclusions on efficacy. Across all other endpoints, the results in this subset were comparable to those of younger women enrolled in this trial. No overall differences in tolerability were observed between older and younger patients.
Reduction in Breast Cancer Incidence in Women at High Risk
In the NSABP P-1 trial, the percentage of women at least 65 years of age was 16%. Women at least 70 years of age accounted for 6% of the participants. A reduction in breast cancer incidence was seen among participants in each of the subsets. A total of 28 and 10 invasive breast cancers were seen among participants 65 and older in the placebo and tamoxifen groups, respectively. Across all other outcomes, the results in this subset reflect the results observed in the subset of women at least 50 years of age. No overall differences in tolerability were observed between older and younger patients.
10. Overdosage
No specific treatment for SOLTAMOX overdosage is recommended, other than symptomatic treatment.
In a study of advanced metastatic cancer patients to determine the maximum tolerated dose of tamoxifen that could reverse multidrug resistance, the following adverse events were observed: acute neurotoxicity manifested by tremor, hyperreflexia, unsteady gait, and dizziness. These symptoms occurred within 3 to 5 days of beginning tamoxifen and cleared within 2 to 5 days after stopping therapy. No permanent neurologic toxicity was noted. One patient experienced a seizure several days after tamoxifen was discontinued and neurotoxic symptoms had resolved. The causal relationship of the seizure to tamoxifen therapy is unknown. Doses given in these patients were all greater than 400 mg/m2 loading dose (at least 6-fold higher than the recommended dosage), followed by maintenance doses of 150 mg/m2 of tamoxifen given twice a day (at least 6-fold higher than the recommended dosage).
In the same study, prolongation of the QT interval on the electrocardiogram was noted when patients were given doses higher than 250 mg/m2 loading dose (at least 6-fold higher than the recommended dose) , followed by maintenance doses of 80 mg/m2 of tamoxifen given twice a day. For a woman with a body surface area of 1.5 m2, the minimal loading dose and maintenance doses at which neurological symptoms and QT changes occurred were at least 6-fold higher in respect to the maximum recommended dose.
Signs observed at the highest doses following studies to determine LD50 in animals were respiratory difficulties and convulsions.
11. Soltamox Description
SOLTAMOX (tamoxifen citrate) oral solution is for oral administration. Tamoxifen citrate is an estrogen agonist/antagonist.
Chemically, tamoxifen is the trans-isomer of a triphenylethylene derivative. The chemical name is (Z)2-[4-(1,2- diphenyl-1-butenyl)phenoxy]-N,N-dimethylethanamine 2-hydroxy-1,2,3- propanetricarboxylate (1:1). The structural formula, empirical formula, and molecular weight are as follows:
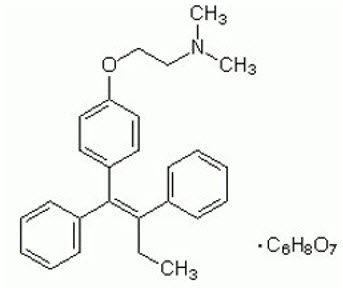 C32H37NO8 M.W. 563.62 |
Tamoxifen citrate has a pKa′ of 8.85. The equilibrium solubility in water at 37°C is 0.5 mg/mL, and is 0.2 mg/mL in 0.02 N HCl at 37°C.
Each 10 mL of SOLTAMOX oral solution contains 20 mg tamoxifen, equivalent to 30.4 mg tamoxifen citrate. The oral solution is a sugar-free, clear colorless liquid, with licorice and aniseed odor and taste supplied in a 150 mL bottle with a dosing cup. SOLTAMOX oral solution contains the following inactive ingredients: ethanol, glycerol, propylene glycol, sorbitol solution, licorice flavor, aniseed flavor, purified water.
12. Soltamox - Clinical Pharmacology
12.1 Soltamox Mechanism of Action
Tamoxifen is an estrogen agonist/antagonist. Tamoxifen competes with estrogen for binding to the estrogen receptor, which can result in a decrease in estrogen receptor signaling-dependent growth in breast tissue. Tamoxifen has demonstrated antitumor activity against human breast cancer cell lines xenografted in mice. The drug has been shown to inhibit the induction of rat mammary carcinoma induced by dimethylbenzanthracene (DMBA) and to cause the regression of already established DMBA-induced tumors.
12.3 Pharmacokinetics
Absorption and Distribution
Following a single oral dose of 20 mg tamoxifen, an average peak plasma concentration of 40 ng/mL (range 35 to 45 ng/mL) occurred approximately 5 hours after dosing. The decline in plasma concentrations of tamoxifen is biphasic with a terminal elimination half-life of about 5 to 7 days. The average peak plasma concentration of N- desmethyl tamoxifen, the major metabolite, is 15 ng/mL (range 10 to 20 ng/mL). Chronic administration of 10 mg tamoxifen given twice daily for 3 months to patients results in average steady-state plasma concentrations of 120 ng/mL (range 67 to 183 ng/mL) for tamoxifen and 336 ng/mL (range 148 to 654 ng/mL) for N-desmethyl tamoxifen. The average steady-state plasma concentrations of tamoxifen and N-desmethyl tamoxifen after administration of 20 mg tamoxifen once daily for 3 months are 122 ng/mL (range 71 to 183 ng/mL) and 353 ng/mL (range 152 to 706 ng/mL), respectively. The steady-state plasma concentrations of endoxifen and 4-hydroxytamoxifen are 29.1 (95% CI 24.6 to 33.6) and 3.7 (95% CI 3.3 to 4.1) ng/mL, respectively. After initiation of therapy, steady-state concentrations for tamoxifen are achieved in about 4 weeks and steady-state concentrations for N-desmethyl tamoxifen are achieved in about 8 weeks, suggesting a half-life of approximately 14 days for this metabolite. In a steady-state, crossover study of 10 mg tamoxifen tablets given twice a day vs. a 20 mg tamoxifen tablet given once daily, the 20 mg tamoxifen tablet was bioequivalent to the 10 mg tamoxifen tablets.
A pharmacokinetic study was performed in healthy perimenopausal and postmenopausal female subjects to evaluate the bioavailability of SOLTAMOX (n=30) in comparison with the commercially available tamoxifen citrate tablets (n=33) under fasting conditions. A third arm evaluated the effect of food on SOLTAMOX (n=16). The rate and extent of absorption of SOLTAMOX was found to be bioequivalent to that of tamoxifen citrate tablets under fasting conditions. There was no difference in bioavailability (Cmax and AUC) of SOLTAMOX oral solution between fed and fasting states, and therefore SOLTAMOX can be given without regard to meals.
Metabolism
Tamoxifen is extensively metabolized by CYP450 enzymes, including CYP3A, CYP2D6, CYP2C9, CYP2C19, and CYP2B6. N-desmethyltamoxifen, formed predominantly by CYP3A, is the major metabolite found in plasma. The pharmacological activity of N-desmethyltamoxifen is similar to that of tamoxifen. Endoxifen and 4-hydroxytamoxifen, identified as minor metabolites, have 100-fold greater affinity for the estrogen receptor and 30 to 100-fold greater potency in suppressing estrogen-dependent cell proliferation than tamoxifen. The polymorphic enzyme CYP2D6 is involved in the formation of endoxifen and 4-hydroxytamoxifen, and it is the key enzyme that catalyzes the formation of endoxifen from N-desmethyltamoxifen. Endoxifen concentrations may differ among patients because of various CYP2D6 genotypes [see Clinical Pharmacology (12.5)]. Phase 2 enzymes, such as SULT1A1, UGT2B7, and UGT1A4, are associated with tamoxifen clearance from plasma.
Excretion
Studies in women receiving 20 mg of 14C tamoxifen showed that approximately 65% of the administered dose was excreted from the body over a period of 2 weeks, with fecal excretion as the primary route of elimination. The drug is excreted mainly as polar conjugates, with unchanged drug and unconjugated metabolites accounting for less than 30% of the total fecal radioactivity.
Specific Populations
The effects of age, gender, and race on the pharmacokinetics of tamoxifen in adults have not been determined. Some data are available on tamoxifen pharmacokinetics in children [see Use in Specific Populations (8.4)]. The effects of reduced liver function on the metabolism and pharmacokinetics of tamoxifen have not been determined.
Drug-Drug Interactions
Aromatase Inhibitors
Anastrozole
In the ATAC trial, coadministration of anastrozole and tamoxifen in breast cancer patients reduced the anastrozole plasma concentration by 27% compared to that achieved with anastrozole alone; however, the coadministration did not affect the pharmacokinetics of tamoxifen or N-desmethyltamoxifen [see Clinical Studies (14.2)].
CYP P450 3A4 Inducers
Rifampin, a cytochrome P450 3A4 inducer, reduced tamoxifen AUC and Cmax by 86% and 55%, respectively.
CYP2D6 Inhibitors
Although concomitant administration of CYP2D6 inhibitors reduces the plasma concentration of endoxifen, a potent metabolite, the clinical significance is not well established [see Drug Interactions (7.4)]. The mean steady-state endoxifen plasma concentration in patients taking CYP2D6 inhibitors was significantly reduced compared to those not taking concomitant CYP2D6 inhibitors (14.8 ± 10.6 versus 26.7 ± 15.4 ng/mL). The mean steady-state plasma concentration of endoxifen in CYP2D6 normal metabolizers who were not receiving CYP2D6 inhibitors was 31.4 ± 14.7 ng/mL compared to 8.8 ± 3.5 ng/mL in CYP2D6 normal metabolizers receiving potent CYP2D6 inhibitors (e.g., paroxetine, fluoxetine) with tamoxifen. The plasma levels of endoxifen in CYP2D6 normal metabolizers taking potent CYP2D6 inhibitors were similar to the levels observed in CYP2D6 poor metabolizers taking no CYP2D6 inhibitors (8.8 versus 7.2 ng/mL).
12.5 Pharmacogenomics
The impact of CYP2D6 polymorphisms on the efficacy of tamoxifen is not well established.
CYP2D6 poor metabolizers carrying two non-functional alleles exhibit significantly lower endoxifen plasma concentrations compared to patients carrying one or more fully functional alleles of CYP2D6.
In patients with estrogen receptor-positive breast cancer who were participating in the WHEL (Women's Health Eating and Living) Study (NCT00003787), the mean (SD) serum concentration of endoxifen was 22.8 (11.3), 15.9 (9.2), 8.1 (4.9) and 5.6 (3.8) ng/mL in 27 ultrarapid, 1,097 normal, 164 intermediate and 82 poor metabolizers (p<0.001), respectively. This finding is consistent with other published studies that report lower endoxifen concentrations in poor metabolizers compared to normal metabolizers.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A carcinogenesis study in rats at doses of 5, 20, and 35 mg/kg/day (approximately 1, 3, and 7× higher than the daily maximum recommended human dose on a mg/m2 basis) administered by oral gavage for up to 2 years resulted in a significant increase in hepatocellular carcinoma at all doses. The incidence of these tumors was significantly greater among rats administered 20 or 35 mg/kg/day (69%) compared to those administered 5 mg/kg/day (14%). In a separate study, rats were administered tamoxifen at 45 mg/kg/day (approximately 10× the daily maximum recommended human dose on a mg/m2 basis); hepatocellular neoplasia was exhibited at 3 to 6 months. In addition, granulosa cell ovarian tumors and interstitial cell testicular tumors were observed in two separate mouse studies. The mice were administered the trans and racemic forms of tamoxifen for 13 to 15 months at doses of 5, 20, and 50 mg/kg/day (approximately 0.5, 2, and 5× the daily recommended human dose on a mg/m2 basis).
Tamoxifen has been found to increase levels of micronucleus formation in vitro in the human lymphoblastoid cell line (MCL-5). No genotoxic potential was found in a conventional battery of in vivo and in vitro assays with prokaryotic and eukaryotic test systems with or without drug metabolizing enzymes; however, increased levels of DNA adducts were observed by 32P post-labeling in DNA from rat liver and cultured human lymphocytes. Based on these findings, tamoxifen is genotoxic.
Tamoxifen produced impairment of fertility and conception in female rats at doses of 0.04 mg/kg/day (about 0.01-fold the daily maximum recommended human dose on a mg/m2 basis) when dosed for two weeks prior to mating through day 7 of pregnancy. At this dose, fertility and reproductive indices were markedly reduced with total fetal mortality.
14. Clinical Studies
14.1 Metastatic Breast Cancer
Premenopausal Women
Three prospective, randomized studies compared another formulation of tamoxifen to ovarian ablation (oophorectomy or ovarian irradiation) in premenopausal women with advanced breast cancer. Although the objective response rate, time to treatment failure, and survival were similar with both treatments, the limited patient accrual prevented a demonstration of equivalence. In an overview analysis of survival data from the 3 studies, the hazard ratio (HR) for death (tamoxifen/ovarian ablation) was 1.00 with two-sided 95% confidence intervals (CI) of 0.73 to 1.37. Limited number of patients with disease progression during tamoxifen therapy responded to subsequent ovarian ablation.
Men with Metastatic Breast Cancer
Published results from 122 patients (119 evaluable) and case reports in 16 patients (13 evaluable) treated with another formulation of tamoxifen have shown that tamoxifen is effective for the palliative treatment of men with metastatic breast cancer. Sixty-six of these 132 evaluable patients responded to tamoxifen, which constitutes a 50% objective response rate.
14.2 Adjuvant Treatment of Breast Cancer
Pooled Studies of Adjuvant Treatment of Breast Cancer
The Early Breast Cancer Trialists' Collaborative Group (EBCTCG) conducted worldwide overviews of systemic adjuvant therapy for early breast cancer in 1985, 1990, 1995, 1998 and 2011.
The 10-year outcome data were reported in 1998 for 36,689 women in 55 randomized trials of another formulation of adjuvant tamoxifen using doses of 20 to 40 mg per day for 1 to 5+ years. Twenty-five percent of patients received 1 year or less of trial treatment, 52% received 2 years, and 23% received about 5 years. Forty-eight percent of tumors were estrogen receptor (ER)- positive (>10 fmol/mg), 21% were ER-poor (<10 fmol/mg), and 31% were ER-unknown. Among 29,441 patients with ER-positive or ER-unknown breast cancer, 58% were entered into trials comparing tamoxifen to no adjuvant therapy and 42% were entered into trials comparing tamoxifen in combination with chemotherapy vs. the same chemotherapy alone. Among these patients, 54% had node-positive disease and 46% had node-negative disease.
In women with ER-positive or ER-unknown breast cancer:
- With positive nodes who received about 5 years of treatment, overall survival at 10 years was 61.4% for tamoxifen vs. 50.5% for control (log-rank 2p <0.00001). The recurrence-free rate at 10 years was 59.7% for tamoxifen vs. 44.5% for control (log-rank 2p <0.00001).
- With negative nodes who received about 5 years of treatment, overall survival at 10 years was 78.9% for tamoxifen vs. 73.3% for control (log-rank 2p <0.00001). The recurrence-free rate at 10 years was 79.2% for tamoxifen vs. 64.3% for control (log-rank 2p <0.00001).
- Who received 1 year or less, 2 years, or about 5 years of tamoxifen, the proportional reductions in mortality were 12%, 17%, and 26%, respectively (2p <0.003). The corresponding reductions in breast cancer recurrence were 21%, 29%, and 47% (2p <0.00001).
Results in patients with ER-poor breast cancer
- Benefit is less clear for women with ER-poor breast cancer in whom the proportional reduction in recurrence was 10% (2p = 0.007) for all durations taken together, or 9% (2p = 0.02) if contralateral breast cancers are excluded. The corresponding reduction in mortality was 6% (not significant).
The effects of about 5 years of tamoxifen on recurrence and mortality were similar regardless of age and concurrent chemotherapy.
The 15-year outcome data were reported in 2011 for 21,457 women in 20 randomized trials, and continued to confirm the earlier overview results. Five-year treatment with adjuvant tamoxifen reduced the 15-year risks of breast cancer recurrence and death.
Node-positive: Individual Studies
Two studies [Hubay and National Surgical Adjuvant Breast and Bowel Project (NSABP) B-09] demonstrated an improved disease-free survival following radical or modified radical mastectomy in postmenopausal women or women 50 years of age or older with surgically curable breast cancer with positive axillary nodes when another formulation of tamoxifen was added to adjuvant cytotoxic chemotherapy. In the Hubay study, tamoxifen was added to "low-dose" CMF (cyclophosphamide, methotrexate, and fluorouracil). In the NSABP B-09 study, tamoxifen was added to melphalan and fluorouracil.
In the Hubay study, patients with a positive (more than 3 fmol) estrogen receptor were more likely to benefit. In the NSABP B-09 study in women age 50 to 59 years, only women with both estrogen and progesterone receptor levels 10 fmol or greater clearly benefited, while survival results were poorer in women with both estrogen and progesterone receptor levels less than 10 fmol. In women age 60 to 70 years, there was an improvement in disease-free survival with tamoxifen without any clear relationship to estrogen or progesterone receptor status.
Three prospective studies (ECOG-1178, Toronto, and Tamoxifen Adjuvant Trial Organization (NATO)] using another formulation of tamoxifen adjuvantly as a single agent demonstrated improved disease-free survival following total mastectomy and axillary dissection for postmenopausal women with positive axillary nodes compared to placebo/no treatment controls. The NATO study also demonstrated an overall survival benefit.
Node–negative: Individual Studies
NSABP B-14, a prospective, double-blind, randomized study, compared another formulation of tamoxifen to placebo as adjuvant therapy in women with axillary node-negative, estrogen-receptor positive (≥10 fmol/mg cytosol protein) breast cancer (following total mastectomy and axillary dissection, or segmental resection, axillary dissection, and breast radiation). After five years of treatment, there was a significant improvement in disease-free survival in women receiving tamoxifen. This benefit was apparent both in women under age 50 and in women at or beyond age 50.
One additional randomized study (NATO) demonstrated improved disease-free survival for another formulation of tamoxifen compared to no adjuvant therapy following total mastectomy and axillary dissection in postmenopausal women with axillary node-negative breast cancer. In this study, the benefits of tamoxifen appeared to be independent of estrogen receptor status.
Duration of Therapy
Available data supported 5 years of adjuvant tamoxifen therapy for patients with breast cancer. In the EBCTCG 1995 overview, the reduction in recurrence and mortality was greater in those studies that used tamoxifen for about 5 years than in those that used tamoxifen for a shorter period of therapy. Although results of the NSABP B-14 study suggest that continuation of therapy beyond five years does not provide additional benefit, a subsequent report of the ATLAS trial suggest that continuing tamoxifen to 10 years further reduced recurrence and mortality.
5 Years vs. Shorter Duration
In the EBCTCG 1995 overview, the reduction in recurrence and mortality was greater in those studies that used tamoxifen for about 5 years than in those that used tamoxifen for a shorter period of therapy.
In a Swedish Breast Cancer Cooperative Group trial of another formulation of adjuvant tamoxifen 40 mg per day (two times the recommended dosage) for 2 or 5 years, overall survival at 10 years was estimated to be 80% in the patients in the 5 year tamoxifen group, compared with 74% among corresponding patients in the 2 year treatment group (p = 0.03). Disease-free survival at 10 years was 73% in the 5 year group and 67% in the 2 year group (p = 0.009). Compared with 2 years of tamoxifen treatment, 5 years of treatment resulted in a slightly greater reduction in the incidence of contralateral breast cancer at 10 years, but this difference was not statistically significant.
5 Years vs. Longer Duration
In the ATLAS trial (Adjuvant Tamoxifen: Longer Against Shorter), 6,846 women with early breast cancer and ER-positive disease who had completed 5 years of treatment with another formulation of tamoxifen were randomly allocated to continue tamoxifen to 10 years or to stop at 5 years. The cumulative risk of recurrence for women randomized to continue tamoxifen was reduced during years 5-9 [recurrence rate ratio of 0.90 (95% CI: 0.79, 1.02)] and further reduced in later years [0.75 (95% CI: 0.62, 0.90)]. Breast cancer mortality in women who received tamoxifen for up to 10 years was also reduced by about 13% [HR = 0.87 (95% CI 0.78, 0.97)].
In the NSABP B-14 trial, in which patients were randomized to another formulation of tamoxifen 20 mg per day for 5 years vs. placebo, those who were disease-free at the end of this 5 year period were offered rerandomization to an additional 5 years of tamoxifen or placebo. With 4 years of follow-up after this rerandomization, 92% of the women that received 5 years of tamoxifen were alive and disease-free, compared to 86% of the women scheduled to receive 10 years of tamoxifen (p = 0.003). Overall survivals were 96% and 94%, respectively (p = 0.08). Results of the B-14 study suggest that continuation of tamoxifen beyond 5 years does not provide additional benefit.
A Scottish trial of 5 years of another formulation of tamoxifen vs. indefinite treatment found a disease-free survival of 70% in the five-year group and 61% in the indefinite group, with 6.2 years median follow-up (HR = 1.27, 95% CI: 0.87 to 1.85).
Contralateral Breast Cancer
The incidence of contralateral breast cancer was reduced in breast cancer patients (premenopausal and postmenopausal) receiving adjuvant tamoxifen compared to placebo. Data on contralateral breast cancer are available from 32,422 out of 36,689 patients in the 1995 overview analysis of the EBCTCG. In clinical trials with tamoxifen of 1 year or less, 2 years, and about 5 years duration, the proportional reductions in the incidence rate of contralateral breast cancer among women receiving tamoxifen were 13% (not significant), 26% (2p = 0.004) and 47% (2p <0.00001), respectively. The reduction in the incidence of contralateral breast cancer was significant with longer tamoxifen duration (2p = 0.008). The proportional reductions in the incidence of contralateral breast cancer were independent of age and ER status of the primary tumor. Treatment with about 5 years of tamoxifen reduced the annual incidence rate of contralateral breast cancer from 7.6 per 1,000 patients in the control group to 3.9 per 1,000 patients in the tamoxifen group.
In a Swedish Breast Cancer Cooperative Group trial where another formulation of adjuvant tamoxifen was given at a dose of 40 mg per day (two times the recommended dosage) for 2 to 5 years, the incidence of second primary breast tumors was reduced 40% (p <0.008) on tamoxifen compared to control. In the NSABP B-14 trial in which patients were randomized to tamoxifen 20 mg per day for 5 years vs. placebo, the incidence of second primary breast cancers was also significantly reduced (p <0.01). The annual rate of contralateral breast cancer was 8.0 per 1,000 patients in the placebo group compared to 5.0 per 1,000 patients in the tamoxifen group, at 10 years after first randomization.
Anastrozole Adjuvant Trial (ATAC: Arimidex, Tamoxifen, Alone or in Combination) – Study of Anastrozole Compared to Tamoxifen for Adjuvant Treatment of Early Breast Cancer
A trial was conducted in 9,366 postmenopausal women with operable breast cancer who were randomized to receive adjuvant treatment with either anastrozole 1 mg daily, another formulation of tamoxifen 20 mg daily, or a combination of these two treatments for 5 years or until recurrence of the disease. At a median follow-up of 33 months, the combination of anastrozole and tamoxifen did not demonstrate any efficacy benefit when compared to tamoxifen alone in all patients, as well as in the hormone receptor-positive subpopulation. The combination treatment arm was discontinued from the trial [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)]. Refer to the full prescribing information for anastrozole tablets for additional information on this trial.
14.3 Ductal Carcinoma in Situ
NSABP B-24 was a double-blind, randomized trial in women with DCIS that compared the addition of another formulation of tamoxifen or placebo to treatment with lumpectomy and radiation therapy. The primary objective was to determine whether 5 years of tamoxifen therapy (20 mg per day) reduced the incidence of invasive breast cancer in the ipsilateral or contralateral breast. In this trial, 1,804 women were randomized to receive either tamoxifen or placebo for 5 years: 902 women were randomized to tamoxifen 10 mg tablets twice a day and 902 women were randomized to placebo. Follow-up data were available for 1,798 women and the median duration of follow-up was 74 months.
The tamoxifen and placebo groups were balanced for baseline demographic and prognostic factors. Over 80% of the tumors were less than or equal to 1 cm in their maximum dimension, were not palpable, and were detected by mammography alone. Over 60% of the study population was postmenopausal. In 16% of patients, the margin of the resected specimen was reported as being positive after surgery. Approximately half of the tumors were reported to contain comedo necrosis.
The incidence of invasive breast cancer was reduced by 43% among women assigned to tamoxifen (44 cases on tamoxifen, 74 cases on placebo; p = 0.004; RR = 0.57, 95% CI: 0.39 to 0.84). No data are available regarding the ER status of the invasive cancers. The stage distribution of the invasive cancers at diagnosis was similar to that reported annually in the SEER data base.
Results are shown in Table 6. At 5 years from study entry, survival was similar in the placebo and tamoxifen groups.
| Type of Event | Lumpectomy, Radiotherapy, and Placebo N=902 | Lumpectomy, Radiotherapy, and Tamoxifen N=902 | RR | 95% CI | ||
|---|---|---|---|---|---|---|
| No. of events | Rate per 1,000 women per year | No. of events | Rate per 1,000 women per year | |||
|
||||||
| Invasive Breast Cancer (Primary Endpoint) | 74 | 16.73 | 44 | 9.60 | 0.57 | 0.39 to 0.84 |
| Ipsilateral | 47 | 10.61 | 27 | 5.90 | 0.56 | 0 |
| Contralateral | 25 | 5.64 | 17 | 3.71 | 0.66 | 0 |
| Side undetermined | 2 | -- | 0 | -- | -- | |
| Secondary Endpoints | ||||||
| DCIS | 56 | 12.66 | 41 | 8.95 | 0.71 | 0 |
| Ipsilateral | 46 | 10.40 | 38 | 8.29 | 0.88 | 0 |
| Contralateral | 10 | 2.26 | 3 | 0.65 | 0.29 | 0 |
| All breast cancer events | 129 | 29.16 | 84 | 18.34 | 0.63 | 0 |
| All ipsilateral events | 96 | 21.70 | 65 | 14.19 | 0.65 | 0 |
| All contralateral events | 37 | 8.36 | 20 | 4.37 | 0.52 | 0 |
| Deaths | 32 | 28 | ||||
| Uterine malignancies* | 4 | 9 | ||||
| Endometrial adenocarcinoma* | 4 | 0.57 | 8 | 1.15 | ||
| Uterine sarcoma* | 0 | 0 | 1 | 0.14 | ||
| Second primary malignancies (other than endometrial and breast) | 30 | 29 | ||||
| Stroke | 2 | 7 | ||||
| Thromboembolic events (DVT, PE) | 5 | 15 | ||||
14.4 Reduction in Breast Cancer Incidence in Women at High Risk
Breast Cancer Prevention Trial (NSABP P-1)
The Breast Cancer Prevention Trial (NSABP P-1) was a double-blind, randomized, placebo-controlled trial with a primary objective to determine whether 5 years of another formulation of tamoxifen therapy (20 mg per day) reduced the incidence of invasive breast cancer in women at high risk for the disease. Secondary objectives included an evaluation of the incidence of ischemic heart disease; the effects on the incidence of bone fractures; and other events associated with the use of tamoxifen, including endometrial cancer, pulmonary embolus, deep vein thrombosis, stroke, and cataract formation and surgery.
The Breast Cancer Assessment Tool1 (the Gail Model) was used to calculate predicted breast cancer risk for women who were less than 60 years of age and did not have lobular carcinoma in situ (LCIS). The following risk factors were used: age; number of first-degree female relatives with breast cancer; previous breast biopsies; presence or absence of atypical hyperplasia; nulliparity; age at first live birth; and age at menarche. A 5-year predicted risk of breast cancer of ≥1.67% was required for entry into the trial.
In this trial, 13,388 women of at least 35 years of age were randomized to receive either tamoxifen or placebo for five years. The median duration of treatment was 3.5 years. Follow-up data were available for 13,114 women. Twenty-seven percent of women randomized to placebo (1,782) and 24% of women randomized to tamoxifen (1,596) completed 5 years of therapy.
Demographics
The demographic characteristics of women on the trial with follow-up data are shown in Table 7.
| Characteristic | Placebo N=6,570 | Tamoxifen N=6,544 | ||
|---|---|---|---|---|
| # | % | # | % | |
|
||||
| Age (yrs.) | ||||
| 35 to 39 | 184 | 3 | 158 | 2 |
| 40 to 49 | 2,394 | 36 | 2,411 | 37 |
| 50 to 59 | 2,011 | 31 | 2,019 | 31 |
| 60 to 69 | 1,588 | 24 | 1,563 | 24 |
| ≥70 | 393 | 6 | 393 | 6 |
| Age at first live birth (yrs.) | ||||
| Nulliparous | 1,202 | 18 | 1,205 | 18 |
| 12 to 19 | 915 | 14 | 946 | 15 |
| 20 to 24 | 2,448 | 37 | 2,449 | 37 |
| 25 to 29 | 1,399 | 21 | 1,367 | 21 |
| ≥30 | 606 | 9 | 577 | 9 |
| Race | ||||
| White | 6,333 | 96 | 6,323 | 96 |
| Black | 109 | 2 | 103 | 2 |
| Other | 128 | 2 | 118 | 2 |
| Age at menarche | ||||
| ≥14 | 1,243 | 19 | 1,170 | 18 |
| 12 to 13 | 3,610 | 55 | 3,610 | 55 |
| ≤11 | 1,717 | 26 | 1,764 | 27 |
| # of first degree relatives with breast cancer | ||||
| 0 | 1,584 | 24 | 1,525 | 23 |
| 1 | 3,714 | 57 | 3,744 | 57 |
| 2+ | 1,272 | 19 | 1,275 | 20 |
| Prior hysterectomy | ||||
| No | 4,173 | 63.5 | 4,018 | 62.4 |
| Yes | 2,397 | 36.5 | 2,464 | 37.7 |
| # of previous breast biopsies | ||||
| 0 | 2,935 | 45 | 2,923 | 45 |
| 1 | 1,833 | 28 | 1,850 | 28 |
| ≥2 | 1,802 | 27 | 1,771 | 27 |
| History of atypical hyperplasia in the breast | ||||
| No | 5,958 | 91 | 5,969 | 91 |
| Yes | 612 | 9 | 575 | 9 |
| History of LCIS at entry | ||||
| No | 6,165 | 94 | 6,135 | 94 |
| Yes | 405 | 6 | 409 | 6 |
| 5-year predicted breast cancer risk (%) * | ||||
| ≤2.00 | 1,646 | 25 | 1,626 | 25 |
| 2.01 to 3.00 | 2,028 | 31 | 2,057 | 31 |
| 3.01 to 5.00 | 1,787 | 27 | 1,707 | 26 |
| ≥5.01 | 1,109 | 17 | 1,162 | 18 |
Results
Results are shown in Table 8. After a median follow-up of 4.2 years, the incidence of invasive breast cancer was reduced by 44% among women assigned to tamoxifen (86 cases on tamoxifen, 156 cases on placebo; p <0.00001; RR = 0.56, 95% CI: 0.43 to 0.72). A reduction in the incidence of breast cancer was seen in each prospectively specified age group (≤49, 50-59, ≥60), in women with or without LCIS, and in each of the absolute risk levels specified in Table 8. A non-significant decrease in the incidence of DCIS was seen (23 tamoxifen, 35 placebo; RR = 0.66, 95% CI: 0.39 to 1.11).
There was no statistically significant difference in the number of myocardial infarctions, severe angina, or acute ischemic cardiac events between the two groups (61 tamoxifen, 59 placebo; RR = 1.04, 95% CI: 0.73 to 1.49).
No overall difference in mortality (53 deaths in tamoxifen group vs. 65 deaths in placebo group) was present. No difference in breast cancer-related mortality was observed (4 deaths in tamoxifen group vs. 5 deaths in placebo group).
Although there was a non-significant reduction in the number of hip fractures (9 on tamoxifen, 20 on placebo) in the tamoxifen group, the number of wrist fractures was similar in the two treatment groups (69 on tamoxifen, 74 on placebo). A subgroup analysis of the trial suggested a difference in effect on bone mineral density (BMD) related to menopausal status in patients receiving tamoxifen. In postmenopausal women, there was no evidence of bone loss of the lumbar spine and hip. Conversely, tamoxifen was associated with significant bone loss of the lumbar spine and hip in premenopausal women.
Tamoxifen increased the risk for endometrial cancer, DVT, PE, stroke, cataract formation, and cataract surgery (see Table 8).
Table 8 summarizes the major outcomes of the NSABP P-1 trial. For most participants, multiple risk factors would have been required for eligibility. This table considers risk factors individually, regardless of other co- existing risk factors, for women who developed breast cancer. The 5-year predicted absolute breast cancer risk accounts for multiple risk factors in an individual and should provide the best estimate of individual benefit.
| Type of Event | # of events | Rate per 1,000 Women per Year | RR | 95% CI | ||
|---|---|---|---|---|---|---|
| Placebo | Tamoxifen | Placebo | Tamoxifen | |||
|
||||||
| Invasive Breast Cancer | 156 | 86 | 6.49 | 3.58 | 0.56 | 0.43 to 0.72 |
| 5-year predicted breast cancer risk (as calculated by the Breast Cancer Assessment Tool) | ||||||
| ≤2.00% | 31 | 13 | 5.36 | 2.26 | 0.42 | 0.22 to 0.81 |
| 2.01 to 3.00% | 39 | 28 | 5.25 | 3.83 | 0.73 | 0.45 to 1.18 |
| 3.01 to 5.00% | 36 | 26 | 5.37 | 4.06 | 0.76 | 0.46 to 1.26 |
| ≥5.00% | 50 | 19 | 13.15 | 4.71 | 0.36 | 0.21 to 0.61 |
| Individual Risk Factors | ||||||
| Age Groups | ||||||
| Age ≤49 | 59 | 38 | 6.34 | 4.11 | 0.65 | 0.43 to 0.98 |
| Age 50 to 59 | 46 | 25 | 6.31 | 3.53 | 0.56 | 0.35 to 0.91 |
| Age ≥60 | 51 | 23 | 7.17 | 3.22 | 0.45 | 0.27 to 0.74 |
| Risk factors for breast cancer history, LCIS | ||||||
| No | 140 | 78 | 6.23 | 3.51 | 0.56 | 0.43 to 0.74 |
| Yes | 16 | 8 | 12.73 | 6.33 | 0.50 | 0.21 to 1.17 |
| History, atypical hyperplasia | ||||||
| No | 138 | 84 | 6.37 | 3.89 | 0.61 | 0.47 to 0.80 |
| Yes | 18 | 2 | 8.69 | 1.05 | 0.12 | 0.03 to 0.52 |
| Number of first degree relatives with breast cancer | ||||||
| 0 | 32 | 17 | 5.97 | 3.26 | 0.55 | 0.30 to 0.98 |
| 1 | 80 | 45 | 5.81 | 3.31 | 0.57 | 0.40 to 0.82 |
| 2 | 35 | 18 | 8.92 | 4.67 | 0.52 | 0.30 to 0.92 |
| ≥3 | 9 | 6 | 13.33 | 7.58 | 0.57 | 0.20 to 1.59 |
| DCIS | 35 | 23 | 1.47 | 0.97 | 0.66 | 0.39 to 1.11 |
| Safety Endpoints | ||||||
| Fractures (protocol-specified sites) | 92 1 | 76 * | 3.87 | 3.20 | 0.61 | 0.83 to 1.12 |
| Hip | 20 | 9 | 0.84 | 0.38 | 0.45 | 0.18 to 1.04 |
| Wrist † | 74 | 69 | 3.11 | 2.91 | 0.93 | 0.67 to 1.29 |
| Total ischemic events | 59 | 61 | 2.47 | 2.57 | 1.04 | 0.71 to 1.51 |
| Myocardial infarction | 27 | 27 | 1.13 | 1.13 | 1.00 | 0.57 to 1.78 |
| Fatal | 8 | 7 | 0.33 | 0.29 | 0.88 | 0.27 to 2.77 |
| Nonfatal | 19 | 20 | 0.79 | 0.84 | 1.06 | 0.54 to 2.09 |
| Angina ‡ | 12 | 12 | 0.50 | 0.50 | 1.00 | 0.41 to 2.44 |
| Acute ischemic syndrome § | 20 | 22 | 0.84 | 0.92 | 1.11 | 0.58 to 2.13 |
| Uterine malignancies ¶ | 17 | 57 | ||||
| Endometrial adenocarcinoma ¶ | 17 | 53 | 0.71 | 2.20 | ||
| Uterine sarcoma ¶ | 1 | 4 | 0.0 | 0.17 | ||
| Stroke # | 24 | 34 | 1.00 | 1.43 | 1.42 | 0.82 to 2.51 |
| Transient ischemic attack | 21 | 18 | 0.88 | 0.75 | 0.86 | 0.43 to 1.70 |
| Pulmonary emboli Þ | 6 | 18 | 0.25 | 0.75 | 3.01 | 1.15 to 9.27 |
| Deep vein thrombosis ß | 19 | 30 | 0.79 | 1.26 | 1.59 | 0.86 to 2.98 |
| Cataracts developing on study à | 483 | 540 | 22.51 | 25.41 | 1.13 | 1.00 to 1.28 |
| Underwent cataract surgery à | 63 | 101 | 2.83 | 4.57 | 1.62 | 1.18 to 2.22 |
| Underwent cataract surgery è | 129 | 201 | 5.44 | 8.56 | 1.58 | 1.26 to 1.97 |
Table 9 describes the characteristics of the breast cancers in the NSABP P-1 trial in women at high risk for breast cancer. Tamoxifen decreased the incidence of small estrogen receptor-positive tumors, but did not alter the incidence of estrogen receptor-negative tumors or larger tumors.
| Staging Parameter | Placebo N=156 | Tamoxifen N=86 |
|---|---|---|
|
||
| Tumor size | ||
| T1 | 117 | 60 |
| T2 | 28 | 20 |
| T3 | 7 | 3 |
| T4 | 1 | 2 |
| Unknown | 3 | 1 |
| Nodal status | ||
| Negative | 103 | 56 |
| 1 to 3 positive nodes | 29 | 14 |
| ≥4 positive nodes | 10 | 12 |
| Unknown | 14 | 4 |
| Stage | ||
| I | 88 | 47 |
| II: node-negative | 15 | 9 |
| II: node-positive | 33 | 22 |
| III | 6 | 4 |
| IV | 2* | 1 |
| Unknown | 12 | 3 |
| Estrogen receptor status | ||
| Positive | 115 | 38 |
| Negative | 27 | 36 |
| Unknown | 14 | 12 |
The 7-year follow-up for the NSABP P-1 trial confirmed the initial findings. The cumulative rates of invasive and non-invasive breast cancer were reduced with tamoxifen. The risks of stroke, deep-vein thrombosis, and cataracts and of ischemic heart disease and death were also similar to those initially reported. Risks of pulmonary embolism were lower than in the original report, and risks of endometrial cancer were higher, but these differences were not statistically significant.
Italian Tamoxifen Prevention Trial and the Royal Marsden Trial
Interim results from two additional trials examining the effects of another formulation of tamoxifen in reducing breast cancer incidence are summarized below.
In the Italian Tamoxifen Prevention trial, 5,408 women without any specific risk factors for breast cancer and who had undergone a total hysterectomy were randomized to receive 20 mg tamoxifen or placebo for 5 years. The primary endpoints were occurrence of, and death from, invasive breast cancer. Hormone replacement therapy (HRT) was used in 14% of participants. The trial closed because of the large number of dropouts during the first year of treatment (26%). After 46 months of follow-up, there were 22 breast cancers in women on placebo and 19 in women on tamoxifen. Although no decrease in breast cancer incidence was observed, there was a trend for reduction in breast cancer among women receiving protocol therapy for at least 1 year (19 placebo, 11 tamoxifen). The small numbers of participants, along with the low level of risk in this otherwise healthy group precluded an adequate assessment of the effect of tamoxifen in reducing the incidence of breast cancer. Tamoxifen is not approved for women who are at low risk for breast cancer.
The Royal Marsden Trial (RMT) was also reported as an interim analysis. The RMT was undertaken as a feasibility study of whether larger scale trials could be mounted. Twenty-four hundred and seventy-one women were enrolled, based on a family history of breast cancer. HRT was used in 40% of participants. In this trial, with a 70-month median follow-up, 34 and 36 breast cancers (8 noninvasive, 4 on each arm) were observed among women on tamoxifen and placebo, respectively. Patients in this trial were younger than those in the NSABP P-1 trial and may have been more likely to develop ER-negative tumors, which are unlikely to be reduced in number by tamoxifen therapy. Although women were selected on the basis of family history and were thought to have a high risk of breast cancer, few events occurred, reducing the statistical power of the study. These factors are potential reasons why the RMT may not have provided an adequate assessment of the effectiveness of tamoxifen in reducing the incidence of breast cancer.
In these two trials, an increased number of cases of deep vein thrombosis, pulmonary embolus, stroke, and endometrial cancer were observed on the tamoxifen arm compared to the placebo arm. The frequencies of these events were consistent with the data observed in the NSABP P-1 trial.
15. References
1Cancer.gov [Internet]. Bethesda (MD):National Cancer Institute [updated 2011 May 16]. Available from: http://www.cancer.gov/bcrisktool/.
16. How is Soltamox supplied
SOLTAMOX oral solution is a sugar-free, clear colorless liquid, with licorice and aniseed odor and taste. It is supplied in a 150 mL bottle with a dosing cup intended for the measurement of SOLTAMOX oral solution only. Each 10 mL of solution contains 20 mg tamoxifen, equivalent to 30.4 mg tamoxifen citrate.
NDC# 51862-682-01
Store at room temperatures 20° to 25°C (68° - 77°F); excursions permitted between 15° to 30°C (59° - 86°F) [see USP Controlled Room Temperature].
DO NOT freeze or refrigerate.
Store in original packaging in order to protect from light. Discard any unused portion after 3 months of first opening of the bottle.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Uterine Malignancies
- Inform women that SOLTAMOX increases the risk of uterine malignancies (endometrial carcinoma and uterine sarcoma), including fatal cases. Advise women to promptly inform a healthcare provider if they experience any abnormal gynecological symptoms (e.g., menstrual irregularities, abnormal vaginal bleeding, changes in vaginal discharge, or pelvic pain or pressure.) Advise women receiving or who have previously received tamoxifen to undergo annual gynecological examinations [see Boxed Warning and Warnings and Precautions (5.1)].
Thromboembolic Events
- Inform patients that SOLTAMOX increases the risk of thromboembolic events (deep vein thrombosis, pulmonary embolism, and stroke), including fatal cases. Advise patients to seek medical attention immediately if signs or symptoms of a thromboembolic event occur [see Boxed Warning and Warnings and Precautions (5.2)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the potential that exposure during pregnancy or within 2 months prior to conception can result in fetal harm, including the potential long-term risk of a DES-like syndrome. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective nonhormonal contraception while taking SOLTAMOX and for 2 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
- Advise women not to breastfeed during treatment and for 3 months after the last dose of SOLTAMOX [see Use in Specific Populations (8.2)].
Effects on the Liver
- Inform patients that SOLTAMOX may increase the risk of liver cancer and liver abnormalities [see Warnings and Precautions (5.4)].
Distributed by:
Mayne Pharma
Greenville, NC 27834
Address medical inquiries to: Mayne Pharma 1-844-825-8500.
F8UD1W4JA2
Medication Guide
Before taking SOLTAMOX, you and your healthcare provider should talk about the possible benefits and risks.
- The benefits and risks are different for women at high risk for breast cancer and women with breast cancer that is only inside the milk ducts (ductal carcinoma in-situ or DCIS) than for women who have been diagnosed with invasive breast cancer.
- For most people who already have invasive breast cancer, the benefits of SOLTAMOX are greater than the risks.
- Talk to your healthcare provider about which risks may affect you.
What is the most important information I should know about SOLTAMOX?
SOLTAMOX can cause serious side effects, including:
-
Uterine cancer. Cancer of the lining of the uterus (endometrial adenocarcinoma) or cancer of the body of the uterus (uterine sarcoma) may happen more often in women who take SOLTAMOX and can lead to death. People who take or have taken SOLTAMOX should have a gynecologic exam every year.
SOLTAMOX can also cause other non-cancer effects on the uterus, including:- overgrowth of the lining of the uterus (hyperplasia) and uterine polyps
- endometriosis
- fibroids
- irregular menstrual periods or no menstrual periods (amenorrhea)
- irregular menstrual periods
- abnormal vaginal bleeding
- a change in your vaginal discharge
- pelvic pain or pressure
-
Blood clots in your veins or lungs. Blood clots in your veins or lungs may happen more often in people who take SOLTAMOX and can lead to death, especially in people who take SOLTAMOX during their treatment with chemotherapy.
Tell your healthcare provider right away if you get any of the following symptoms of a blood clot during treatment with SOLTAMOX:- sudden chest pain, shortness of breath, or coughing up blood
- pain, tenderness, or swelling in one or both of your legs
-
Stroke. Stroke can cause serious problems and can lead to death. Get medical help right away if you get any of these symptoms of a stroke:
- sudden weakness, tingling, or numbness of your face, arm or leg, especially on one side of your body
- sudden confusion, trouble speaking or understanding
- sudden trouble seeing in one or both eyes
- sudden trouble walking, dizziness, loss of balance or coordination
- sudden severe headache with no known cause
What is SOLTAMOX?
SOLTAMOX is a prescription medicine used:
- to treat adults with estrogen receptor-positive breast cancer that has spread to other parts of the body (metastatic)
- to treat adults with early stage estrogen receptor-positive breast cancer after surgery and radiation for breast cancer
- to reduce the chance of developing breast cancer in the other breast in adults after surgery and radiation for breast cancer
- to reduce the risk of invasive breast cancer in adult women with DCIS, after breast surgery and radiation treatment
- to reduce the risk of getting breast cancer in women with a high risk of getting breast cancer
It is not known if SOLTAMOX is safe and effective in children.
Do not take SOLTAMOX if you:
- have had a serious allergic reaction to tamoxifen or any other ingredient in SOLTAMOX. Ask your healthcare provider if you are not sure.
- have DCIS or a high-risk of breast cancer, and you
- need to take the blood thinner medicine warfarin, or
- have a history of a blood clot in your veins or lungs
Before taking SOLTAMOX, tell your healthcare provider about all of your medical conditions, including if you:
- have uterine cancer or other problems with your uterus. See "What is the most important information I should know about SOLTAMOX?"
- have irregular menstrual periods, no menstrual periods, or abnormal vaginal bleeding
- have or had a blood clot in your veins or lungs
- have had a stroke
- are pregnant or plan to become pregnant. SOLTAMOX can harm your unborn baby. You should not become pregnant during treatment with SOLTAMOX or within 2 months after you stop taking it.
Females who are able to become pregnant:- Before you start treatment with SOLTAMOX, your healthcare provider should do a pregnancy test to make sure that you are not pregnant.
- You should use effective birth control (contraception) during treatment with SOLTAMOX and for 2 months after the last dose. Talk to your healthcare provider about birth control options that you may use during this time. You should not use hormonal contraceptives such as birth control pills, patches, implants, or IUDs that contain hormones.
- Tell your healthcare provider right away if you become pregnant during treatment with SOLTAMOX.
- are breastfeeding or plan to breastfeed. It is not known if SOLTAMOX passes into your breast milk. You should not breastfeed during treatment with SOLTAMOX and for 3 months after the last dose.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Know the medicines you take. Keep a list of them to show to your healthcare provider and pharmacist when you get a new medicine.
How should I take SOLTAMOX?
- Take SOLTAMOX exactly as your healthcare provider tells you to take it.
- Your healthcare provider will tell you how much SOLTAMOX to take and when to take it.
- Your healthcare provider will decide how long you should continue to take SOLTAMOX, depending on your medical condition.
- Do not stop taking SOLTAMOX without first talking with your healthcare provider.
- Use the dosing cup that comes with SOLTAMOX to measure the correct amount for each dose. Ask your healthcare provider or pharmacist if you are not sure how to measure your dose.
- If you forget a dose of SOLTAMOX, take it when you remember, then take the next dose at your usual time. If it is almost time for your next dose, skip the missed dose. Just take the next dose at your regular time. Do not take 2 doses at the same time. If you are not sure about your dosing, call your healthcare provider.
- If you take too much SOLTAMOX, call your healthcare provider right away.
What are the possible side effects of SOLTAMOX?
SOLTAMOX can cause serious side effects, including:
- See "What is the most important information I should know about SOLTAMOX?"
- Liver problems, including liver cancer. SOLTAMOX can cause changes in liver function blood tests, and sometimes can cause liver cancer and other serious liver problems that can lead to death. Your healthcare provider should do blood tests to check you for these problems during treatment with SOLTAMOX.
- Other cancers.
- High levels of calcium in the blood (hypercalcemia). People with breast cancer that has spread to their bones may develop hypercalcemia. Hypercalcemia can happen within a few weeks after you start taking SOLTAMOX. Your healthcare provider should check you for this problem. If it is severe, your healthcare provider may tell you to stop taking SOLTAMOX.
- Decreased blood cell counts. SOLTAMOX can cause a decrease in platelet counts, red blood cell counts, and white blood cell counts that can be severe. Your healthcare provider should check your blood counts during treatment with SOLTAMOX.
- Eye problems. SOLTAMOX can increase your chance of developing cataracts and other eye problems. Tell your healthcare provider about any changes in your vision or other eye symptoms during treatment with SOLTAMOX.
The most common side effects of SOLTAMOX include:
- hot flashes
- mood changes
- vaginal discharge
- vaginal bleeding. See "What is the most important information I should know about SOLTAMOX?"
- nausea
- swelling (fluid retention)
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of SOLTAMOX.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store SOLTAMOX?
- Store SOLTAMOX at room temperature between 68°F to 77°F (20°C to 25°C). Do not freeze or refrigerate.
- Keep SOLTAMOX in the original bottle to protect it from light.
- Keep the bottle tightly closed.
- Use within 3 months of opening. Throw away any SOLTAMOX remaining in the bottle after 3 months.
Keep SOLTAMOX and all medicines out of the reach of children.
General information about the safe and effective use of SOLTAMOX.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use SOLTAMOX for a condition for which it was not prescribed. Do not give SOLTAMOX to other people, even if they have the same symptoms that you have. It may harm them. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about SOLTAMOX that is written for health professionals.
What are the ingredients in SOLTAMOX?
Active ingredient: tamoxifen citrate
Inactive ingredients: ethanol, glycerol, propylene glycol, sorbitol solution, licorice flavor, aniseed flavor, purified water.
Distributed by:
Mayne Pharma
Greenville, NC 27834
The brands listed (other than SOLTAMOX®) are registered trademarks of their respective owners.
For more information, call 1-844-825-8500.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: 11/2020
F8UD1W4JB2
| SOLTAMOX
tamoxifen citrate liquid |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Mayne Pharma (867220261) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Rosemont Pharmaceuticals Ltd | 212997852 | MANUFACTURE(51862-682) , ANALYSIS(51862-682) , LABEL(51862-682) , PACK(51862-682) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Herd Mundy Richardson Ltd | 345833292 | ANALYSIS(51862-682) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pharma Packaging Solutions LLC | 928861723 | LABEL(51862-682) , PACK(51862-682) | |
Frequently asked questions
- How do you relieve joint pain associated with tamoxifen or Aromasin?
- Is it common to lose hair AFTER stopping tamoxifen?
More about Soltamox (tamoxifen)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: hormones/antineoplastics
- Breastfeeding
- En español