Varithena: Package Insert / Prescribing Info
Package insert / product label
Generic name: polidocanol
Dosage form: injection
Drug class: Sclerosing agents
Medically reviewed by Drugs.com. Last updated on Oct 15, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
VARITHENA (polidocanol injectable foam), for intravenous use
Initial U.S. Approval: 2013
Indications and Usage for Varithena
VARITHENA (polidocanol injectable foam) is a sclerosing agent indicated for the treatment of incompetent great saphenous veins, accessory saphenous veins, and visible varicosities of the great saphenous vein (GSV) system above and below the knee. VARITHENA improves the symptoms of superficial venous incompetence and the appearance of visible varicosities. (1).
Varithena Dosage and Administration
Incompetent great saphenous or accessory saphenous veins: Use Varithena 1% (CEAP Class 2-6 Disease). (2).
For intravenous use which should be performed under ultrasound guidance when treating the GSV.
Use up to 5 mL per injection and 15 mL per treatment session. (2)
Separate treatment sessions by a minimum of 5 days. (2)
Dosage Forms and Strengths
Warnings and Precautions
Adverse Reactions/Side Effects
In clinical trials, the most common related adverse events (occurring in ≥3% of patients treated with VARITHENA) were pain/discomfort in extremity, infusion site thrombosis (retained coagulum), injection site hematoma or pain, thrombophlebitis superficial, and extravasation.(6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Biocompatibles, Inc. at 1-855-971-VEIN (1-855-971-8346) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
There are no known drug interactions with VARITHENA. (7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2019
Full Prescribing Information
1. Indications and Usage for Varithena
VARITHENA (polidocanol injectable foam) is indicated for the treatment of incompetent great saphenous veins, accessory saphenous veins, and visible varicosities of the great saphenous vein (GSV) system above and below the knee. VARITHENA improves the symptoms of superficial venous incompetence and the appearance of visible varicosities.
2. Varithena Dosage and Administration
For intravenous use only.
VARITHENA is intended for intravenous injection using ultrasound guidance, administered via a single cannula into the lumen of the target incompetent trunk veins or by direct injection into varicosities. Use up to 5 mL per injection and no more than 15 mL per session.
Physicians administering VARITHENA must be experienced with venous procedures and be trained in the administration of VARITHENA.
Activate VARITHENA using the VARITHENA oxygen canister and polidocanol canister (see Instructions for Use). Once a VARITHENA transfer unit is in place, foam can be generated and transferred to a syringe. Discard the syringe contents if there are any visible bubbles. Administer the injectable foam within 75 seconds of extraction from the canister to maintain injectable foam properties. Use a new sterile syringe after each injection. Use a new VARITHENA transfer unit for each treatment session.
Local anesthetic may be administered prior to cannula insertion but neither tumescent anesthesia nor patient sedation is required. Cannulate the vein to be treated using ultrasound guidance to confirm venous access.
Inject freshly generated VARITHENA injectable foam slowly (approximately 1 mL/second in the GSV and 0.5 mL/second in accessory veins or varicosities) while monitoring using ultrasound. Confirm venospasm of the treated vein using ultrasound.
When treating the proximal GSV, stop the injection when VARITHENA is 3-5 cm distal to the saphenofemoral junction (SFJ).
Apply compression bandaging and stockings and have the patient walk for at least 10 minutes, while being monitored. Maintain compression for 2 weeks after treatment.
Repeat treatment may be necessary if the size and extent of the veins to be treated require more than 15 mL of VARITHENA. Separate treatment sessions by a minimum of 5 days.
Retained coagulum may be removed by aspiration (microthrombectomy) to improve comfort and reduce skin staining.
3. Dosage Forms and Strengths
VARITHENA is available in the following presentations:
- 180 mg/18 mL (10 mg/mL)
- 77.5 mg/7.75 mL (10 mg/mL)
Once activated, VARITHENA is a white, injectable foam delivering a 1% polidocanol solution.
Each mL of VARITHENA injectable foam contains 1.3 mg of polidocanol
4. Contraindications
The use of VARITHENA is contraindicated in patients with:
- known allergy to polidocanol [see Warnings and Precautions (5.1)]
- acute thromboembolic disease
5. Warnings and Precautions
5.1 Anaphylaxis
Severe allergic reactions have been reported following administration of liquid polidocanol, including anaphylactic reactions, some of them fatal. Observe patients for at least 10 minutes following injection and be prepared to treat anaphylaxis appropriately.
5.2 Tissue Ischemia and Necrosis
Intra-arterial injection or extravasation of polidocanol can cause severe necrosis, ischemia or gangrene Patients with underlying arterial disease, such as marked peripheral arteriosclerosis or thromboangiitis obliterans (Buerger’s Disease) may be at increased risk for tissue ischemia. If intra-arterial injection of polidocanol occurs, consult a vascular surgeon immediately.
5.3 Venous Thrombosis
VARITHENA can cause venous thrombosis [see Adverse Reactions (6)]. Follow administration instructions closely and monitor for signs of venous thrombosis after treatment. Patients with reduced mobility, history of deep vein thrombosis or pulmonary embolism, or recent (within 3 months) major surgery, prolonged hospitalization, or pregnancy are at increased risk for developing thrombosis.
6. Adverse Reactions/Side Effects
6.1 Clinical Trials Experience
Because clinical trials are conducted under controlled but widely varying conditions, adverse reaction rates observed in clinical trials of VARITHENA cannot be directly compared to rates in the clinical trials of other drugs or procedures and may not reflect the rates observed in practice.
A total of 1333 patients with GSVI in 12 clinical trials were evaluated for safety when treated with VARITHENA at dose concentrations of 0.125%, 0.5%, 1.0%, or 2.0%, including 437 patients treated with VARITHENA in placebo-controlled clinical trials.
Adverse reactions occurring in 3% more patients receiving VARITHENA 1% than receiving placebo are shown in Table 1.
| a Retained coagulum.
b Common femoral vein thrombus extension (non-occlusive thrombi starting in the superficial vein and extending into the common femoral vein). |
||
| Table 1: Treatment-emergent adverse reactions (3% more on VARITHENA 1% than on placebo) through Week 8 (n=588) |
||
| Adverse Reaction | Placebo (N=151) | VARITHENA 1.0% (N=149) |
| Pain in extremity | 14 (9.3) | 25 (16.8) |
| Infusion site thrombosisb | 0 | 24 (16.1) |
| Contusion/injection site hematoma | 9 (6.0) | 23 (15.4) |
| Limb discomfort | 5 (3.3) | 18 (12.1) |
| Tenderness/injection site pain | 5 (3.3) | 16 (10.7) |
| Venous thrombosis limbc | 0 | 12 (8.1) |
| Thrombophlebitis superficial | 2 (1.3) | 8 (5.4) |
| Deep vein thrombosis | 0 | 7 (4.7) |
In VARITHENA-treated patients, 80% of pain events in the treated extremity resolved within 1 week.
Proximal symptomatic venous thrombi occurred in <1% of patients treated with VARITHENA. Approximately half of patients with thrombi received treatment with anticoagulants.
Since VARITHENA induces thrombosis in the treated superficial veins, D-dimer is commonly elevated post-treatment and is not useful diagnostically to assess patients for venous thrombus following treatment with VARITHENA.
Neurologic adverse events (cerebrovascular accident, migraines) have been reported in patients following administration of physician compounded foam sclerosants. None of the 1333 patients in the VARITHENA trials experienced clinically important neurological or visual adverse events suggestive of cerebral gas embolism. The incidence of neurologic and visual adverse events within 1 day of treatment in the placebo-controlled studies was 2.7% in the pooled VARITHENA group and 4.0% in the placebo groups.
Skin discoloration adverse events were reported in 1.1% of the pooled VARITHENA group and 0.7% of the placebo group in the placebo-controlled studies.
Related/similar drugs
7. Drug Interactions
No specific drug interaction studies have been performed. There are no known drug interactions with VARITHENA.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Few published case reports with use of polidocanol-containing products, including VARITHENA, in pregnant women have not identified any drug-associated risk for major birth defects, miscarriage, or adverse maternal or fetal outcomes. Although no risks have been identified, there is minimal benefit in treating lower extremity varicosities during pregnancy and lower extremity varicosities that develop during pregnancy as they may spontaneously regress postpartum. In animal reproduction studies, no adverse developmental effects were observed with intravenous administration of polidocanol to pregnant rats and rabbits during organogenesis at dose levels up to approximately 13.5 and 12 times, respectively, the proposed maximum human dose of 15 mL of 1% VARITHENA based on body surface area (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Developmental reproductive toxicity testing was performed in rats and rabbits using intravenous administration of polidocanol solution. In rabbits, dose levels up to and including 10 mg/kg/day (approximately 12 times the proposed maximum human dose of 15 mL of 1% VARITHENA based on body surface area) did not produce any indication of adverse effects on embryo-fetal mortality, fetal weight, or the incidences of fetal abnormalities and variants. In rats administered 27 mg/kg/day of polidocanol solution (approximately 13.5 times the human dose based on body surface area), there were no adverse effects on pregnancy performance or fetal development. In a peri-natal and post-natal study in rats, dose levels of polidocanol up to 9 mg/kg/day (approximately 4.5 times the human dose based on body surface area) were without effects on the development of the conceptus and offspring, and at a dose level of 27 mg/kg/day of polidocanol solution (approximately 13.5 times the human dose based on body surface area), effects were confined to an equivocal reduction in body weights of first-generation males, and an associated equivocal delay in the age of preputial separation.
8.2 Lactation
Risk Summary
There are no data on the presence of polidocanol in human milk, the effects on the breastfed infant, or the effects on milk production. A lactating woman may consider interrupting breastfeeding and pumping anddiscarding breast milk up to 8 hours after VARITHENA administration in order to minimize exposure to a breastfed infant.
10. Overdosage
There are no known cases of overdosage with VARITHENA. In clinical studies, total volumes of up to 60 mL of VARITHENA per treatment session have been administered.
11. Varithena Description
VARITHENA injectable foam contains the sclerosant, polidocanol. It is intended for intravenous use only.
Chemically, polidocanol is polyoxyl lauryl ether. The structural formula is represented below:
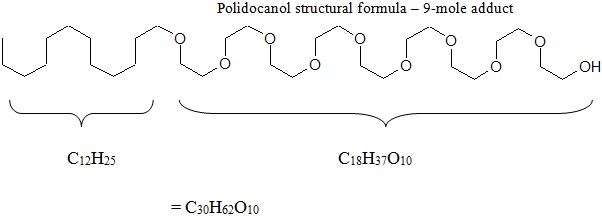
Polidocanol has the molecular formula CH3(CH2)11(OCH2CH2)nOH and a molecular weight of 582.9 when the average ethylene glycol moieties is nine (n=9). Polidocanol is a white to almost white, waxy, hygroscopic solid that is soluble in water and alcohol and melts at temperatures above 20°C.
VARITHENA is a sterile, injectable foam of an aqueous polidocanol solution (1%) containing the following inactive ingredients: ethanol (4.2% w/w), disodium hydrogen phosphate dihydrate (0.24% w/w), and potassium dihydrogen phosphate (0.085% w/w) with pH adjustment using 0.1 M sodium hydroxide solution and 0.1 M hydrochloric acid solution to achieve a pH of 6.0-7.5.
The injectable foam is generated after activation of the polidocanol canister with oxygen from a second aluminum canister, resulting in a final gas mixture of oxygen:carbon dioxide in a ratio of 65:35 with low (<0.8%) nitrogen content. At the time of use, VARITHENA is generated as an injectable foam of controlled density and bubble size. The foam is then transferred to a syringe through the VARITHENA transfer unit. The injectable foam has a liquid to gas ratio of approximately 1:7 by volume. The median bubble diameter is less than 100 µm and no bubbles are greater than 500 µm.
12. Varithena - Clinical Pharmacology
12.1 Mechanism of Action
VARITHENA is a drug/device combination product that generates injectable foam. The injectable foam is composed of a liquid and gas phase, both of which are necessary to have its therapeutic effect. VARITHENA is intended to act as follows: (1) the foam displaces blood from the vein to be treated, and (2) the polidocanol then scleroses the endothelium.
The active pharmaceutical ingredient of VARITHENA is polidocanol, a non-ionic surfactant sclerosing agent. The hydrophobic pole of the polidocanol molecule attaches to the lipid cell membrane of the venous endothelium, resulting in disruption of the osmotic barrier, destruction of the venous endothelium, and vasospasm. Following exposure to polidocanol, the interior surface of the vein becomes thrombogenic, which leads to thrombus formation and venous occlusion. The occluded vein is eventually replaced by fibrous connective tissue. Polidocanol is deactivated upon contact with blood, thus limiting the sclerosant action to the endothelium near the site of injection.
12.2 Pharmacodynamics
The active pharmaceutical ingredient in VARITHENA is polidocanol. Polidocanol damages the endothelium of blood vessels.
12.3 Pharmacokinetics
The pharmacokinetics of VARITHENA (as a weighted sum of 4 oligomers: E5, E9, E12 and E14) were evaluated at two concentrations (1% and 2%) randomly assigned within gender in 20 patients with GSV incompetence.
When administered as an intravenous injectable foam as two fixed 5 mL doses separated by 10 minutes, polidocanol was rapidly detected in plasma, reaching maximum concentration of drug in the body after dosing (Cmax) within 15 minutes of the first injection and within 5 minutes of receiving the second injection of VARITHENA 1% or VARITHENA 2%. The mean volume of distribution (Vd) of polidocanol ranged from 35 to 82 L.
Mean systemic clearance (CL) of polidocanol ranged from 0.2 to 0.4 L/min. The clearance of E5 was significantly greater than that of longer oligomers. Mean terminal elimination half-life (t1/2) ranged from 102 to 153 minutes, with most plasma samples below the limit of quantitation (BLQ) at the end of the 8-hour collection period. The increase in plasma polidocanol concentrations was less than proportional with increasing VARITHENA concentration. Weight-normalized data demonstrated no consistent differences in Cmax or AUC between males and females.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals have not been performed to evaluate carcinogenic potential of VARITHENA. No mutagenic activity was observed in the in vitro bacterial reverse mutation assay at non-toxic concentrations. No mutagenic activity was observed in the in vitro mouse lymphoma assay in the absence of S9 mix and was weakly mutagenic in the presence of S9 close to the limit of acceptance for the accompanying level of toxicity. No micronucleus induction was detected in the in vivo assay on mouse bone marrow cells up to the maximum tolerated dose of 80 mg/kg.
There was no adverse effect on fertility in both male and female rats at 27 mg/kg/day. This dose level is approximately 13.5 times the proposed maximum human dose based on body surface area.
13.2 Animal Toxicology and/or Pharmacology
The pharmacological effects of polidocanol solution on the renal function of the rat were evaluated and at the highest dose tested (10 mg/kg) hematuria occurred in 67% of animals. This dose is 5 times higher than the proposed maximum human dose based on body surface area. Blood was no longer detectable in urine 24 hours after dosing. In the 28-day repeated dose toxicity study in rat blood, pigments were noted in the urine for animals in all treatment groups, including male controls, at the end of the 4-week treatment period with up to 27 mg polidocanol/kg/day. Following the 2-week recovery period, there was still evidence that blood pigments were present in the urine but the incidence and severity was decreased when compared to the main study animals. There were no histopathological findings in the urinary bladder in any study animals.
In a cardiovascular pharmacology study in the anesthetized dog at 20 mg/kg (approximately 33 times the human dose based on body surface area), statistically significantly higher values for P-Q interval were measured before and during dosing and at all time points up to 30 minutes after dosing. An increase in QRS interval was also measured after dosing of 20 mg/kg and at 5 and 10 minutes after dosing. This effect was short lived and was no longer seen at 15 minutes after dosing. In addition, there was an increase in diastolic pressure with increasing dose of polidocanol. This increase became significantly greater (p<0.05) than baseline before injection of the final and highest dose (20 mg/kg).
In a further cardiovascular pharmacology study conducted with a once weekly, for four weeks, intravenous bolus injection of VARITHENA in the conscious dog, dose levels of up to 8.0 mL/kg (approximately 17 times the human dose based on body surface area) to beagle dogs caused only a transient, but consistent, effect on respiration, evidenced by a decrease in tidal volume and RMV at 15 minutes post-dose, resolving by one hour post-dose. Histopathology of the lung at the end of the 3-month follow-up period showed no abnormalities.
14. Clinical Studies
VARITHENA was evaluated in two randomized, blinded, multicenter clinical trials designed to assess the efficacy and safety of VARITHENA 0.5%, 1.0%, and 2.0% (VANISH-1) and VARITHENA 0.5% and 1.0% (VANISH-2) compared with placebo in the treatment of both symptoms and appearance in patients with SFJ incompetence as evidenced by reflux of the GSV or major accessory veins. In both studies, a VARITHENA 0.125% treatment group was included as a control for blinding of the duplex ultrasound assessment. Patients with history of deep vein thrombosis or pulmonary embolism; inability to comply with post-treatment compression due to severe peripheral arterial disease or leg obesity; incompetence of the small saphenous vein or deep venous reflux as a major source of reflux; or reduced mobility, major surgery, pregnancy, or prolonged hospitalization within 3 months were excluded. Patients were randomized in an equal distribution to each treatment group; the primary time point for analyses of the primary, secondary, and tertiary efficacy endpoints was Week 8.
In these clinical trials, the maximum volume of injectable foam or placebo to be administered per treatment session was 15 mL.
In VANISH-1, patients received one blinded treatment and in VANISH-2, patients received one blinded treatment with an option for a second blinded treatment 1 week later. In VANISH-2, patients in the VARITHENA 1.0% treatment group received an average of 1.4 blinded treatments. All patients received post-procedure compression therapy for 14 days following treatment.
Of the 519 patients randomized into VANISH-1 and VANISH-2, a total of 511 were treated with either VARITHENA 0.5% (n=111), 1.0% (n=110), or 2.0% (n=63), VARITHENA 0.125% as control (n=114), or placebo (n=113). Ninety-nine percent of the patients in VANISH-1 and VANISH-2 completed the blinded treatment period.
In the VARITHENA 1% group in VANISH-2, 23 of 58 patients received an additional blinded treatment. Two of these patients had retreatment of veins treated in the initial treatment session. The remaining 21 patients received treatment for additional veins not treated in the initial treatment session.
The mean age was approximately 50 years and approximately three-fourths of the patients were women. The mean BMI was similar in VANISH-1 and VANISH-2, at 28 kg/m2 (range 16 to 44 kg/m2) and 30 kg/m2 (range 17 to 48 kg/m2), respectively. The mean baseline GSV diameter was also similar in VANISH-1 and VANISH-2, at 7.6 mm (range 1.5 to 25.9 mm) and 8.7 mm (range 3.1 to 19.4 mm), respectively. Overall, 22% of patients in VANISH-1 and 25% of patients in VANISH-2 reported one or more prior varicose vein procedures in the leg to be treated.
For both clinical trials, the primary efficacy endpoint was improvement in patient symptoms, as measured by the change from baseline to Week 8 in the 7-day average electronic daily diary VVSymQ® score. The VVSymQ® score is a patient-reported outcome measure based on daily patient assessment of the varicose vein symptoms determined to be most important to patients: heaviness, achiness, swelling, throbbing, and itching. VVSymQ® scores range from 0 to 25, where 0 represents no symptoms and 25 represents all 5 symptoms experienced all of the time. Results are shown in Table 2.
For both VANISH-1 and VANISH-2, treatment with 1.0% was superior to placebo in improving symptoms as measured by VVSymQ®, when either a duration or an intensity scale was used to measure patients’ symptoms.
| *Percent of patients who reported their symptoms had "moderately improved" or "much improved" compared with baseline. | ||||||
| Table 2: Improvement in Symptoms of Varicose Veins as Measured by VVSymQ® at Week 8, VANISH-1 and VANISH-2 | ||||||
| VVSymQ® | ||||||
| VANISH-1 | VANISH-2 | |||||
| Placebo | VARITHENA 1.0% | Placebo | VARITHENA 1.0% | |||
| N | 55 | 50 | 54 | 57 | ||
| Baseline score, mean | 8.60 | 8.82 | 9.26 | 7.82 | ||
| Adjusted mean change from baseline at week 8 | -2.13 | -4.87 | -2.00 | -5.06 | ||
| Clinically meaningful improvement in symptoms at week 8* | 5.4% (n=56) | 64.7% (n=51) | 19.6% (n=56) | 75.9% (n=58) |
||
| Comparison vs. Placebo at week 8, p-value, adjusted mean change | <0.0001 | <0.0001 | ||||
The co-secondary endpoints in VANISH-1 and VANISH-2 were the improvement in appearance of visible varicosities from baseline to Week 8 as measured by 1) patients scoring the appearance of their varicose veins in the medial view of their study leg (PA-V3 score) from "Not at all noticeable" (a score of 0) to "Extremely noticeable" (a score of 4); and 2) an independent photography review panel rating the severity of the patient's varicose vein appearance in standardized digital photographs of the medial view of each patient's study leg (IPR-V3 score) from "None" (a score of 0) to "Very severe" (a score of 4). Results are shown in Table 3.
| †Percent who reported the appearance of varicose veins had “moderately improved” or “much improved” compared with baseline. |
||||
| Table 3: Improvement in Appearance of Visible Varicosities as Measured by IPR-V3and PA-V3at Week 8, VANISH-1 and VANISH-2 |
||||
| VANISH-1 | VANISH-2 | |||
| Placebo | VARITHENA 1.0% | Placebo | VARITHENA 1.0% |
|
| IPR-V3 | ||||
| n | 55 | 49 | 56 | 57 |
| Baseline score, mean | 1.82 | 1.98 | 2.18 | 2.02 |
| Adjusted mean change from baseline at week 8 | -0.01 | -0.76 | -0.07 | -0.83 |
| Clinically meaningful improvement in appearance at week 8† | 8.9% (n=56) | 70.6% (n=51) | 0 (n=56) | 70.7% (n=58) |
| Comparison vs. Placebo, p-value at week 8, adjusted mean change | <0.0001 | <0.0001 | ||
| PA-V3 | ||||
| N | 55 | 50 | 56 | 57 |
| Baseline score, mean | 3.49 | 3.46 | 3.30 | 3.49 |
| Adjusted mean change from baseline at week 8 | -0.15 | -1.60 | -0.32 | -1.79 |
| Clinically meaningful improvement in appearance at week 8† | 3.6% (n=56) | 54.9% (n=51) | 7.1% (n=56) | 69.0% (n=58) |
| Comparison vs. Placebo, p-value at week 8, adjusted mean change | <0.0001 | <0.0001 | ||
Tertiary endpoints in VANISH-1 and VANISH-2 included response to treatment as determined by change from baseline in Venous Clinical Severity Score (VCSS), by duplex ultrasound, and by change from baseline in Venous Insufficiency Epidemiologic and Economic Study – Quality of Life/Symptoms (VEINES-QOL) score.
VCSS is a clinician rating of severity of chronic venous insufficiency ranging from 0 to 30, where higher scores indicate more severe venous disease. In VANISH-1 and VANISH-2, the adjusted mean changes from baseline in VCSS in the 1% VARITHENA treatment groups were 3.70 and 5.05, respectively, at Week 8 compared with 0.75 and 1.52 points in the placebo groups, respectively. For both studies, the differences between these improvements are statistically significant (P<0.0001).
The physiological response to treatment as measured by duplex ultrasound (duplex response) was defined as elimination of reflux through the SFJ and/or complete occlusion of all incompetent GSV and major accessory veins at baseline. The primary comparison for duplex response in both studies was the pooled VARITHENA groups versus the VARITHENA 0.125% (control) group. Results are shown in Table 4.
| *In VANISH-1, a significant dose-response trend was evident between the percent of responders and the dose concentration of VARITHENA (P<0.0001). | |||
| Table 4: Response to Treatment as Measured by Duplex Ultrasound at Week 8, VANISH-1 and VANISH-2 | |||
| Parameter | Treatment Group, % | ||
| Placebo | VARITHENA 0.125% (control) | VARITHENA 1.0% | |
| Responders, VANISH-1* | 5.4% (n=56) | 42.1% (n=57) | 80.4% (n=51) |
| Responders, VANISH-2 | 1.8% (n=56) | 59.6% (n=57) | 86.2% (n=58) |
VEINES-QOL is a disease-specific quality of life instrument, ranging from 0 (worst possible quality of life) to 100 (best possible quality of life). In VANISH-1 and VANISH-2, the adjusted mean changes from baseline in VEINES-QOL in the pooled VARITHENA treatment groups were 21.2 and 21.6, respectively, at Week 8 compared with 7.7 and 7.4 points in the placebo groups, respectively. For both studies, the differences between these improvements are statistically significant (P<0.0001).
For efficacy endpoints, VARITHENA treatment effects were consistent across subgroups of age, sex, BMI (up to 48 kg/m2), CEAP clinical class, GSV diameter (up to 25.9 mm), and VCSS.
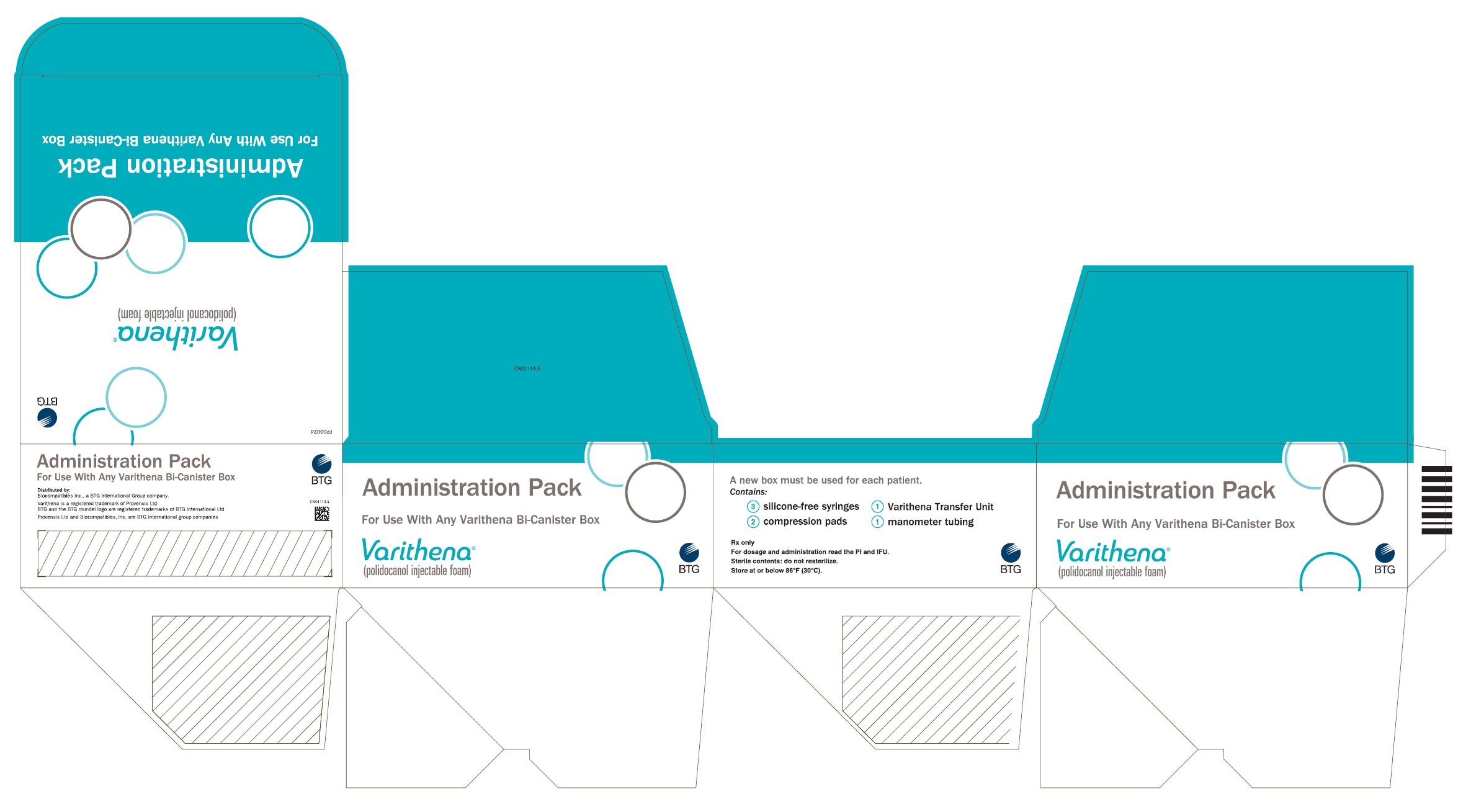
16. How is Varithena supplied
16.1 How Supplied
VARITHENA (polidocanol injectable foam) product is available in four configurations, each containing two sterile, connected, 303-mL aluminum alloy cylinders, one containing polidocanol solution (10 mg/mL) under carbon dioxide, and the other containing pressurized oxygen.
| *Ancillary Pack includes three 10-mL syringes, one 20-inch manometer tube, and two compression pads. | ||||
| Polidocanol mg | Usable foam mL | Administration Pack | NDC | |
| Transfer units | Ancillary Pack* | |||
| 77.5 | 15 | 0 | 0 | 60635-107-01 PD Canister - 60635-007-01 |
| 1 | 1 | 60635-111-01 PD Canister - 60635-007-01 |
||
| 180 | 45 | 0 | 0 | 60635-118-01 PD Canister - 60635-018-01 |
| 3 | 3 | 60635-133-01 PD Canister - 60635-018-01 |
||
16.2 Storage and Handling
Do not shake VARITHENA canisters.
Avoid contact with eyes.
Store the VARITHENA Bi-Canister or convenience box at or below 86°F (30°C);
Do not refrigerate or freeze.
Unused, non-activated VARITHENA canisters may be stored in the flat or upright position.
Contains gas under pressure: May explode if heated. Store in a well-ventilated place. Store the canisters away from sources of heat including strong light conditions.
Pressurized Oxygen: May cause or intensify fire; oxidizer. Store away from combustible materials.
Once activated, the canister of 180 mg/18 mL (10 mg/mL) VARITHENA must be used within thirty (30) days.
Once activated, the canister of 77.5mg/7.75mL (10mg/mL) VARITHENA must be used within thirty (30) days.
Store activated canisters of VARITHENA upright, with the VARITHENA transfer unit attached, under the same temperature conditions as the VARITHENA Bi-Canister or convenience box. Use a new VARITHENA transfer unit for each treatment session.
Discard aerosol canisters after use in accordance with state and local requirements.
For more information, please refer to the IFU.
17. Patient Counseling Information
Advise the patient to keep post-treatment bandages dry and in place for 48 hours and to wear compression stockings on the treated legs continuously for 2 weeks. Compression stockings should be thigh high or knee high, depending upon the area treated, in order to provide adequate coverage. Advise the patient to walk for at least 10 minutes immediately after the procedure and daily for the next month. Following treatment, advise the patient to avoid heavy exercise for 1 week and extended periods of inactivity for 1 month.
If you would like more information, please talk with your doctor. For more information about VARITHENA, you can also call us at 1-855-971-VEIN (1-855-971-8346) or go to www.VARITHENA.com.
Manufactured for Provensis Ltd by:
Biocompatibles UK Ltd
Chapman House, Weydon Lane, Farnham, UK, GU9 8QL
Distributed by:
Biocompatibles Inc.,
Provensis Ltd, Biocompatibles UK Ltd, and Biocompatibles, Inc. are BTG International group companies
VARITHENA and VVSymQ are registered trademarks of Provensis Ltd
BTG and the BTG roundel logo are registered trademarks of BTG International Ltd
Tyvek is a registered trademark of E. I. du Pont de Nemours and Company





| VARITHENA
polidocanol kit |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| VARITHENA
polidocanol kit |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| VARITHENA
polidocanol kit |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| VARITHENA
polidocanol kit |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| VARITHENA
polidocanol kit |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Biocompatibles, Inc. (024194234) |
| Registrant - Provensis Ltd (236996703) |
More about Varithena (polidocanol)
- Compare alternatives
- Pricing & coupons
- Reviews (1)
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: sclerosing agents
- En español