Voluven: Package Insert / Prescribing Info
Package insert / product label
Generic name: hydroxyethyl starch 130/0.4
Dosage form: injection, solution
Drug class: Plasma expanders
Medically reviewed by Drugs.com. Last updated on Dec 9, 2024.
The Voluven brand name has been discontinued in the U.S. If generic versions of this product have been approved by the FDA, there may be generic equivalents available.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
VOLUVEN ®(6% hydroxyethyl starch 130/0.4 in 0.9% sodium chloride injection), for administration by intravenous infusion
Initial U.S. Approval: 2007
WARNING: MORTALITY
RENAL REPLACEMENT THERAPY
See full prescribing information for complete boxed warning.
-
In critically ill adult patients, including patients with sepsis, use of hydroxyethyl starch (HES) products, including Voluven
®, increases risk of
- Mortality
- Renal replacement therapy
- Do not use HES products, including Voluven ®, in critically ill adult patients, including patients with sepsis.
Indications and Usage for Voluven
Voluven ® is a plasma volume substitute indicated for the treatment and prophylaxis of hypovolemia in adults and children. ( 1)
Voluven Dosage and Administration
Administer by intravenous infusion only.
-
Daily dose and rate of infusion depend on the patient’s blood loss, hemodynamics and on the hemodilution effects. ( 2)
|
| Recommended Daily Dose
|
| Adults (
2.1)
| Up to 50 mL/kg body weight
|
|
Recommended Daily Dose |
Mean Daily Dose ± SD in Clinical Trials (2.2) |
|
|
Pediatric age groups ( 2.2) |
Up to 50 mL/kg body weight in all age groups |
- |
|
< 2 years |
16 ± 9 mL/kg body weight |
|
|
2 – 12 years |
36 ± 11 mL/kg body weight |
|
|
> 12 years |
- |
Dosage Forms and Strengths
500 mL freeflex® flexible plastic intravenous solution container. Each 100 mL contains 6 g hydroxyethyl starch 130/0.4 in isotonic sodium chloride injection. ( 3)
Contraindications
- Do not use hydroxyethyl starch (HES) products, including Voluven ®, in critically ill adult patients, including patients with sepsis due to increased risk of mortality and renal replacement therapy. ( 4)
- Do not use HES products, including Voluven ®, in patients with severe liver disease. ( 4)
- Do not use HES products, including Voluven ®, in patients with known hypersensitivity to hydroxyethyl starch. ( 4)
- Do not use HES products in clinical conditions with volume overload. ( 4)
- Do not use HES products in patients with pre-existing coagulation or bleeding disorders. ( 4)
- Do not use HES products in patients with renal failure with oliguria or anuria not related to hypovolemia. ( 4)
- Do not use HES products in patients receiving dialysis. ( 4)
- Do not use HES products in patients with severe hypernatremia or severe hyperchloremia. ( 4)
- Do not use HES products in patients with intracranial bleeding. ( 4)
Warnings and Precautions
- Anaphylactoid and hypersensitivity reactions. ( 5.1, 6)
- Avoid use in patients with pre-existing renal dysfunction. ( 5.2)
- Discontinue use of Voluven ® at the first sign of renal injury. ( 5.2)
- Continue to monitor renal function in hospitalized patients for at least 90 days as use of renal replacement therapy has been reported up to 90 days after administration of HES products. ( 5.2)
- Monitor the coagulation status of patients undergoing open heart surgery in association with cardiopulmonary bypass as excess bleeding has been reported with HES solutions in this population. Discontinue use of Voluven ® at the first sign of coagulopathy. ( 5.3)
- Avoid fluid overload; adjust dosage in patients with cardiac or renal dysfunction. ( 5.4)
- In severe dehydration, a crystalloid solution should be given first. ( 5.4)
- Monitor liver function in patients receiving HES products, including Voluven ®. ( 5.5)
- Monitor kidney function, fluid balance and serum electrolytes ( 5.5)
- Elevated serum amylase values may occur and interfere with the diagnosis of pancreatitis ( 5.5)
- High dosages may cause dilution of blood components ( 5.5)
Adverse Reactions/Side Effects
- Serious adverse reactions reported in clinical trials were increased mortality and need for renal replacement therapy in critically ill patients including sepsis.
Most common adverse reactions (incidence >1%) are pruritus, elevated serum amylase, hemodilution (resulting in dilution of blood components, e.g., coagulation factors and other plasma proteins, and in a decrease in hematocrit).
Anaphylactoid/hypersensitivity reactions can occur. ( 6)
To report SUSPECTED ADVERSE REACTIONS, contact Hospira Inc. at 1-800-441-4100 or electronically at ProductComplaintsPP@hospira.com or FDA at 1-800-FDA-1088 or at www.fda.gov/medwatch.
Drug Interactions
No interactions with other drugs or nutritional products are known. ( 7)
The safety and compatibility of additives have not been established.
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2015
Full Prescribing Information
WARNING:MORTALITY RENAL REPLACEMENT THERAPY
-
In critically ill adult patients, including patients with sepsis, use of hydroxyethyl starch (HES) products, including Voluven
®, increases risk of
- Mortality
- Renal replacement therapy
- Do not use HES products, including Voluven ®, in critically ill adult patients, including patients with sepsis.
1. Indications and Usage for Voluven
Voluven ® (6% hydroxyethyl starch 130/0.4 in 0.9% sodium chloride injection) is indicated for the treatment and prophylaxis of hypovolemia in adults and children. It is not a substitute for red blood cells or coagulation factors in plasma.
2. Voluven Dosage and Administration
Voluven ® is administered by intravenous infusion only. The daily dose and rate of infusion depend on the patient’s blood loss, on the maintenance or restoration of hemodynamics and on the hemodilution (dilution effect). Voluven ® can be administered repetitively over several days. [see Warnings and Precautions (5)]
The initial 10 to 20 mL should be infused slowly, keeping the patient under close observation due to possible anaphylactoid reactions. [see General Warnings and Precautions (5.1)]
2.1 Adult Dose
Up to 50 mL of Voluven ® per kg of body weight per day (equivalent to 3 g hydroxyethyl starch and 7.7 mEq sodium per kg of body weight). This dose is equivalent to 3500 mL of Voluven ® for a 70 kg patient.
2.2 Pediatric Dose
The dosage in children should be adapted to the individual patient colloid needs, taking into account the disease state, as well as the hemodynamic and hydration status.
In 41 newborns to infants (< 2 years), a mean dose of 16 ± 9 mL/kg was administered. In 31 children from 2 to 12 years of age a mean dose of 36 ± 11 mL/kg was administered. The dose in adolescents > 12 is the same as the adult dose. [see Pediatric Use (8.4)]
2.3 Directions for Use of Voluven
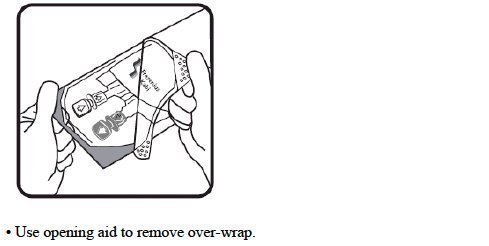
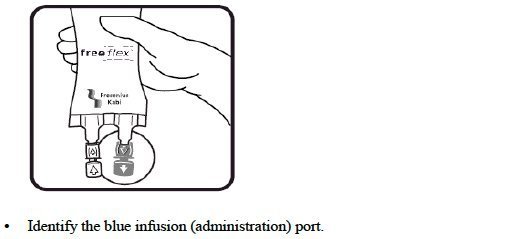
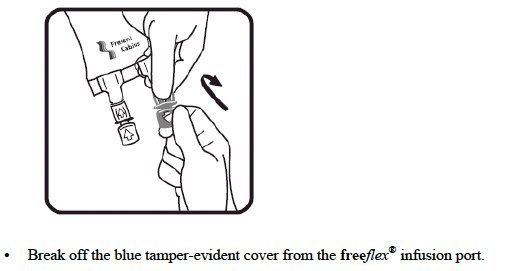
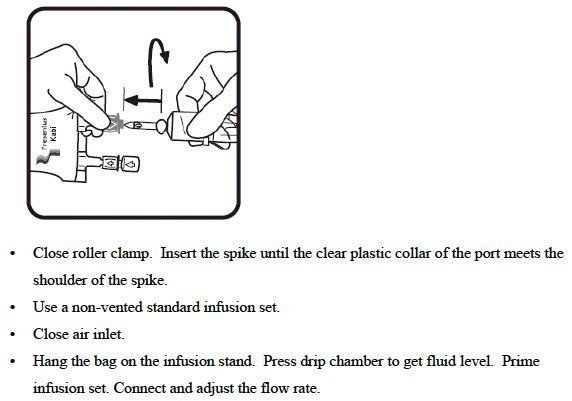
- Do not remove the freeflex ® IV container from its overwrap until immediately before use.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- Do not administer unless the solution is clear, free from particles and the freeflex® IV container is undamaged.
- Voluven ® should be used immediately after insertion of the administration set.
- Do not vent.
- If administered by pressure infusion, air should be withdrawn or expelled from the bag through the medication/administration port prior to infusion.
- Discontinue the infusion if an adverse reaction occurs.
- It is recommended that administration sets be changed at least once every 24 hours.
- For single use only. Discard unused portion.
3. Dosage Forms and Strengths
500 mL freeflex® flexible plastic intravenous solution container are available. Each 100 mL contains 6 g hydroxyethyl starch 130/0.4 in isotonic sodium chloride injection.
4. Contraindications
- Do not use hydroxyethyl starch (HES) products, including Voluven ®, in critically ill adult patients, including patients with sepsis, due to increased risk of mortality and renal replacement therapy (RRT).
- Do not use HES products, including Voluven ®, in patients with severe liver disease.
- Do not use HES products, including Voluven ®, in patients with known hypersensitivity to hydroxyethyl starch [see General Warnings and Precautions (5.1)]
- Do not use HES products in clinical conditions with volume overload.
- Do not use HES products in patients with pre-existing coagulation or bleeding disorders.
- Do not use HES products in patients with renal failure with oliguria or anuria not related to hypovolemia.
- Do not use HES products in patients receiving dialysis treatment.
- Do not use HES products in patients with severe hypernatremia or severe hyperchloremia.
- Do not use HES products in patients with intracranial bleeding.
5. Warnings and Precautions
5.1 Anaphylactoid Reactions
Anaphylactoid reactions (mild influenza-like symptoms, bradycardia, tachycardia, bronchospasm, non-cardiac pulmonary edema) have been reported with solutions containing hydroxyethyl starch. If a hypersensitivity reaction occurs, administration of the drug should be discontinued immediately and the appropriate treatment and supportive measures should be undertaken until symptoms have resolved. [see Adverse Reactions (6)]
5.2 Renal Dysfunction
Avoid use in patients with pre-existing renal dysfunction.
Discontinue use of Voluven ® at the first sign of renal injury.
Continue to monitor renal function in hospitalized patients for at least 90 days as use of RRT has been reported up to 90 days after administration of HES products.
5.3 Coagulopathy
Monitor the coagulation status of patients undergoing open heart surgery in association with cardiopulmonary bypass as excess bleeding has been reported with HES solutions in this population. Discontinue use of Voluven ® at the first sign of coagulopathy.
5.4 Fluid Equilibrium
Avoid fluid overload; adjust dosage in patients with cardiac or renal dysfunction. Fluid status and rate of infusion should be assessed regularly during treatment, especially in patients with cardiac insufficiency or severe kidney dysfunction.
In cases of severe dehydration, a crystalloid solution should be given first. Generally, sufficient fluid should be administered in order to avoid dehydration.
5.5 Monitoring: Laboratory Tests
Clinical evaluation and periodic laboratory determinations are necessary to monitor fluid balance, serum electrolyte concentrations, kidney function, acid-base balance, and coagulation parameters during prolonged parenteral therapy or whenever the patient’s condition warrants such evaluation. Monitor liver function in patients receiving HES products, including Voluven ®.
5.6 Interference with Laboratory Tests
Elevated serum amylase levels may be observed temporarily following administration of the product and can interfere with the diagnosis of pancreatitis.
At high dosages the dilutional effects may result in decreased levels of coagulation factors and other plasma proteins and a decrease in hematocrit.
6. Adverse Reactions/Side Effects
6.1 Overall Adverse Reaction Profile
Serious adverse reactions reported in clinical trials include increased mortality and increased use of RRT in critically ill subjects, including subjects with sepsis.
The most common adverse reactions after administration of Voluven ® occurring in more than 1% of patients are: pruritus (itching; ≥1% to <10%), elevation of serum amylase (≥1% to <10%; interference with the diagnosis of pancreatitis), and dilutional effects that may result in decreased levels of coagulation factors and other plasma proteins and in a decrease of hematocrit (≥1% to <10%).
Anaphylactoid reactions occur rarely in <0.1% after administration of hydroxyethyl starch solutions. Disturbances of blood coagulation beyond dilution effects can occur rarely in <0.1% depending on the dosage with the administration of hydroxyethyl starch solutions. 1
6.2 Adverse Reactions in Clinical Trials
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug or another patient population and may not reflect the rates observed in clinical practice.
During clinical development, a total of 899 subjects received the hydroxyethyl starch 130/0.4 drug substance contained in Voluven ® at different concentrations (2%, 4%, 6%, or 10%) and at cumulative doses of several mL up to 66 L. 2 Of these 899 subjects, 602 were exposed to Voluven ® (i.e., 6% hydroxyethyl startch 130/0.4). The mean duration of treatment with hydroxyethyl starch 130/0.4 was 3.7 ± 3.1 days, mean cumulative doses were 3185 ± 3498 mL, and the longest follow-up period was 90 days.
In a randomized controlled trial (RCT) of subjects (N=100) undergoing elective orthopedic surgery, Voluven ® (N=49) or hetastarch (6% hydroxyethyl starch in 0.9% sodium chloride injection; N=51) were administered for intraoperative volume replacement. 3 Mean infusion volumes were 1613 ± 778 mL for Voluven ® and 1584 ± 958 mL for hetastarch.
Adverse reactions observed in at least 1% of subjects: in the orthopedic surgery trial conducted in the U.S., no significant differences in serious adverse reactions were noted overall between the two treatment arms. A possible relationship to Voluven ® was reported in five cases among three subjects (aPTT elevated, PT prolonged, wound hemorrhage, anemia, pruritus); a possible relationship to hetastarch was reported in five subjects (three cases of coagulopathy; two cases of pruritus). The three coagulopathy cases in the hetastarch group were serious and occurred in subjects receiving more than the labeled ceiling dose (20 mL/kg), which is known to increase the risk of bleeding, whereas no serious coagulopathy occurred in the Voluven ® group. Since calculated red blood cell loss for the two treatment arms was not statistically different (95% confidence interval included unity), the difference observed for Factor VIII (see Table 1, below) must be interpreted with caution. An exploratory analysis of total erythrocyte volume transfused (8.0 mL/kg vs. 13.8 mL/kg, Voluven ® vs. hetastarch, respectively) also must be viewed with caution.
Table 1: Safety Variables for the Orthopedic Surgery Trial conducted in the US
|
|
Mean |
Ratio Voluven/Hetastarch |
||
|
Variable |
Voluven N=49 |
Hetastarch N=51 |
Estimate |
95% Cl |
| Calculated red blood cell loss [L]*
|
1.17 |
1.31 |
0.910 |
[0.720; 1.141] |
| Factor VIII [%]*
|
100.5 |
81.4 |
1.244 |
[1.000; 1.563] |
| von Willebrand factor [%]*
|
97.7 |
88.7 |
1.128 |
[0.991; 1.285] |
| Fresh frozen plasma [mL]*
|
72 |
144 |
0.723 |
[0.000; 2.437] |
* Exploratory analyses
A safety profile for Voluven ® at least as favorable as for pentastarch was also demonstrated in studies where Voluven ® was administered at doses higher (up to 50 mL/kg or 3 g/kg) than for pentastarch (up to 33 mL/kg or 2 g/kg) in clinical settings where large or repetitive doses were administered.
Trials in critically ill adult subjects
Three RCTs followed critically ill adult subjects treated with different HES products for 90 days.
One trial using Voluven ® in severe sepsis subjects (N=196) reported no difference in mortality (relative risk, 1.20; 95% CI, 0.83 to 1.74; p=0.33) and a trend for increased use of RRT (relative risk, 1.83; 95% CI, 0.93 to 3.59; p=0.06) in HES subjects. 4
Another trial using Voluven ® in a heterogeneous population of critically ill subjects admitted to the ICU (N=7000) reported no difference in mortality (relative risk, 1.06; 95% CI, 0.96 to 1.18; p=0.26) but increased use of RRT (relative risk, 1.21; 95% CI, 1.00 to 1.45; p=0.04) in HES subjects. 5 A third trial in severe sepsis subjects (N=804) using an HES product not licensed in the U.S. (HES 130/0.42) reported increased mortality (relative risk, 1.17; 95% CI, 1.01 to 1.36; p=0.03) and increased use of RRT (relative risk, 1.35; 95% CI, 1.01 to 1.80; p=0.04) in HES subjects. 6
6.3 Postmarketing Experience
Because adverse reactions are reported voluntarily post-approval from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to product exposure.
Among the very rarely occurring serious adverse drug reactions in patients treated with Voluven ®, anaphylactic/anaphylactoid/hypersensitivity reactions or hypotension/shock/ circulatory collapse were most frequently reported.
The following adverse reactions have been identified and reported during the post-approval use of different HES products in critically ill adult patients, including patients with sepsis:
Mortality
Renal: use of RRT
Related/similar drugs
7. Drug Interactions
The safety and compatibility of other additives have not been established.
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Category C. Voluven ® has been shown to cause embryocidal or other adverse effects in rats and rabbits when given in doses 1.7 times the human dose.
The type of hydroxyethyl starch present in Voluven ® had no teratogenic properties in rats or rabbits. At 5 g/kg of body weight per day, administered as a bolus injection, fetal retardations and embryolethal effects were observed in rats and rabbits, respectively. In rats, a bolus injection of this dose during pregnancy and lactation reduced body weight of offspring and induced developmental delays. All adverse effects were seen exclusively at maternal toxic doses due to fluid overload. [see Animal Pharmacology and/or Toxicology (13.2)]
Fertility studies on directly exposed animals have not been conducted.
There are no adequate and well-controlled studies in pregnant women. Voluven ® should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
8.2 Labor and Delivery
Information on the use of Voluven ® during labor or delivery is unknown. Use if clearly needed.
8.3 Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when Voluven ® is administered to a nursing woman.
8.4 Pediatric Use
In one trial, newborns and infants < 2 years of age undergoing elective surgery were randomized to receive Voluven ® (N=41) or 5% albumin (N=41). The mean dose of Voluven ® administered was 16 ± 9 mL/kg. 7
In an additional trial, children from 2 - 12 years of age undergoing cardiac surgery were randomized to receive Voluven ® (N=31) or 5% albumin (N=30). The mean dose administered was 36 ± 11 mL/kg.
Use of Voluven ® in adolescents > 12 years is supported by evidence from adequate and well-controlled studies of Voluven ® in adults.
Dosage in children should be adapted to individual patient colloid needs, taking into account underlying disease, hemodynamics and hydration status.
Studies conducted in children have not been of sufficient size or follow-up duration to assess the risks of renal injury and mortality in this patient population. [See Pediatric Dose (2.2)]
8.5 Geriatric Use
Of the total number of subjects in clinical studies of Voluven ® (N=471), 32% were ≥ 65 years old while 7% were ≥ 75 years old. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
Voluven ® is mainly excreted by the kidneys, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Volume status, infusion rate, and urine output should be closely monitored. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection. [see Pharmacokinetics (12.3)]
10. Overdosage
Overdosage can lead to overloading of the circulatory system (e.g., pulmonary edema). In this case, the infusion should be stopped immediately and if necessary, a diuretic should be administered. [see General Warnings and Precautions (5.1)]
11. Voluven Description
Voluven ® (6% hydroxyethyl starch 130/0.4 in 0.9% sodium chloride injection) is a clear to slightly opalescent, colorless to slightly yellow, sterile, non-pyrogenic, isotonic solution for intravenous administration using sterile equipment.
Each 100 mL of the solution contains: 6 g of Hydroxyethyl Starch 130/0.4 and 900 mg of Sodium Chloride USP in Water for Injection USP.
In addition, sodium hydroxide, USP, or Hydrochloric acid, USP, has been added to adjust the final pH so the final solution pH is 4.0 to 5.5.
The electrolyte composition is as follows (mEq/L): Sodium 154, Chloride 154.
The calculated osmolarity is 308 mOsmol/L.
The hydroxyethyl starch contained in Voluven ® is a synthetic colloid for use in plasma volume replacement. The chemical name of hydroxyethyl starch is poly(O-2-hydroxyethyl) starch. The structural formula of hydroxyethyl starch is
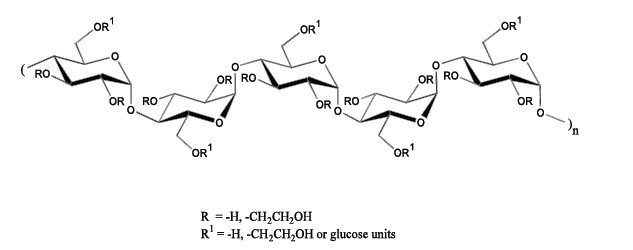
Voluven ® is packaged in 500 mL flexible plastic containers ( freeflex®). Freeflex® is a flexible container made from coextruded polyolefin and is free of PVC, plasticizers, adhesives or latex (Non-DEHP, Latex-free). The freeflex® container offers an air-closed system and can be used with non-vented IV sets which prevent external air contamination. Freeflex® is collapsible and can be used in emergency cases for pressure infusion.
12. Voluven - Clinical Pharmacology
12.1 Mechanism of Action
Voluven ® contains hydroxyethyl starch in a colloidal solution which expands plasma volume when administered intravenously. This effect depends on the mean molecular weight (130,000 daltons; range 110,000 – 150,000 daltons), the molar substitution by hydroxyethyl groups (0.4; range 0.38 – 0.45) on glucose units of the starch, the pattern of hydroxyethyl substitution (C 2/C 6 ratio) of approximately 9:1, and the concentration (6%), as well as the dosage and infusion rate.
Hydroxyethyl starch is a derivative of thin boiling waxy corn starch, which mainly consists of a glucose polymer (amylopectin) predominantly composed of α-1-4-connected glucose units with several α-1-6-branches. Substitution of hydroxyethyl groups on the glucose units of the polymer reduces the normal degradation of amylopectin by α-amylase in the body. The low molar substitution (0.4) is the main pharmacological determinant for the beneficial effects of Voluven ® on pharmacokinetics, intravascular volume and hemodilution 8. To describe the molecular weight and molar substitution characteristics of the hydroxyethyl starch in Voluven ®, the compound is designated as hydroxyethyl starch 130/0.4.
12.2 Pharmacodynamics
After isovolemic exchange of blood with 500 mL of Voluven ® in healthy volunteers, blood volume is maintained for at least 6 hours.
12.3 Pharmacokinetics
The pharmacokinetic profile of hydroxyethyl starch is complex and largely dependent on its molar substitution as well as its molecular weight. 8 When administered intravenously, molecules smaller than the renal threshold (60,000-70,000 daltons) are readily and rapidly excreted in the urine, while molecules with higher molecular weights are metabolized by plasma α-amylase prior to excretion via the renal route.
The mean in vivo molecular weight of Voluven ® in plasma is 70,000 – 80,000 daltons immediately following infusion and remains above the renal threshold throughout the treatment period.
Following intravenous administration of 500 mL Voluven ® to healthy volunteers, plasma levels of Voluven ® remain at 75% of peak concentration at 30 minutes post-infusion and decrease to 14% at 6 hours post-infusion. Plasma levels of Voluven ® return to baseline levels 24 hours following infusion. Plasma clearance, volume of distribution, and elimination half-life of Voluven ® in healthy volunteers following IV administration of 500 mL were 31.4 mL/min, 5.9 liters, and 12 hours, respectively. Approximately 62% of Voluven ® was excreted as hydroxyethyl starch molecules in urine within 72 hours.
The pharmacokinetics of Voluven ® are similar following single and multiple dose administration. No significant plasma accumulation occurred after daily administration of 500 mL of a 10% solution containing hydroxyethyl starch 130/0.4 over a period of 10 days. Approximately 70% of Voluven ® was excreted as hydroxyethyl starch molecules in urine within 72 hours.
Renal Impairment:
Following a single intravenous administration of Voluven ® (500 mL) in subjects with varying degrees of renal dysfunction, the AUC and clearance of Voluven ® increased by 73% and decreased by 42% in subjects, respectively, with creatinine clearance < 50 mL/min as compared to subjects with creatinine clearance > 50 mL/min. However, terminal half-life and peak hydroxyethyl starch concentration were not affected by renal impairment. Plasma levels of Voluven ® returned to baseline levels 24 hours following infusion. Approximately 59 % and 51 % of Voluven ® were excreted as hydroxyethyl starch molecules in urine within 72 hours in subjects with creatinine clearance ≥30 mL/min and <30 mL/min, respectively 9.
There are no data available on the use of Voluven ® in subjects undergoing hemodialysis.
Pharmacokinetic data in patients with hepatic insufficiency or in pediatric or geriatric patients are not available. Effects of gender or race on the pharmacokinetics of Voluven ® have not been studied.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to evaluate the carcinogenic potential of Voluven ® have not been performed. No mutagenic effects were observed with hydroxyethyl starch 130/0.4 (10% solution) in the following tests on mutagenic activity: Salmonella typhimurium reverse mutation assay ( in vitro), mammalian cells in the in vitro gene mutation assay, assessment of the clastogenic activity in cultured human peripheral lymphocytes ( in vitro), bone marrow cytogenetic test in Sprague-Dawley rats.
Fertility studies on directly exposed animals have not been performed.
13.2 Animal Pharmacology and/or Toxicology
Toxicology
Three-month repeat infusion toxicology studies were conducted in rats and dogs in which three groups of animals were administered daily intravenous infusion over three hours. Dosing volumes of either 60 or 90 mL/kg body weight of hydroxyethyl starch 130/0.4 (10% solution) or 90 mL/kg 0.9% sodium chloride injection were studied. Observed toxicity following repeat infusion of hydroxyethyl starch is consistent with the oncotic properties of the solution resulting in hypervolemia in the animals. There were no gender-related effects on toxicity following repeat administration of hydroxyethyl starch 130/0.4 in rats or dogs.
In reproduction studies in rats and rabbits, hydroxyethyl starch 130/0.4 (10% solution) had no teratogenic properties. Embryolethal effects were observed in rabbits at 5 g/kg body weight/day. In rats, bolus injection of this dose during pregnancy and lactation reduced body weight of offspring and induced developmental delays. Signs of fluid overload were seen in the dams. Hydroxyethyl starch 130/0.4 (10% solution) had no effect in studies assessing skin sensitization, antigenicity, and blood compatibility.
Pharmacology
The pharmacodynamic effect of Voluven ® was examined in a hemorrhagic shock model in conscious rats and a hemodilution model in dogs. In both studies the control group received pentastarch (6% hydroxyethyl starch 200/0.5).
Voluven ® was as effective as pentastarch in maintaining cardiopulmonary function during isovolemic hemodilution in beagle dogs. In the three-hour follow-up period no additional administration of colloid was necessary.
There were no differences in long-term survival of rats after a single administration of Voluven ® and pentastarch solutions following induced hemorrhagic shock (67% and 50% blood loss). In the 67% induced bleeding group receiving Voluven ® (N=6), the survival rate was 83% which is within the normal range for this type of experiment. In the corresponding pentastarch group, survival was 100%. Infusion of Ringer's lactate resulted in a 50% survival rate after a 50% blood loss and a 0% survival after a 67% blood loss.
After multiple intravenous infusions of 0.7 g per kg body weight per day of 10% hydroxyethyl starch 130/0.4 or 10% hydroxyethyl starch 200/0.5 solution during 18 consecutive days, the plasma hydroxyethyl starch concentration in rats treated with hydroxyethyl starch 130/0.4 was lower compared to rats treated with hydroxyethyl starch 200/0.5. Hydroxyethyl starch 130/0.4 was eliminated faster than hydroxyethyl starch 200/0.5. In both groups, clear signs of hydroxyethyl starch tissue storage were detected in lymph nodes and spleen. Numerous empty vacuoles in macrophages were observed. Only minimal cellular vacuolization was found in the liver and kidney. Histochemical differences between the groups were not observed.
A study with 10% radiolabeled 14C-hydroxyethyl starch 130/0.4 and 10% 14C- hydroxyethyl starch 200/0.5 solutions was carried out. 10 In animals treated with hydroxyethyl starch 130/0.4, radioactivity decreased from 4.3% of the total administered dose (2.6 g hydroxyethyl starch 130/0.4 per animal) on day 3 to 0.65% on day 52. In animals treated with hydroxyethyl starch 200/0.5, the 14C-activity decreased from 7.7% of the total administered dose (2.7 g hydroxyethyl starch 200/0.5 per animal) on day 3 to 2.45% on day 52. These results confirm the faster elimination and lower persistence of hydroxyethyl starch 130/0.4 in tissue.
14. Clinical Studies
Voluven ® was studied in controlled clinical trials among adult and pediatric subjects undergoing various types of surgery (orthopedic, urologic, cardiac) in which hypovolemia was treated (pre-, intra-, and postoperatively) or prevented (autologous blood donation, acute normovolemic hemodilution, hypervolemic hemodilution before cardiac surgery). Adult subjects in intensive care units also were studied. The safety and efficacy of Voluven ® were compared with other colloidal plasma substitutes [pentastarch (6% hydroxyethyl starch 200/0.5), hetastarch (6% hydroxyethyl starch 450/0.7), gelatin solution or human serum albumin]. Perioperative fluid administration of Voluven ® ranged from 500 to 4500 mL/day in surgical subjects, and cumulatively, from 6 to 66 L in intensive care unit subjects following traumatic brain injury.
Orthopedic surgery trial
A multicenter, double-blind RCT in subjects (N=100) undergoing elective orthopedic surgery was conducted in the U.S. to evaluate Voluven ® (N=49) compared with hetastarch (6% hydroxyethyl starch in 0.9% sodium chloride injection) (N =51) for intraoperative volume replacement therapy 3. The primary efficacy variable, total volume of colloid solution required for intraoperative volume replacement therapy, was equivalent for the two treatment groups. Mean volume infused was 1613 ± 778 mL for Voluven ® and 1584 ± 958.4 mL for hetastarch. The Voluven ®/hetastarch ratio estimate of 1.024 with a 95% CI of 0.84 to 1.25 was within the equivalence range (0.55 to 1.82) prespecified in the study protocol. This indicated that Voluven ® and hetastarch have similar efficacy as intraoperative volume replacement therapy in major orthopedic surgery.
A second objective of the trial was to show superiority for safety between Voluven ® and hetastarch. Four safety endpoints were prospectively defined and compared in a sequential manner (in order to preserve the type-1 error rate, i.e., observing a difference where none actually exists). Per protocol, if no difference was found between treatment arms for the first safety endpoint (calculated red blood cell loss), the remaining endpoints were to be considered exploratory analyses requiring additional studies for confirmation. [see Adverse Reactions in Clinical Trials (6.2)]
There was no statistically significant difference between the two treatment groups with respect to the secondary efficacy endpoints of hemodynamic stability, body temperature, hemodynamic parameters, blood pressure, central venous pressure, heart rate, fibrinogen and platelet count.
In addition to the U.S. trial, three non-U.S. trials were conducted with the primary objective of showing equivalency (based on mean difference rather than mean ratio as in the U.S. study) between Voluven ® and pentastarch in maintaining or restoring hemodynamic parameters. The largest of the three trials (N=100) met the prespecified boundary (-500 mL to 500 mL), but the two smaller studies (N=52 and N=59) did not.
In exploratory analyses, the effect of Voluven ® on coagulation parameters (von Willebrand factor, Factor VIII, and Ristocetin cofactor) was shown to be significantly lower than pentastarch at one or more time points (U.S. and non-U.S. trials). These findings are consistent with the lower molar substitution, lower average molecular weight and narrower molecular weight distribution of Voluven ® as compared to pentastarch resulting in a lower in vivo molecular weight and increased elimination from the circulation.
Trials in critically ill adult subjects
A multicenter, double-blind RCT of subjects (N=196) with severe sepsis ≥ 18 years old compared Voluven ® (n=100) with 0.9% sodium chloride (n=96) infused over a maximum of 4 days for volume replacement therapy. 4 The primary endpoint was volume of study drug required to achieve initial hemodynamic stabilization (HDS). HDS was based on prespecified criteria for MAP, CVP, urine output and central venous oxygen saturation. Acute renal failure was prospectively defined as use of RRT or doubling of baseline serum creatinine at some point during the 90-day observation period. Two scoring systems based on changes in serum creatinine concentration and urine output used to assess severity of acute kidney injury, AKIN (Stage 1, Stage 2, Stage 3) and RIFLE (Risk, Injury, Failure, Loss, End-stage kidney disease), also were evaluated.
In subjects achieving HDS (N=88 vs. 86), significantly less Voluven ® was required to reach HDS compared with 0.9% sodium chloride: 1379 mL ± 886 (Voluven ®) vs. 1709 ± 1164 mL (0.9% sodium chloride), representing a mean difference of 331 mL (95% CI, -640 mL to -21 mL, p=0.02). Less time was needed from start of study drug to achievement of HDS in the Voluven ® group compared with the 0.9% sodium chloride group (11.8 hours ±10.1 hours vs. 14.3 hours ± 11.1 hours, mean ± SD).
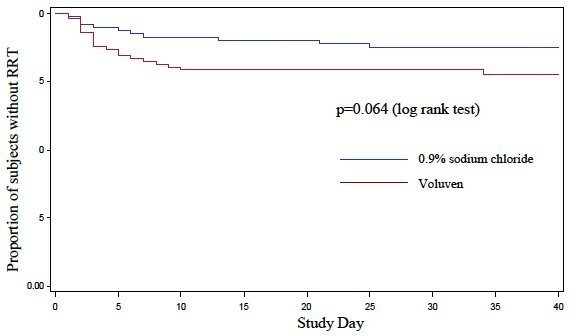
The number of treatment emergent serious adverse events (SAEs)(53 vs. 44) and the number of treatment emergent SAEs leading to death (38 vs. 32) in the Voluven ® and normal saline treatment arms, respectively, during the 90-day observation period did not reach statistical significance. More Voluven ® subjects than 0.9% sodium chloride subjects were treated with RRT: 21 (21%) vs. 11 (12%)(relative risk, 1.83; 95% CI, 0.95 to 3.58; p=0.08). Kaplan-Meier curves for time-to-RRT (Figure 1, below) showed a trend against Voluven ® (p=0.064, long-rank test). The number of subjects meeting AKIN Stage 1, Stage 2, and Stage 3 criteria was 21 (21%) vs. 21 (22%), 5 (5%) vs. 6 (6%) and 22 (22%) vs. 17 (18%), Voluven ® vs. 0.9% sodium chloride, respectively. Corresponding data for RIFLE stages Risk, Injury, and Failure were 13 (13%) vs. 11 (12%), 4 (4%) vs. 5 (5%), and 5 (%) vs. 7 (7%), repsectively. [see 6. Adverse Reactions]
Figure 1: Kaplan-Meier Curves for time- to- RRT
Another multicenter, double-blind RCT in a heterogeneous adult ICU population (N=7000) that included subjects with sepsis as well as trauma subjects and postoperative elective surgery subjects, compared Voluven ® with 0.9% sodium chloride for volume replacement therapy. 5 The primary endpoint was death within 90 days. Secondary outcomes included incidence of acute kidney injury defined by RIFLE criteria, use of RRT, and new organ failure based on SOFA score. 90-day mortality was not different overall (597 (18%) Voluven ® subjects vs. 566 (17%) 0.9% sodium chloride subjects; relative risk, 1.06; 95% CI, 0.96 to 1.18; p=0.26) or in six pre-defined subgroups including subjects with sepsis. Significantly more Voluven ® subjects were treated with RRT: 235 (7.0%) vs. 196 (5.8%) (relative risk, 1.21; 95% CI, 1.00 to 1.45; p=0.04). In a post hoc analysis, use of Voluven ® during the first seven days of therapy was associated with increased serum creatinine levels and decreased urine output; in a pre-planned analysis the number of subjects with RIFLE stages Risk and Injury was less frequent in the Voluven ® vs. 0.9% sodium chloride group:1788 (54.0%) vs. 1912 (57.3%; p=0.007) and 1130 (34.6%) vs. 1253 (38.0%; p=0.005), respectively. There was no statistical difference in RIFLE stage F: 336 (10.4%) vs. 301 (9.2%), Voluven ® vs. 0.9% sodium chloride, respectively. Voluven ® subjects experienced lower rates of new cardiovascular organ failure (hypotension) (36.5% vs. 39.9%; relative risk, 0.91; 95% CI, 0.84 to 0.99; p=0.03) but an increased SOFA score for hepatic function (bilirubinemia) (1.9% vs. 1.2%; relative risk, 1.56; 95% CI, 1.03 to 2.36; p=0.03). [see Adverse Reactions in Clinical Trials (6.2)]
A third multicenter, double-blind RCT performed in severe sepsis subjects (N=804) compared HES 130/0.42 in Ringer’s acetate injection (RA), an HES solution not approved in the U.S. with modified RA for volume replacement therapy. The composite primary endpoint was mortality or dialysis-dependence up to 90 days after randomization. Secondary endpoints included death at 28 days; acute kidney injury defined as use of RRT or a renal SOFA score ≥3 in subjects with renal SOFA ≤2 (serum creatinine <2.0 mg/dL or urinary output <171 mL per day) at randomization; and doubling of serum creatinine. At the end of the 90 day study period, both mortality and use of RRT in the ITT population (N=798) were significantly higher in the HES 130/0.42 treatment arm: 201 (51%) vs. 172 (43%)(relative risk, 1.17; 95% CI, 1.01 to 1.36; p=0.03) and 87 (22%) vs. 65 (16%)(relative risk, 1.35; 95% CI, 1.01 to 1.80; p=0.04), respectively; one subject in each treatment cohort was dependent on dialysis on day 90. Mortality at 28 days was not different between groups (154 (39%) vs. 144 (36%); relative risk, 1.08; 95% CI, 0.90 to 1.28; p=0.43); use of RRT or renal SOFA score ≥3, and doubling of serum creatinine, were numerically higher in the HES 130/0.42 cohort: 129 (32%) vs. 108 (27%)(relative risk, 1.20; 95% CI, 0.97 to 1.48; p=0.10) and 148 (41%) vs. 127 (35%)(relative risk, 1.18; 95% CI, 0.98 to 1.43; p=0.08), respectively. In a post hoc analysis, the number of subjects with RIFLE Risk, Injury, or Failure was 52 (14%) vs. 73 (20%), 62 (17%) vs. 53 (15%) and 84 (23%) vs. 67 (18%), HES 130/0.42 vs. RA, respectively. [see Adverse Reactions in Clinical Trials (6.2)]
15. References
- Kozek-Langenecker S. Effects of hydroxyethyl starch solutions on hemostasis. Anesthesiology 2005; 103 (3): 654-60.
- Neff TA, Doelberg M, Jungheinrich C, et al. Repetitive large-dose infusion of the novel hydroxyethyl starch HES 130/0.4 in patients with severe head injury. Anest Analg 2003; 96 (5): 1453-9.
- Gandhi SD, Weiskopf RB, Jungheinrich C et al. Volume replacement therapy during major orthopedic surgery using Voluven ® (hydroxyethyl starch 130/0.4) or hetastarch. Anesthesiology 2007; 106(6):1120-7.
- Guidet B, Martinet O, Boulain T, et al. Assessment of hemodynamic efficacy and safety of 6% hydroxyethylstarch 130/0.4 versus 0.9% NaCl fluid replacement in patients with severe sepsis: The CRYSTMAS study. Crit Care 2012; 16(3): R94.
- Myburgh JA, Finfer S, Bellomo R, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367(20): 1901-11.
- Perner A, Haase N, Guttormsen AB, et al. Hydroxyethyl Starch 130/0.42 versus Ringer's Acetate in Severe Sepsis. N Eng J Med 2012; 367(2): 124-34.
- Standl T, Lochbuehler H, Galli C, et al. HES 130/0.4 (Voluven ®) or human albumin in children younger than 2 yr undergoing non-cardiac surgery. A prospective, randomized, open label, multicentre trial. Eur J Anaesthesiol 2008; 25(6): 437-45.
- Jungheinrich C, Neff T. Pharmacokinetics of hydroxyethyl starch. Clin Pharmacokinetik 2005; 44 (7): 681-99.
- Jungheinrich C, Scharpf R, Wargenau M, et al. The pharmacokinetics and tolerability of an intravenous infusion of the new hydroxyethyl starch 130/0.4 (6%, 500 mL) in mild-to-severe renal impairment. Anesth Analg 2002; 95 (3): 544-51.
- Leuschner J, Opitz J, Winkler A, Scharpf R, Bepperling F. Tissue storage of 14C-labeled hydroxyethyl starch (HES) 130/0.4 and HES 200/0.5 after repeated intravenous administration to rats. Drugs R D 2003; 4 (6): 331-8.
16. How is Voluven supplied
16.1 How Supplied
Voluven ® (6% hydroxyethyl starch 130/0.4 in 0.9% sodium chloride injection) for intravenous infusion is supplied in the following primary container and carton sizes:
Polyolefin bag ( freeflex®) with overwrap: 500 mL NDC 0409-1029-11
Carton of 15 x 500 mL NDC 0409-1029-01
Carton of 20 x 500 mL NDC 0409-1029-02
17. Patient Counseling Information
Because this product is not used directly by patients, patient counseling or instructions for use by patients is not considered necessary.
Manufactured by:

Fresenius Kabi Norge AS
P.O. Box 430,
NO-1753 Halden, Norway
Distributed by:
![]()
Hospira, Inc.
275 North Field Drive
Lake Forest, Illinois 60045 USA
Made in Norway
Issued: 08/2014

| VOLUVEN
hydroxyethyl starch 130/0.4 injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Hospira, Inc. (141588017) |
| Registrant - Fresenius Kabi Deutschland GmbH (315520085) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Fresenius Kabi Austria GmbH | 303448575 | manufacture(0409-1029) , api manufacture(0409-1029) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Fresenius Kabi Norge AS | 731170932 | analysis(0409-1029) , manufacture(0409-1029) | |
More about Voluven (hydroxyethyl starch)
- Compare alternatives
- Latest FDA alerts (2)
- Side effects
- Dosage information
- During pregnancy
- Drug class: plasma expanders