Vaprisol: Package Insert / Prescribing Info
Package insert / product label
Generic name: conivaptan hydrochloride
Dosage form: injection, solution
Drug class: Vasopressin antagonists
Medically reviewed by Drugs.com. Last updated on Jun 5, 2025.
On This Page
Vaprisol Description
VAPRISOL® (conivaptan hydrochloride injection) is a nonpeptide, dual antagonist of arginine vasopressin (AVP) V1A and V2 receptors.
Conivaptan hydrochloride is chemically [1,1'-biphenyl]-2-carboxamide, N-[4-[(4,5-dihydro-2-methylimidazo[4,5-d][1]benzazepin-6(1H)-yl)carbonyl]phenyl]-, monohydrochloride, having a molecular weight of 535.04 and molecular formula C32H26N4O2 .HCl. The structural formula of conivaptan hydrochloride is:
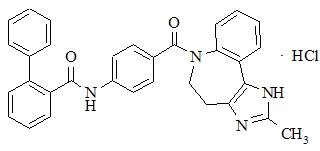
Conivaptan hydrochloride is a white to off-white or pale orange-white powder that is very slightly soluble in water (0.15 mg/mL at 23°C). Conivaptan hydrochloride injection is supplied as a sterile liquid in an ampule and as a sterile premixed solution with dextrose in a flexible plastic container.
VAPRISOL (conivaptan hydrochloride injection) Ampule
Each ampule will deliver 20 mg conivaptan hydrochloride, 1.2 g propylene glycol, 0.4 g ethanol and Water for Injection, q.s. Lactic acid is added for pH adjustment to 3.0.
VAPRISOL (conivaptan hydrochloride injection) Premixed in 5% Dextrose
Each container contains a clear, colorless, sterile, non-pyrogenic solution of conivaptan hydrochloride in dextrose. Each 100 mL, single-use premixed INTRAVIA Container contains 20 mg of conivaptan hydrochloride and 5 g of Dextrose Hydrous, USP. Lactic Acid, USP is added for pH adjustment to pH 3.4 to 3.8. The flexible plastic container is fabricated from a specially designed multilayer plastic (PL 2408). Solutions in contact with the plastic container leach out certain of the chemical components from the plastic in very small amounts; however, biological testing was supportive of the safety of the plastic container materials. The flexible container has a foil overwrap. Water can permeate the plastic into the overwrap, but the amount is insufficient to significantly affect the premixed solution.
Vaprisol - Clinical Pharmacology
Pharmacodynamics
Conivaptan hydrochloride is a dual AVP antagonist with nanomolar affinity for human V1A and V2 receptors in vitro. The level of AVP in circulating blood is critical for the regulation of water and electrolyte balance and is usually elevated in both euvolemic and hypervolemic hyponatremia. The AVP effect is mediated through V2 receptors, which are functionally coupled to aquaporin channels in the apical membrane of the collecting ducts of the kidney. These receptors help to maintain plasma osmolality within the normal range. The predominant pharmacodynamic effect of conivaptan hydrochloride in the treatment of hyponatremia is through its V2 antagonism of AVP in the renal collecting ducts, an effect that results in aquaresis, or excretion of free water. The pharmacodynamic effects of conivaptan hydrochloride include increased free water excretion (i.e., effective water clearance [EWC]) generally accompanied by increased net fluid loss, increased urine output, and decreased urine osmolality. Studies in animal models of hyponatremia showed that conivaptan hydrochloride prevented the occurrence of hyponatremia-related physical signs in rats with the syndrome of inappropriate antidiuretic hormone secretion.
Pharmacokinetics
The pharmacokinetics of conivaptan have been characterized in healthy subjects, special populations and patients following both oral and intravenous dosing regimens. The pharmacokinetics of conivaptan following intravenous infusion (40 mg/day to 80 mg/day) and oral administration are non-linear, and inhibition by conivaptan of its own metabolism seems to be the major factor for the non-linearity. The intersubject variability of conivaptan pharmacokinetics is high (94% CV in CL).
The pharmacokinetics of conivaptan and its metabolites were characterized in healthy male subjects administered conivaptan hydrochloride as a 20 mg loading dose (infused over 30 minutes) followed by a continuous infusion of 40 mg/day for 3 days. Mean Cmax for conivaptan was 619 ng/mL and occurred at the end of the loading dose. Plasma concentrations reached a minimum at approximately 12 hours after start of the loading dose, then gradually increased over the duration of the infusion to a mean concentration of 188 ng/mL at the end of the infusion. The mean terminal elimination half-life after conivaptan infusion was 5.0 hours, and the mean clearance was 15.2 L/h.
In an open-label safety and efficacy study, the pharmacokinetics of conivaptan were characterized in hypervolemic or euvolemic hyponatremia patients (ages 20-92 years) receiving conivaptan hydrochloride as a 20 mg loading dose (infused over 30 minutes) followed by a continuous infusion of 20 or 40 mg/day for 4 days. The median-plasma conivaptan concentrations are shown in Figure 1 and pharmacokinetic parameters are summarized in Table 1.
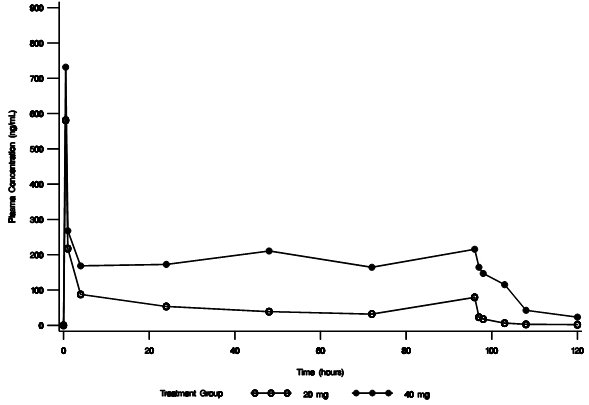
Figure 1. Median Plasma Concentration-Time Profiles From Rich PK Sampling After 20 mg Loading Dose and 20 mg/day (open circle) or 40 mg/day (closed circle) Infusion for 4 Days
|
Parameter |
IV Conivaptan 20 mg/day |
IV Conivaptan 40 mg/day |
|
Conivaptan concentration at the end of loading dose (ng/mL, at 0.5 hours) Median (range) |
659.4 (144.5-1587.6) |
679.5 (0.0-1910.8) |
|
Conivaptan concentration at the end of infusion (ng/mL, at 96 hours) Median (range) |
117.6 (4.9-938.3) |
215.7 (2.1-1999.3) |
|
Elimination half-life (hr) Median (range) |
5.3 (3.3-9.3) |
8.1 (4.1-22.5) |
|
Clearance (L/hr) Median (range) |
16.1 (7.2-37.6) |
8.73 (2.1-20.9) |
Distribution
Conivaptan is extensively bound to human plasma proteins, being 99% bound over the concentration range of approximately 10 to 1000 ng/mL.
Metabolism and Excretion
CYP3A4 was identified as the sole cytochrome P450 isozyme responsible for the metabolism of conivaptan. Four metabolites have been identified. The pharmacological activity of the metabolites at V1A and V2 receptors ranged from approximately 3-50% and 50-100% that of conivaptan, respectively. The combined exposure of the metabolites following intravenous administration of conivaptan is approximately 7% that of conivaptan and hence, their contribution to the clinical effect of conivaptan is minimal.
After intravenous (10 mg) or oral (20 mg) administration of conivaptan hydrochloride in a mass balance study, approximately 83% of the dose was excreted in feces as total radioactivity and 12% in urine over several days of collection. Over the first 24 hours after dosing, approximately 1% of the intravenous dose was excreted in urine as intact conivaptan.
Special Populations
Hepatic Impairment
The effect of hepatic impairment (including ascites, cirrhosis, or portal hypertension) on the elimination of conivaptan after intravenous administration has not been systematically evaluated. However, increased systemic exposures after administration of oral conivaptan (up to a mean 2.8-fold increase) have been seen in patients with stable cirrhosis and moderate hepatic impairment. Intravenous VAPRISOL resulted in higher conivaptan exposure than did oral conivaptan, in study subjects without hepatic function impairment. Caution should be exercised when administering VAPRISOL to patients with impaired hepatic function.
Renal Impairment
The effect of renal impairment on the elimination of conivaptan after intravenous administration has not been evaluated. However, following administration of oral conivaptan, the AUC for conivaptan was up to 80% higher in patients with renal impairment (CLcr < 60 mL/min/1.73 m2) as compared to those with normal renal function. Intravenous VAPRISOL resulted in higher conivaptan exposure than did oral conivaptan, in study subjects without renal function impairment. Caution should be exercised when administering VAPRISOL to patients with impaired renal function.
Geriatric Patients
Following a single oral dose of conivaptan hydrochloride (15, 30 or 60 mg), drug exposure (AUC) in elderly male and female volunteers (65 to 90 years of age) compared to that seen in young male subjects was similar for the 15 and 30 mg doses but increased nearly 2-fold at the 60 mg dose.
In an open label study to assess the safety and efficacy of conivaptan, a subset of geriatric hypervolemic or euvolemic hyponatremia patients (65 to 92 years of age) received a 20 mg intravenous loading dose followed by a 20 mg/day (N = 27) or 40 mg/day (N = 135) intravenous infusion for 4 days. The median conivaptan plasma concentration in these patients at the end of the loading dose infusion was 654 ng/mL. The median conivaptan plasma concentrations at the end of the 4-day continuous infusion were 118 and 215 ng/mL for the 20 mg/day and 40 mg/day regimens, respectively.
CYP3A4
Conivaptan is a sensitive substrate of CYP3A4. The effect of ketoconazole, a potent CYP3A4 inhibitor, on the pharmacokinetics of intravenous conivaptan has not been evaluated. Coadministration of oral conivaptan hydrochloride 10 mg with ketoconazole 200 mg resulted in 4- and 11-fold increases in Cmax and AUC of conivaptan, respectively.
Conivaptan is a potent inhibitor of CYP3A4. The effect of conivaptan on the pharmacokinetics of CYP3A4 substrates has been evaluated with the coadministration of conivaptan with midazolam, simvastatin, and amlodipine. Intravenous conivaptan hydrochloride 40 mg/day increased the mean AUC values by approximately 2- and 3-fold for 1 mg intravenous or 2 mg oral doses of midazolam, respectively. Intravenous conivaptan hydrochloride 30 mg/day resulted in a 3-fold increase in the AUC of simvastatin. Oral conivaptan hydrochloride 40 mg twice daily resulted in a 2-fold increase in the AUC and half-life of amlodipine.
Digoxin
Coadministration of a 0.5-mg dose of digoxin, a P-glycoprotein substrate, with oral conivaptan hydrochloride 40 mg twice daily resulted in a 30% reduction in clearance and 79% and 43% increases in digoxin Cmax and AUC values, respectively.
Warfarin
The effect of intravenous conivaptan on warfarin pharmacokinetics or pharmacodynamics has not been evaluated. The potential drug-drug interaction of oral conivaptan with warfarin, which undergoes major metabolism by CYP2C9 and minor metabolism by CYP3A4, was investigated in a clinical study.
The effects of oral conivaptan hydrochloride 40 mg twice daily on prothrombin time was assessed in patients receiving stable oral warfarin therapy. After 10 days of oral conivaptan administration, the S- and R-warfarin concentrations were 90% and 98%, respectively, of those prior to conivaptan administration. The corresponding prothrombin time values after 10 days of oral conivaptan administration were 95% of baseline. No effect of oral conivaptan on the pharmacokinetics or pharmacodynamics of warfarin was observed.
Captopril and Furosemide
The effects of captopril (25 mg) on the pharmacokinetics of conivaptan hydrochloride (30 mg) and furosemide (40 mg or 80 mg once daily for 6 days) on the pharmacokinetics of conivaptan hydrochloride (20 mg and 40 mg) were assessed in separate studies. The pharmacokinetics of conivaptan were unchanged with coadministration of either captopril or furosemide.
Electrophysiology
The effect of VAPRISOL 40 mg IV and 80 mg IV on the QT interval was evaluated after the first dose (Day 1) and at the last day during treatment (Day 4) in a randomized, single-blind, parallel group, placebo- and positive-controlled (moxifloxacin 400 mg IV) study in healthy male and female volunteers aged 18 to 45 years. Digital ECGs were obtained at baseline and on Days 1 and 4. The placebo-corrected changes from baseline in individualized QT correction (QTcI) in the VAPRISOL 40 mg and 80 mg dose groups on Day 1 were -3.5 msec and -2.9 msec, respectively, on Day 1, and -2.1 msec for both dose groups on Day 4. Similar results were obtained using either the Bazett's or Fridericia's correction methods. Moxifloxacin elicited placebo-corrected changes from baseline in QTcI of +7 to +10 msec on Days 1 and 4, respectively.
|
Drug and Dose |
QTcI |
|
Placebo |
-3 msec |
|
Vaprisol 40 mg IV |
-5.1 msec |
|
Vaprisol 80 mg IV |
-5.1 msec |
|
Moxifloxacin 400 mg IV |
+7.4 msec |
The results of the central tendency analysis of QTc indicate that VAPRISOL had no effect on cardiac repolarization.
Clinical Studies
In a double-blind, placebo-controlled, randomized, multicenter study, 84 patients with euvolemic or hypervolemic hyponatremia (serum sodium 115-130 mEq/L) due to a variety of underlying causes (malignant or nonmalignant diseases of the central nervous system, lung, or abdomen; congestive heart failure [CHF]; hypertension; myocardial infarction; diabetes; osteoarthritis; or idiopathic) were treated for 4 days with VAPRISOL or placebo. All patients received standard care for hyponatremia, primarily fluid restriction (daily fluid intake restricted to less than or equal to 2.0 liters). Study participants were randomized to receive either placebo IV (N = 29), or VAPRISOL 40 mg/day IV (N = 29), or VAPRISOL 80 mg/day IV (N = 26). VAPRISOL was administered as a continuous infusion following a 30 minute IV infusion of a 20 mg loading dose on the first treatment day. Serum or plasma sodium concentrations were assessed at predose (Hour 0) and at 4, 6, 10, and 24 hours post dose on all treatment days. Mean serum sodium concentration was 123.3 mEq/L at study entry.
The mean change in serum sodium concentration from baseline over the 4-day treatment period is shown in Figure 2.
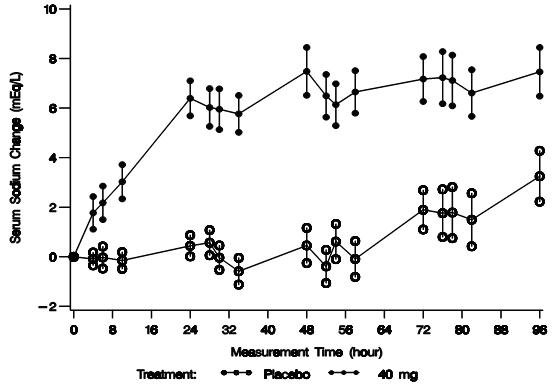
Following treatment with 40 mg/day of intravenous VAPRISOL, 79% of patients achieved an increase of ≥ 4 mEq/L in serum sodium concentration. The mean change from baseline in serum sodium concentration at the end of 2 days of treatment with VAPRISOL was 5.3 mEq/L (mean concentration 128.6 mEq/L). At the end of the 4-day treatment period, the mean change from baseline was 6.5 mEq/L (mean concentration 129.8 mEq/L). In addition, after 2 days and 4 days of treatment with VAPRISOL, 41% (after 2 days) and 69% (after 4 days) of patients achieved a ≥ 6 mEq/L increase in serum sodium concentration or a normal serum sodium of ≥ 135 mEq/L. Although 80 mg/day was also studied, it was not significantly more effective than 40 mg/day. The maximum daily dose of VAPRISOL (after the loading dose) is 40 mg/day. Additional efficacy data are summarized in Table 3.
|
Efficacy Variable |
Placebo
|
VAPRISOL 40 mg/day N = 29 |
||
|
Day 2* |
Day 4 |
Day 2* |
Day 4 |
|
|
Baseline adjusted serum Na+ AUC over duration of treatment (mEq·hr/L) Mean (SD) |
|
|
|
|
|
Number of patients (%) and median event time (h) from first dose of study medication to a confirmed ≥ 4 mEq/L increase from Baseline in serum Na+, [95% CI] |
2 (7%) Not estimable Not estimable |
9 (31%) Not estimable Not estimable |
22 (76%) 23.7† [10, 2] |
23 (79%) 23.7†
|
|
Total time (h) from first dose of study medication to Day 2 or Day 4 end of treatment during which patients had a confirmed ≥ 4 mEq/L increase in serum Na+ from Baseline |
|
|
|
|
|
Serum Na+ (mEq/L) |
0.2 (2.5) |
|
|
|
|
Number (%) of patients who obtained a confirmed ≥ 6 mEq/L increase from Baseline in serum Na+ or a normal serum Na+ concentration ≥ 135 mEq/L during treatment |
0 (0) |
6 (21%) |
12 (41%)† |
20 (69%)† |
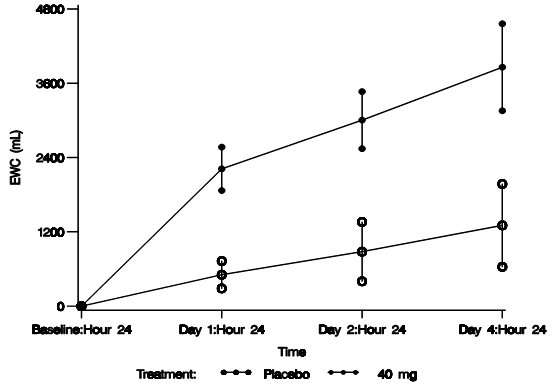
The aquaretic effect of VAPRISOL is shown in Figure 3. VAPRISOL produced a baseline-corrected cumulative increase in effective water clearance of over 3800 mL compared to approximately 1300 mL with placebo by Day 4.
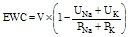
, where V is urine volume (mL/d), UNa is urine sodium concentration, UK is urine potassium concentration, PNa is plasma/serum sodium concentration, and PK is plasma/serum potassium concentration.
In an open-label study in patients with euvolemic or hypervolemic hyponatremia, 251 patients were treated for 4 days with VAPRISOL 20 or 40 mg/day IV as a continuous infusion following a 30 minute IV infusion of a 20 mg loading dose on the first treatment day. The results are shown in Table 4.
|
Primary Efficacy Endpoint |
20 mg/day N = 37 |
40 mg/day N = 214 |
|
Baseline adjusted serum Na+ AUC over duration of treatment (mEq·hr/L) Mean (SD) |
|
|
|
Secondary Efficacy Endpoints | ||
|
Number of patients (%) and median event time (h) from first dose of study medication to a confirmed ≥ 4 mEq/L increase from Baseline in serum Na+, [95% CI] |
29 (78%)
|
178 (83%)
|
|
Total time (h) from first dose of study medication to end of treatment during which patients had a confirmed ≥ 4 mEq/L increase in serum Na+ from Baseline Mean (SD) |
|
|
|
Serum Na+ (mEq/L) Mean (SD) at Follow-up Day 11 Mean Change (SD) from Baseline to Follow-up Day 11 Mean (SD) at Follow-up Day 34 Mean Change (SD) from Baseline to Follow-up Day 34 |
131.8 (3.9) 9.4 (5.3) 129.9 (6.2) 7.1 (8.2) 134.3 (4.5) 11.5 (7.3) |
132.5 (4.6) 8.8 (5.4) 131.8 (5.8) 8.0 (6.5) 134.3 (5.2) 10.7 (6.7) |
|
Number (%) of patients who obtained a confirmed ≥ 6 mEq/L increase from Baseline in serum Na+ or a normal serum Na+ concentration ≥ 135 mEq/L during treatment |
|
|
The effectiveness of VAPRISOL for the treatment of congestive heart failure has not been established.
Indications and Usage for Vaprisol
VAPRISOL is indicated for the treatment of euvolemic and hypervolemic hyponatremia in hospitalized patients.
Important Limitation:
VAPRISOL is not indicated for the treatment of congestive heart failure. VAPRISOL should only be used for the treatment of hyponatremia in patients with underlying heart failure when the expected clinical benefit of raising serum sodium outweighs the increased risk of adverse events for heart failure patients. (See PRECAUTIONS and ADVERSE REACTIONS)
Contraindications
VAPRISOL is contraindicated in patients with hypovolemic hyponatremia.
The coadministration of VAPRISOL with potent CYP3A4 inhibitors, such as ketoconazole, itraconazole, clarithromycin, ritonavir, and indinavir, is contraindicated. (See PRECAUTIONS: Drug Interactions for details and other important considerations)
VAPRISOL (conivaptan hydrochloride injection) Premixed in 5% Dextrose
Solutions containing dextrose may be contraindicated in patients with known allergy to corn or corn products.
Precautions
Congestive Heart Failure
The number of heart failure patients with hypervolemic hyponatremia who have been treated with intravenous VAPRISOL is too small to establish safety in patients with underlying congestive heart failure. (See ADVERSE REACTIONS)
Overly Rapid Correction of Serum Sodium
An overly rapid increase in serum sodium concentration (>12 mEq/L/24 hours) may result in serious sequelae. In controlled clinical trials of VAPRISOL, about 9% of patients who received VAPRISOL in doses of 20-40 mg/day IV met laboratory criteria for overly rapid correction of serum sodium, but none of these patients had permanent neurologic sequelae. Although not observed in the clinical studies with VAPRISOL, osmotic demyelination syndrome has been reported following rapid correction of low serum sodium concentrations. Serum sodium concentration and neurologic status should be monitored appropriately during VAPRISOL administration, and VAPRISOL administration should be discontinued if the patient develops an undesirably rapid rate of rise of serum sodium. If the serum sodium concentration continues to rise, VAPRISOL should not be resumed. If hyponatremia persists or recurs (after initial discontinuation of VAPRISOL for an undesirably rapid rate of rise of serum sodium concentration), and the patient has had no evidence of neurologic sequelae of rapid rise in serum sodium, VAPRISOL may be resumed at a reduced dose.
Hepatic Impairment
The use of VAPRISOL in patients with hepatic impairment (including ascites, cirrhosis, or portal hypertension) has not been systematically evaluated.
Increased systemic exposures after oral administration of conivaptan have been seen in patients with stable cirrhosis and moderate hepatic impairment. Intravenous VAPRISOL resulted in higher conivaptan exposure than did oral conivaptan, in study subjects without hepatic function impairment. Caution should be used when administering VAPRISOL to patients with hepatic impairment.
Renal Impairment
The effect of renal impairment on the elimination of conivaptan after intravenous administration has not been evaluated. However, following oral administration of conivaptan, the AUC for conivaptan was up to 80% higher after a single oral dose and 35% higher with repeated oral dosing in patients with renal impairment (CLcr < 60 mL/min/1.73 m2) as compared to those with normal renal function. Intravenous VAPRISOL resulted in higher conivaptan exposure than did oral conivaptan, in study subjects without renal function impairment. Caution should be used when administering VAPRISOL to patients with renal impairment.
Injection Site Reactions
Conivaptan may cause significant injection site reactions, even with proper dilution and infusion rates (seeADVERSE REACTIONS). The VAPRISOL ampule must only be administered when properly prepared and diluted (see Preparation). VAPRISOL should be administered via large veins, and the infusion site should be rotated every 24 hours (see DOSAGE AND ADMINISTRATION).
CYP3A4
Conivaptan is a substrate of CYP3A4. Coadministration of VAPRISOL with CYP3A4 inhibitors could lead to an increase in conivaptan concentrations. The consequences of increased conivaptan concentrations are unknown. Concomitant use of VAPRISOL with potent CYP3A4 inhibitors such as ketoconazole, itraconazole, clarithromycin, ritonavir, and indinavir is contraindicated.
Conivaptan is a potent inhibitor of CYP3A4. VAPRISOL may increase plasma concentrations of coadministered drugs that are primarily metabolized by CYP3A4. In clinical trials of oral conivaptan hydrochloride, two cases of rhabdomyolysis occurred in patients who were also receiving a CYP3A4-metabolized HMG-CoA reductase inhibitor. Concomitant use of VAPRISOL with drugs that are primarily metabolized by CYP3A4 should be closely monitored or the combination should be avoided. If a clinical decision is made to discontinue concomitant medications at recommended doses, allow an appropriate amount of time (at least 24 hours) following the end of VAPRISOL administration before resuming these medications.
Digoxin
Coadministration of digoxin, a P-glycoprotein substrate, with oral conivaptan resulted in a reduction in clearance and an increases in digoxin Cmax and AUC values. Therefore, if digoxin is administered with VAPRISOL, the clinician should be alert to the possibility of increases in digoxin levels.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Standard lifetime (104 week) carcinogenicity bioassays were conducted in mice and rats. Mice were given oral doses of 3, 10 or 30 mg/kg/day in males and 1, 3 or 10 mg/kg/day in females by gavage. Rats were given oral doses of 0.3, 1, 3 or 10 mg/kg/day in males and 1, 3, 10 or 30 mg/kg/day in females by gavage. No increased incidence of tumors was observed at doses up to 30 mg/kg/day in mice (6 times human systemic exposure of an IV bolus of 20 mg on Day 1 followed by IV infusion 40 mg/day for 3 days based on AUC comparison) or rats (2 times human systemic exposure of an IV bolus of 20 mg on Day 1 followed by IV infusion 40 mg/day for 3 days based on AUC comparison).
Conivaptan was not mutagenic or clastogenic with or without metabolic activation in the Ames test with Salmonella typhimurium and Escherichia coli, in human peripheral blood lymphocytes, or in vivo rat micronucleus assay.
In fertility studies after 4 weeks treatment by intravenous bolus at 0.5, 1.25 or 2.5 mg/kg/day, male fertility was unaffected. However, in females given IV bolus conivaptan 15 days before mating through gestation day 7 there was prolonged diestrus, decreased fertility and increased pre- and post-implantation loss at 2.5 mg/kg/day (systemic exposures less than the therapeutic dose).
Pregnancy
Pregnancy Category C
Conivaptan has been shown to have adverse effects on the fetus when given to animals during pregnancy at systemic exposures less than those achieved at a therapeutic dose based on AUC comparisons. There are no adequate and well-controlled studies in pregnant women. VAPRISOL should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. The patient should be apprised of the potential hazard to the fetus. Conivaptan crosses the placenta and is found in fetal tissue in rats. Fetal tissue levels were <10% of maternal plasma concentrations while placental levels were 2.2-fold higher than maternal plasma concentrations indicating that conivaptan can be transferred to the fetus. Conivaptan that is taken up by fetal tissue is slowly cleared, suggesting that fetal accumulation is possible. Milk levels were up to 3 times higher than maternal plasma levels following an intravenous dose of 1 mg/kg (systemic exposures less than therapeutic based on AUC comparisons).
In female rats given an intravenous bolus dose of 0.5, 1.25 or 2.5 mg/kg/day conivaptan hydrochloride before mating and continuing through gestation day 7, prolonged diestrus, decreased fertility and increased pre- and post-natal implantation loss occurred at 2.5 mg/kg/day (systemic exposures less than the therapeutic dose).
In pregnant rats given intravenous doses of 0.5, 1.25 or 2.5 mg/kg/day from gestation day 7 through 17 (organogenesis), no significant maternal or fetal effects were observed at systemic exposures less than therapeutic exposure based on AUC comparisons.
Pregnant rats were administered intravenous conivaptan hydrochloride at a dose of 2.5 mg/kg/day (systemic exposures less than therapeutic based on AUC) from gestation day 7 through lactation day 20 (weaning), and the pups showed decreased neonatal viability, weaning indices, delayed growth and physical development (including sexual maturation), and delayed reflex development. No discernible changes were seen in pups from dams administered conivaptan hydrochloride at 0.5 or 1.25 mg/kg/day from this same period. No maternal adverse effects were seen with conivaptan hydrochloride administration (0.5, 1.25, or 2.5 mg/kg/day from gestation day 7 through lactation day 20; systemic exposures less than therapeutic dose based on AUC comparisons).
In pregnant rabbits given intravenous doses of 3, 6 or 12 mg/kg/day from gestation day 6 through 18 (organogenesis) there were no fetal findings; however, maternal toxicity was observed in all groups (systemic exposures less than the therapeutic dose).
In bolus intravenous postnatal rat studies, decreased neonatal viability, decreased weaning indices, delayed growth/physical development and delayed sexual maturation of offspring were observed at 2.5 mg/kg/day (systemic exposures less than the therapeutic dose).
Labor and Delivery
The effect of conivaptan on labor and delivery in humans has not been studied. Conivaptan hydrochloride delayed delivery in rats dosed orally at 10 mg/kg/day by oral gavage (systemic exposures equivalent to the therapeutic dose based on AUC comparisons). Administration of conivaptan hydrochloride at 2.5 mg/kg/day intravenously increased peripartum pup mortality (systemic exposures were less than the therapeutic dose based on AUC comparisons). These effects may be associated with conivaptan activity on oxytocin receptors in the rat. The relevance to humans is unclear.
Lactating Women
It is not known whether conivaptan is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when VAPRISOL is administered to a lactating woman. Conivaptan is excreted in milk and detected in neonates when given by intravenous administration to lactating rats. Milk levels of conivaptan in rats reached maximal levels at 1 hour post dose following intravenous administrations and were up to 3 times greater than maternal plasma levels. Administration of conivaptan hydrochloride at 2.5 mg/kg/day intravenously increased peripartum pup mortality; systemic exposures were less than the therapeutic dose based on AUC comparisons.
Geriatric Use
In clinical studies of intravenous VAPRISOL administered as a 20 mg IV loading dose followed by 20 mg/day or 40 mg/day IV for 2 to 4 days, 89% (20 mg/day regimen) and 60% (40 mg/day regimen) of participants were greater than or equal to 65 years of age and 60% (20 mg/day regimen) and 40% (40 mg/day regimen) were greater than or equal to 75 years of age. In general, the adverse event profile in elderly patients was similar to that seen in the general study population.
Adverse Reactions/Side Effects
The most common adverse reactions reported with VAPRISOL administration were infusion site reactions. In studies in patients and healthy volunteers, infusion site reactions occurred in 73% and 63% of subjects treated with VAPRISOL 20 mg/day and 40 mg/day, respectively, compared to 4% in the placebo group. Infusion site reactions were the most common type of adverse event leading to discontinuation of VAPRISOL. Discontinuations from treatment due to infusion site reactions were more common among VAPRISOL-treated patients (3%) than among placebo-treated patients (0%). Some serious infusion site reactions did occur. (See DOSAGE AND ADMINISTRATION)
The adverse reactions presented in Table 5 are derived from 72 healthy volunteers and 243 patients with euvolemic or hypervolemic hyponatremia who received VAPRISOL 20 mg IV as a loading dose followed by 40 mg/day IV for 2 to 4 days, from 37 patients with euvolemic or hypervolemic hyponatremia who received VAPRISOL 20 mg IV as a loading dose followed by 20 mg/day IV for 2 to 4 days in an open-label study, and from 40 healthy volunteers and 29 patients with euvolemic or hypervolemic hyponatremia who received placebo. The adverse reactions occurred in at least 5% of patients treated with VAPRISOL and at a higher incidence for VAPRISOL-treated patients than for placebo-treated patients.
| Adapted from MedDRA version 6.0 | |||
|
Term |
Placebo (N = 69) N (%) |
20 mg (N = 37) N (%) |
40 mg (N = 315) N (%) |
|
Blood and Lymphatic System Disorders |
|||
|
Anemia NOS |
2 (3%) |
2 (5%) |
18 (6%) |
|
Cardiac Disorders |
|||
|
Atrial Fibrillation |
0 (0%) |
2 (5%) |
7 (2%) |
|
Gastrointestinal Disorders |
|||
|
Constipation |
2 (3%) |
3 (8%) |
20 (6%) |
|
Diarrhea NOS |
0 (0%) |
0 (0%) |
23 (7%) |
|
Nausea |
3 (4%) |
1 (3%) |
17 (5%) |
|
Vomiting NOS |
0 (0%) |
2 (5%) |
23 (7%) |
|
General Disorders and Administration Site Conditions |
|||
|
Edema Peripheral |
1 (1%) |
1 (3%) |
24 (8%) |
|
Infusion Site Erythema |
0 (0%) |
0 (0%) |
18 (6%) |
|
Infusion Site Pain |
1 (1%) |
0 (0%) |
16 (5%) |
|
Infusion Site Phlebitis |
1 (1%) |
19 (51%) |
102 (32%) |
|
Infusion Site Reaction |
0 (0%) |
8 (22%) |
61 (19%) |
|
Pyrexia |
0 (0%) |
4 (11%) |
15 (5%) |
|
Thirst |
1 (1%) |
1 (3%) |
19 (6%) |
|
Infections and Infestations |
|||
|
Pneumonia NOS |
0 (0%) |
2 (5%) |
7 (2%) |
|
Urinary Tract Infection NOS |
2 (3%) |
2 (5%) |
14 (4%) |
|
Injury, Poisoning and Procedural Complications |
|||
|
Post Procedural Diarrhea |
0 (0%) |
2 (5%) |
0 (0%) |
|
Investigations |
|||
|
Electrocardiogram ST Segment Depression |
0 (0%) |
2 (5%) |
0 (0%) |
|
Metabolism and Nutrition Disorders |
|||
|
Hypokalemia |
2 (3%) |
8 (22%) |
30 (10%) |
|
Hypomagnesemia |
0 (0%) |
2 (5%) |
6 (2%) |
|
Hyponatremia |
1 (1%) |
3 (8%) |
20 (6%) |
|
Nervous System Disorders |
|||
|
Headache |
2 (3%) |
3 (8%) |
32 (10%) |
|
Psychiatric Disorders |
|||
|
Confusional State |
2 (3%) |
0 (0%) |
16 (5%) |
|
Insomnia |
0 (0%) |
2 (5%) |
12 (4%) |
|
Respiratory, Thoracic and Mediastinal Disorders |
|||
|
Pharyngolaryngeal Pain |
3 (4%) |
2 (5%) |
3 (1%) |
|
Skin and Subcutaneous Tissue Disorders |
|||
|
Pruritus |
0 (0%) |
2 (5%) |
2 (1%) |
|
Vascular Disorders |
|||
|
Hypertension NOS |
0 (0%) |
3 (8%) |
20 (6%) |
|
Hypotension NOS |
2 (3%) |
3 (8%) |
16 (5%) |
|
Orthostatic Hypotension |
0 (0%) |
5 (14%) |
18 (6%) |
Although a dose of 80 mg/day of intravenous VAPRISOL was also studied, it was associated with a higher incidence of infusion site reactions and a higher rate of discontinuation due to adverse events than was the 40 mg/day intravenous VAPRISOL dose. The maximum daily dose of VAPRISOL (after the loading dose) is 40 mg/day.
Related/similar drugs
Congestive Heart Failure
In clinical trials where intravenous VAPRISOL was administered to 79 hypervolemic hyponatremic patients with underlying heart failure and intravenous placebo administered to 10 patients, adverse cardiac failure events, atrial dysrhythmias, and sepsis occurred more frequently among patients treated with VAPRISOL (32%, 5% and 8% respectively) than among patients treated with placebo (20%, 0% and 0% respectively). The number of heart failure patients with hypervolemic hyponatremia who have been treated with intravenous VAPRISOL is too small to establish safety in this specific population. VAPRISOL should only be used in patients with underlying heart failure when the expected clinical benefit of raising serum sodium outweighs the risk of adverse events.
In ten Phase 2/pilot heart failure studies, VAPRISOL did not show statistically significant improvement for heart failure outcomes, including such measures as length of hospital stay, changes in categorized physical findings of heart failure, change in ejection fraction, change in exercise tolerance, change in functional status, or change in heart failure symptoms, as compared to placebo. In these studies, the changes in the physical findings and heart failure symptoms were no worse in the VAPRISOL-treated group (N = 818) compared to the placebo group (N = 290).
Drug Abuse and Dependence
VAPRISOL does not have known potential for psychogenic drug abuse and/or dependence.
Overdosage
Although no data on overdosage in humans are available, VAPRISOL has been administered as a 20 mg loading dose on Day 1 followed by continuous infusion of 80 mg/day for 4 days in hyponatremia patients and up to 120 mg/day for 2 days in CHF patients. No new toxicities were identified at these higher doses, but adverse events related to the pharmacologic activity of VAPRISOL, e.g. hypotension and thirst, occurred more frequently at these higher doses.
In case of overdose, based on expected exaggerated pharmacological activity, symptomatic treatment with frequent monitoring of vital signs and close observation of the patient is recommended.
Vaprisol Dosage and Administration
VAPRISOL is for intravenous use only.
VAPRISOL is for use in hospitalized patients only.
Administration of VAPRISOL through large veins and change of the infusion site every 24 hours are recommended to minimize the risk of vascular irritation.
VAPRISOL therapy should begin with a loading dose of 20 mg IV administered over 30 minutes.
The loading dose should be followed by 20 mg of VAPRISOL administered in a continuous intravenous infusion over 24 hours. Following the initial day of treatment, VAPRISOL is to be administered for an additional 1 to 3 days in a continuous infusion of 20 mg/day. If serum sodium is not rising at the desired rate, VAPRISOL may be titrated upward to a dose of 40 mg daily, again administered in a continuous intravenous infusion.
The total duration of infusion of VAPRISOL (after the loading dose) should not exceed four days. The maximum daily dose of VAPRISOL (after the loading dose) is 40 mg/day.
Patients receiving VAPRISOL must have frequent monitoring of serum sodium and volume status. An overly rapid rise in serum sodium (>12 mEq/L/24 hours) may result in serious neurologic sequelae. For patients who develop an undesirably rapid rate of rise of serum sodium, VAPRISOL should be discontinued, and serum sodium and neurologic status should be carefully monitored. If the serum sodium continues to rise, VAPRISOL should not be resumed. If hyponatremia persists or recurs, and the patient has had no evidence of neurologic sequelae of rapid rise in serum sodium, VAPRISOL may be resumed at a reduced dose. (See PRECAUTIONS: Overly Rapid Correction of Serum Sodium)
For patients who develop hypovolemia or hypotension while receiving VAPRISOL, VAPRISOL should be discontinued, and volume status and vital signs should be frequently monitored. Once the patient is again euvolemic and is no longer hypotensive, VAPRISOL may be resumed at a reduced dose if the patient remains hyponatremic.
Preparation
Compatibility and Stability
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If particulate matter, discoloration or cloudiness is observed, the drug solution should not be used.
VAPRISOL (conivaptan hydrochloride injection) Ampule
Caution: VAPRISOL ampule should be diluted only with 5% Dextrose Injection.
The VAPRISOL ampule is compatible with 5% Dextrose Injection and is stable for up to 24 hours after mixing. The VAPRISOL ampule should not be mixed or administered with Lactated Ringer's Injection or 0.9% Sodium Chloride Injection. Compatibility with other drugs has not been studied; therefore, VAPRISOL should not be combined with any other product in the same intravenous line or container.
The diluted solution of VAPRISOL should be used immediately and administration completed within 24 hours of mixing.
Loading Dose
Withdraw 4 mL (20 mg) of VAPRISOL (4 mL of conivaptan hydrochloride injection) and add to an infusion bag containing 100 mL of 5% Dextrose Injection, USP. Gently invert the bag several times to ensure complete mixing of the solution. The contents of the IV bag should be administered over 30 minutes.
Continuous Infusion
To prepare a continuous IV infusion containing 20 mg conivaptan hydrochloride, withdraw 4 mL (20 mg) from a single ampule of VAPRISOL and dilute into an IV bag containing 250 mL of 5% Dextrose Injection, USP. Gently invert the bag several times to ensure complete mixing of the solution. The contents of the IV bag should be administered over 24 hours.
To prepare a continuous IV infusion containing 40 mg conivaptan hydrochloride, withdraw 4 mL (20 mg) from each of two ampules of VAPRISOL (8 mL [40 mg] of conivaptan hydrochloride injection) and dilute into an IV bag containing 250 mL of 5% Dextrose Injection, USP. Gently invert the bag several times to ensure complete mixing of the solution. The contents of the IV bag should be administered over 24 hours.
The VAPRISOL ampule is for single use only. Discard unused contents of the ampule.
VAPRISOL (conivaptan hydrochloride injection) Premixed in 5% Dextrose
VAPRISOL is supplied in a single-use 100 mL flexible INTRAVIA container containing a sterile premixed dilute, ready-to-use, nonpyrogenic solution of conivaptan hydrochloride, 0.2 mg per mL (20 mg/100 mL) in 5% dextrose. NO FURTHER DILUTION OF THIS PREPARATION IS NECESSARY.
VAPRISOL is compatible with 5% Dextrose Injection. VAPRISOL should not be administered with Lactated Ringer's Injection. VAPRISOL should not be combined with any other product in the same intravenous line or container.
Continuous Infusion
For patients requiring 20 mg conivaptan hydrochloride injection per day, administer one 20 mg/100 mL VAPRISOL flexible plastic container over 24 hours.
For patients requiring 40 mg conivaptan hydrochloride injection per day, administer two consecutive 20 mg/100 mL VAPRISOL flexible plastic containers over 24 hours.
Since the flexible container is for single-use only, any unused portion should be discarded.
CAUTION: Do not use plastic containers in series connections. Such use could result in air embolism due to residual air being drawn from the primary container before administration of the fluid from the secondary container is completed.
Directions for VAPRISOL (conivaptan hydrochloride injection) Premixed in 5% Dextrose:
Do not remove container from overwrap until ready for use. The overwrap is a moisture and light barrier. The inner container maintains the sterility of the product.
Tear overwrap down side at slit and remove solution container. Some opacity of the plastic due to moisture absorption during the sterilization process may be observed. This is normal and does not affect the solution quality or safety. The opacity will diminish gradually. After removing overwrap, check for minute leaks by squeezing inner container firmly. If leaks are found, discard solution as sterility may be impaired. Do not use if the solution is cloudy or a precipitate is present.
DO NOT ADD SUPPLEMENTARY MEDICATION.
Preparation for Administration:
- 1.
- Suspend container from eyelet support.
- 2.
- Remove protector from outlet port at bottom of container.
- 3.
- Attach administration set. Refer to complete directions accompanying set.
Storage and Handling
VAPRISOL (conivaptan hydrochloride injection) Ampule
Store at 25°C (77° F); excursions permitted to 15-30°C (59-86° F), controlled room temperature (in accordance with USP). Do not store below 15°C (59° F). Ampules should be stored in their cardboard container protected from light until ready for use.
VAPRISOL (conivaptan hydrochloride injection) Premixed in 5% Dextrose
VAPRISOL in INTRAVIA Plastic Containers should be stored at 25°C (77° F); however, brief exposure up to 40°C (104° F) does not adversely affect the product. Avoid excessive heat. Protect from freezing. Protect from light until ready to use.
How is Vaprisol supplied
VAPRISOL (conivaptan hydrochloride injection) ampule is supplied in 4 mL clear glass, one-point cut ampules. Each ampule contains 20 mg conivaptan hydrochloride.
10 ampules/carton (NDC 0469-1601-04)
VAPRISOL (conivaptan hydrochloride injection) in 100 mL INTRAVIA Plastic Containers is supplied as a single-use, premixed solution, containing 20 mg of conivaptan hydrochloride in 5% Dextrose.
10 containers/carton (NDC 0469-1602-11)
Rx Only
VAPRISOL is a registered trademark of Astellas Pharma US, Inc.
INTRAVIA is a registered trademark of Baxter International, Inc.
Marketed by:
Astellas Pharma US, Inc.
Deerfield, IL 60015-2548
VAPRISOL Ampule Manufactured by:
Astellas Tokai Co., Ltd.
Yaizu Plant
Shizuoka 425-0072, Japan
VAPRISOL in INTRAVIA Plastic Container Manufactured by:
Baxter Healthcare Corporation
Deerfield, IL 60015
Revision Date: October 2008
07-19-53-243




| VAPRISOL
conivaptan hydrochloride solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| VAPRISOL
conivaptan hydrochloride liquid |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Astellas Pharma US, Inc. (605764828) |
More about Vaprisol (conivaptan)
- Check interactions
- Compare alternatives
- Reviews (1)
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: vasopressin antagonists
- En español