ProHance: Package Insert / Prescribing Info
Package insert / product label
Generic name: gadoteridol
Dosage form: injection, solution
Drug class: Magnetic resonance imaging contrast media
Medically reviewed by Drugs.com. Last updated on Mar 25, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
PROHANCE MULTIPACK (gadoteridol injection), for intravenous use PHARMACY BULK PACKAGE -- NOT FOR DIRECT INFUSION
Initial U.S. Approval: 2003
WARNING: RISK ASSOCIATED WITH INTRATHECAL USE and NEPHROGENIC SYSTEMIC FIBROSIS
See full prescribing information for complete boxed warning
- Intrathecal administration of gadolinium based contrast agents (GBCAs) can cause serious adverse reactions including death, coma, encephalopathy, and seizures. ProHance is not approved for intrathecal use. (5.1)
-
GBCAs increase the risk for nephrogenic systemic fibrosis
(NSF) among patients with impaired elimination of the drugs. Avoid
use of ProHance in these patients unless the diagnostic information
is essential and not available with non-contrasted MRI or other modalities.
NSF may result in fatal or debilitating systemic fibrosis affecting
the skin, muscle and internal organs. The risk for NSF appears highest
among patients with:
- chronic, severe kidney disease (GFR less than 30 mL/min/1.73m2), or
- acute kidney injury
Indications and Usage for ProHance
ProHance Multipack is a gadolinium-based contrast agent indicated for magnetic resonance imaging (MRI) to visualize:
- lesions with disrupted blood brain barrier and/or abnormal vascularity in the brain (intracranial lesions), spine and associated tissues in adults and pediatric patients, including term neonates (1.1)
- lesions in the head and neck in adults (1.2)
ProHance Dosage and Administration
- Dispense multiple single doses into separate sterile syringes for intravenous administration (2.3)
- Recommended dose in adult and pediatric patients is 0.2 mL/kg (0.1 mmol/kg) body weight administered as rapid intravenous infusion or bolus (2.1)
- Follow injection with a saline flush of at least 5 mL normal saline (2.1)
Dosage Forms and Strengths
Contraindications
Allergic or hypersensitivity reactions to ProHance (4).
Warnings and Precautions
- Hypersensitivity: anaphylactic/anaphylactoid reactions with cardiovascular, respiratory and cutaneous manifestations, ranging from mild to severe reactions including shock can occur. Monitor patients closely for need of emergency cardiorespiratory support (5.3).
- Gadolinium is retained for months or years in brain, bone, and other organs. (5.4)
Adverse Reactions/Side Effects
The most commonly reported adverse reactions are nausea and taste perversion with an incidence ≥ 0.9% (6.1)
To report SUSPECTED ADVERSE REACTIONS, Contact Bracco Diagnostics Inc. at 1-800-257-5181 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Use In Specific Populations
Pregnancy: Use only if imaging is essential during pregnancy and cannot be delayed. (8.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2025
Full Prescribing Information
WARNING: RISK ASSOCIATED WITH INTRATHECAL USE and NEPHROGENIC SYSTEMIC FIBROSIS
Risk Associated with Intrathecal Use
Intrathecal administration of gadolinium-based
contrast agents (GBCAs) can cause serious adverse reactions including
death, coma, encephalopathy, and seizures. ProHance is not approved
for intrathecal use [see Warnings and Precautions (5.1)].
Nephrogenic Systemic Fibrosis
GBCAs increase the risk for nephrogenic
systemic fibrosis (NSF) among patients with impaired elimination of
the drugs. Avoid use of ProHance in these patients unless the diagnostic
information is essential and not available with non-contrasted MRI
or other modalities. NSF may result in fatal or debilitating systemic
fibrosis affecting the skin, muscle and internal organs.
The risk for NSF appears highest among patients with:
- chronic, severe kidney disease (GFR less than 30 mL/min/1.73m2), or
- acute kidney injury
Screen patients for acute kidney injury and other conditions that may reduce renal function. For patients at risk for chronically reduced renal function (e.g. age greater than 60 years, hypertension or diabetes), estimate the glomerular filtration rate (GFR) through laboratory testing.
For patients at highest risk for NSF, do not exceed the recommended ProHance dose and allow a sufficient period of time for elimination of the drug from the body prior to re-administration [see Warnings and Precautions (5.2)].
Indications and Usage for ProHance
1.1 MRI of the Central Nervous System (CNS)
ProHance is indicated for magnetic resonance imaging (MRI) in adults and pediatric patients including term neonates to visualize lesions with disrupted blood brain barrier and/or abnormal vascularity in the brain (intracranial lesions), spine and associated tissues.
ProHance Dosage and Administration
2.1 Recommended Dose
The recommended dose for adult and pediatric patients, including term neonates, is 0.2 mL/kg (0.1 mmol/kg) administered as a rapid intravenous infusion (10 mL/min to 60 mL/min) or bolus (greater than 60 mL/min). Table 1 provides weight-adjusted recommended dose volumes.
| Table 1: Recommended Volume of ProHance Injection by Body Weight | |
| Body Weight (kg) | Volume to be Administered (mL) |
| 2.5 | 0.5 |
| 5 | 1 |
| 10 | 2 |
| 20 | 4 |
| 30 | 6 |
| 40 | 8 |
| 50 | 10 |
| 60 | 12 |
| 70 | 14 |
| 80 | 16 |
| 90 | 18 |
| 100 | 20 |
| 110 | 22 |
| 120 | 24 |
| 130 | 26 |
| 140 | 28 |
| 150 | 30 |
- A supplementary dose of 0.4 mL/kg (0.2 mmol/kg) may be given up to 30 minutes after the first dose in adult patients with normal renal function suspected of having poorly visualized CNS lesions, in the presence of negative or equivocal scans
- The safety and efficacy of supplementary dosing have not been established in pediatric patients
2.2 Administration
- Visually inspect ProHance for particulate matter and discoloration prior to use
- Do not administer the solution if it is discolored or particulate matter is present
- Concurrent medications or parenteral nutrition should not be physically mixed with contrast agents and should not be administered in the same intravenous line because of the potential for chemical incompatibility
- Inject at least a 5 mL normal saline flush immediately after ProHance injection to ensure complete administration
- Imaging procedures should be completed within 1 hour
2.3 Directions for Proper Use of Pharmacy Bulk Package
NOT FOR DIRECT INFUSION
The pharmacy bulk package is used as a multiple dose container with an appropriate transfer device to fill empty sterile syringes. Use the following procedures when transferring ProHance from the pharmacy bulk package to individual syringes:
- Use of this product is restricted to a suitable work area, such as a laminar flow hood, utilizing aseptic technique
- Prior to entering the vial, remove the seal and cleanse the rubber closure with a suitable antiseptic agent
- The container closure may be penetrated only one time, utilizing a suitable transfer device or dispensing set that allows measured dispensing of the contents
- Once the pharmacy bulk package is punctured, it should not be removed from the aseptic work area during the entire period of use
- Withdrawal of container contents should be accomplished without delay. A maximum time of 8 hours from initial closure entry is permitted to complete fluid transfer operations
- Any unused contents must be discarded by 8 hours after initial puncture of the bulk package
- Once drawn into syringe, administer transferred agent promptly for single-dose administration
Dosage Forms and Strengths
ProHance Multipack is supplied as a sterile, nonpyrogenic, and colorless to slightly yellow solution available in 50 mL and 100 mL pharmacy bulk packages for intravenous administration. Each mL contains 279.3 mg (0.5 mmol/mL) of gadoteridol for injection.
Contraindications
ProHance is contraindicated in patients with known allergic or hypersensitivity reactions to ProHance [see Warnings and Precautions (5.3)].
Warnings and Precautions
5.1 Risk Associated with Intrathecal Use
Intrathecal administration of GBCAs can cause serious adverse reactions including death, coma, encephalopathy, and seizures. The safety and effectiveness of ProHance have not been established with intrathecal use. ProHance is not approved for intrathecal use [see Dosage and Administration (2.1)].
5.2 Nephrogenic Systemic Fibrosis
GBCAs increase the risk for nephrogenic systemic fibrosis (NSF) among patients with impaired elimination of the drugs. Avoid use of ProHance among these patients unless the diagnostic information is essential and not available with non-contrast MRI or other modalities. The GBCA-associated NSF risk appears highest for patients with chronic, severe kidney disease (GFR less than 30 mL/min/1.73m2) as well as patients with acute kidney injury. The risk appears lower for patients with chronic, moderate kidney disease (GFR 30-59 mL/min/1.73m2) and little, if any, for patients with chronic, mild kidney disease (GFR 60-89 mL/min/1.73m2). NSF may result in fatal or debilitating fibrosis affecting the skin, muscle and internal organs. Report any diagnosis of NSF following ProHance Multipack administration to Bracco Diagnostics (1-800-257-5181) or FDA (1-800-FDA-1088 or www.fda.gov/medwatch).
Screen patients for acute kidney injury and other conditions that may reduce renal function. Features of acute kidney injury consist of rapid (over hours to days) and usually reversible decrease in kidney function, commonly in the setting of surgery, severe infection, injury or drug-induced kidney toxicity. Serum creatinine levels and estimated GFR may not reliably assess renal function in the setting of acute kidney injury. For patients at risk for chronically reduced renal function (for example, age greater than 60 years, diabetes mellitus or chronic hypertension), estimate the GFR through laboratory testing.
Among the factors that may increase the risk for NSF are repeated or higher than recommended doses of a GBCA and the degree of renal impairment at the time of exposure. Record the specific GBCA and the dose administered to a patient. For patients at highest risk for NSF, do not exceed the recommended ProHance dose and allow a sufficient period of time for elimination of the drug prior to re-administration. For patients receiving hemodialysis, physicians may consider the prompt initiation of hemodialysis following the administration of a GBCA in order to enhance the contrast agent’s Page 4 elimination. The usefulness of hemodialysis in the prevention of NSF is unknown [see Clinical Pharmacology (12)].
5.3 Hypersensitivity Reactions
Anaphylactic and anaphylactoid reactions have been reported, involving cardiovascular, respiratory, and/or cutaneous manifestations. Some patients experienced circulatory collapse and died. In most cases, initial symptoms occurred within minutes of ProHance administration and resolved with prompt emergency treatment.
Prior to ProHance administration, ensure the availability of trained personnel and medications to treat hypersensitivity reactions. Consider the risk for hypersensitivity reactions, especially in patients with a history of hypersensitivity reactions or a history of asthma or other allergic disorders. If such a reaction occurs, stop ProHance and immediately begin appropriate therapy. Observe patients for signs and symptoms of a hypersensitivity reaction during and for up to 2 hours after ProHance administration.
5.4 Gadolinium Retention
Gadolinium is retained for months or years in several organs. The highest concentrations (nanomoles per gram of tissue) have been identified in the bone, followed by other organs (e.g. brain, skin, kidney, liver, and spleen). The duration of retention also varies by tissue and is longest in bone. Linear GBCAs cause more retention than macrocyclic GBCAs. At equivalent doses, retention varies among the linear agents with Omniscan (gadodiamide) and Optimark (gadoversetamide) causing greater retention than other linear agents [Eovist (gadoxetate disodium), Magnevist (gadopentetate dimeglumine), MultiHance (gadobenate dimeglumine)]. Retention is lowest and similar among the macrocyclic GBCAs [Dotarem (gadoterate meglumine), Gadavist (gadobutrol), ProHance (gadoteridol)].
Consequences of gadolinium retention in the brain have not been established. Pathologic and clinical consequences of GBCA administration and retention in skin and other organs have been established in patients with impaired renal function [see Warnings and Precautions (5.2)]. There are rare reports of pathologic skin changes in patients with normal renal function. Adverse events involving multiple organ systems have been reported in patients with normal renal function without an established causal link to gadolinium retention [see Adverse Reactions (6.2)].
While clinical consequences of gadolinium retention have not been established in patients with normal renal function, certain patients might be at higher risk. These include patients requiring multiple lifetime doses, pregnant and pediatric patients, and patients with inflammatory conditions. Consider the retention characteristics of the agent when choosing a GBCA for these patients. Minimize repetitive GBCA imaging studies, particularly closely spaced studies when possible.
Adverse Reactions/Side Effects
The following serious adverse reactions are discussed in greater detail in other sections of the prescribing information:
- Nephrogenic systemic fibrosis [see Boxed Warning and Warnings and Precautions (5.2)]
- Hypersensitivity reactions [see Contraindications (4) and Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The adverse events described in this section were observed in clinical trials involving 3174 subjects (including 2896 adults and 278 pediatric subjects ages 0 to 17 years) exposed to ProHance. Approximately 48% of the subjects were men and ethnic distribution was 78% Caucasian, 6% Black, 3% Hispanic, 6% Asian, and 2% other. In 5% of the subjects, race was not reported. Average age was 47 years (range from 1 day to 91 years) and the exposure ranged from 0.03 to 0.3 mmol/kg.
Overall, approximately 5.8% of subjects reported one or more adverse reactions during a follow-up period that ranged from 24 hours to 7 days after ProHance administration.
| Table 2: More frequent adverse reactions in clinical trials | |
| Reaction | Rate (%) N = 3174 |
| Nausea | 1.4% |
| Dysgeusia | 0.9% |
| Headache | 0.7% |
| Dizziness | 0.4% |
| Urticaria | 0.4% |
| The following additional adverse events occurred in fewer than 0.4% of the subjects: | |
| General disorders and administration site conditions: | Asthenia; chest discomfort, facial edema, feeling hot, injection site coldness, injection site erythema, injection site pain, injection site warmth, pain, pyrexia |
| Cardiac: | Angina pectoris, palpitations, atrio-ventricular block first degree |
| Ear and labyrinth disorders: | Ear discomfort, tinnitus |
| Eye disorders: | Eye pruritis, lacrimation increased |
| Gastrointestinal disorders: | Abdominal discomfort, abdominal pain, diarrhea, dry mouth, gingival pain, oral pruritis, swollen tongue, vomiting |
| Infections and infestations: | Gingivitis, rhinitis |
| Investigations: | Alanine aminotransferase increased, aspartate aminotransferase increased, blood chloride increased, blood pressure immeasurable, blood urea decreased, hemoglobin decreased, heart rate increased |
| Metabolism and nutrition disorders: | Decreased appetite, hypoglycemia |
| Musculoskeletal and connective tissue disorders: | Back pain, musculoskeletal stiffness |
| Nervous system disorders: | Formication, hypoesthesia, hypokinesia, lethargy, loss of consciousness, migraine, paresthesia, presyncope, seizure, syncope, taste disorder |
| Psychiatric disorders: | Anxiety, mental status changes |
| Respiratory, thoracic and mediastinal disorders: | Cough, dry throat, dyspnea, nasal discomfort, throat irritation |
| Skin and subcutaneous tissue disorders: | Hyperhidrosis, pruritis, rash, rash morbilliform |
| Vascular disorders: | Flushing, hypotension, peripheral coldness, vascular rupture, vasodilatation, vasospasm |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of ProHance or other GBCAs that were not observed in the clinical trials. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
| * Cases of acute renal failure have been reported in patients with pre-existing severe renal impairment. | |
| The following adverse drug reactions have also been reported: | |
| General Disorders and Administration Site Conditions: | Adverse events with variable onset and duration have been reported after GBCA administration [see Warnings and Precautions (5.4)]. These include fatigue, asthenia, pain syndromes, and heterogeneous clusters of symptoms in the neurological, cutaneous, and musculoskeletal systems. |
| Cardiac disorders: | Cardiac arrest, bradycardia, hypertension |
| Gastrointestinal disorders: | Acute pancreatitis with onset within 48 hours after GBCA administration |
| Immune system disorders: | Hypersensitivity/anaphylactoid reactions including cardiac arrest, cyanosis, pharyngeal edema, laryngospasm, bronchospasm, angioedema, cough, sneezing, conjunctivitis, eyelid edema, hyperhidrosis, urticaria [see Warnings and Precautions (5.3)]. |
| Nervous system disorders: | Coma, loss of consciousness, vasovagal reaction, tremor |
| Respiratory, thoracic and mediastinal disorders: | Respiratory arrest, acute respiratory distress syndrome, pulmonary edema |
| Renal and urinary system disorders: | Acute renal failure * |
Related/similar drugs
Use In Specific Populations
8.1 Pregnancy
Risk Summary
GBCAs cross the placenta
and result in fetal exposure and gadolinium retention. The human data
on the association between GBCAs and adverse fetal outcomes are limited
and inconclusive (see Data). Because of the potential
risks of gadolinium to the fetus, use ProHance only if imaging is
essential during pregnancy and cannot be delayed.
In animal reproduction studies in rats, gadoteridol doubled the incidence of post-implantation loss at up to 16 times the recommended human dose (RHD). There were no adverse developmental effects observed in rabbits with intravenous administration of gadoteridol during organogenesis at doses up to 19 times the recommended human dose of 0.1 mmol/kg (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and is 15 to 20%, respectively.
Data
Human Data
Contrast agent is visualized
in the placenta and fetal tissues after maternal GBCA administration.
Cohort studies and case reports on exposure to GBCAs during pregnancy
have not reported a clear association between GBCAs and adverse effects
in the exposed neonates. However, a retrospective cohort study, comparing
pregnant women who had a GBCA MRI to pregnant women who did not have
an MRI, reported a higher occurrence of stillbirths and neonatal deaths
in the group receiving GBCA MRI. Limitations of this study include
a lack of comparison with non-contrast MRI and lack of information
about the maternal indication for MRI.
Animal Data
Gadolinium
Retention
GBCAs administered to pregnant non-human primates
(0.1 mmol/kg on gestational days 85 and 135) result in measurable
gadolinium concentration in the offspring in bone, brain, skin, liver,
kidney, and spleen for at least 7 months. GBCAs administered to pregnant
mice (2 mmol/kg daily on gestational days 16 through 19) result in
measurable gadolinium concentrations in the pups in bone, brain, kidney,
liver, blood, muscle, and spleen at one-month postnatal age.
Reproductive Toxicology
Gadoteridol was administered in intravenous doses of 0, 0.375, 1.5,
6.0, and 10 mmol/kg/day [0.6, 2.4, 9.7, and 16 times the recommended
human dose (RHD) based on body surface area (BSA)] to female rats
from gestational day (GD)6 until GD17. Gadoteridol at 10 mmol/kg/day
for 12 days during gestation doubled the incidence of post-implantation
loss. When rats were administered 6.0 or 10.0 mmol/kg/day for 12 days,
an increase in spontaneous locomotor activity was observed in the
offspring. Pregnant rabbits were administered gadoteridol in intravenous
doses of 0, 0.4, 1.5, and 6 mmol/kg/day (1.3, 4.8, and 19.4 times
the RHD based on BSA) from GD6 to GD18. Gadoteridol increased the
incidence of spontaneous abortion and early delivery in rabbits administered
6 mmol/kg/day for 13 days during gestation.
8.2 Lactation
Risk Summary
There are no data
on the presence of gadoteridol in human milk, the effects on the breastfed
infant, or the effects on milk production. However, published lactation
data on other GBCAs indicate that 0.01 to 0.04% of the maternal gadolinium
dose is present in breast milk and there is limited GBCA gastrointestinal
absorption in the breast-fed infant. Gadoteridol is present in rat
milk (see Data). The developmental and health benefits of breastfeeding
should be considered along with the mother’s clinical need for ProHance
and any potential adverse effects on the breastfed infant from ProHance
or from the underlying maternal condition.
Data
ProHance
excretion in the milk of lactating rats was evaluated at 30 minutes,
6 and 24 hours after intravenous administration of 0.1 mmol/kg of 153Gd-gadoteridol to nursing mothers. Small amounts
of compound were found in milk immediately after injection (0.14%
of the ID), with the amount declining to a low level 24 hours after
injection (<0.01% of the ID).
8.4 Pediatric Use
The safety and effectiveness of ProHance have been established for use with MRI to visualize lesions with abnormal blood brain barrier or abnormal vascularity of the brain, spine, and associated tissues in pediatric patients from birth, including term neonates, to 17 years of age. Pediatric use is based on evidence of effectiveness in adults and in 103 pediatric patients 2 years of age and older, in addition to experience in 125 pediatric patients birth to less than 2 years of age that supported extrapolation from adult data [see Clinical Studies (14)]. Adverse reactions in pediatric patients were similar to those reported in adults [see Adverse Reactions (6.1)].
The safety and efficacy of > 0.1 mmol/kg, and sequential and/or repeat procedures have not been studied in pediatric patients [see Indications and Usage (1) and Dosage and Administration (2)].
No case of NSF associated with ProHance or any other GBCA has been identified in pediatric patients ages 6 years and younger. Pharmacokinetic studies suggest that weight normalized clearance of ProHance is similar in pediatric patients and adults, including pediatric patients age younger than 2 years. Normal estimated GFR (eGFR) is around 30 mL/min/1.73m2 at birth and increases to mature levels around 1 year of age, reflecting growth in both glomerular function and relative body surface area. Clinical studies in pediatric patients younger than 1 year of age have been conducted in patients with the following minimum eGRF; 59.37 mL/min/1.73m2 (age just after birth to < 30 days), 118.84 mL/min/1.73m2 (age 30 days to < 6 months), 140.44 mL/min/1.73m2 (age 6 to 12 months).
8.5 Geriatric Use
Of the total number of 2673 adult subjects in clinical studies of ProHance, 22% were 65 and over. No overall differences in safety were observed between these elderly subjects and the younger subjects.
ProHance is known to be substantially excreted by the kidneys, and the risk of toxic reactions from ProHance may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, it may be useful to monitor renal function.
Overdosage
Clinical consequences of overdose with ProHance have not been reported. The safety of ProHance has been tested in clinical studies using doses up to 0.3 mmol/kg and no clinical consequences related to increasing dose have been observed to date. ProHance can be removed by hemodialysis [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
ProHance Description
ProHance, a gadolinium-based paramagnetic MRI contrast agent, is a colorless to slightly yellow aqueous, sterile, nonpyrogenic injectable solution. Each mL contains 279.3 mg (0.5 mmol/mL) gadoteridol, 0.23 mg calteridol calcium, 1.21 mg tromethamine and water for injection; pH adjusted with hydrochloric acid and/or sodium hydroxide. ProHance contains no antimicrobial preservative.
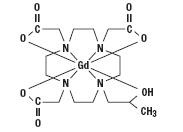
Gadoteridol is the gadolinium complex of 10-(2-hydroxy-propyl)-1,4,7,10-tetraazacyclododecane-1,4,7- triacetic acid with a molecular weight of 558.7, an empirical formula of C17H29N4O7Gd and has the following structural formula:
ProHance has a pH of 6.5 to 8.0. Pertinent physiochemical parameters are provided below:
| Osmolality | 630 mOsmol/kg water at 37 °C |
| Viscosity | 1.3 cP at 37 °C |
| Density | 1.137 g/mL at 25 °C |
ProHance has an osmolality that is 2.2 times that of plasma (285 mOsmol/kg water) and is hypertonic under conditions of use.
ProHance - Clinical Pharmacology
12.1 Mechanism of Action
Gadoteridol is a paramagnetic agent and, as such, develops a magnetic moment when placed in a magnetic field. The relatively large magnetic moment produced by the paramagnetic agent results in a relatively large local magnetic field, which can enhance the relaxation rates of water protons in the vicinity of the paramagnetic agent.
In MRI, visualization of normal and pathologic brain tissue depends, in part, on variations in the radiofrequency signal intensity that occur with: 1) differences in proton density; 2) differences of the spin-lattice or longitudinal relaxation times (T1); and 3) differences in the spin-spin or transverse relaxation time (T2). When placed in a magnetic field, gadoteridol decreases T1 relaxation times in the target tissues. At recommended doses, the effect is observed with greatest sensitivity in the T1-weighted sequences.
12.2 Pharmacodynamics
Gadoteridol affects proton relaxation times and consequently the MR signal. Signal intensity is affected by the dose and relaxivity of the gadoteridol molecule. Consistently, for all gadolinium based contrast agents, the relaxivity of gadoteridol decreases with the increase of the magnetic field strength used in clinical MRI (0.2 – 3.0T).
Disruption of the blood-brain barrier or abnormal vascularity allows accumulation of gadoteridol in lesions such as neoplasms, abscesses, and subacute infarcts. The pharmacokinetics of gadoteridol in various lesions is not known.
12.3 Pharmacokinetics
The pharmacokinetics of intravenously administered gadoteridol in normal subjects conforms to a two-compartment open model.
Distribution
After intravenous administration,
gadoteridol is rapidly distributed in the extracellular space. The
plasma distribution volume (mean ± SD) for the non-renally impaired
adults was 0.205 ± 0.025 L/kg. It is unknown if protein binding of
gadoteridol occurs in vivo.
Following GBCA administration, gadolinium is present for months or years in brain, bone, skin, and other organs [see Warnings and Precautions (5.4)].
Metabolism
It is unknown if biotransformation
or decomposition of gadoteridol occur in vivo.
Elimination
Gadoteridol is eliminated unchanged via the kidneys.
The elimination half-life (mean ± SD) is about 1.57 ± 0.08 hours.
Within 24 hours post-injection, 94.4 ± 4.8% of the dose is excreted
in the urine. The renal and plasma clearance rates (1.41 ± 0.33 mL/
min/kg and 1.50 ± 0.35 mL/ min/kg, respectively) of gadoteridol are
essentially identical, indicating no alteration in elimination kinetics
on passage through the kidneys and that the drug is essentially cleared
through the kidney. The volume of distribution (204 ± 58 mL/kg) is
equal to that of extracellular water, and clearance is similar to
that of substances which are subject to glomerular filtration.
Specific Populations
Gender
Gender has no clinically
relevant effect on the pharmacokinetics of gadoteridol.
Geriatric
There were 7 elderly subjects receiving 0.1 (n = 3) and 0.3 mmol/kg
(n = 4) dose of ProHance. The clearance was slightly lower in elderly
subjects as compared to non-elderly subjects. [see Use in
Specific Populations (8.5)].
Pediatric
A population pharmacokinetic analysis incorporated data
from 79 subjects, 45 males and 34 females. Among 79 subjects, 41 were
healthy subjects including 28 pediatric subjects between 5 years and
15 years of age. The pediatric subjects received a single intravenous
dose of 0.1 mmol/kg of ProHance. From population PK model, the mean
Cmax was 0.66 ± 0.21 mmol/L in pediatric subjects
2 years to 6 years of age, 0.58 ± 0.06 mmol/L in pediatric subjects
6 years to 12 years of age, and 0.68 ± 0.12 mmol/L in adolescent subjects
older than 12 years. The mean AUC 0-∞ was 0.74
± 0.20 mmol/L⋅h in pediatric subjects 2 years to 6 years of age, 0.74
± 0.09 mmol/L⋅h in pediatric subjects 6 years to 12 years of age,
and 0.98 ± 0.09 mmol/L⋅h in adolescent subjects older than 12 years
of age. The mean distribution half- life (t1/2,alpha) was 0.14 ± 0.04 hours in pediatric subjects 2 years to 6 years
of age, 0.18 ± 0.07 hours in pediatric subjects 6 years to 12 years
of age, and 0.20 ± 0.07 hours in adolescent subjects older than 12
years of age. The mean elimination half-life (t1/2,beta) was 1.32 ± 0.006 hours in pediatric subjects 2 years to 6 years,
1.32 ± 0.07 hours in pediatric subjects 6 years to 12 years of age,
and 1.61 ± 0.19 hours in adolescent subjects older than 12 years of
age. There was no significant gender-related difference in the pharmacokinetic
parameters in the pediatric patients. Over 80% of the dose was recovered
in urine for pediatric subjects after 10 hours. Pharmacokinetic simulations
indicate similar half-life, AUC, and Cmax values
for ProHance in pediatric subjects less than 2 years of age when compared
to those reported for adults; no age-based dose adjustment is necessary
for this pediatric population.
Renal Impairment
In patients with impaired renal function, the serum half-life of
gadoteridol is prolonged. After intravenous injection of 0.1 mmol/kg,
the elimination half-life of gadoteridol was 10.65 ± 0.60 hours in
mild to moderately impaired patients (creatinine clearance 30 to 60
mL/min) and 9.10±0.26 hours in severely impaired patients not on dialysis
(creatinine clearance 10 to 30 mL/min). The mean serum clearance of
gadoteridol in patients with normal renal function was 116.14 ± 26.77
mL/min, compared to 37.2 ± 16.4 mL/min in patients with mild to moderate
renal impairment and 16.0 ± 3.0 mL/min in patients with severe renal
impairment.
In patients with moderately and severely impaired renal function about 97% and 76% of the administered dose was recovered in the urine within 7 days and 14 days, respectively.
For patients receiving hemodialysis, physicians may consider the prompt initiation of hemodialysis following the administration of ProHance in order to enhance the contrast agent’s elimination. Seventy- two percent (72%) of gadoteridol is removed from the body after the first dialysis, 91% after the second dialysis, and 98% after the third dialysis session. [See Warnings and Precautions (5.2) and Use in Specific Populations (8.6).]
Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies have been performed to evaluate the carcinogenic potential of gadoteridol.
No changes in reproductive performance and outcome of pregnancy were caused in rats and rabbits by daily intravenous administration of ProHance to parent animals before and during gestation up to 1.5 mmol/kg/day (15 times the recommended human dose).
Gadoteridol did not demonstrate genotoxic activity in: bacterial reverse mutation assays using Salmonella typhimurium and Escherichia coli; a mouse lymphoma forward mutation assay; an in vitro cytogenetic assay measuring chromosomal aberration frequencies in Chinese hamster ovary cells; and an in vivo mouse micronucleus assay at intravenous doses up to 5.0 mmol/kg.
Clinical Studies
14.1 MRI of the CNS
ProHance was evaluated in two multicenter trials of 310 evaluable patients suspected of having neurological pathology. After the administration of ProHance 0.1 mmol/kg IV, the results were similar to those described below [see Clinical Studies (14.2)].
In another multicenter study of 49 evaluable adult patients with known intracranial tumor with high suspicion of having cerebral metastases, two doses of ProHance were administered. First ProHance 0.1 mmol/kg was injected followed 30 minutes later with 0.2 mmol/kg. In comparison to the 0.1 mmol/kg dose alone, the addition of the 0.2 mmol/kg dose improved visualization in 67% and improved border definition in 56% of patients. In comparison to non-contrast MRI, the number of lesions after 0.1 mmol/kg increased in 34% of patients. After ProHance 0.2 mmol/kg, this increased to 44%.
Pediatric Patients
ProHance was
evaluated in a multicenter study of 103 patients undergoing brain
or spine MRI. Among these patients, the age range was 2 to 20 years;
54 were between 2 and 12 years of age; 74% were Caucasian, 11% Black,
12% Hispanic, 2% Asian, and 2% other. ProHance was given in one single
0.1 mmol/kg dose. Repeat dosing was not studied. The results of the
non-contrast and ProHance MRI scans were compared. In this database,
MRI enhancement was noted in approximately 60% of the scans and additional
diagnostic information in 30 to 95% of the scans.
A prospectively planned study of 125 pediatric patients younger than 2 years of age retrospectively selected was performed. These patients (70 boys and 55 girls) had an age range of 1 day to 24 months old; 17 were less than 1 month of age, 40 were between 1 month and 6 months of age, 29 were between 6 months and 12 months of age, and 39 were between 12 months and 24 months of age; 56% were Caucasian, 25% Black, 5% Asian, and 14% other. ProHance was given in one single 0.1 mmol/kg dose. Repeat dosing was not studied. Three independent, blinded readers evaluated pre-contrast MRI image sets and paired pre-plus-post-contrast MRI image sets using ProHance and rated the images according to three co-primary visualization endpoints: lesion border delineation, visualization of lesion internal morphology, and lesion contrast enhancement. All three blinded readers reported improvement in the paired image sets for each of the three co-primary endpoints.
14.2 MRI of the Head and Neck
ProHance was evaluated in two blinded read studies in a total of 133 adults who had an indication for head and neck extracranial or extraspinal MRI. These 133 adults (74 men, 59 women) had a mean age of 53 with a range of 19 to 76 years. Of these patients, 85% were Caucasian, 13% Black, 2% Asian, and less than 1% other. The results of the non-contrast and contrast MRI scans were compared. Approximately 75-82% of the scans were enhanced, 45-48% of the scans provided additional diagnostic information, and 8-25% of the diagnoses were changed. The relevance of the findings to disease sensitivity and specificity has not been fully evaluated.
How is ProHance supplied
How Supplied
ProHance Multipack is supplied as a sterile, nonpyrogenic, and colorless to slightly yellow solution containing 279.3 mg/mL (0.5 mmol/mL) of gadoteridol in rubber stoppered vials. ProHance Multipack is supplied in boxes of five 50 mL pharmacy bulk packages (NDC 0270-1111-70) and boxes of five 100 mL pharmacy bulk packages (NDC-0270-1111-85).
Storage and Handling
Store at 25°C (77° F). Excursions permitted to 15°C to 30°C (59°F to 86°F) [See USP Controlled Room Temperature]. Protect from light. DO NOT FREEZE. Should freezing occur in the vial, ProHance Multipack should be brought to room temperature before use. If allowed to stand at room temperature for a minimum of 60 minutes, ProHance Multipack should return to a clear, colorless to slightly yellow solution. Before use, examine the product to assure that all solids are redissolved and that the container and closure have not been damaged. Should solids persist, discard vial.
Patient Counseling Information
- Advise the patient to read the FDA-approved patient labeling (Medication Guide) found at www.prohancemedicationguide.com.
Nephrogenic Systemic
Fibrosis
Instruct patients to inform their
physician if they:
- have a history of kidney disease
- have recently received a GBCA
GBCAs increase the risk for NSF in patients with impaired elimination of the drugs. To counsel patients at risk for NSF:
- describe the clinical manifestations of NSF
- describe procedures to screen for the detection of renal impairment
Instruct patients to contact their physician if they develop signs or symptoms of NSF following ProHance administration, such as burning, itching, swelling, scaling, hardening and tightening of the skin; red or dark patches on the skin; stiffness in joints with trouble moving, bending or straightening the arms, hands, legs or feet; pain in the hip bones or ribs; or muscle weakness.
- Pregnancy: Advise a pregnant woman of the potential risk of fetal exposure to ProHance [see Use in Specific Populations (8.1)]
- Gadolinium retention: Advise patients that gadolinium is retained for months or years in brain, bone, skin, and other organs in patients with normal renal function. The clinical consequences of retention are unknown. Retention depends on multiple factors and is greater following administration of linear GBCAs than following administration of macrocyclic GBCAs [see Warnings and Precautions (5.4)].
Manufactured
for:
Bracco Diagnostics Inc.
Princeton, NJ 08540
| This Medication Guide has been approved by the U.S. Food and Drug Administration | Revised: 03/2025 |
| MEDICATION GUIDE PROHANCE®(prō-ˈhan(t)s) (Gadoteridol injection) for intravenous use |
|
What is the
most important information I should know about PROHANCE?
|
|
What is PROHANCE?
|
|
| Do not receive PROHANCE if you have had a severe allergic reaction to PROHANCE. | |
Before receiving
PROHANCE, tell your healthcare provider about all your medical conditions,
including if you:
|
|
What are
the possible side effects of PROHANCE?
These are not all the possible side effects of PROHANCE. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
| General information
about the safe and effective use of PROHANCE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your healthcare provider for information about PROHANCE that is written for health professionals. |
|
| What are
the ingredients in PROHANCE?
Active ingredient: gadoteridol Inactive ingredients: calteridol calcium, tromethamine Manufactured by: BIPSO GmbH-78224 Singen (Germany) Manufactured for: Bracco Diagnostics Inc., Princeton, NJ 08540 For more information, go to www.imaging.bracco.com or call 1-800-257-5181. |
|


| PROHANCE
gadoteridol injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Bracco Diagnostics Inc (849234661) |
| Registrant - Bracco Diagnostics Inc (849234661) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| BRACCO IMAGING SPA | 434384007 | API MANUFACTURE(0270-1111) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| BIPSO GmbH | 342104149 | MANUFACTURE(0270-1111) , ANALYSIS(0270-1111) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Labor LS SE & Co. KG | 314929072 | ANALYSIS(0270-1111) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| BioChem Labor für biologishe und chemische | 318354230 | ANALYSIS(0270-1111) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bracco Imaging SPA | 434384008 | ANALYSIS(0270-1111) | |
More about Prohance (gadoteridol)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- Drug class: magnetic resonance imaging contrast media
- Breastfeeding
- En español