Ferriprox: Package Insert / Prescribing Info
Package insert / product label
Generic name: deferiprone
Dosage form: tablet, film coated
Drug classes: Antidotes, Chelating agents
Medically reviewed by Drugs.com. Last updated on Apr 22, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
FERRIPROX® (deferiprone) tablets, for oral use
Initial U.S. Approval: 2011
WARNING: AGRANULOCYTOSIS AND NEUTROPENIA
See full prescribing information for complete boxed warning.
• FERRIPROX can cause agranulocytosis that can lead to serious infections and death. Neutropenia may precede the development of agranulocytosis. (5.1)
• Measure the absolute neutrophil count (ANC) before starting FERRIPROX and monitor regularly while on therapy. (5.1)
• Interrupt FERRIPROX therapy if neutropenia develops. (5.1)
• Interrupt FERRIPROX if infection develops and monitor the ANC more frequently. (5.1)
• Advise patients taking FERRIPROX to report immediately any symptoms indicative of infection. (5.1)
Recent Major Changes
| Warnings and Precautions, Agranulocytosis and Neutropenia (5.1) | 3/2025 |
Indications and Usage for Ferriprox
FERRIPROX Tablets are an iron chelator indicated for the treatment of transfusional iron overload in adult and pediatric patients 8 years of age and older with thalassemia syndromes, sickle cell disease or other anemias. (1)
Limitations of Use
Safety and effectiveness have not been established for the treatment of transfusional iron overload in patients with myelodysplastic syndrome or in patients with Diamond Blackfan anemia.
Ferriprox Dosage and Administration
- FERRIPROX Tablets are available in three formulations. Two different 1,000 mg formulations, and a 500 mg formulation, which have different dosing regimens to achieve the same total daily dosage. (2.1)
- To prevent medication errors, before prescribing and dispensing, ensure that the tablet formulation is appropriate for the dosing regimen. Each tablet has distinct identifying characteristics. (2.1, 3)
- FERRIPROX Tablets (twice a day), 1,000 mg:
• Starting oral dosage: 75 mg/kg/day (actual body weight) in two divided doses (2.2)
• Maximum oral dosage: 99 mg/kg/day (actual body weight) in two divided doses (2.2)
- FERRIPROX Tablets (three times a day), 1,000 mg:
○ Starting oral dosage: 75 mg/kg/day (actual body weight) in three divided doses (2.2)
○ Maximum oral dosage: 99 mg/kg/day (actual body weight) in three divided doses (2.2)
- FERRIPROX Tablets (three times a day), 500 mg:
○ Starting oral dosage: 75 mg/kg/day (actual body weight) in three divided doses (2.2)
○ Maximum oral dosage: 99 mg/kg/day (actual body weight) in three divided doses (2.2)
Dosage Forms and Strengths
Contraindications
Hypersensitivity to deferiprone or to any of the excipients in the formulations. (4)
Warnings and Precautions
Adverse Reactions/Side Effects
- The most common adverse reactions in patients with thalassemia (incidence ≥ 6%) are nausea, vomiting, abdominal pain, arthralgia, ALT increased and neutropenia. (6)
- The most common adverse reactions in patients with sickle cell disease or other anemias (incidence ≥6%) are pyrexia, abdominal pain, bone pain, headache, vomiting, pain in extremity, sickle cell anemia with crisis, back pain, ALT increased, AST increased, arthralgia, oropharyngeal pain, nasopharyngitis, neutrophil count decreased, cough and nausea. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Chiesi USA, Inc. at 1-888-661-9260 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Drug Interactions
- Drugs Associated with Neutropenia or Agranulocytosis: Avoid co-administration. If co-administration is unavoidable, closely monitor the absolute neutrophil count. (7.1)
- UGT1A6 Inhibitors: Avoid co-administration. (7.2)
- Polyvalent Cations: Allow at least a 4-hour interval between administration of FERRIPROX and drugs or supplements containing polyvalent cations (e.g., iron, aluminum, or zinc). (2.2, 7.2)
Use In Specific Populations
- Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2025
Full Prescribing Information
WARNING: AGRANULOCYTOSIS AND NEUTROPENIA
• FERRIPROX can cause agranulocytosis that can lead to serious infections and death. Neutropenia may precede the development of agranulocytosis. [see Warnings and Precautions (5.1)]
• Measure the absolute neutrophil count (ANC) before starting FERRIPROX therapy and monitor regularly while on therapy.
• Interrupt FERRIPROX therapy if neutropenia develops. [see Warnings and Precautions (5.1)]
• Interrupt FERRIPROX if infection develops, and monitor the ANC more frequently. [see Warnings and Precautions (5.1)]
• Advise patients taking FERRIPROX to report immediately any symptoms indicative of infection. [see Warnings and Precautions (5.1)]
1. Indications and Usage for Ferriprox
FERRIPROX Tablets are indicated for the treatment of transfusional iron overload in adult and pediatric patients 8 years of age and older with thalassemia syndromes, sickle cell disease or other anemias.
Limitations of Use
• Safety and effectiveness have not been established for the treatment of transfusional iron overload in patients with myelodysplastic syndrome or in patients with Diamond Blackfan anemia.
2. Ferriprox Dosage and Administration
2.1 Important Dosage and Administration Information
FERRIPROX Tablets are available in two different 1,000 mg formulations and a 500 mg formulation, which have different oral dosing regimens to achieve the same total daily dosage.
- FERRIPROX Tablets (twice a day) - 1,000 mg - given two times a day [see Dosage and Administration (2.2)]
- FERRIPROX Tablets (three times a day) - 1,000 mg - given three times a day [see Dosage and Administration (2.3)]
- FERRIPROX Tablets - 500 mg - given three times a day [see Dosage and Administration (2.4)]
To prevent medication errors, before prescribing and dispensing, ensure that the tablet formulation is appropriate for the dosing regimen. Each tablet has distinct identifying characteristics [see Dosage Forms and Strengths (3)].
For patients who have trouble swallowing tablets, consider the use of FERRIPROX Oral Solution (see the prescribing information for FERRIPROX Oral Solution).
Monitoring for Safety
Due to the risk of agranulocytosis, monitor ANC before and during FERRIPROX therapy.
Test ANC prior to start of FERRIPROX therapy and monitor on the following schedule during treatment:
• First six months of therapy: Monitor ANC weekly;
• Next six months of therapy: Monitor ANC once every two weeks;
• After one year of therapy: Monitor ANC every two to four weeks (or at the patient’s blood transfusion interval in patients that have not experienced an interruption due to any decrease in ANC [see Warnings and Precautions (5.1)].
Due to the risk of hepatic transaminase elevations, monitor ALT before and monthly during FERRIPROX therapy [see Warnings and Precautions (5.2)].
Due to the risk of zinc deficiency, monitor zinc levels before and regularly during FERRIPROX therapy [see Warnings and Precautions (5.3)].
2.2 Recommended Dosage for 1,000 mg FERRIPROX Tablets (twice a day) for Adult and Pediatric Patients with Transfusional Iron Overload due to Thalassemia Syndromes, Sickle Cell Disease or Other Anemias
Starting Dosage for Twice a Day Tablets
The recommended starting oral dosage of FERRIPROX Tablets (twice a day) is 75 mg/kg/day (actual body weight) in two divided doses per day (taken approximately 12 hours apart), with food. Round the total daily dose to the nearest 500 mg (half-tablet). Table 1 describes the number of FERRIPROX Tablets (twice a day) needed to achieve the 75 mg/kg/day total starting daily dosage.
| Table 1: Number of FERRIPROX 1,000 mg Tablets (twice a day) Needed to Achieve the Total Starting Daily Dosage of 75 mg/kg (rounded to the nearest half-tablet) | ||
| Body Weight
(kg) | Morning | Evening |
| 20 | 0.5 | 1 |
| 30 | 1 | 1.5 |
| 40 | 1.5 | 1.5 |
| 50 | 2 | 2 |
| 60 | 2 | 2.5 |
| 70 | 2.5 | 3 |
| 80 | 3 | 3 |
| 90 | 3.5 | 3.5 |
To minimize gastrointestinal upset when first starting therapy, dosing can start at 45 mg/kg/day and increase weekly by 15 mg/kg/day increments until the full prescribed dose is achieved.
Dosage Adjustments for Twice a Day Tablets
Tailor dosage adjustments of FERRIPROX Tablets (twice a day) to the individual patient’s response and therapeutic goals (maintenance or reduction of body iron burden). The maximum total daily oral dosage is 99 mg/kg (actual body weight) divided into two doses taken approximately 12 hours apart with food. Table 2 describes the number of FERRIPROX Tablets (twice a day) needed to achieve the 99 mg/day total maximum daily dosage.
| Table 2: Number of FERRIPROX 1,000 mg Tablets (twice a day) Needed to Achieve a Total Maximum Recommended Daily Dosage of 99 mg/kg (rounded to the nearest half-tablet) | ||
| Body Weight
(kg) | Morning | Evening |
| 20 | 1 | 1 |
| 30 | 1.5 | 1.5 |
| 40 | 2 | 2 |
| 50 | 2.5 | 2.5 |
| 60 | 3 | 3 |
| 70 | 3.5 | 3.5 |
| 80 | 4 | 4 |
| 90 | 4.5 | 4.5 |
2.3 Recommended Dosage for 1,000 mg FERRIPROX Tablets (three times a day) for Adult and Pediatric Patients with Transfusional Iron Overload due to Thalassemia Syndromes, Sickle Cell Disease or Other Anemias
Starting Dosage for Three Times a Day Tablets
The recommended starting oral dosage of FERRIPROX Tablets (three times a day) is 75 mg/kg/day (actual body weight), in three divided doses per day. Table 3 describes the number of FERRIPROX Tablets (three times a day) needed to achieve the 75 mg/kg/day total starting dosage). Round dose to the nearest 500 mg (half-tablet).
| Table 3: Number of FERRIPROX 1,000 mg Tablets (three times a day) Needed to Achieve the Total Starting Daily Dosage of 75 mg/kg (rounded to the nearest half-tablet) | |||
| Body Weight
(kg) | Morning | Midday | Evening |
| 20 | 0.5 | 0.5 | 0.5 |
| 30 | 1 | 0.5 | 1 |
| 40 | 1 | 1 | 1 |
| 50 | 1.5 | 1 | 1.5 |
| 60 | 1.5 | 1.5 | 1.5 |
| 70 | 2 | 1.5 | 2 |
| 80 | 2 | 2 | 2 |
| 90 | 2.5 | 2 | 2.5 |
To minimize gastrointestinal upset when first starting therapy, dosing can start at 45 mg/kg/day and increase weekly by 15 mg/kg/day increments until the full prescribed dose is achieved.
Dosage Adjustments for Three Times Daily Tablets
Tailor dosage adjustments for FERRIPROX Tablets (three times a day) to the individual patient’s response and therapeutic goals (maintenance or reduction of body iron burden). The maximum oral dosage is 99 mg/kg/day (actual body weight), in three divided doses per day. Table 4 describes the number of FERRIPROX Tablets (three times a day) needed to achieve the 99 mg/day total maximum daily dosage.
| Table 4: Number of FERRIPROX 1,000 mg Tablets (three times a day) Needed to Achieve the Maximum Total Daily Dosage of 99 mg/kg (rounded to the nearest half-tablet) | |||
| Body Weight
(kg) | Morning | Midday | Evening |
| 20 | 0.5 | 0.5 | 1 |
| 30 | 1 | 1 | 1 |
| 40 | 1.5 | 1 | 1.5 |
| 50 | 1.5 | 1.5 | 2 |
| 60 | 2 | 2 | 2 |
| 70 | 2.5 | 2 | 2.5 |
| 80 | 2.5 | 2.5 | 3 |
| 90 | 3 | 3 | 3 |
2.4 Recommended Dosage for 500 mg FERRIPROX Tablets (three times a day) for Adult and Pediatric Patients with Transfusional Iron Overload due to Thalassemia Syndromes, Sickle Cell Disease or Other Anemias
Starting Dosage for Three Times a Day Tablets
The recommended starting oral dosage of FERRIPROX Tablets (three times a day) is 75 mg/kg/day (actual body weight), in three divided doses per day. Table 5 describes the number of FERRIPROX Tablets (three times a day) needed to achieve the 75 mg/kg/day total starting dosage. Round dose to the nearest 250 mg (half-tablet).
| Table 5: Number of FERRIPROX 500 mg Tablets (three times a day) Needed to Achieve the Total Starting Daily Dosage of 75 mg/kg dose (rounded to the nearest half-tablet) | |||
| Body Weight
(kg) | Morning | Midday | Evening |
| 20 | 1 | 1 | 1 |
| 30 | 1.5 | 1.5 | 1.5 |
| 40 | 2 | 2 | 2 |
| 50 | 2.5 | 2.5 | 2.5 |
| 60 | 3 | 3 | 3 |
| 70 | 3.5 | 3.5 | 3.5 |
| 80 | 4 | 4 | 4 |
| 90 | 4.5 | 4.5 | 4.5 |
To minimize gastrointestinal upset when first starting therapy, dosing can start at 45 mg/kg/day and increase weekly by 15 mg/kg/day increments until the full prescribed dose is achieved.
Dosage Adjustments
Tailor dosage adjustments for FERRIPROX Tablets (three times a day) to the individual patient’s response and therapeutic goals (maintenance or reduction of body iron burden). The maximum oral dosage is 99 mg/kg/day (actual body weight), in three divided doses per day. Table 6 describes the number of FERRIPROX Tablets (three times a day) needed to achieve the 99 mg/day total maximum daily dosage.
| Table 6: Number of FERRIPROX 500 mg Tablets (three times a day) Needed to Achieve the Maximum Total Daily Dosage of 99 mg/kg dose (rounded to the nearest half-tablet) | |||
| Body Weight
(kg) | Morning | Midday | Evening |
| 20 | 1.5 | 1 | 1.5 |
| 30 | 2 | 2 | 2 |
| 40 | 3 | 2 | 3 |
| 50 | 3.5 | 3 | 3.5 |
| 60 | 4 | 4 | 4 |
| 70 | 5 | 4.5 | 4.5 |
| 80 | 5.5 | 5 | 5.5 |
| 90 | 6 | 6 | 6 |
2.5 Monitoring Ferritin Levels to Assess Efficacy
Monitor serum ferritin concentration every two to three months to assess the effect of FERRIPROX on body iron stores. If the serum ferritin is consistently below 500 mcg/L, consider temporarily interrupting FERRIPROX therapy until serum ferritin rises above 500 mcg/L.
3. Dosage Forms and Strengths
- Tablets (twice a day): 1,000 mg, capsule-shaped, white to off-white tablets with functional scoring, engraved “FPX” bisect “DR” on one side, “APO” bisect “1000” on the other”.
- Tablets (three times a day): 1,000 mg film-coated, capsule-shaped, white to off-white tablets with functional scoring, and imprinted with “APO” score “1000” on one side and plain on the other.
- Tablets: 500 mg film-coated, capsule-shaped, white to off-white tablets with functional scoring, and imprinted with “APO” score “500” on one side and plain on the other.
4. Contraindications
FERRIPROX is contraindicated in patients with known hypersensitivity to deferiprone or to any of the excipients in the formulations. The following reactions have been reported in association with the administration of deferiprone: Henoch-Schönlein purpura; urticaria; and periorbital edema with skin rash [see Adverse Reactions (6.2)].
5. Warnings and Precautions
5.1 Agranulocytosis and Neutropenia
Fatal agranulocytosis can occur with FERRIPROX use. FERRIPROX can also cause neutropenia, which may foreshadow agranulocytosis. Measure the absolute neutrophil count (ANC) before starting FERRIPROX therapy and monitor it regularly while on therapy [see Dosage and Administration (2.1)].
Reduction in the frequency of ANC monitoring should be considered on an individual patient basis, according to the health care provider’s assessment of the patient’s understanding of the risk minimization measures required during therapy.
Interrupt FERRIPROX therapy if neutropenia develops (ANC < 1.5 x 109/L).
Interrupt FERRIPROX if infection develops and monitor the ANC frequently.
Advise patients taking FERRIPROX to immediately interrupt therapy and report to their physician if they experience any symptoms indicative of infection.
The incidence of agranulocytosis was 1% of patients in pooled clinical trials of 642 patients with thalassemia syndromes and 0.5% of patients in pooled clinical trials of 196 patients with sickle cell disease or other anemias. The mechanism of FERRIPROX-associated agranulocytosis is unknown. Agranulocytosis and neutropenia usually resolve upon discontinuation of FERRIPROX, but there have been reports of agranulocytosis leading to death.
Implement a plan to monitor for and to manage agranulocytosis and neutropenia prior to initiating FERRIPROX treatment.
For agranulocytosis (ANC < 0.2 x 109/L) and severe neutropenia (0.2 x 109/L ≤ ANC < 0.5 x 109/L):
Consider hospitalization and other management as clinically appropriate.
Do not resume FERRIPROX in patients who have developed agranulocytosis unless potential benefits outweigh potential risks. Do not rechallenge patients who have developed neutropenia with FERRIPROX unless potential benefits outweigh potential risks.
For neutropenia (ANC < 1.5 x 109/L and ≥ 0.5 x 109/L):
Instruct the patient to immediately discontinue FERRIPROX and all other medications with a potential to cause neutropenia.
Obtain a complete blood cell (CBC) count, including a white blood cell (WBC) count corrected for the presence of nucleated red blood cells, an absolute neutrophil count (ANC), and a platelet count daily until recovery (ANC ≥ 1.5 x 109/L).
5.2 Liver Enzyme Elevations
In pooled clinical trials, 7.5% of 642 patients with thalassemia syndromes treated with FERRIPROX developed increased ALT values. Four (0.62%) FERRIPROX-treated subjects discontinued the drug due to increased serum ALT levels and 1 (0.16%) due to an increase in both ALT and AST. In pooled clinical trials, 7.7% of 196 patients with sickle cell disease or other anemias treated with FERRIPROX developed increased ALT values.
Monitor serum ALT values monthly during therapy with FERRIPROX and consider interruption of therapy if there is a persistent increase in the serum transaminase levels [see Dosage and Administration (2.1)].
5.3 Zinc Deficiency
Decreased plasma zinc concentrations have been observed on FERRIPROX therapy. Monitor plasma zinc annually, and supplement in the event of a deficiency [see Dosage and Administration (2.1)].
5.4 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies and evidence of genotoxicity, FERRIPROX can cause fetal harm when administered to a pregnant woman. The available data on the use of FERRIPROX in pregnant women are insufficient to inform risk. In animal studies, administration of deferiprone during the period of organogenesis resulted in embryo-fetal death and malformations at doses lower than equivalent human clinical doses. Advise pregnant women and females of reproductive potential of the potential risk to the fetus [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use an effective method of contraception during treatment with FERRIPROX and for at least six months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with FERRIPROX and for at least three months after the last dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described below and elsewhere in the labeling:
- Agranulocytosis and Neutropenia [see Warnings and Precautions (5.1)]
- Liver Enzyme Elevations [see Warnings and Precautions (5.2)]
- Zinc Deficiency [see Warnings and Precautions (5.3)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
FERRIPROX Tablets (twice a day) were evaluated in trials in healthy subjects. FERRIPROX Tablets (twice a day) contain deferiprone, the same active ingredient as FERRIPROX Tablets (deferiprone) (three times a day) and FERRIPROX Oral Solution (deferiprone).
The following adverse reaction information represents the pooled data collected from single arm or active-controlled clinical trials with FERRIPROX Tablets (deferiprone) (three times a day) or FERRIPROX Oral Solution (deferiprone).
Thalassemia Syndromes
The safety of FERRIPROX was evaluated in the pooled clinical trial database [see Clinical Studies (14.1)]. Patients received FERRIPROX Tablets (three times a day) or FERRIPROX Oral Solution . FERRIPROX was administered orally three times a day (total daily dose either 50, 75, or 99 mg/kg), N=642. Among 642 patients receiving FERRIPROX, 492 (76.6%) were exposed for 6 months or longer and 365 (56.9%) were exposed for greater than one year.
The median age of patients who received FERRIPROX was 19 years (range 1, 77 years); 50.2% female; 71.2% White, 17.8% Asian, 9.2% Unknown, 1.2% Multi-racial and 0.6% Black.
The most serious adverse reaction reported in clinical trials with FERRIPROX was agranulocytosis [see Warnings and Precautions (5.1)].
The most common adverse reactions (≥6%) reported during clinical trials were nausea, vomiting, abdominal pain, arthralgia, alanine aminotransferase increased and neutropenia.
The table below lists the adverse drug reactions that occurred in at least 1% of patients treated with FERRIPROX in clinical trials in patients with thalassemia syndromes.
| Body System | (N=642) |
| Adverse Reaction | % Patients |
| BLOOD AND LYMPHATIC SYSTEM DISORDERS | |
| Neutropenia* | 7 |
| Agranulocytosis† | 1 |
| GASTROINTESTINAL DISORDERS | |
| Nausea | 13 |
| Abdominal pain/discomfort | 10 |
| Vomiting | 10 |
| Diarrhea | 3 |
| Dyspepsia | 2 |
| INVESTIGATIONS | |
| Alanine aminotransferase increased | 7 |
| Weight increased | 2 |
| Aspartate aminotransferase increased | 1 |
| METABOLISM AND NUTRITION
DISORDERS | |
| Increased appetite | 4 |
| Decreased appetite | 1 |
| MUSCULOSKELETAL AND
CONNECTIVE TISSUE DISORDERS | |
| Arthralgia | 10 |
| Back pain | 2 |
| Pain in extremity | 2 |
| Arthropathy | 1 |
| NERVOUS SYSTEM DISORDERS | |
| Headache | 2 |
| *Neutropenia includes events of severe neutropenia (ANC ≥0.2 x 109/L and <0.5 x 109/L). †Agranulocytosis (ANC< 0.2 x 109/L) |
|
Gastrointestinal symptoms such as nausea, vomiting, and abdominal pain were the most frequent adverse reactions reported by patients participating in clinical trials and led to the discontinuation of FERRIPROX therapy in 1.6% of patients.
Chromaturia (reddish/brown discoloration of the urine) is a result of the excretion of iron in the urine.
Sickle Cell Disease or Other Anemias
The safety of FERRIPROX compared to deferoxamine was evaluated in LA38-0411 [see Clinical Studies (14.2)]. Patients received FERRIPROX Tablets or FERRIPROX Oral Solution orally three times a day (total daily dose 75-99 mg/kg/day) n=152) or the control arm, deferoxamine, 20-40 mg/kg/day (children) or 40-50 mg/kg/day (adults), by subcutaneous infusion for 5 – 7 days per week, n=76. Among 152 patients receiving FERRIPROX, 120 (78.9%) were exposed for 6 months or longer and 17 (11.2%) were exposed for greater than one year.
The median age of patients who received FERRIPROX was 15 years (range 3, 59 years); 54.6% male; 78.9% White, 15.1% Black and 5.9% Multi-racial.
The most common adverse reactions (≥6%) reported during clinical trials in patients with SCD or other anemias were pyrexia, abdominal pain, bone pain, headache, vomiting, pain in extremity, sickle cell anemia with crisis, back pain, alanine aminotransferase (ALT) increased, aspartate aminotransferase (AST) increased, arthralgia, oropharyngeal pain, nasopharyngitis, neutrophil count decreased, cough and nausea.
The table below lists the adverse reactions (irrespective of a causal assessment; adverse events) of interest that occurred in patients treated with FERRIPROX in clinical trials in subjects with sickle cell disease or other anemias.
| Body System
Adverse Reaction | FERRIPROX (N=152)
% Patients | DEFEROXAMINE (N=76)
% Patients |
| BLOOD AND LYMPHATIC SYSTEM DISORDERS | ||
| Sickle cell anemia with crisis | 17 | 13 |
| GASTROINTESTINAL DISORDERS | ||
| Abdominal pain* | 26 | 13 |
| Vomiting | 19 | 11 |
| Nausea | 7 | 9 |
| Diarrhea | 5 | 8 |
| GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONS | ||
| Pyrexia | 28 | 33 |
| Pain | 5 | 4 |
| INFECTIONS AND INFESTATIONS | ||
| Nasopharyngitis | 9 | 12 |
| Upper respiratory tract infection | 5 | 3 |
| INVESTIGATIONS | ||
| Alanine aminotransferase increased | 12 | 0 |
| Aspartate aminotransferase increased | 11 | 0 |
| Neutrophil count decreased | 8 | 4 |
| MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERS | ||
| Bone pain | 25 | 34 |
| Pain in extremity | 18 | 15 |
| Back pain | 13 | 18 |
| Arthralgia | 10 | 8 |
| NERVOUS SYSTEM DISORDERS | ||
| Headache | 20 | 13 |
| RESPIRATORY, THORACIC AND MEDIASTINAL DISORDERS | ||
| Oropharyngeal pain | 10 | 15 |
| Cough | 8 | 15 |
*Grouped term
Clinically relevant adverse reactions in <5% of patients include neutropenia and agranulocytosis.
Pediatric Patients
FERRIPROX has been studied in 86 pediatric patients with sickle cell disease or other anemias. Pediatric patients (<17 years) had an increase in the following adverse reactions as compared to adults: abdominal pain, neutrophil count decreased, bone pain and oropharyngeal pain.
6.2 Postmarketing Experience
The following additional adverse reactions have been reported in patients receiving FERRIPROX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or to establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: thrombocytosis, pancytopenia.
Cardiac disorders: atrial fibrillation, cardiac failure.
Congenital, familial and genetic disorders: hypospadias.
Eye disorders: diplopia, papilledema, retinal toxicity.
Gastrointestinal disorders: enterocolitis, rectal hemorrhage, gastric ulcer, pancreatitis, parotid gland enlargement.
General disorders and administration site conditions: chills, edema peripheral, multi-organ failure.
Hepatobiliary disorders: jaundice, hepatomegaly.
Immune system disorders: anaphylactic shock, hypersensitivity.
Infections and infestations: cryptococcal cutaneous infection, enteroviral encephalitis, pharyngitis, pneumonia, sepsis, furuncle, infectious hepatitis, rash pustular, subcutaneous abscess.
Investigations: blood bilirubin increased, blood creatinine phosphokinase increased.
Metabolism and nutrition disorders: metabolic acidosis, dehydration.
Musculoskeletal and connective tissue disorders: myositis, chondropathy, trismus.
Nervous system disorders: cerebellar syndrome, cerebral hemorrhage, convulsion, gait disturbance, intracranial pressure increased, psychomotor skills impaired, pyramidal tract syndrome, somnolence.
Psychiatric disorders: bruxism, depression, obsessive-compulsive disorder.
Renal disorders: glycosuria, hemoglobinuria.
Respiratory, thoracic and mediastinal disorders: acute respiratory distress syndrome, epistaxis, hemoptysis, pulmonary embolism.
Skin, subcutaneous tissue disorders: hyperhidrosis, periorbital edema, photosensitivity reaction, pruritis, urticaria, rash, Henoch-Schönlein purpura.
Vascular disorders: hypotension, hypertension.
Related/similar drugs
7. Drug Interactions
7.1 Drugs Associated with Neutropenia or Agranulocytosis
Avoid co-administration of FERRIPROX with other drugs known to be associated with neutropenia or agranulocytosis. If co-administration is unavoidable, closely monitor the absolute neutrophil count [see Warnings and Precautions (5.1)].
7.2 Effect of Other Drugs on FERRIPROX
UDP-Glucuronosyltransferases (UGT)
Avoid use of UGT1A6 inhibitors (e.g., diclofenac, probenecid, or silymarin (milk thistle)) with FERRIPROX [see Dosage and Administration (2.2), Adverse Reactions (6.1), Clinical Pharmacology (12.3)].
Polyvalent Cations
Deferiprone has the potential to bind polyvalent cations (e.g., iron, aluminum, and zinc); allow at least a 4-hour interval between FERRIPROX and other medications (e.g., antacids), or supplements containing these polyvalent cations [see Dosage and Administration (2.2)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
In animal reproduction studies, oral administration of deferiprone to pregnant rats and rabbits during organogenesis at doses 33% and 49%, respectively, of the maximum recommended human dose (MRHD) resulted in structural abnormalities, embryo-fetal mortality and alterations to growth (see Data). The limited available data from deferiprone use in pregnant women are insufficient to inform a drug-associated risk of major birth defects and miscarriage. Based on evidence and developmental toxicity in animal studies, FERRIPROX can cause fetal harm when administered to a pregnant woman. Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and of miscarriage is 2-4% and 15-20%, respectively.
Data
Human Data
Post-marketing data available from 39 pregnancies of deferiprone-treated patients and 10 pregnancies of partners of deferiprone-treated patients are as follows:
Of the 39 pregnancies in deferiprone-treated patients, 23 resulted in healthy newborns, 6 ended in spontaneous abortion, 9 had unknown outcomes, and 1 infant was born with anal atresia, nephroptosis, ventricular septal defect, hemivertebra and urethral fistula.
Of the 10 pregnancies in partners of deferiprone-treated patients, 5 resulted in healthy newborns, 1 resulted in a healthy newborn with slight hypospadias, 1 was electively terminated, 1 resulted in the intrauterine death of twins, and 2 had unknown outcomes.
Animal Data
During organogenesis, pregnant rats and rabbits received deferiprone at oral doses of 0, 30, 80 or 200 mg/kg/day, and 0, 10, 50, or 150 mg/kg/day, respectively. The daily dose was administered as two equal divided doses approximately 7 hours apart. Doses of 200 mg/kg/day in rats and 150 mg/kg/day in rabbits, approximately 33% and 49% of the MRHD, respectively, resulted in increased post-implantation loss and reduced fetal weights in the presence of maternal toxicity (reduced maternal body weight and body weight gain in both rats and rabbits; abnormal large placenta at low incidence in rats). The 200 mg/kg/day dose in rats resulted in external, visceral and skeletal fetal malformations such as cranial malformations, cleft palate, limb malrotation, anal atresia, internal hydrocephaly, anophthalmia and fused bones. The dose of 150 mg/kg/day in rabbits resulted in external fetal malformations (partially opened eyes) and minor blood vessel and skeletal variations.
In rats, malformations including micrognathia and persistent ductus arteriosus could be observed in the absence of maternal toxicity at doses equal to or greater than 30 and 80 mg/kg/day, approximately 5% and 13% of the MHRD, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of deferiprone in human milk, the effects on the breastfed child, or the effects on milk production.
Because of the potential for serious adverse reactions in the breastfed child, including the potential for tumorigenicity shown for deferiprone in animal studies, advise patients that breastfeeding is not recommended during treatment with FERRIPROX, and for at least 2 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiating FERRIPROX.
Contraception
Females
FERRIPROX can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise female patients of reproductive potential to use effective contraception during treatment with FERRIPROX and for at least 6 months after the last dose.
Males
Based on genotoxicity findings, advise males with female partners of reproductive potential to use effective contraception during treatment with FERRIPROX and for at least 3 months after the last dose [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of FERRIPROX for the treatment of transfusional iron overload due to thalassemia syndromes have been established in pediatric patients 8 years of age and older. Use of FERRIPROX for this indication is supported by evidence of efficacy from clinical trials in adult patients with thalassemia and evidence of safety in pediatric patients with sickle cell disease.
The safety and effectiveness of FERRIPROX for the treatment of transfusional iron overload due to sickle cell disease or other anemias have been established in 86 pediatric patients 3 to 16 years of age, among the 152 patients treated with FERRIPROX Tablets or Oral Solution in an adequate and well-controlled study [see Adverse Reactions (6.1) and Clinical Studies (14.2)]. The study included 56 patients 3 to <12 years of age and 30 patients 12 to 16 years of age. Seventy-six percent of these patients had sickle cell disease. The recommended starting dose and dose-modifications are the same for children and adults [see Indications and Usage (1), Dosage and Administration (2.1), and Clinical Studies (14)].
Fourteen patients with spherocytosis (including hereditary) (ages 3-15), two patients with pyruvate kinase deficiency (ages 4 and 6), two patients with dyserythropoietic anemia (ages 10-12) and two patients with hemolytic anemia (ages 8 and 10 years old) were treated with FERRIPROX in the clinical trial, LA38-0411.
A US registry established from December 2011 through December 2019, contains 125 patients from 4 to < 17 years old who have received FERRIPROX and have sickle cell disease. The adverse reactions, including agranulocytosis, seen in the 8 year period of the registry are similar to those seen in the most recent clinical studies.
Safety and effectiveness of FERRIPROX Tablets have not been established in pediatric patients with chronic iron overload due to blood transfusions who are less than 8 years of age.
8.5 Geriatric Use
Clinical studies of deferiprone did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
10. Overdosage
No cases of acute overdose have been reported. There is no specific antidote to FERRIPROX overdose.
Neurological disorders such as cerebellar symptoms, diplopia, lateral nystagmus, psychomotor slowdown, hand movements and axial hypotonia have been observed in children treated with 2.5 to 3 times the recommended dose for more than one year. The neurological disorders progressively regressed after deferiprone discontinuation.
11. Ferriprox Description
FERRIPROX Tablets (deferiprone) contain 1,000 mg or 500 mg deferiprone (3-hydroxy-1,2-dimethylpyridin-4-one), a synthetic, orally active, iron-chelating agent. The molecular formula for deferiprone is C7H9NO2 and its molecular weight is 139.15 g/mol. Deferiprone has the following structural formula:
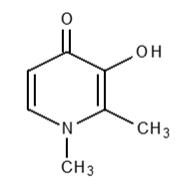
Deferiprone is a white to pinkish-white powder. It is sparingly soluble in deionized water (14.3 mg/mL) and has a melting point range of 272 °C - 278 °C.
FERRIPROX Tablets (twice a day), 1,000 mg
White to off-white, capsule-shaped tablets, and imprinted with “FPX” score “DR” on one side and “APO” score “1000” on the other. The tablets can be broken in half along the score line. Each tablet contains 1,000 mg deferiprone and the following inactive ingredients: Tablet core - hypromellose acetate succinate, magnesium oxide, colloidal silicon dioxide and magnesium stearate; Coating - triethyl citrate, talc, titanium dioxide, and methacrylic acid and ethyl acrylate copolymer.
FERRIPROX Tablets (three times a day), 1,000 mg
White to off-white, capsule-shaped tablets, and imprinted with “APO” score “1000” on one side and plain on the other. The tablets can be broken in half along the score line. Each tablet contains 1,000 mg deferiprone and the following inactive ingredients: Tablet core - methylcellulose, crospovidone, and magnesium stearate; Coating - hypromellose, hydroxypropyl cellulose, macrogol, and titanium dioxide.
FERRIPROX Tablets, 500 mg
White to off-white, capsule-shaped tablets, and imprinted with “APO” score “500” on one side and plain on the other. The tablets can be broken in half along the score line. Each tablet contains 500 mg deferiprone and the following inactive ingredients: Tablet core - microcrystalline cellulose, magnesium stearate, colloidal silicon dioxide; Coating - hypromellose, polyethylene glycol, titanium dioxide.
12. Ferriprox - Clinical Pharmacology
12.1 Mechanism of Action
Deferiprone is a chelating agent with an affinity for ferric ions (iron III). Deferiprone binds with ferric ions to form neutral 3:1 (deferiprone:iron) complexes that are stable at physiological pH.
12.2 Pharmacodynamics
No clinical studies were performed to assess the relationship between the dose of deferiprone and the amount of iron eliminated from the body.
Cardiac Electrophysiology
At the maximum approved recommended dose, deferiprone does not prolong the QT interval to any clinically relevant extent.
12.3 Pharmacokinetics
FERRIPROX Tablets (twice a day), 1,000 mg
In healthy subjects, the mean ± SD Cmax of deferiprone in serum was 6 ± 2 mcg/mL, and the mean ± SD AUC was 28 ± 7 mcg∙h/mL following oral administration of a 1,000 mg dose of FERRIPROX Tablets (twice a day) with food.
Absorption
Peak serum concentrations of deferiprone occur approximately 2 hours after a single dose of FERRIPROX Tablets (twice a day) in fasted healthy subjects.
Effect of Food
Following the administration of FERRIPROX Tablets (twice a day) to healthy volunteers, the Cmax and the AUC of deferiprone remain unchanged after a high-fat meal (approximately 1,000 calories, 53% fat, 33% carbohydrates, and 14% protein) compared to fasted conditions.
Effect of Alcohol
At 40% (v/v) alcohol concentration in vitro dissolution studies, there was 88% release of deferiprone from a 1,000 mg FERRIPROX tablet (twice a day) within two hours compared to 4% release of deferiprone within 2 hours in the absence of alcohol.
Distribution
The apparent mean ± SD volume of distribution (V/F) of deferiprone was 97 ± 28 L following oral administration of a 1,000 mg dose of FERRIPROX Tablets (twice a day) with food.
Elimination
The mean ± SD elimination half-life of deferiprone is 1.8 ± 0.3 hours following the administration of FERRIPROX Tablets (twice a day).
Metabolism
Deferiprone is metabolized primarily by UGT1A6. The major metabolite of deferiprone is the 3-O-glucuronide, which lacks iron- binding capability.
Excretion
Following oral administration, 75% to 90% of the administered dose was recovered in urine (primarily as metabolite) in the first 24 hours.
FERRIPROX Tablets (three times a day), 1,000 mg and 500 mg
The mean Cmax and AUC of deferiprone was 20 mcg/mL and 50 mcg∙h/mL, respectively, in healthy subjects. The dose proportionality of deferiprone over the approved recommended dosage range is unknown.
Absorption
Deferiprone appeared in the blood within 5 to 10 minutes after oral administration. Peak serum concentration of deferiprone was reached approximately 1 to 2 hours after a single dose.
Effect of Food
No clinically significant differences in the pharmacokinetics of deferiprone were observed following administration with food.
Elimination
The elimination half-life of deferiprone is approximately 2 hours.
Metabolism
Deferiprone is metabolized primarily by UGT1A6. The major metabolite of deferiprone is the 3-O-glucuronide, which lacks iron binding capability.
Excretion
Following oral administration, 75% to 90% of the administered dose was recovered in urine (primarily as metabolite) in the first 24 hours.
Specific Populations
No clinically significant differences in the pharmacokinetics of deferiprone were observed based on sex, race/ethnicity, body weight, mild to severe (eGFR 15 to 89 mL/min/1.73 m2) renal impairment, or mild (Child Pugh Class A) to moderate (Child Pugh Class B) hepatic impairment. The effect of age, including geriatric or pediatric populations, end stage renal disease or severe (Child Pugh Class C) hepatic impairment on the pharmacokinetics of deferiprone is unknown.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with deferiprone. However, in view of the genotoxicity results, and the findings of mammary gland hyperplasia and mammary gland tumors in rats treated with deferiprone in the 52-week toxicology study, tumor formation in carcinogenicity studies must be regarded as likely.
Deferiprone was positive in a mouse lymphoma cell assay in vitro. Deferiprone was clastogenic in an in vitro chromosomal aberration test in mice and in a chromosomal aberration test in Chinese Hamster Ovary cells. Deferiprone given orally or intraperitoneally was clastogenic in a bone marrow micronucleus assay in non-iron-loaded mice. A micronucleus test was also positive when mice predosed with iron dextran were treated with deferiprone. Deferiprone was not mutagenic in the Ames bacterial reverse mutation test.
A fertility and early embryonic development study of deferiprone was conducted in rats. Sperm counts, motility and morphology were unaffected by treatment with deferiprone. There were no effects observed on male or female fertility or reproductive function at the highest dose which was 25% of the MRHD.
14. Clinical Studies
FERRIPROX Tablets (twice a day) were evaluated in trials in healthy subjects. FERRIPROX Tablets (twice a day) contain deferiprone, the same active ingredient as FERRIPROX Tablets and FERRIPROX Oral Solution. The following information is based on studies with FERRIPROX Tablets (deferiprone) (three times a day) and FERRIPROX Oral Solution (deferiprone).
14.1 Transfusional Iron Overload in Patients with Thalassemia Syndromes
In a prospective, planned, pooled analysis of patients with thalassemia syndromes from several studies, the efficacy of deferiprone was assessed in transfusion-dependent iron overload patients in whom previous iron chelation therapy had failed or was considered inadequate due to poor tolerance. The main criterion for chelation failure was serum ferritin > 2,500 mcg/L before treatment with deferiprone. Deferiprone therapy (35-99 mg/kg/day) was considered successful in individual patients who experienced a ≥ 20% decline in serum ferritin within one year of starting therapy.
Data from a total of 236 patients were analyzed. Of the 224 patients with thalassemia who received deferiprone monotherapy and were eligible for serum ferritin analysis, 105 (47%) were male and 119 (53%) were female. The mean age of these patients was 18.2 years (range 2 to 62; 91 patients were <17).
For the patients in the analysis, the endpoint of at least a 20% reduction in serum ferritin was met in 50% (of 236 subjects), with a 95% confidence interval of 43% to 57%.
A small number of patients with thalassemia and iron overload were assessed by measuring the change in the number of milliseconds (ms) in the cardiac MRI T2* value before and after treatment with deferiprone for one year. There was an increase in cardiac MRI T2* from a mean at baseline of 11.8 ± 4.9 ms to a mean of 15.1 ± 7.0 ms after approximately one year of treatment. The clinical significance of this observation is not known.
14.2 Transfusional Iron Overload in Patients with Sickle Cell Disease and other Anemias
Study LA38-0411, an actively-controlled non-inferiority study compared the efficacy of FERRIPROX to that of deferoxamine in patients with sickle cell disease and other transfusion-dependent anemias by evaluating liver iron concentration (LIC). The efficacy of FERRIPROX was established based upon the change in LIC from baseline after 12 months of FERRIPROX (75 or 99 mg/kg/day) compared to deferoxamine (20 or 40 mg/kg (pediatric patients); 40 or 50 mg/kg (adult patients)). Patient enrollment was stopped following an interim analysis. After adjusting for the type I (alpha) error, the non-inferiority criterion was established as the upper limit of the 96.01% confidence interval for the difference between treatments being ≤2 mg/g dry weight (dw).
Data from 185 patients (122 on FERRIPROX and 63 on deferoxamine) were available. Among the 122 FERRIPROX treated patients, the mean age was 15.9 years (range 3-46); 57.4% were male; 75.4% were White, 17.2% were Black and 7.4% were Multi-racial; 85% were diagnosed with Sickle Cell Disease and 15% with other anemias. Over 12 months, the Least Squares estimate of mean decrease from baseline in LIC was 4.13 ± 0.50 mg/g dw for FERRIPROX and 4.38 ± 0.59 mg/g dw for deferoxamine, and the non-inferiority criterion was met.
Upon completion of the first year of therapy in the non inferiority study, 89 patients from the FERRIPROX group opted to continue with treatment and 45 from the deferoxamine group opted to switch to ferriprox treatment. This group continued for up to an additional 2 years. LIC continued to decrease over time, with the mean value dropping from 14.93 mg/g dw at baseline to 12.30 mg/g dw after one year of treatment, to 11.19 mg/g dw after two years of treatment, and to 10.45 mg/g dw after three years of treatment.
16. How is Ferriprox supplied
FERRIPROX Tablets (twice a day), 1,000 mg
FERRIPROX® Tablets (deferiprone) (twice a day) are white to off-white capsule-shaped, beveled edge, biconvex coated tablets, and have a functional score engraved “FPX” bisect “DR” on one side, “APO” bisect “1000” on the other. They are supplied in child-resistant blister packs or HDPE bottles.
1,000 mg tablets, carton of 5 x 10-count blister packs NDC 10122-104-01
1,000 mg tablets, bottle of 50 tablets NDC 10122-104-05
1,000 mg tablets, bottle of 500 tablets NDC 10122-104-50
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
FERRIPROX Tablets (three times a day), 1,000 mg
FERRIPROX® Tablets (deferiprone) (three times a day) are white to off-white capsule-shaped tablets, film-coated, and have a functional score imprinted with “APO” score “1000” on one side and are plain on the other. They are provided in HDPE bottles.
1,000 mg film-coated tablets, 50 tablets NDC 10122-103-05
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Keep the bottle tightly closed to protect from moisture.
FERRIPROX Tablets, 500 mg
FERRIPROX® Tablets (deferiprone) are white to off-white capsule-shaped tablets, film-coated, and have a functional score imprinted with “APO” score “500” on one side and are plain on the other. They are provided in HDPE bottles.
500 mg film-coated tablets, 100 tablets NDC 10122-100-10
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide)
- Instruct patients and their caregivers to store FERRIPROX at 68°F to 77°F (20°C to 25°C); excursions permitted to 59°F to 86°F (15°C to 30°C) [see USP Controlled Room Temperature].
-
FERRIPROX Tablets (twice a day), 1,000 mg:
Advise patients to take the first dose of FERRIPROX Tablets (twice a day) in the morning and the second in the evening.
Advise patients to take FERRIPROX Tablets (twice a day) with food to reduce the risk of nausea and vomiting.
Advise patients to avoid alcohol while taking FERRIPROX Tablets (twice a day). Consumption of alcohol while taking FERRIPROX Tablets (twice a day) may result in more rapid release of deferiprone. -
FERRIPROX Tablets (three times a day), 1,000 mg:
Store in the originally supplied bottle, closed tightly to protect from moisture.
Advise patients to take the first dose of FERRIPROX in the morning, the second dose at midday, and the third dose in the evening. Clinical experience suggests that taking FERRIPROX with meals may reduce nausea. -
FERRIPROX Tablets, 500 mg:
Store in the originally supplied bottle, closed tightly to protect from moisture.
Advise patients to take the first dose of FERRIPROX in the morning, the second dose at midday, and the third dose in the evening. Clinical experience suggests that taking FERRIPROX with meals may reduce nausea. - If a dose of this medicine has been missed, take it as soon as possible. However, if it is almost time for the next dose, skip the missed dose and go back to the regular dosing schedule. Do not catch-up or double doses.
- Inform patients of the risks of developing agranulocytosis and the need for regular blood testing before and during their treatment to monitor for decreases in their ANC. Instruct them to immediately interrupt therapy and report to their physician if they experience any symptoms of infection such as fever, sore throat or flu-like symptoms [see Dosage and Administration (2.1) and Warnings and Precautions (5.1)] in order to check their ANC within 24 hours. Advise them if they are unable to reach their physician, seek care from another provider so as not to delay medical care.
- Inform patients of the risk of abnormal liver transaminases and the need for regular blood testing before and during their treatment to monitor for increases in ALT [see Dosage and Administration (2.1) and Warnings and Precautions (5.2)].
- Inform patients of the risk of zinc deficiency and the need for regular blood testing before and during their treatment to monitor for reductions in zinc [see Dosage and Administration (2.1) and Warnings and Precautions (5.3)].
- Advise patients to contact their physician in the event of overdose.
- Inform patients that their urine might show a reddish/brown discoloration due to the excretion of the iron-deferiprone complex. This is a very common sign of the desired effect, and it is not harmful.
Embryo-Fetal toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1)]. Advise female patients of reproductive potential to use effective contraception during treatment with FERRIPROX and for at least six months after the last dose [see Use in Specific Populations (8.1, 8.3)]. Advise males with female partners of reproductive potential to use effective contraception during treatment with FERRIPROX and for at least three months after the last dose [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
Lactation
Advise females not to breastfeed during treatment with FERRIPROX and for at least 2 weeks after the last dose [see Use in Specific Populations (8.2)].
Distributed by Chiesi USA, Inc., Cary, NC 27518. Manufactured by Apotex Inc., Toronto, Ontario, Canada, M9L 1T9.
US-618-s1-W
Medication Guide
| Medication Guide
FERRIPROX (Feh ri prox) Tablets (deferiprone) |
||||||
| What is the most important information I should know about FERRIPROX Tablets?
FERRIPROX Tablets can cause serious side effects, including a very low white blood cell count. One type of white blood cell that is important for fighting infections is called a neutrophil. If your neutrophil count is low (neutropenia), you may be at risk of developing a serious infection that can lead to death. Neutropenia is common with FERRIPROX Tablets and can become severe in some people. Severe neutropenia is known as agranulocytosis. If you develop agranulocytosis, you will be at risk of developing serious infections that can lead to death. Your healthcare provider will do a blood test before you start FERRIPROX Tablets and regularly during treatment to check your neutrophil count. If you develop neutropenia, your healthcare provider should check your blood counts every day until your white blood cell count improves. Your healthcare provider may temporarily stop treatment with FERRIPROX Tablets if you develop neutropenia or infection. Stop taking FERRIPROX Tablets and call your healthcare provider or get medical help right away if you develop any of these symptoms of infection:
See “What are the possible side effects of FERRIPROX Tablets?” for more information about side effects. |
||||||
| What is FERRIPROX Tablets?
FERRIPROX Tablets is a prescription medicine used to treat iron overload from blood transfusions in adults and children 8 years of age and older with:
|
||||||
| Do not take FERRIPROX Tablets if you are allergic to deferiprone or any of the ingredients in FERRIPROX Tablets. See the end of this Medication Guide for a complete list of ingredients in FERRIPROX Tablets. | ||||||
Before taking FERRIPROX Tablets, tell your healthcare provider about all of your medical conditions, including if you:
○ Your healthcare provider should do a pregnancy test before you start treatment with FERRIPROX Tablets. ○ You should use effective birth control during treatment with FERRIPROX Tablets and for at least 6 months after the last dose. Males with female partners who are able to become pregnant: ○ You should use effective birth control during treatment with FERRIPROX Tablets and for at least 3 months after the last dose.
|
||||||
How should I take FERRIPROX Tablets?
|
||||||
| FERRIPROX Tablets
1,000 mg Twice-a-Day 2 times each day with food | FERRIPROX Tablets
1,000 mg 3 times each day | FERRIPROX Tablets
500 mg 3 times each day |
||||
| Take your first dose in the morning and the second dose in the evening, about 12 hours apart. | Take your first dose in the morning, the second dose at mid day, and the third dose in the evening. | Take your first dose in the morning, the second dose at mid-day, and the third dose in the evening. |
||||
|
||||||
What should I avoid during treatment with FERRIPROX Tablets?
|
||||||
| What are the possible side effects of FERRIPROX Tablets?
FERRIPROX Tablets can cause serious side effects, including:
|
||||||
| The most common side effects of FERRIPROX Tablets in people with thalassemia include: | ||||||
|
|
|
||||
| The most common side effects of FERRIPROX Tablets in people with sickle cell disease or other anemias include: | ||||||
|
|
|
||||
| FERRIPROX Tablets may cause a change in urine color to reddish-brown. This is not harmful and is expected during treatment with FERRIPROX Tablets. These are not all of the possible side effects of FERRIPROX Tablets. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||||||
| How should I store FERRIPROX Tablets? | ||||||
| FERRIPROX Tablets
1,000 mg Twice-a-Day 2 times each day with food | FERRIPROX Tablets
1,000 mg 3 times each day | FERRIPROX Tablets
500 mg 3 times each day |
||||
|
|
|
||||
| Keep FERRIPROX Tablets and all medicines out of the reach of children. | ||||||
| General information about the safe and effective use of FERRIPROX Tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use FERRIPROX Tablets for a condition for which it was not prescribed. Do not give FERRIPROX Tablets to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about FERRIPROX Tablets that is written for health professionals. |
||||||
| What are the ingredients in FERRIPROX Tablets? | ||||||
| FERRIPROX Tablets
1,000 mg Twice-a-Day 2 times each day with food | FERRIPROX Tablets
1,000 mg 3 times each day | FERRIPROX Tablets
500 mg 3 times each day |
||||
| Active ingredient: deferiprone Inactive ingredients: Tablet core: hypromellose acetate succinate, magnesium oxide, colloidal silicon dioxide and magnesium stearate. Coating: triethyl citrate, talc, titanium dioxide, and methacrylic acid and ethyl acrylate copolymer. | Active ingredient: deferiprone Inactive ingredients: Tablet core: methylcellulose, crospovidone, and magnesium stearate. Coating: hypromellose, hydroxypropyl cellulose, macrogol, and titanium dioxide. | Active ingredient: deferiprone Inactive ingredients: Tablet core: microcrystalline cellulose, magnesium stearate, colloidal silicon dioxide. Coating: hypromellose, polyethylene glycol, and titanium dioxide. |
||||
| Distributed by: Chiesi USA, Inc., Cary, NC 27518. Manufactured by: Apotex Inc., Toronto, Ontario, Canada, M9L 1T9. CTFD-033-0521-00-SPL-1 For more information, call 1-888-661-9260. |
||||||
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 11/2021
PRINCIPAL DISPLAY PANEL
Chiesi USA, Inc. NDC 10122-104-01
Ferriprox (deferiprone) tablets
1,000 mg
Rx only
50 Tablets
PRINCIPAL DISPLAY PANEL
Chiesi USA, Inc. NDC 10122-104-05
Ferriprox (deferiprone) tablets
1,000 mg
Rx only
50 Tablets



| FERRIPROX
deferiprone tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| FERRIPROX
deferiprone tablet, film coated |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| FERRIPROX
deferiprone tablet, film coated |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Chiesi USA, Inc. (088084228) |
More about Ferriprox (deferiprone)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Imprints, shape & color data
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- FDA approval history
- Drug class: antidotes
- Breastfeeding
- En español