Givlaari: Package Insert / Prescribing Info
Package insert / product label
Generic name: givosiran sodium
Dosage form: injection, solution
Drug class: Miscellaneous metabolic agents
J Code (medical billing code): J0223 (0.5 mg, injection)
Medically reviewed by Drugs.com. Last updated on May 16, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
GIVLAARI (givosiran) injection, for subcutaneous use
Initial U.S. Approval: 2019
Recent Major Changes
| Warnings and Precautions, Pancreatitis (5.6) | 4/2024 |
Indications and Usage for Givlaari
GIVLAARI is an aminolevulinate synthase 1-directed small interfering RNA indicated for the treatment of adults with acute hepatic porphyria (AHP). (1)
Givlaari Dosage and Administration
The recommended dose of GIVLAARI is 2.5 mg/kg once monthly by subcutaneous injection. (2.1)
Dosage Forms and Strengths
Injection: 189 mg/mL in a single-dose vial. (3)
Contraindications
Severe hypersensitivity to givosiran. (4)
Warnings and Precautions
- Anaphylactic Reaction: Ensure that medical support is available to appropriately manage anaphylactic reactions when administering GIVLAARI. Monitor for signs and symptoms. If anaphylaxis occurs, discontinue GIVLAARI and administer appropriate medical treatment. (5.1)
- Hepatic Toxicity: Measure liver function at baseline and periodically during treatment with GIVLAARI. Interrupt or discontinue treatment with GIVLAARI for severe or clinically significant transaminase elevations. (2.1, 5.2)
- Renal Toxicity: Monitor renal function during treatment with GIVLAARI as clinically indicated. (5.3)
- Injection Site Reactions: May occur, including recall reactions. Monitor for reactions and manage clinically as needed. (5.4)
- Blood Homocysteine Increased: Measure blood homocysteine at baseline and monitor for changes during treatment with GIVLAARI. In patients with elevated blood homocysteine, consider supplementation with vitamin B6 (as monotherapy or multivitamin). (5.5)
- Pancreatitis: Consider acute pancreatitis as a potential diagnosis in GIVLAARI-treated patients with acute upper abdominal pain, clinically significant elevation of pancreatic enzymes and/or imaging findings of acute pancreatitis, to ensure appropriate management. (5.6)
Adverse Reactions/Side Effects
The most common adverse reactions (≥20% of patients) included nausea and injection site reactions. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Alnylam Pharmaceuticals at 1-877-256-9526 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Sensitive CYP1A2 and CYP2D6 Substrates: Avoid concomitant use with CYP1A2 and CYP2D6 substrates for which minimal concentration changes may lead to serious or life-threatening toxicities. (7.1)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2024
Full Prescribing Information
1. Indications and Usage for Givlaari
GIVLAARI is indicated for the treatment of adults with acute hepatic porphyria (AHP).
2. Givlaari Dosage and Administration
2.1 Recommended Dosage
The recommended dose of GIVLAARI is 2.5 mg/kg administered via subcutaneous injection once monthly. Dosing is based on actual body weight.
Missed Dose
Administer GIVLAARI as soon as possible after a missed dose. Resume dosing at monthly intervals following administration of the missed dose.
Dose Modification for Adverse Reactions
In patients with severe or clinically significant transaminase elevations, who have dose interruption and subsequent improvement, reduce the dose to 1.25 mg/kg once monthly [see Warnings and Precautions (5.2)]. In patients who resume dosing at 1.25 mg/kg once monthly without recurrence of severe or clinically significant transaminase elevations, the dose may be increased to the recommended dose of 2.5 mg/kg once monthly.
2.2 Administration Instructions
Ensure that medical support is available to appropriately manage anaphylactic reactions when administering GIVLAARI [see Warnings and Precautions (5.1)].
GIVLAARI is intended for subcutaneous use by a healthcare professional only.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. GIVLAARI is a sterile, preservative-free, clear, colorless-to-yellow solution. It is supplied in a single-dose vial, as a ready-to-use solution that does not require additional reconstitution or dilution prior to administration.
Use aseptic technique.
- Calculate the required volume of GIVLAARI based on the recommended weight-based dosage [see Dosage and Administration (2.1)].
- Withdraw the indicated injection volume of GIVLAARI using a 21-gauge or larger needle.
- Divide doses requiring volumes greater than 1.5 mL equally into multiple syringes.
- Replace the 21-gauge or larger needle with either a 25-gauge or 27-gauge needle with 1/2" or 5/8" needle length.
- Avoid having GIVLAARI on the needle tip until the needle is in the subcutaneous space.
- Administer injection into the abdomen, the back or side of the upper arms, or the thighs. Rotate injection sites. An injection should never be given into scar tissue or areas that are reddened, inflamed, or swollen.
- If injecting into the abdomen, avoid a 5 cm diameter circle around the navel.
- If more than one injection is needed for a single dose of GIVLAARI, the injection sites should be at least 2 cm apart from previous injection locations.
- Discard unused portion of the drug.
3. Dosage Forms and Strengths
Injection: 189 mg/mL clear, colorless-to-yellow solution in a single-dose vial
4. Contraindications
GIVLAARI is contraindicated in patients with known severe hypersensitivity to givosiran. Reactions have included anaphylaxis [see Warnings and Precautions (5.1)].
5. Warnings and Precautions
5.1 Anaphylactic Reaction
Anaphylaxis has occurred with GIVLAARI treatment (<1% of patients in clinical trials) [see Adverse Reactions (6.1)]. Ensure that medical support is available to appropriately manage anaphylactic reactions when administering GIVLAARI. Monitor for signs and symptoms of anaphylaxis. If anaphylaxis occurs, immediately discontinue administration of GIVLAARI and institute appropriate medical treatment.
5.2 Hepatic Toxicity
Transaminase elevations (ALT) of at least 3 times the upper limit of normal (ULN) were observed in 15% of patients treated with GIVLAARI in the placebo-controlled trial [see Adverse Reactions (6.1)]. Transaminase elevations primarily occurred between 3 to 5 months following initiation of treatment.
Measure liver function tests prior to initiating treatment with GIVLAARI, repeat every month during the first 6 months of treatment, and as clinically indicated thereafter. Interrupt or discontinue treatment with GIVLAARI for severe or clinically significant transaminase elevations. For resumption of dosing after interruption, see Dosage and Administration (2.1).
5.3 Renal Toxicity
Increases in serum creatinine levels and decreases in estimated glomerular filtration rate (eGFR) have been reported during treatment with GIVLAARI [see Adverse Reactions (6.1)]. In the placebo-controlled study, 15% of the patients in the GIVLAARI arm experienced a renally-related adverse reaction. The median increase in creatinine at Month 3 was 0.07 mg/dL. Monitor renal function during treatment with GIVLAARI as clinically indicated.
5.4 Injection Site Reactions
Injection site reactions have been reported in 25% of patients receiving GIVLAARI in the placebo-controlled trial. Symptoms included erythema, pain, pruritus, rash, discoloration, or swelling around the injection site. Among 12 patients with reactions, the highest severity of the reaction was mild among 11 (92%) patients and moderate in one (8%) patient. One (2%) patient experienced a single, transient, recall reaction of erythema at a prior injection site with a subsequent dose administration [see Adverse Reactions (6.1)].
5.5 Blood Homocysteine Increased
Increases in blood homocysteine levels have occurred in patients receiving GIVLAARI [see Adverse Reactions (6.1)]. In the ENVISION study, during the open label extension, adverse reactions of blood homocysteine increased were reported in 15 of 93 (16%) patients treated with GIVLAARI. The clinical relevance of the elevations in blood homocysteine during treatment with GIVLAARI is unknown. Measure blood homocysteine levels prior to initiating treatment and monitor for changes during treatment with GIVLAARI. In patients with elevated blood homocysteine levels, assess folate, vitamins B12 and B6. Consider treatment with a supplement containing vitamin B6 (as monotherapy or a multivitamin preparation).
5.6 Pancreatitis
Cases of acute pancreatitis, some severe, have been reported in GIVLAARI-treated patients.
Consider acute pancreatitis as a potential diagnosis in GIVLAARI-treated patients with signs/symptoms of acute pancreatitis including acute upper abdominal pain, clinically significant elevation of pancreatic enzymes, and/or imaging findings of acute pancreatitis, to ensure appropriate management. Consider interruption and/or discontinuation of GIVLAARI treatment for severe cases.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Anaphylactic Reaction [see Warnings and Precautions (5.1)]
- Transaminase Elevations [see Warnings and Precautions (5.2)]
- Serum Creatinine Increase [see Warnings and Precautions (5.3)]
- Injection Site Reactions [see Warnings and Precautions (5.4)]
- Blood Homocysteine Increased [see Warnings and Precautions (5.5)]
- Pancreatitis [see Warnings and Precautions (5.6)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In the pivotal placebo-controlled, double-blind study (ENVISION), 48 patients received 2.5 mg/kg GIVLAARI and 46 patients received placebo, administered once monthly via subcutaneous injection for up to 6 months. Patients received GIVLAARI for a median of 5.5 months (range 2.7-6.4 months). Of these, 47 patients received ≥5 months of treatment. The most frequently occurring (≥20% incidence) adverse reactions reported in patients treated with GIVLAARI were nausea (27%) and injection site reactions (25%). Permanent discontinuation occurred in one patient due to elevated transaminases.
| Adverse Reaction | GIVLAARI N=48 N (%) | Placebo N=46 N (%) |
|---|---|---|
| Nausea | 13 (27) | 5 (11) |
| Injection site reactions | 12 (25) | 0 |
| Rash* | 8 (17) | 2 (4) |
| Serum creatinine increase† | 7 (15) | 2 (4) |
| Transaminase elevations | 6 (13) | 1 (2) |
| Fatigue | 5 (10) | 2 (4) |
Adverse reactions observed at a lower frequency occurring in placebo-controlled and open-label clinical studies included anaphylactic reaction (one patient, 0.9%) and hypersensitivity (one patient, 0.9%).
In the ENVISION study, during the open label extension, adverse reactions of blood homocysteine increased were reported in 15 of 93 (16%) patients treated with GIVLAARI [see Warnings and Precautions (5.5)].
6.2 Immunogenicity
As with all oligonucleotides, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In placebo-controlled and open-label clinical studies, 1 of 111 patients with AHP (0.9%) developed treatment-emergent anti-drug antibodies (ADA) during treatment with GIVLAARI. No clinically significant differences in the clinical efficacy, safety, pharmacokinetic, or pharmacodynamic profiles of GIVLAARI were observed in the patient who tested positive for anti-givosiran antibodies.
6.3 Postmarketing Experience
The following additional adverse reactions have been reported during post-approval use. Because these events are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal Disorders: Acute pancreatitis
Related/similar drugs
7. Drug Interactions
7.1 Effect of GIVLAARI on Other Drugs
Sensitive CYP1A2 and CYP2D6 Substrates
Concomitant use of GIVLAARI increases the concentration of CYP1A2 or CYP2D6 substrates [see Clinical Pharmacology (12.3)], which may increase adverse reactions of these substrates. Avoid concomitant use of GIVLAARI with CYP1A2 or CYP2D6 substrates, for which minimal concentration changes may lead to serious or life-threatening toxicities. If concomitant use is unavoidable, decrease the CYP1A2 or CYP2D6 substrate dosage in accordance with approved product labeling.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
In animal reproduction studies, subcutaneous administration of givosiran to pregnant rabbits during the period of organogenesis resulted in adverse developmental outcomes at doses that produced maternal toxicity (see Data).
There are no available data with GIVLAARI use in pregnant women to evaluate a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Consider the benefits and risks of GIVLAARI for the mother and potential adverse effects to the fetus when prescribing GIVLAARI to a pregnant woman.
The estimated background risk of major birth defects and miscarriage in the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Porphyria attacks during pregnancy, often triggered by hormonal changes, occur in 24% to 95% of AHP patients, with maternal mortality ranging from 2% to 42%. Pregnancy in AHP patients is associated with higher rates of spontaneous abortion, hypertension and low birth weight infants.
Data
Animal Data
In an embryo-fetal development study in pregnant rabbits, givosiran was administered subcutaneously at doses of 0.5, 1.5, and 5 mg/kg/day during organogenesis (gestational days 7-19) or 20 mg/kg as a single administration on gestation day 7. Administration of givosiran was maternally toxic based on decreased body weight gain at all dose levels tested and resulted in increased postimplantation loss starting at 1.5 mg/kg/day. An increased incidence of skeletal variations of the sternebrae was observed at 20 mg/kg. The 1.5 mg/kg/day dose in rabbits is 5 times the maximum recommended human dose (MRHD) of 2.5 mg/kg/month normalized to 0.089 mg/kg/day, based on body surface area. In a combined fertility and embryo-fetal development study in female rats, givosiran was administered subcutaneously at doses of 0.5 to 5 mg/kg/day during organogenesis (gestational days 6-17). The 5 mg/kg/day dose (9 times the normalized MRHD based on body surface area) was associated with a skeletal variation (incomplete ossification of pubes) and produced maternal toxicity.
In a pre- and postnatal development study, givosiran administered subcutaneously to pregnant rats on gestation days 7, 13, and 19 and postnatal days 6, 12, and 18 at doses up to 30 mg/kg did not produce maternal toxicity or developmental effects in the offspring.
8.2 Lactation
Risk Summary
There are no data on the presence of GIVLAARI in human milk, the effects on the breastfed child, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for GIVLAARI and any potential adverse effects on the breastfed child from GIVLAARI or from the underlying maternal condition.
11. Givlaari Description
GIVLAARI is an aminolevulinate synthase 1-directed small interfering RNA (siRNA), covalently linked to a ligand containing three N-acetylgalactosamine (GalNAc) residues to enable delivery of the siRNA to hepatocytes.
The structural formulas of the givosiran drug substance in its sodium form, and the ligand (L96), are presented below.
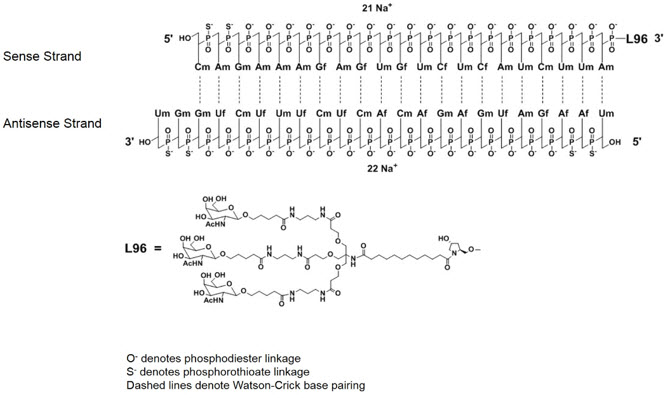
Abbreviations: Af = adenine 2'-F ribonucleoside; Cf = cytosine 2'-F ribonucleoside; Uf = uracil 2'-F ribonucleoside; Am = adenine 2'-OMe ribonucleoside; Cm = cytosine 2'-OMe ribonucleoside; Gf = guanine 2'-F ribonucleoside; Gm = guanine 2'-OMe ribonucleoside; Um = uracil 2'-OMe ribonucleoside; L96 = triantennary GalNAc (N-acetylgalactosamine)
GIVLAARI is supplied as a sterile, preservative-free, 1-mL colorless-to-yellow solution for subcutaneous injection containing 189 mg givosiran in a single-dose, 2-mL Type 1 glass vial with a fluoropolymer-coated rubber stopper and a flip-off aluminum seal. GIVLAARI is available in cartons containing one single-dose vial each. GIVLAARI is formulated in Water for Injection. Sodium hydroxide and/or phosphoric acid may have been added for pH adjustment during product manufacturing.
The molecular formula of givosiran sodium is C524 H651 F16 N173 Na43 O316 P43 S6 with a molecular weight of 17,245.56 Da.
The molecular formula of givosiran (free acid) is C524 H694 F16 N173 O316 P43 S6 with a molecular weight of 16,300.34 Da.
12. Givlaari - Clinical Pharmacology
12.1 Mechanism of Action
Givosiran is a double-stranded small interfering RNA that causes degradation of aminolevulinate synthase 1 (ALAS1) mRNA in hepatocytes through RNA interference, reducing the elevated levels of liver ALAS1 mRNA. This leads to reduced circulating levels of neurotoxic intermediates aminolevulinic acid (ALA) and porphobilinogen (PBG), factors associated with attacks and other disease manifestations of AHP.
12.2 Pharmacodynamics
The pharmacodynamic effects of GIVLAARI were evaluated in chronic high excreters treated with 0.035 to 2.5 mg/kg single dose and AHP patients treated with 2.5 to 5 mg/kg once monthly and 2.5 to 5 mg/kg once quarterly dose via subcutaneous injection. Dose-dependent reduction in urinary ALAS1 mRNA, ALA and PBG levels was observed over the 0.035 to 5 mg/kg dose range (0.14 to 2-fold the approved recommended dosage). Median reductions from baseline in urinary ALA and PBG of 83.7% and 75.1%, respectively, were observed 14 days after the first dose of GIVLAARI 2.5 mg/kg once monthly in AHP patients. Maximal reductions in ALA and PBG levels were achieved around Month 3, with median reductions from baseline of 93.8% for ALA and 94.5% for PBG, and were sustained thereafter with repeated once monthly dosing.
Cardiac Electrophysiology
The effect of GIVLAARI on the QTc interval was evaluated in a double-blind, placebo-controlled study and the open-label extension in 94 patients. No large mean increase in QTc (i.e. >20 ms) was detected at the 2.5 mg/kg once monthly dose level. A dedicated thorough QT study has not been conducted with GIVLAARI.
12.3 Pharmacokinetics
The pharmacokinetics of givosiran and its active metabolite [AS(N-1)3′ givosiran] were evaluated following single and multiple dosing in chronic high excreter subjects and AHP patients as summarized in Table 2.
| Givosiran | AS(N-1)3′ Givosiran | ||
|---|---|---|---|
|
|||
| General Information | |||
| Steady-State Exposure | Cmax [Mean (CV%)] | 321 ng/mL (51%) | 123 ng/mL (64%) |
| AUC24 [Mean (CV%)] | 4130 ng∙h/mL (43%) | 1930 ng∙h/mL (63%) | |
| Dose Proportionality |
|
||
| Accumulation |
|
||
| Absorption | |||
| Tmax [Median (range)] | 3 (0.5-8) hours | 7 (1.5-12) hours | |
| Distribution | |||
| Apparent Central Volume of Distribution (Vz/F) [Mean (RSE%)]* | 10.4 L (2.3%) | ||
| Protein Binding | 90%† | Not evaluated | |
| Organ Distribution | Givosiran and AS(N-1)3′ givosiran distribute primarily to the liver after subcutaneous dosing. | ||
| Elimination | |||
| Half-Life [Mean (CV%)] | 6 hours (46%) | 6 hours (41%) | |
| Apparent Clearance [Mean (CV%)]* | 35.1 L/hr (18%) | 64.7 L/hr (33%) | |
| Metabolism | |||
| Primary Pathway | Givosiran is metabolized by nucleases to oligonucleotides of shorter lengths. Givosiran is not a substrate of CYP enzymes‡. | ||
| Active Metabolite | The active metabolite, AS(N-1)3′ givosiran, is equipotent to givosiran in plasma and the AUC0-24 represents 45% of givosiran AUC, at the approved recommended givosiran dosage. | ||
| Excretion | |||
| Primary Pathway | The dose recovered in urine was 5%-14% as givosiran and 4%-13% as AS(N-1)3′ givosiran§. | ||
Specific Populations
No clinically meaningful differences in givosiran pharmacokinetics or pharmacodynamics (percent reduction in urinary ALA and PBG) were observed based on age (19 to 65 years), sex, race/ethnicity, mild, moderate or severe renal impairment (eGFR ≥15 to <89 mL/min/1.73m2 estimated by the Modification of Diet in Renal Disease [MDRD] formula), and mild hepatic impairment (bilirubin ≤1×ULN and AST >1×ULN, or bilirubin >1×ULN to 1.5×ULN).The effect of end-stage renal disease (eGFR <15 mL/min/1.73m2), and moderate to severe hepatic impairment on givosiran pharmacokinetics is unknown.
Drug Interaction Studies
Clinical Studies
Effect of givosiran on CYP1A2 Substrates: Concomitant use of a single subcutaneous dose of givosiran 2.5 mg/kg increased caffeine (sensitive CYP1A2 substrate) AUC by 3.1-fold and Cmax by 1.3-fold [see Drug Interactions (7.1)].
Effect of givosiran on CYP2D6 Substrates: Concomitant use of a single subcutaneous dose of givosiran 2.5 mg/kg increased dextromethorphan (sensitive CYP2D6 substrate) AUC by 2.4-fold and Cmax by 2.0-fold [see Drug Interactions (7.1)].
Effect of givosiran on other CYP450 Substrates: Concomitant use of a single subcutaneous dose of givosiran 2.5 mg/kg increased losartan (CYP2C9 substrate) AUC by 1.1-fold with no change in Cmax; increased omeprazole (sensitive CYP2C19 substrate) AUC by 1.6-fold and Cmax by 1.1-fold; increased midazolam (sensitive CYP3A4 substrate) AUC by 1.5-fold and Cmax by 1.2-fold. These changes in exposure were not considered clinically relevant.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a carcinogenicity study in Sprague Dawley rats administered 25, 50, or 100 mg/kg givosiran by subcutaneous injection every 28 days for up to 89 weeks (males) or 85 weeks (females), givosiran doses in rats were 2, 3, and 6 times, respectively, the MRHD based on body surface area. A statistically significant increase in hepatocellular adenomas occurred in males at 100 mg/kg/month, the clinical significance of which is uncertain.
In a carcinogenicity study in male and female Tg.rasH2 mice administered givosiran by subcutaneous injection every 28 days for 26 weeks, up to 1500 mg/kg, givosiran was not carcinogenic.
Givosiran was not genotoxic in the in vitro bacterial reverse mutation (Ames) assays, an in vitro chromosomal aberration assay in cultured human peripheral blood lymphocytes or the in vivo micronucleus assay in rats.
In fertility and early embryonic development studies, givosiran administered subcutaneously once weekly at doses up to 30 mg/kg in male and female rats prior to and during mating, and continuing in females throughout organogenesis, resulted in no adverse effects on fertility or reproductive function in male or female animals.
14. Clinical Studies
The efficacy of GIVLAARI in patients with acute hepatic porphyria was evaluated in the ENVISION trial (NCT03338816), a randomized, double-blind, placebo-controlled, multinational study.
ENVISION enrolled 94 patients with acute hepatic porphyria (AHP) (89 patients with AIP, 2 patients with variegate porphyria [VP], 1 patient with hereditary coproporphyria [HCP], and 2 patients with no identified mutation). Eligible patients were randomized 1:1 to receive once monthly subcutaneous injections of GIVLAARI 2.5 mg/kg or placebo during the 6-month double-blind period. In this study, inclusion criteria specified a minimum of 2 porphyria attacks requiring hospitalization, urgent healthcare visit, or intravenous hemin administration at home in the 6 months prior to study entry. After the 6 month treatment period patients were enrolled in an open label extension period for up to 30 months. Ninety-three patients were enrolled in the open label extension period. Hemin use during the study was permitted for the treatment of acute porphyria attacks.
The median age of patients studied was 37.5 years (range 19 to 65 years), 89% of patients were female, and 78% were white. GIVLAARI and placebo arms were balanced with respect to historical porphyria attack rate, hemin prophylaxis prior to study entry, use of opioid medications, and patient-reported measures of pain symptoms between attacks.
Efficacy in the 6-month double-blind period was measured by the rate of porphyria attacks that required hospitalizations, urgent healthcare visit, or intravenous hemin administration at home.
Efficacy results for GIVLAARI are provided in Table 3. On average, AHP patients on GIVLAARI experienced 70% (95% CI: 60%, 80%) fewer porphyria attacks compared to placebo.
| Patients with AHP | ||
|---|---|---|
| GIVLAARI (N=48) | Placebo (N=46) |
|
| Mean Rate (95% CI) of Porphyria Attacks | 1.9 (1.3, 2.8) | 6.5 (4.5, 9.3) |
| Rate Ratio† (95% CI) (GIVLAARI/placebo) | 0.3‡ (0.2, 0.4) | |
| Mean Days (95% CI) of Hemin Use | 4.7 (2.8, 7.9) | 12.8 (7.6, 21.4) |
| Ratio† (95% CI) (GIVLAARI/placebo) | 0.3§ (0.1, 0.5) | |
GIVLAARI also resulted in a reduction in hemin use, urinary ALA, and urinary PBG.
16. How is Givlaari supplied
17. Patient Counseling Information
Advise patients of the potential risks of GIVLAARI treatment:
- Anaphylactic Reaction: Inform patients about the risk and possible symptoms of severe hypersensitivity reactions that could occur [see Warnings and Precautions (5.1)].
- Hepatic Toxicity: Inform patients that transaminase elevations may occur, and that laboratory testing will be conducted in the first 6 months of treatment and as clinically indicated thereafter [see Warnings and Precautions (5.2)].
- Renal Toxicity: Inform patients that increases in serum creatinine and decreases in eGFR have been reported and that laboratory testing will be conducted as clinically indicated [see Warnings and Precautions (5.3)].
- Injection Site Reactions: Inform patients of the signs and symptoms of injection site reactions (examples include redness, pain, itching, rash, discoloration, or localized swelling) [see Warnings and Precautions (5.4)].
- Blood Homocysteine Increased: Inform patients that increases in blood homocysteine levels have been reported when using GIVLAARI, and that laboratory testing will be conducted prior to and during treatment with GIVLAARI. Vitamin supplementation may be considered for elevated blood homocysteine levels [see Warnings and Precautions (5.5)].
| GIVLAARI
givosiran sodium injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Alnylam Pharmaceuticals, Inc. (115524410) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Ajinomoto Althea, Inc. | 023050730 | MANUFACTURE(71336-1001) | |
More about Givlaari (givosiran)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: miscellaneous metabolic agents
- En español