Rydapt: Package Insert / Prescribing Info
Package insert / product label
Generic name: midostaurin
Dosage form: capsule, liquid filled
Drug class: Multikinase inhibitors
Medically reviewed by Drugs.com. Last updated on Jun 23, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
RYDAPT® (midostaurin) capsules, for oral use
Initial U.S. Approval: 2017
Indications and Usage for Rydapt
RYDAPT is a kinase inhibitor indicated for the treatment of adult patients with:
- Newly diagnosed acute myeloid leukemia (AML) that is FLT3 mutation-positive as detected by an FDA-approved test, in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation. (1.1)
Limitations of Use: RYDAPT is not indicated as a single-agent induction therapy for the treatment of patients with AML. - Aggressive systemic mastocytosis (ASM), systemic mastocytosis with associated hematological neoplasm (SM-AHN), or mast cell leukemia (MCL). (1.2)
Rydapt Dosage and Administration
Dosage Forms and Strengths
Capsules: 25 mg (3)
Contraindications
Hypersensitivity to midostaurin or any of the excipients. (4)
Warnings and Precautions
- Embryo-Fetal Toxicity: RYDAPT may cause fetal harm when administered to a pregnant woman. Advise of the potential risk to a fetus. (5.1, 8.1)
- Pulmonary Toxicity: Monitor for symptoms of interstitial lung disease or pneumonitis. Discontinue RYDAPT in patients with signs or symptoms of pulmonary toxicity. Fatal cases have occurred. (5.2)
Adverse Reactions/Side Effects
- AML: The most common adverse reactions (≥ 20%) were febrile neutropenia, nausea, mucositis, vomiting, headache, petechiae, musculoskeletal pain, epistaxis, device-related infection, hyperglycemia, electrocardiogram (ECG) QT prolonged, and upper respiratory tract infection. (6.1)
- ASM, SM-AHN, or MCL: The most common adverse reactions (≥ 20%) were nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, constipation, pyrexia, headache, and dyspnea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 and/or at https://report.novartis.com/ or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Strong CYP3A4 Inhibitors: Strong CYP3A4 inhibitors may increase exposure to midostaurin and its active metabolites. Consider alternative therapies that do not strongly inhibit CYP3A4 or monitor for increased risk of adverse reactions. (7.1)
- Strong CYP3A4 Inducers: Avoid concomitant use as strong CYP3A4 inducers decrease exposure to midostaurin and its active metabolites. (7.1)
- CYP2B6, BCRP, OATP1B1 Substrates: Dose adjustments for coadministered CYP2B6, BCRP, and OATP1B1 substrates may be necessary with RYDAPT. (7.2)
Use In Specific Populations
Lactation: Advise females not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2023
Full Prescribing Information
1. Indications and Usage for Rydapt
1.1 Acute Myeloid Leukemia
RYDAPT is indicated in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation chemotherapy, for the treatment of adult patients with newly diagnosed acute myeloid leukemia (AML) who are FLT3 mutation-positive, as detected by an FDA approved test [see Dosage and Administration (2.1), Clinical Studies (14.1)].
Limitations of Use
RYDAPT is not indicated as a single-agent induction therapy for the treatment of patients with AML.
2. Rydapt Dosage and Administration
2.1 Patient Selection
Select patients for the treatment of AML with RYDAPT based on the presence of FLT3 mutation positivity [see Clinical Studies (14)]. Information on FDA-approved tests for the detection of FLT3 mutation in AML is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage in Acute Myeloid Leukemia
The recommended dose of RYDAPT for patients with AML is 50 mg orally twice daily with food on Days 8 to 21 of each cycle of induction with cytarabine and daunorubicin and on Days 8 to 21 of each cycle of consolidation with high-dose cytarabine. For a description of the experience with single-agent treatment with RYDAPT beyond induction and consolidation [see Clinical Studies (14.1)].
2.3 Recommended Dosage in ASM, SM-AHN, and MCL
The recommended dose of RYDAPT for patients with ASM, SM-AHN, and MCL is 100 mg orally twice daily with food. Continue treatment until disease progression or unacceptable toxicity occurs. Table 1 provides recommendations for dose modifications of RYDAPT in patients with ASM, SM-AHN, and MCL. Monitor patients for toxicity at least weekly for the first 4 weeks, every other week for the next 8 weeks, and monthly thereafter while on treatment.
| Criteria | RYDAPT Dosing |
|---|---|
| ANC less than 1 x 109/L attributed to RYDAPT in patients without MCL, or ANC less than 0.5 x 109/L attributed to RYDAPT in patients with baseline ANC value of 0.5-1.5 x 109/L | Interrupt RYDAPT until ANC greater than or equal to 1 x 109/L, then resume RYDAPT at 50 mg twice daily, and if tolerated, increase to 100 mg twice daily. Discontinue RYDAPT if low ANC persists for greater than 21 days and is suspected to be related to RYDAPT. |
| Platelet count less than 50 x 109/L attributed to RYDAPT in patients without MCL, or platelet count less than 25 x 109/L attributed to RYDAPT in patients with baseline platelet count of 25-75 x 109/L | Interrupt RYDAPT until platelet count greater than or equal to 50 x 109/L, then resume RYDAPT at 50 mg twice daily, and if tolerated, increase to 100 mg twice daily. Discontinue if low platelet count persists for greater than 21 days and is suspected to be related to RYDAPT. |
| Hemoglobin less than 8 g/dL attributed to RYDAPT in patients without MCL, or life-threatening anemia attributed to RYDAPT in patients with baseline hemoglobin value of 8 to 10 g/dL | Interrupt RYDAPT until hemoglobin greater than or equal to 8 g/dL, then resume RYDAPT at 50 mg twice daily, and if tolerated, increase to 100 mg twice daily. Discontinue if low hemoglobin persists for greater than 21 days and is suspected to be related to RYDAPT. |
| Grade 3/4 nausea and/or vomiting despite optimal anti-emetic therapy | Interrupt RYDAPT for 3 days (6 doses), then resume RYDAPT at 50 mg twice daily, and if tolerated, increase to 100 mg twice daily. |
| Other Grade 3/4 non-hematological toxicities | Interrupt RYDAPT until event has resolved to less than or equal to Grade 2, then resume RYDAPT at 50 mg twice daily, and if tolerated, increase to 100 mg twice daily. |
| Abbreviations: ANC, absolute neutrophil count; MCL, mast cell leukemia. National Cancer Institute Common Terminology for Adverse Events (NCI CTCAE) severity: Grade 1 = mild symptoms; 2 = moderate symptoms; 3 = severe symptoms; 4 = life-threatening symptoms. |
|
2.4 Recommended Administration
- Administer prophylactic anti-emetics before treatment with RYDAPT to reduce the risk of nausea and vomiting.
- Administer RYDAPT orally with food, twice daily at approximately 12-hour intervals [see Clinical Pharmacology (12.3)]. Do not open or crush RYDAPT capsules.
- If a dose of RYDAPT is missed or vomited, do not make up the dose; take the next dose at the usual scheduled time.
- Consider interval assessments of QT by electrocardiogram (ECG) if RYDAPT is taken concurrently with medications that can prolong the QT interval.
3. Dosage Forms and Strengths
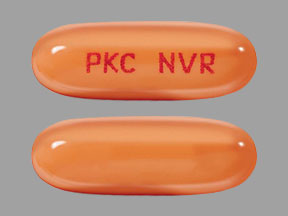
25 mg capsules: pale orange oblong soft capsule with red ink imprint ‘PKC NVR’.
4. Contraindications
RYDAPT is contraindicated in patients with hypersensitivity to midostaurin or to any of the excipients [see Description (11)]. Hypersensitivity reactions have included anaphylactic shock, dyspnea, flushing, chest pain, and angioedema (e.g., swelling of the airways or tongue, with or without respiratory impairment) [see Adverse Reactions (6.1)].
5. Warnings and Precautions
5.1 Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal reproduction studies, RYDAPT may cause fetal harm when administered to pregnant women. In animal studies, midostaurin caused embryo-fetal toxicities, including late embryo-fetal death and reduced fetal birth weight, with delays in fetal growth at doses lower than the recommended human dose. Advise pregnant women of the potential risk to the fetus. Verify the pregnancy status of females of reproductive potential within 7 days prior to initiating RYDAPT therapy. Advise females of reproductive potential to use effective contraception during treatment with RYDAPT and for 4 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with RYDAPT and for 4 months after the last dose [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
5.2 Pulmonary Toxicity
Cases of interstitial lung disease and pneumonitis, some fatal, have occurred in patients treated with RYDAPT as monotherapy or with chemotherapy.
Monitor patients for pulmonary symptoms. Discontinue RYDAPT in patients who experience signs or symptoms of interstitial lung disease or pneumonitis without an infectious etiology.
5.3 Risk of Prolonged Severe Neutropenia and Thrombocytopenia in Pediatric Patients Treated With Combination Chemotherapy
Prolonged Grade 4 neutropenia and thrombocytopenia occurred in two pediatric patients with AML who received an unapproved formulation of midostaurin in combination with chemotherapy, including anthracyclines, fludarabine, and cytarabine; these two patients were coadministered an azole antifungal (a strong CYP3A4 inhibitor), which may increase midostaurin concentrations and subsequently, the risk of toxicity [see Drug Interactions (7.1), Use in Specific Populations (8.4)]. The safety and effectiveness of RYDAPT in pediatric patients have not been established.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described elsewhere in the labeling:
- Pulmonary Toxicity [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Acute Myeloid Leukemia
The safety evaluation of RYDAPT (50 mg twice daily with food) in patients with newly diagnosed FLT3 mutated AML is based on a randomized, double-blind, trial of RYDAPT (n = 345) or placebo (n = 335) with chemotherapy [see Clinical Studies (14.1)]. The overall median duration of exposure was 42 days (range, 2 to 576 days) for patients in the RYDAPT plus chemotherapy arm versus 34 days (range, 1 to 465 days) for patients in the placebo plus chemotherapy arm. On the RYDAPT plus chemotherapy arm, 35% of patients completed induction and consolidation therapy, compared to 25% of patients on the placebo plus chemotherapy arm.
The most frequent (incidence greater than or equal to 20%) adverse drug reactions (ADRs) in the RYDAPT plus chemotherapy arm were febrile neutropenia, nausea, mucositis, vomiting, headache, petechiae, musculoskeletal pain, epistaxis, device-related infection, hyperglycemia, ECG QT prolonged, and upper respiratory tract infections. The most frequent Grade 3/4 adverse reactions (incidence ≥ 10%) were febrile neutropenia, device-related infection, and mucositis.
The most frequent serious adverse reaction (≥ 10%) in patients in the RYDAPT plus chemotherapy arm was febrile neutropenia (16%), which occurred at a similar rate in the placebo arm (16%).
Discontinuation due to any adverse reaction occurred in 9% of patients in the RYDAPT arm versus 6% in the placebo arm. The most frequent (> 1%) Grade 3/4 adverse reactions leading to discontinuation in the RYDAPT arm was renal insufficiency (1%).
Excluding deaths due to disease progression, no fatal adverse reactions occurred in the study. Overall, the most frequent non-treatment related cause of death in the RYDAPT plus chemotherapy arm was sepsis (2%) and occurred at a similar rate in the placebo arm (2%).
Table 2 presents the frequency category of adverse reactions reported in the randomized trial in patients with newly diagnosed FLT3 mutated AML. Adverse reactions are listed according to body system. Within each body system, the adverse reactions are ranked by frequency, with the most frequent reactions first. Table 3 presents the key laboratory abnormalities from the same randomized trial in patients with newly diagnosed FLT3 mutated AML.
| All Grades
| Grades ≥ 3 | |||
| Adverse Reaction | RYDAPT + chemo n = 2291 % | Placebo + chemo n = 2261 % | RYDAPT + chemo n = 3451 % | Placebo + chemo n = 3351 % |
| Gastrointestinal disorders | ||||
| Nausea | 83 | 70 | 6 | 10 |
| Mucositisa | 66 | 62 | 11 | 13 |
| Vomiting | 61 | 53 | 3 | 5 |
| Hemorrhoids | 15 | 11 | 1 | 0 |
| Blood and lymphatic system disorders | ||||
| Febrile neutropenia | 83 | 81 | 84 | 83 |
| Petechiae | 36 | 27 | 1 | 1 |
| Nervous system disorders | ||||
| Headachea | 46 | 38 | 3 | 3 |
| Musculoskeletal and connective tissue disorders | ||||
| Musculoskeletal paina | 33 | 31 | 5 | 2 |
| Arthralgia | 14 | 8 | < 1 | < 1 |
| Respiratory, thoracic, and mediastinal disorders | ||||
| Epistaxis | 28 | 24 | 3 | 1 |
| Infections and infestations | ||||
| Device-related infection | 24 | 17 | 16 | 10 |
| Upper respiratory tract infectiona | 20 | 15 | 4 | 3 |
| Investigations | ||||
| Hyperglycemiaa | 20 | 17 | 7 | 6 |
| Electrocardiogram QT prolonged | 20 | 17 | 6 | 5 |
| Activated partial thromboplastin time prolonged | 13 | 8 | 3 | 2 |
| Skin and subcutaneous tissue disorders | ||||
| Hyperhidrosis | 14 | 8 | 0 | 0 |
| Renal and urinary disorders | ||||
| Renal insufficiencya | 12 | 9 | 5 | 3 |
| Psychiatric disorders | ||||
| Insomnia | 12 | 8 | 0 | < 1 |
| 1For trial sites in North America, only Grades 3 and 4 were collected. aGrouped terms: • Upper respiratory tract infections: e.g., nasopharyngitis, upper respiratory tract infections, sinusitis. • Mucositis: e.g., radiation mucositis, stomatitis, laryngeal pain. • Musculoskeletal pain: e.g., back pain, bone pain, pain in extremity. • Renal insufficiency: e.g., blood creatinine increased, renal failure, acute kidney injury. • Hyperglycemia: mainly hyperglycemia. |
||||
Other notable adverse reactions occurring in < 10% of patients treated with RYDAPT but at least 2% more frequently than in the placebo group included:
- Infections and infestations: Cellulitisa (7%), fungal infectiona (7%)
- Metabolism and nutrition disorders: Hyperuricemia (8%)
- Nervous system disorders: Tremor (4%)
- Eye disorders: Eyelid edema (3%)
- Cardiac disorders: Hypertensiona (8%), pericardial effusion (4%)
- Respiratory, thoracic, and mediastinal disorders: Pleural effusion (6%)
- Skin and subcutaneous tissue disorders: Dry skin (7%)
- General disorders and administration-site conditions: Thrombosisa (5%)
- Investigations: Weight increased (7%), hypercalcemia (3%)
aGrouped terms:
- Thrombosis: e.g., thrombosis in device, thrombosis.
- Cellulitis: e.g., cellulitis, erysipelas.
- Fungal infection: e.g., bronchopulmonary aspergillosis, pneumonia fungal, splenic infection fungal, hepatic candidiasis.
Other clinically important adverse reactions (All Grades) at ≥ 10% that did not meet criteria for Table 2:
- Respiratory, thoracic, and mediastinal disorders: Pneumonitis (11%)
| Laboratory Abnormality | RYDAPT (50 mg twice daily) N = 345 All Grades % | RYDAPT (50 mg twice daily) N = 345 Grade 3/4 % | Placebo N = 335 All Grades % | Placebo N = 335 Grade 3/4 % |
|---|---|---|---|---|
| Alanine aminotransferase increased | 71 | 20 | 69 | 16 |
| Hypernatremia | 21 | 1 | 15 | 2 |
| Hypocalcemia | 74 | 7 | 70 | 8 |
In Study 1, 205 patients (120 in RYDAPT arm and 85 in placebo arm) who remained in remission following completion of consolidation continued to receive either single agent RYDAPT or placebo for a median of 11 months (range, 0.5 to 17 months) with 69 in the RYDAPT arm and 51 in the placebo completing 12 treatment cycles. Common adverse reactions (greater than or equal to 5% difference between the RYDAPT and placebo arms) reported for these patients included nausea (47% vs 18%), hyperglycemia (20% vs 13%), and vomiting (19% vs 5%).
Systemic Mastocytosis
Two single-arm, open-label multicenter trials (Study 2 and Study 3) evaluated the safety of RYDAPT (100 mg twice daily with food) as a single agent in 142 adult patients total with ASM, SM-AHN, or MCL. The median age was 63 (range, 24 to 82), 63% had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and 75% had no hepatic impairment (bilirubin and AST ≤ upper limit of normal (ULN)] at baseline. The median duration of exposure to RYDAPT was 11.4 months (range, 0 to 81 months), with 34% treated for ≥ 24 months.
The most frequent adverse reactions (≥ 20%), excluding laboratory terms, were nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, constipation, pyrexia, headache, and dyspnea (Table 4). Grade ≥ 3 adverse reactions reported in ≥ 5%, excluding laboratory terms, were fatigue, sepsis, gastrointestinal hemorrhage, pneumonia, diarrhea, febrile neutropenia, edema, dyspnea, nausea, vomiting, abdominal pain, and renal insufficiency (Table 4).
Adverse reactions led to dose modifications (interruption or reduction) in 56% of patients. Among these, the most frequent adverse reactions (> 5%) were gastrointestinal symptoms, QT prolongation, neutropenia, pyrexia, thrombocytopenia, gastrointestinal hemorrhage, lipase increase, and fatigue. The median time to first dose modification for toxicity was 1.6 months, with 75% of dose modifications first occurring within 5 months of starting treatment.
Treatment discontinuation due to adverse reactions occurred in 21% of patients. The most frequent adverse reactions causing treatment discontinuation included infection, nausea or vomiting, QT prolongation, and gastrointestinal hemorrhage.
Serious adverse reactions were reported in 68% of patients, most commonly (≥ 20%) due to infections and gastrointestinal disorders.
On-treatment deaths unrelated to the underlying malignancy occurred in 16 patients (11%), most commonly from infection (sepsis or pneumonia), followed by cardiac events. Of the on-treatment deaths from disease progression, 4 were also attributable to infection.
Table 4 summarizes the adverse reactions reported in ≥ 10% of the patients with advanced SM.
| RYDAPT (100 mg twice daily)
N = 142 |
||
|---|---|---|
| Adverse Reactiona | All Grades % | Grade ≥ 3 % |
| Gastrointestinal disorders | ||
| Nausea | 82 | 6 |
| Vomiting | 68 | 6 |
| Diarrheaa | 54 | 8 |
| Abdominal paina | 34 | 6 |
| Constipation | 29 | < 1 |
| Gastrointestinal hemorrhagea | 14 | 9 |
| General disorders and administration-site conditions | ||
| Edemaa | 40 | 7 |
| Fatiguea | 34 | 9 |
| Pyrexia | 27 | 4 |
| Infections and infestations | ||
| Upper respiratory tract infectiona | 30 | 1 |
| Urinary tract infectiona | 16 | 3 |
| Pneumoniaa | 10 | 8 |
| Herpesvirus infectiona | 10 | 1 |
| Musculoskeletal and connective tissue disorders | ||
| Musculoskeletal paina | 35 | 4 |
| Arthralgia | 19 | 2 |
| Nervous system disorders | ||
| Headachea | 26 | 1 |
| Dizziness | 13 | 0 |
| Respiratory, thoracic, and mediastinal disorders | ||
| Dyspneaa | 23 | 7 |
| Cougha | 18 | < 1 |
| Pleural effusion | 13 | 4 |
| Epistaxis | 12 | 3 |
| Skin and subcutaneous disorders | ||
| Rasha | 14 | 3 |
| Investigations | ||
| QT prolonged | 11 | < 1 |
| Psychiatric disorders | ||
| Insomnia | 11 | 0 |
| Renal disorders | ||
| Renal insufficiencya | 11 | 5 |
| Toxicity was graded per National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 3. Represents adverse reactions, excluding laboratory terms, occurring up to 28 days after last midostaurin dose, regardless of baseline grade. aGrouped terms:
|
||
Gastrointestinal Toxicities Leading to Treatment Modification: In patients with advanced SM, the median time to onset of nausea was 9 days, with 75% of cases beginning within the first 3 months. The median time to onset of vomiting was 1 month.
Other clinically significant adverse reactions occurring in ≤ 10% of patients included:
Infections and infestations: Sepsis (9%)a, bronchitis (6%), cellulitis or erysipelas (5%)
Blood and lymphatic system disorders: Febrile neutropenia (8%)
Cardiac disorders: Cardiac failure (6%), myocardial infarction, or ischemia (4%)a
Immune system disorders: Hypersensitivity (4%)a
Nervous system disorders: Disturbance in attention (7%), tremor (6%), mental status changes (4%)
Ear and labyrinth disorders: Vertigo (5%)
Vascular disorders: Hypotension (9%), hematoma (6%)
Respiratory, thoracic, and mediastinal disorders: Oropharyngeal pain (4%), pulmonary edema (3%)a, interstitial lung disease (1%), pneumonitis (<1%)
Gastrointestinal disorders: Dyspepsia (6%), gastritis (3%)a
General disorders and administration site conditions: Chills (5%)
Investigations: Weight increased (6%)
Injury, poisoning, and procedural complications: Contusion (6%)
aGrouped terms:
- Sepsis: e.g., sepsis, staphylococcal/Enterobacter/Escherichia sepsis
- Hypersensitivity: includes one report of anaphylactic shock
- Myocardial infarction or ischemia: e.g., myocardial infarction and acute myocardial infarction, angina pectoris
- Gastritis: gastritis, gastritis erosive, gastritis hemorrhagic
- Pulmonary edema: pulmonary edema, pulmonary congestion
Table 5 summarizes new or worsening laboratory abnormalities. Common (≥ 10%) Grade 3 or higher non-hematologic laboratory abnormalities were hyperglycemia (non-fasting), lipase increase, and hyperuricemia. The most common (≥ 20%) Grade 3 or higher hematologic laboratory abnormalities were lymphopenia, anemia, thrombocytopenia, and neutropenia. Grade 4 hematologic abnormalities occurring in ≥ 5% were thrombocytopenia (13%), neutropenia (8%), anemia (6%), and lymphopenia (6%).
| RYDAPT (100 mg twice daily)
N = 142 |
||
|---|---|---|
| Abbreviations: Alk phos, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma glutamyltransferase. Includes abnormalities occurring up to 28 days after last midostaurin dose, if new or worsened from baseline or if baseline was unknown. aNon-fasting. bAmong 116 evaluable patients. |
||
| Test | All Grades % | Grade ≥ 3 % |
| Hematology | ||
| Lymphopenia | 66 | 42 |
| Leukopenia | 61 | 19 |
| Anemia | 60 | 38 |
| Thrombocytopenia | 50 | 27 |
| Neutropenia | 49 | 22 |
| Chemistry | ||
| Hyperglycemiaa | 80 | 18 |
| Alk phos increase | 39 | 9 |
| Hypocalcemia | 39 | 2 |
| Lipase increase | 37 | 18 |
| Hyperuricemia | 37 | 11 |
| GGT increaseb | 35 | 9 |
| Hyponatremia | 34 | 5 |
| AST increase | 32 | 3 |
| ALT increase | 31 | 4 |
| Hyperbilirubinemia | 29 | 4 |
| Hypoalbuminemia | 27 | 1 |
| Hypokalemia | 25 | 6 |
| Creatinine increase | 25 | < 1 |
| Hyperkalemia | 23 | 4 |
| Hypophosphatemia | 22 | 1 |
| Amylase increase | 20 | 7 |
| Hypomagnesemia | 20 | 0 |
6.2 Postmarketing Experience
The following adverse drug reactions have been derived from postmarketing experience with RYDAPT via spontaneous case reports and literature cases. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Respiratory, thoracic, and mediastinal disorders: Interstitial lung disease
- Skin and subcutaneous tissue disorders: Acute febrile neutrophilic dermatosis (Sweet syndrome)
Related/similar drugs
7. Drug Interactions
7.1 Effect of Other Drugs on RYDAPT
Table 6 lists the potential effects of the coadministration of strong CYP3A modulators on RYDAPT.
| Strong CYP3A Inhibitors | |
|---|---|
| aThe effect of grapefruit juice varies widely among brands and is concentration-, dose-, and preparation-dependent. Studies have shown that it can be classified as a “strong CYP3A inhibitor” when a certain preparation was used (e.g., high dose, double strength) or as a “moderate CYP3A inhibitor” when another preparation was used (e.g., low dose, single strength). bThe induction potency of St. John’s wort may vary widely based on preparation. |
|
| Clinical Impact |
|
| Prevention or Management |
|
| Examples | Boceprevir, clarithromycin, cobicistat, conivaptan, danoprevir and ritonavir, diltiazem, elvitegravir and ritonavir, grapefruit juicea, idelalisib, indinavir and ritonavir, itraconazole, ketoconazole, lopinavir and ritonavir, nefazodone, nelfinavir, paritaprevir and ritonavir and (ombitasvir and/or dasabuvir), posaconazole, ritonavir, saquinavir and ritonavir, tipranavir and ritonavir, troleandomycin, voriconazole |
| Strong CYP3A Inducers | |
| Clinical Impact |
|
| Prevention or Management | Avoid coadministration of RYDAPT with strong CYP3A4 inducers. |
| Examples | Carbamazepine, enzalutamide, mitotane, phenytoin, rifampin, St. John’s wortb |
7.2 Effect of RYDAPT on Other Drugs
CYP2B6 Substrates
RYDAPT decreased the systemic exposure of a sensitive CYP2B6 substrate [see Clinical Pharmacology (12.3)]. Dose adjustments for the coadministered CYP2B6 substrate may be necessary with RYDAPT.
Substrates of Transporters
Coadministration of RYDAPT increased the exposure of a breast cancer resistance protein (BCRP) and organic anion transporter polypeptide (OATP)1B1 substrate [see Clinical Pharmacology (12.3)]. Dose adjustments for the coadministered BCRP or OATP1B1 substrates may be necessary with RYDAPT.
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to RYDAPT during pregnancy. Females who may have been exposed to RYDAPT during pregnancy directly or through a male partner receiving RYDAPT therapy should contact the Novartis Pharmaceuticals Corporation at 1-888-669-6682 and/or at https://report.novartis.com/.
Risk Summary
Based on mechanism of action and findings in animal reproduction studies, RYDAPT may cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on RYDAPT use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. In animal reproduction studies, oral administration of midostaurin to pregnant rats and rabbits during organogenesis caused embryo-fetal toxicities, including late embryo-fetal death and reduced fetal birth weight, with delays in fetal growth at doses lower than the recommended human dose (see Data). Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population are unknown. Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
When midostaurin was administered to female rats prior to mating and through the first week of pregnancy at a dose of 60 mg/kg/day [approximately 0.1 times the human exposure at the recommended dose based on area under the curve (AUC)], there were increases in pre- and post-implantation loss, including total litter loss, resulting in a reduction in the number of live embryos.
During organogenesis, midostaurin administered at oral doses greater than or equal to 3 mg/kg/day (approximately 0.004 times the human exposure at the recommended dose by AUC) to pregnant female rats caused late embryo-fetal death. Dilated lateral brain ventricles were observed in offspring of rats given doses greater than or equal to 3 mg/kg/day. Extra rib and reduced fetal birth weight with effects on fetal growth (severe renal pelvic cavitation and widened anterior fontanelle) were observed in the absence of maternal toxicity at the highest dose of 30 mg/kg/day (approximately 0.05 times the human exposure at the recommended dose by AUC). Midostaurin administered orally to pregnant rabbits during organogenesis led to maternal toxicity with spontaneous abortions and some delay in fetal growth (reduced fetal birth weight) at doses greater than or equal to 10 mg/kg/day (approximately 0.01 times the human exposure at the recommended dose by AUC).
In an oral pre- and postnatal development study in the rat, adverse effects upon maternal performance included dams with signs of dystocia and a lower live litter size at 30 mg/kg/day (approximately 0.05 times the human exposure at the recommended dose by AUC). For the F1 offspring, lower body weights, accelerated complete eye opening and delayed auricular startle ontogeny were noted at 30 mg/kg/day.
8.2 Lactation
Risk Summary
There are no data on the presence of midostaurin or its active metabolites in human milk, the effect on the breastfed infant, or the effect on milk production. Orally administered midostaurin and its active metabolites pass into the milk of lactating rats within 1 hour of a 30 mg/kg/day dose, with approximately 5 times more in the milk of lactating rats compared to plasma. Because of the potential for serious adverse reactions in breastfed infants from RYDAPT, advise women not to breastfeed during treatment with RYDAPT and for 4 months after the last dose.
8.3 Females and Males of Reproductive Potential
RYDAPT may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential within seven days prior to initiating RYDAPT.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with RYDAPT and for 4 months after the last dose.
Males
Advise males with female partners of reproductive potential to use effective contraception during RYDAPT treatment and for 4 months after stopping treatment with RYDAPT [see Nonclinical Toxicology (13.1)].
Infertility
Based on findings in animals, RYDAPT may impair fertility in females and males of reproductive potential. It is not known whether these effects on fertility are reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of RYDAPT have not been established in pediatric patients.
The safety and effectiveness of an unapproved midostaurin formulation was investigated, but not established in two open-label studies: a study in 22 pediatric patients aged 6 months to 17 years who received midostaurin as a single agent for relapsed or refractory leukemia [NCT00866281] and a study in 4 patients aged 8 to 14 years who received midostaurin in combination with chemotherapy for newly diagnosed FLT3-mutated AML [NCT03591510]. Prolonged Grade 4 neutropenia and thrombocytopenia occurred in 2 of the 4 pediatric patients with AML who received midostaurin in combination with chemotherapy, including anthracyclines, fludarabine, and cytarabine [see Warnings and Precautions (5.3)]. Grade 4 thrombocytopenia lasted for 44 days in one patient and Grade 4 thrombocytopenia and Grade 4 neutropenia lasted for 51 days and 46 days, respectively, in the other patient. These two patients were coadministered an azole antifungal (a strong CYP3A4 inhibitor), which may increase midostaurin concentrations and subsequently, the risk of toxicity [see Drug Interactions (7.1)].
8.5 Geriatric Use
Of the 142 patients with advanced SM in clinical studies of RYDAPT, 64 (45%) were aged 65 and over, and 16 (11%) were aged 75 years and over. No overall differences in safety or response rate were observed between the subjects aged 65 and over compared with younger subjects. Greater sensitivity of older individuals cannot be ruled out.
Clinical studies in AML with RYDAPT did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
In general, administration for elderly patients should be cautious, based on patient’s eligibility for concomitant chemotherapy and reflecting the greater frequency of concomitant disease or other drug therapy.
11. Rydapt Description
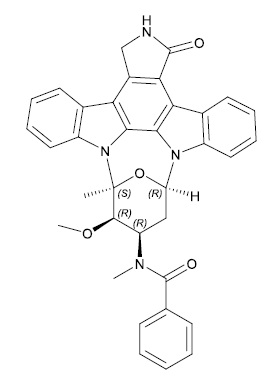
Midostaurin is a kinase inhibitor for oral use. The molecular formula for midostaurin is C35H30N4O4. The molecular weight is 570.65 g/mol. The chemical name of midostaurin is Benzamide, N-[(9S,10R,11R,13R)-2,3,10,11,12,13-hexahydro-10-methoxy-9-methyl-1-oxo-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiazonin-11-yl]-N-methyl-. The chemical structure of midostaurin is shown below:
RYDAPT is supplied as a soft capsule containing 25 mg of midostaurin. The capsule contains carmine, corn oil mono-di-triglycerides, dehydrated alcohol, ferric oxide red, ferric oxide yellow, gelatin, glycerin 85%, hypromellose 2910, polyethylene glycol 400, polyoxyl 40 hydrogenated castor oil, propylene glycol, purified water, titanium dioxide, and vitamin E.
12. Rydapt - Clinical Pharmacology
12.1 Mechanism of Action
Midostaurin is a small molecule that inhibits multiple receptor tyrosine kinases. In vitro biochemical or cellular assays have shown that midostaurin or its major human active metabolites CGP62221 and CGP52421 inhibit the activity of wild type FLT3, FLT3 mutant kinases (ITD and TKD), KIT (wild type and D816V mutant), PDGFRα/β, as well as members of the serine/threonine kinase PKC (protein kinase C) family.
Midostaurin demonstrated the ability to inhibit FLT3 receptor signaling and cell proliferation, and it induced apoptosis in leukemic cells expressing ITD and TKD mutant FLT3 receptors or overexpressing wild type FLT3 and PDGF receptors. Midostaurin also demonstrated the ability to inhibit KIT signaling, cell proliferation and histamine release and induce apoptosis in mast cells.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of RYDAPT (75 mg twice daily for 3 days) on the QTc interval was evaluated in a randomized, placebo and moxifloxacin controlled, multiple-dose, blinded, parallel study. There was no clinically significant prolongation of QTc interval or relationship between changes in QTc and concentrations for midostaurin and its active metabolites (CGP62221 and CGP52421). The study duration was not long enough to estimate the effects of the metabolite CGP52421 on the QT/QTc interval.
In pooled clinical studies in patients with advanced SM, 4.7% patients had a post-baseline QTcF > 480 ms, no patients had a QTcF > 500 ms, and 6.3% patients had a QTcF > 60 ms compared to baseline.
In a randomized placebo-controlled study in patients with AML, the proportion of patients with QTc prolongation was higher in patients randomized to midostaurin as compared to placebo (QTcF > 480 ms: 10.1% vs 5.7%; QTcF > 500 ms: 6.2% vs 2.6%; QTcF > 60 ms change from baseline: 18.4% vs 10.7%).
12.3 Pharmacokinetics
Midostaurin exhibits time-dependent pharmacokinetics with an initial increase in minimum concentrations (Cmin) that reach the highest Cmin concentrations during the first week followed by a decline to a steady-state after approximately 28 days. The pharmacokinetics of the CGP62221 showed a similar trend. The plasma concentrations of CGP52421 continued to increase after one month of treatment.
The highest Cmin and steady-state of midostaurin, CGP62221, and CGP52421 were similar when RYDAPT was administered with food at a dose of 50 mg twice daily or 100 mg twice daily.
Absorption
The time to maximal concentrations (Tmax) occurred between 1 to 3 hours post dose in the fasted state.
Effect of Food
Midostaurin exposure, represented by AUC over time to infinity (AUCinf), increased 1.2-fold when RYDAPT was coadministered with a standard meal (457 calories, 50 g fat, 21 g proteins, and 18 g carbohydrates) and 1.6-fold when coadministered with a high-fat meal (1007 calories, 66 g fat, 32 g proteins, and 64 g carbohydrates) compared to when RYDAPT was administered in a fasted state. Midostaurin maximum concentrations (Cmax) were reduced by 20% with a standard meal and by 27% with a high-fat meal compared to a fasted state. Tmax was delayed when RYDAPT was administered with a standard meal or a high-fat meal (median Tmax = 2.5 hrs to 3 hrs) [see Dosage and Administration (2.5)].
Distribution
Midostaurin has an estimated geometric mean volume of distribution (% coefficient of variation) of 95.2 L (31%). Midostaurin and its metabolites are distributed mainly in plasma in vitro. Midostaurin, CGP62221, and CGP52421 are greater than 99.8% bound to plasma protein in vitro. Midostaurin is mainly bound to α1-acid glycoprotein in vitro.
Elimination
The geometric mean terminal half-life (% coefficient of variation) is 19 hours (39%) for midostaurin, 32 hours (31%) for CGP62221 and 482 hours (25%) for CGP52421.
Metabolism
Midostaurin is primarily metabolized by CYP3A4. CGP62221 and CGP52421 (mean ± standard deviation) account for 28 ± 2.7% and 38 ± 6.6% respectively of the total circulating radioactivity.
Excretion
Fecal excretion accounted for 95% of the recovered dose with 91% of the recovered dose excreted as metabolites and 4% of the recovered dose as unchanged midostaurin. Only 5% of the recovered dose was found in urine.
Specific Populations
Age (20 to 94 years), sex, race, mild (total bilirubin greater than 1.0 to 1.5 times the ULN or aspartate aminotransferase (AST) greater than the ULN) or moderate (total bilirubin 1.5 to 3.0 times the ULN and any value for AST) hepatic impairment, and renal impairment (creatinine clearance (CLCr) ≥ 30 mL/min) did not have clinically meaningful effects on the pharmacokinetics of midostaurin, CGP62221, or CGP52421. The pharmacokinetics of midostaurin in patients with baseline severe hepatic impairment (total bilirubin greater than 3.0 times the ULN and any value for AST) or severe renal impairment (CLCr 15 to 29 mL/min) is unknown.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Effect of Strong CYP3A4 Inhibitors on Midostaurin
Coadministration of ketoconazole (a strong CYP3A4 inhibitor) with a single dose of RYDAPT (50 mg) increased AUCinf of midostaurin by 10.4-fold and CGP62221 by 3.5-fold and area under the curve over time to last measurable concentrations (AUC0-t) of CGP52421 by 1.2-fold compared to a single RYDAPT dose coadministered with placebo [see Drug Interactions (7.1)].
Coadministration of itraconazole (a strong CYP3A4 inhibitor) with multiple doses of RYDAPT (100 mg twice daily on Days 1 to 2 and 50 mg twice daily on Days 3 to 28) increased Day 28 Cmin concentrations of midostaurin by 2.1-fold, CGP62221 by 1.2-fold, and CGP52421 by 1.3-fold compared to the respective Day 21 Cmin concentrations with RYDAPT alone [see Drug Interactions (7.1)].
Effect of Strong CYP3A4 Inducers on Midostaurin
Coadministration of rifampicin (a strong CYP3A4 inducer) with a single dose of RYDAPT (50 mg) decreased AUCinf of midostaurin by 96% and CGP62221 by 92% and AUC0-t of CGP52421 by 59% [see Drug Interactions (7.1)].
Effect of Midostaurin on CYP2B6 Substrates
Coadministration of multiple doses of RYDAPT (50 mg twice daily) at steady-state with a single dose of bupropion (a sensitive CYP2B6 substrate) decreased the AUCinf of bupropion by 48% and hydroxybupropion by 65% [see Drug Interactions (7.2)].
Effect of Midostaurin on CYP3A, CYP2C8, CYP2D6 Substrates
Coadministration of multiple doses of RYDAPT (50 mg twice daily) at steady-state with single doses of midazolam (a sensitive CYP3A substrate) or pioglitazone (a moderate sensitive CYP2C8 substrate) did not affect AUCinf of midazolam or pioglitazone. However, the effect of multiple doses of RYDAPT on sensitive substrates of CYP3A and CYP2C8 during the first week, when midostaurin trough concentrations are highest, is unknown.
Coadministration of a single dose of RYDAPT (100 mg) with a single dose of dextromethorphan (a sensitive CYP2D6 substrate) did not affect the AUCinf of dextromethorphan. The effect of multiple doses of RYDAPT on dextromethorphan is unknown.
Coadministration of multiple doses of RYDAPT (50 mg twice daily) at steady-state with a single dose of a hormonal contraceptive containing ethinyl estradiol and levonorgestrel (CYP3A4 substrates) increased the area under the curve over time to the last measurable concentration (AUClast) of ethinyl estradiol by 10% and levonorgestrel by 42%. However, the effect of multiple doses of RYDAPT on ethinyl estradiol and levonorgestrel during the first week, when midostaurin trough concentrations are highest, is unknown.
Effect of Midostaurin on P-gp, BCRP, and OATP1B1 Substrates
Coadministration of a single dose of RYDAPT (100 mg) with a single dose of rosuvastatin (BCRP and OATP1B1 substrate) increased the AUClast of rosuvastatin by 48%. RYDAPT 50 mg twice daily at steady state is predicted to increase the AUC of an OATP1B1 substrate up to 2-fold, with unknown effect on a BCRP substrate [see Drug Interactions (7.2)].
Coadministration of a single dose of RYDAPT (100 mg) with a single dose of digoxin (sensitive P-gp substrate) did not affect the AUCinf of digoxin. The effect of multiple doses of RYDAPT on digoxin is unknown.
In Vitro Studies
Effect of Midostaurin on CYP Enzymes
Midostaurin inhibits CYP1A2 and CYP2E1; CGP62221 inhibits CYP1A2 in vitro. Midostaurin, CGP52421, and CGP62221 induce CYP1A2 in vitro.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with midostaurin.
Midostaurin was not mutagenic in vitro in the bacterial reverse mutation assay (Ames test) or in Chinese hamster V97 cells. Midostaurin increased the frequency of polyploidy cells in an in vitro chromosomal aberrations assay in Chinese hamster ovary cells, but was not clastogenic in an in vivo rat bone marrow micronucleus assay when tested to the maximum tolerated dose (MTD) of 200 mg/kg (1,200 mg/m2). This dose was approximately 20-fold the recommended human dose, based on body surface area.
Reproductive toxicity was observed in a fertility study, in male and female rats given oral doses of midostaurin at 10, 30 and 60 mg/kg/day (approximately 0.01, 0.05, and 0.1 times, respectively, the AUC at the recommended human dose). In males, testicular degeneration and atrophy was observed at doses greater than or equal to 10 mg/kg/day and reduced sperm count and motility, and a decrease in reproductive organ weights were observed at 60 mg/kg/day. In females, increased resorptions, decreased pregnancy rate, and decreased number of implants and live embryos were observed at 60 mg/kg/day. In a 3-month toxicology study in dogs, there was inhibition of spermatogenesis at doses greater than or equal to 3 mg/kg/day (approximately 0.01 times the exposure at the recommended human dose).
14. Clinical Studies
14.1 Acute Myeloid Leukemia
Study 1
RYDAPT in combination with chemotherapy was investigated in a randomized, double-blind placebo-controlled trial of 717 patients with newly-diagnosed FLT3-mutated AML. In this study, FLT3 mutation status was determined prospectively with a clinical trial assay and verified retrospectively using the companion diagnostic LeukoStrat® CDx FLT3 Mutation Assay, which is an FDA-approved test for selection of patients with AML for RYDAPT treatment. Patients were stratified by FLT3 mutation status: TKD, ITD with allelic ratio less than 0.7, and ITD with allelic ratio greater than or equal to 0.7. Patients with acute promyelocytic leukemia or therapy-related AML were not eligible. Patients were randomized (1:1) to receive RYDAPT 50 mg twice daily (n = 360) or placebo (n = 357) with food on Days 8 to 21 in combination with daunorubicin (60 mg/m2 daily on Days 1 to 3) /cytarabine (200 mg/m2 daily on Days 1 to 7) for up to two cycles of induction and high dose cytarabine (3 g/m2 every 12 hours on Days 1, 3, and 5) for up to four cycles of consolidation, followed by continuous RYDAPT or placebo treatment according to initial assignment for up to 12 additional 28-day cycles. There was no re-randomization at the start of post consolidation therapy. Patients who proceeded to hematopoietic stem cell transplantation (SCT) stopped receiving study treatment.
The randomized patients had a median age of 47 years (range, 18 to 60 years), 44% were male, and 88% had a performance status of 0-1. AML was de novo onset in 95%. The percentage of patients with FLT3-ITD allelic ratio < 0.7, FLT3-ITD allelic ratio ≥ 0.7, and FLT3-TKD mutations were identical (per randomized FLT3 stratum) on both arms (48%, 30%, and 23%, respectively). Of the 563 patients with NPM1 testing, 58% had an NPM1 mutation. The two treatment groups were generally balanced with respect to the baseline demographics and disease characteristics, except that the placebo arm had a higher percentage of females (59%) than in the midostaurin arm (52%). NPM1 mutations were identified in 55% of patients tested on the midostaurin arm and 60% of patients tested on the placebo arm.
A second course of induction was administered to 25% of the patients, 62% initiated at least one cycle of consolidation, 29% initiated maintenance, and 17% completed all 12 planned cycles of maintenance; 21% of the patients underwent SCT in first CR. The overall rate of SCT (induction failure, first CR or salvage after relapse) was 59% (214/360) of patients in the RYDAPT plus standard chemotherapy arm versus 55% (197/357) in the placebo plus standard chemotherapy arm. All patients were followed for survival.
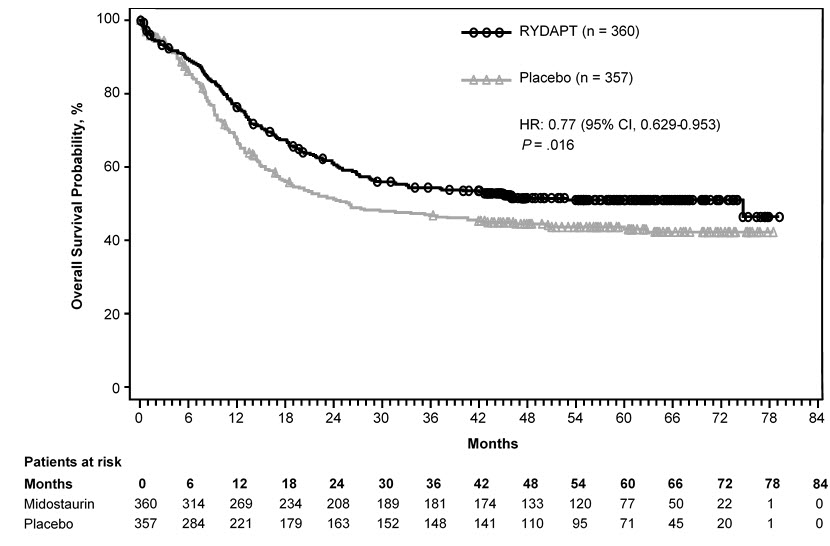
Efficacy was established on the basis of overall survival (OS), measured from the date of randomization until death by any cause. The primary analysis was conducted after a minimum follow-up of approximately 3.5 years after the randomization of the last patient. RYDAPT plus standard chemotherapy was superior to placebo plus standard chemotherapy in OS [HR 0.77; 95% confidence interval (CI) 0.63, 0.95; 2-sided p = 0.016] (Figure 1). Because survival curves plateaued before reaching the median, median survival could not be reliably estimated.
Figure 1: Kaplan-Meier Curve for Overall Survival in Study 1
The analysis of event-free survival (EFS), defined as a failure to obtain a complete remission (CR) within 60 days of initiation of protocol therapy, or relapse, or death from any cause, showed a statistically significant improvement with a median of 8.2 months for RYDAPT plus standard chemotherapy versus 3.0 months for placebo plus standard chemotherapy with HR 0.78 (95% CI 0.66, 0.93) and 2-sided p = 0.005. In an exploratory analysis of EFS defined as a failure to obtain a CR any time during induction, or relapse, or death from any cause with failures assigned as an event on study Day 1, the median EFS was 10.6 months for RYDAPT plus standard chemotherapy versus 5.6 months for placebo plus standard chemotherapy with HR 0.72 (95% CI 0.61, 0.86).
14.2 Systemic Mastocytosis
Study 2
A single-arm, open-label, multicenter trial evaluated the efficacy of RYDAPT as a single agent in ASM, SM-AHN, and MCL, collectively referred to as advanced SM. The study enrolled 116 adult patients with relapse or progression to 0, 1, or 2 prior regimens for SM. The study excluded patients with serum creatinine > 2.0 mg/dL, hepatic transaminases > 2.5 x ULN or > 5 x ULN if disease-related, total bilirubin > 1.5 x ULN or > 3 x ULN if disease-related, QTc > 450 msec, cardiovascular disease, including left-ventricular ejection fraction < 50%, or any pulmonary infiltrates. In addition, the study excluded patients with acute-stage or life-threatening AHN. Patients received RYDAPT 100 mg orally twice daily in 28-day cycles until disease progression or intolerable toxicity.
Of the 116 patients treated, a study steering committee identified 89 patients who had measurable C-findings and were evaluable for response. The median age in this group was 64 years (range, 25 to 82), 64% of patients were male, and nearly all patients (97%) were Caucasian. Among these patients, 36% had prior therapy for SM, and 82% had the KIT D816V mutation detected at baseline. Their median duration of treatment was 11 months (range, < 1 to 68 months), with treatment ongoing in 17%.
Efficacy was established on the basis of confirmed complete remission (CR) plus incomplete remission (ICR) by 6 cycles of RYDAPT by modified Valent criteria for ASM and SM-AHN (Table 7). Table 7 shows responses to RYDAPT according to modified Valent criteria. Confirmed major or partial responses occurred in 46 of 73 patients with a documented KIT D816V mutation, 7 of 16 with wild-type or unknown status with respect to KIT D816V mutation, and 21 of 32 having prior therapy for SM.
| Modified Valent Criteria | All Patients Evaluatede
(N = 89) | ASM (N = 16) | SM-AHN (N = 57) | MCL (N = 16) |
|---|---|---|---|---|
| CR + ICR by 6 cycles, na,b
(95% CI, %) | 19 (21%) (13, 31) | 6 (38%) (15, 65) | 9 (16%) (7, 28) | 4 (25%) (7, 52) |
| Median duration of CR + ICR (months)c
(95% CI) Ranged | NR (24.1, NE) 6.6+, 65.8+ | NR (24.1, NE) 12.1+, 36.8+ | NR (7.4, NE) 6.6+, 52.1+ | NR (NE, NE) 19.1+, 65.8+ |
| Median time to CR + ICR (months)
Range | 0.5 0.1, 3.0 | 0.7 0.3, 1.9 | 0.5 0.1, 3.0 | 0.3 0.1, 0.5 |
| Abbreviations: ASM, aggressive systemic mastocytosis; CI, confidence interval; CR, complete remission; ICR, incomplete remission; MCL, mast cell leukemia; NE, not estimated; NR, not reached; SM-AHN, systemic mastocytosis with associated hematological neoplasm. aPer Study Steering Committee. Response confirmation after ≥ 8 weeks was required. No CRs were reported. bPatients who received concomitant high-dose corticosteroids were considered unevaluable and were excluded from response assessment. cAmong patients with a response of CR or ICR. The estimated median follow-up for duration of response (DOR) was 35.4 months overall. dA "+" sign indicates a censored value. e25 patients were not assessable for the presence of MCL on central histopathology review, and 11 patients with unconfirmed presence of AHN were regarded as not having AHN. |
||||
As a post-hoc exploratory analysis, efficacy was also assessed using modified 2013 International Working Group-Myeloproliferative Neoplasms Research and Treatment-European Competence Network on Mastocytosis (IWG-MRT-ECNM) consensus criteria. Response after 6 cycles of RYDAPT was determined using a computational algorithm. The efficacy of RYDAPT for MCL was based on the CR results by these criteria. There were 115 patients evaluable for response assessment, of whom 47 (41%) had prior therapy for SM, and 93 (81%) had a documented D816V mutation at baseline. Table 8 provides the results of this analysis. Overall response by modified IWG-MRT-ECNM criteria was reported for 16 (17%) of 93 patients with a documented D816V mutation, and in 8 (17%) of 47 patients having prior therapy for SM.
| All Patients Evaluated (N = 115)b,c | ASM (N = 16) | SM-AHN (N = 72) | MCL (N = 21) | Subtype not Established (N = 6) |
|
|---|---|---|---|---|---|
| Overall response in 6 cycles, na
(95% CI) | 19 (17%) (10, 25) | 5 (31%) (11, 59) | 8 (11%) (5, 21) | 4 (19%) (5, 42) | 2 (33%) (4, 78) |
| Best overall response, n
Complete remission Partial remission | 2 (2%) 17 (15%) | 1 (6%) 4 (25%) | 0 (0%) 8 (11%) | 1 (5%) 3 (14%) | 0 (0%) 2 (33%) |
| Duration of response (months)d
Rangee | 6.8+, 60.5+ | 10.2+, 36.4+ | 6.8+, 51.8+ | 8.6+, 55.9+ | 27.3+, 60.5+ |
| Abbreviations: ASM, aggressive systemic mastocytosis; IWG-MRT-ECNM, international working group-myeloproliferative neoplasms research and treatment-european competence network on mastocytosis; MCL, mast cell leukemia; SM-AHN, systemic mastocytosis with associated hematological neoplasm. aDetermined with 12-week confirmation. Patients who received high-dose corticosteroids were considered evaluable for response. bMedian exposure to midostaurin was 11.5 (range, 0.3, 68.3) months. c31 patients were not assessable for MCL on central review, and 15 patients with unconfirmed AHN were classified as not having AHN. dMedian duration of response (DOR) was not reached in any subtype. Median follow up for DOR, among all responders, was 35.0 months. eA “+” sign indicates a censored value. |
|||||
Study 3
Study 3 was a single-arm, multicenter, open-label trial of 26 patients with advanced SM. RYDAPT was administered orally at 100 mg twice daily with food. The median age in this group was 64 years, 58% of patients were male and most were Caucasian (81%). Eligibility criteria were similar to Study 2. By Valent criteria per investigator assessment, of 17 patients with SM-AHN, 10 achieved a response (1 partial, 9 major) by 2 cycles that was sustained for at least 8 weeks. Patients who received concomitant corticosteroids were included. Of the 6 patients with MCL, 1 achieved partial response and 1 achieved major response. Median duration of response (DOR) for either group had not been reached, with DOR ranging from 3.4+ to 79.2+ months in patients with SM-AHN and 28.6+ to 32.1+ months in patients with MCL. The subtype of SM in the remaining 3 patients was unconfirmed.
16. How is Rydapt supplied
RYDAPT 25 mg capsules
Pale orange oblong soft capsule with red ink imprint ‘PKC NVR’; available in:
56 soft capsules………………………………………………………………………………………NDC 0078-0698-99
Contents: Each carton contains two inner packs, each with 28 capsules (7 blister cards with 4 capsules each)
112 soft capsules……………………………………………………………………………………..NDC 0078-0698-19
Contents: Each carton contains four inner packs, each with 28 capsules (7 blister cards with 4 capsules each)
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature]. Store in the original container to protect from moisture.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Pulmonary Adverse Reactions
Inform patients to seek medical attention for new cough, chest discomfort, or shortness of breath [see Warnings and Precautions (5.2)].
Gastrointestinal Adverse Reactions
Inform patients that RYDAPT can cause nausea, vomiting, and diarrhea. Advise patients to contact their healthcare provider if these symptoms occur or are persisting despite supportive medications [see Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with RYDAPT and for 4 months after the last dose. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)].
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment with RYDAPT and for 4 months after the last dose [see Use in Specific Populations (8.3)].
- Advise females who may have been exposed to RYDAPT during pregnancy directly or through male partner receiving RYDAPT therapy to contact the Novartis Pharmaceuticals Corporation at 1-888-669-6682 and/or at https://report.novartis.com/ [see Use in Specific Populations (8.1)].
Lactation
Advise women not to breastfeed during treatment with RYDAPT and for 4 months after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise females and males of reproductive potential that RYDAPT may impair fertility [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Distributed by:
Novartis Pharmaceuticals Corporation
One Health Plaza
East Hanover, New Jersey 07936
T2023-25
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: November 2021 |
| PATIENT INFORMATION
RYDAPT® (rye-dapt) (midostaurin) capsules |
|
| What is RYDAPT?
RYDAPT is a prescription medicine used to treat adults:
|
|
Do not take RYDAPT if you are allergic to midostaurin or any of the ingredients in RYDAPT. See the end of this leaflet for a complete list of ingredients in RYDAPT.
|
|
Before you take RYDAPT, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. |
|
How should I take RYDAPT?
|
|
| What are the possible side effects of RYDAPT?
RYDAPT may cause serious side effects, including:
The most common side effects of RYDAPT in people with AML include: |
|
|
|
| The most common side effects of RYDAPT in people with ASM, SM-AHN, or MCL include: | |
|
|
| Call or inform your healthcare provider if nausea, vomiting or diarrhea occurs, gets worse or does not go away. RYDAPT may cause fertility problems in females and males, which may affect your ability to have children. Talk to your healthcare provider if you have concerns about fertility. Your healthcare provider may tell you to decrease your dose, temporarily stop, or completely stop taking RYDAPT if you develop certain side effects during treatment with RYDAPT. Your healthcare provider will do blood tests to check you for side effects during treatment with RYDAPT. These are not all of the possible side effects of RYDAPT. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store RYDAPT?
Keep RYDAPT and all medicines out of the reach of children. |
|
| General information about the safe and effective use of RYDAPT.
Medicines are sometimes prescribed for conditions not listed in the Patient Information leaflet. Do not take RYDAPT for a condition for which it was not prescribed. Do not give RYDAPT to other people, even if they have the same condition or symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about RYDAPT that is written for healthcare professionals. |
|
| What are the ingredients in RYDAPT?
Active ingredient: midostaurin Inactive ingredients: carmine, corn oil mono-di-triglycerides, dehydrated alcohol, ferric oxide red, ferric oxide yellow, gelatin, glycerin 85%, hypromellose 2910, polyethylene glycol 400, polyoxyl 40 hydrogenated castor oil, propylene glycol, purified water, titanium dioxide, and vitamin E. Distributed by: Novartis Pharmaceuticals Corporation, One Health Plaza, East Hanover, New Jersey 07936 © Novartis For more information about RYDAPT, visit www.US.RYDAPT.com. |
|
T2021-150

| RYDAPT
rydapt capsule, liquid filled |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |
Frequently asked questions
More about Rydapt (midostaurin)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (2)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- FDA approval history
- Drug class: multikinase inhibitors
- Breastfeeding
- En español