Mylotarg: Package Insert / Prescribing Info
Package insert / product label
Generic name: gemtuzumab ozogamicin
Dosage form: injection, powder, lyophilized, for solution
Drug class: CD33 monoclonal antibodies
J Code (medical billing code): J9203 (0.1 mg, injection)
Medically reviewed by Drugs.com. Last updated on Aug 6, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
MYLOTARG™ (gemtuzumab ozogamicin) for injection, for intravenous use
Initial U.S. Approval: 2000
Recent Major Changes
|
Dosage and Administration, Instructions for Reconstitution, Dilution, and Administration (2.4) |
08/2021 |
Indications and Usage for Mylotarg
MYLOTARG is a CD33-directed antibody and cytotoxic drug conjugate indicated for:
Mylotarg Dosage and Administration
- •
- Newly-diagnosed, de novo AML (combination regimen)
Adults: - Pediatric patients 1 month and older:
- •
- Newly-diagnosed AML (single-agent regimen):
Adults:- -
- Induction: 6 mg/m2 (not limited to one 4.5 mg vial) on Day 1 and 3 mg/m2 (not limited to one 4.5 mg vial) on Day 8 (2.2).
- -
- Continuation: For patients without evidence of disease progression following induction, up to 8 continuation courses of MYLOTARG 2 mg/m2 (not limited to one 4.5 mg vial) on Day 1 every 4 weeks (2.2).
- •
- Relapsed or refractory AML (single-agent regimen):
- Adults and pediatric patients 2 years and older:
- -
- 3 mg/m2 (up to one 4.5 mg vial) on Days 1, 4, and 7 (2.2).
- •
- Premedicate with a corticosteroid, antihistamine, and acetaminophen (2.1).
Dosage Forms and Strengths
For Injection: 4.5 mg as a lyophilized cake or powder in a single-dose vial for reconstitution and dilution (3).
Contraindications
Hypersensitivity to MYLOTARG or any of its components (4).
Warnings and Precautions
- •
- Infusion-related reactions (including anaphylaxis): Premedicate with a corticosteroid, acetaminophen, and diphenhydramine. Monitor patients during and for at least 1 hour after the end of the infusion. Interrupt the infusion, administer steroids or antihistamines, or permanently discontinue treatment as necessary (2.1, 5.2, 6).
- •
- Hemorrhage: Severe, including fatal, hemorrhage may occur when MYLOTARG is used at recommended doses. Monitor platelet counts frequently (5.3, 6.1).
- •
- Embryo-fetal toxicity: Can cause fetal harm. Advise patients of reproductive potential of the potential risk to a fetus and to use effective contraception (5.6, 8.1, 8.3).
Adverse Reactions/Side Effects
The most common adverse reactions (greater than 15%) were hemorrhage, infection, fever, nausea, vomiting, constipation, headache, increased AST, increased ALT, rash, mucositis, febrile neutropenia, and decreased appetite (6).
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2021
Full Prescribing Information
WARNING: HEPATOTOXICITY
Hepatotoxicity, including severe or fatal hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS), has been reported in association with the use of MYLOTARG as a single agent, and as part of a combination chemotherapy regimen. Monitor frequently for signs and symptoms of VOD after treatment with MYLOTARG. (5.1 and 6.1)
1. Indications and Usage for Mylotarg
2. Mylotarg Dosage and Administration
2.1 Premedication and Special Considerations
- •
- Premedicate adults with acetaminophen 650 mg orally and diphenhydramine 50 mg orally or intravenously 1 hour prior to MYLOTARG dosing and 1 mg/kg methylprednisolone or an equivalent dose of an alternative corticosteroid within 30 minutes prior to infusion of MYLOTARG.
- •
- Premedicate pediatric patients 1 month and older with acetaminophen 15 mg/kg (maximum of 650 mg) and diphenhydramine 1 mg/kg (maximum of 50 mg) 1 hour prior to MYLOTARG dosing, and 1 mg/kg methylprednisolone orally or intravenously within 30 minutes prior to infusion of MYLOTARG; additional doses of acetaminophen and diphenhydramine may be administered every 4 hours after the initial pretreatment dose. Repeat with the same dose of methylprednisolone or an equivalent corticosteroid for any sign of an infusion reaction, such as fever, chills, hypotension, or dyspnea during the infusion or within 4 hours afterwards [see Warnings and Precautions (5.2)].
- •
- Use appropriate measures to prevent tumor lysis syndrome.
- •
- For patients with hyperleukocytosis (leukocyte count greater than or equal to 30 Gi/L), cytoreduction is recommended prior to administration of MYLOTARG.
2.2 Recommended Dosage
Newly-Diagnosed De Novo CD33-positive AML (Combination Regimen)
Adults
The recommended dose of MYLOTARG in adults is 3 mg/m2. A treatment course including MYLOTARG in combination therapy for adults with newly-diagnosed de novo CD33-positive AML consists of 1 induction cycle and 2 consolidation cycles [see Clinical Studies (14.1)].
For the induction cycle, the recommended dose of MYLOTARG is 3 mg/m2 (up to one 4.5 mg vial) on Days 1, 4, and 7 in combination with daunorubicin and cytarabine. For patients requiring a second induction cycle, do NOT administer MYLOTARG during the second induction cycle.
For the consolidation cycles, the recommended dose of MYLOTARG is 3 mg/m2 on Day 1 (up to one 4.5 mg vial) in combination with daunorubicin and cytarabine.
Pediatric Patients 1 Month and Older
The recommended dose of MYLOTARG in pediatric patients 1 month and older is:
- •
- 3 mg/m2 for patients with body surface area (BSA) greater than or equal to 0.6 m2
- •
- 0.1 mg/kg for patients with BSA less than 0.6 m2
For Induction 1, MYLOTARG is given once in combination with standard chemotherapy. No MYLOTARG is given in the second induction cycle [see Clinical Studies (14.1)].
No MYLOTARG is given in the first or third intensification cycles. For Intensification 2, MYLOTARG is given once in combination with standard chemotherapy. Consider the risks and potential benefits before giving MYLOTARG during Intensification 2 [see Adverse Reactions (6.1)].
Newly-Diagnosed CD33-positive AML (Single-agent Regimen)
A treatment course of MYLOTARG as a single agent for adults with newly-diagnosed CD33-positive AML consists of 1 cycle of induction and up to 8 cycles of continuation therapy [see Clinical Studies (14.1)].
For the induction cycle, the recommended dose of MYLOTARG is 6 mg/m2 (not limited to one 4.5 mg vial) as a single agent on Day 1, and 3 mg/m2 (not limited to one 4.5 mg vial) on Day 8.
For continuation, the recommended dose of MYLOTARG is 2 mg/m2 (not limited to one 4.5 mg vial) as a single agent on Day 1 every 4 weeks.
Relapsed or Refractory CD33-positive AML (Single-agent Regimen)
The recommended dose of MYLOTARG as a single agent for treatment for adults and pediatric patients 2 years and older with relapsed or refractory CD33-positive AML is 3 mg/m2 (up to one 4.5 mg vial) on Days 1, 4, and 7. Treatment in the relapsed or refractory setting consists of a single course of MYLOTARG [see Clinical Studies (14.2)].
2.3 Dosage Modifications for Toxicities
Monitor blood counts frequently through resolution of cytopenias. Monitor blood counts and chemistries at least three times per week through recovery from treatment-related toxicities. Management of some adverse reactions [see Warnings and Precautions (5), Adverse Reactions (6)] may require dose interruptions or permanent discontinuation of MYLOTARG. Table 1 shows the dose modification guidelines for hematologic and nonhematologic toxicities.
| Hematologic and Nonhematologic Toxicities | Recommended Action |
|---|---|
| Abbreviations: ALT=alanine aminotransferase; AST=aspartate aminotransferase; VOD=veno-occlusive disease; ULN=upper limit of normal. | |
|
For patients receiving MYLOTARG in combination therapy |
|
|
Persistent thrombocytopenia |
|
|
Persistent neutropenia |
|
|
For all patients receiving MYLOTARG (Monotherapy or in Combination) |
|
|
VOD |
|
|
Total bilirubin greater than 2 × ULN, or AST and/or ALT greater than 2.5 × ULN |
|
|
Infusion-related reactions |
|
|
Other severe or life-threatening non-hematologic toxicities |
|
2.4 Instructions for Reconstitution, Dilution, and Administration
Use appropriate aseptic technique for the reconstitution and dilution procedures. Protect the reconstituted and diluted MYLOTARG solution from light.
Reconstitution
- •
- MYLOTARG is a cytotoxic drug. Follow applicable special handling and disposal procedures.1
- •
- Calculate the dose (mg) and number of vials of MYLOTARG required.
- •
- Prior to reconstitution, allow drug product vials to reach room temperature (up to 30°C) for approximately 5 minutes.
- •
- Reconstitute each vial with 5 mL of Sterile Water for Injection, USP to obtain a concentration of 1 mg/mL of MYLOTARG that delivers 4.5 mL (4.5 mg).
- •
- Gently swirl the vial to aid dissolution. DO NOT SHAKE.
- •
- Inspect the reconstituted solution for particulates and discoloration. The reconstituted solution may contain small white to off-white, opaque to translucent, and amorphous to fiber-like particles.
- •
- MYLOTARG contains no bacteriostatic preservatives.
- •
-
If the reconstituted solution cannot be used immediately, it may be stored in the original vial for up to 16 hours in a refrigerator (2°C to 8°C; 36°F to 46°F) or up to 3 hours at room temperature (up to 30°C).
PROTECT FROM LIGHT. DO NOT FREEZE.
Dilution
- •
- Calculate the required volume of the reconstituted solution needed to obtain the appropriate dose according to patient body surface area. Withdraw this amount from the vial(s) using a syringe. PROTECT FROM LIGHT. Discard any unused reconstituted solution left in the vial.
Doses must be mixed to a concentration between 0.075 mg/mL to 0.234 mg/mL according to the following instructions:
- •
- Doses less than 3.9 mg must be prepared for administration by syringe. Add the reconstituted MYLOTARG solution to a syringe with 0.9% Sodium Chloride Injection to a final concentration between 0.075 mg/mL to 0.234 mg/mL. PROTECT FROM LIGHT.
- •
- Doses greater than or equal to 3.9 mg are to be diluted in a syringe or a polyvinyl chloride (PVC) with di(2-ethylhexyl)phthalate (DEHP), non-PVC polyolefin, or ethylene vinyl acetate intravenous infusion bag in an appropriate volume of 0.9% Sodium Chloride Injection to ensure a final concentration between 0.075 mg/mL to 0.234 mg/mL. PROTECT FROM LIGHT.
- •
- Gently invert the infusion container to mix the diluted solution. DO NOT SHAKE.
- •
- Following dilution with 0.9% Sodium Chloride Injection, MYLOTARG solution should be infused immediately. If not used immediately, the diluted solution may be stored up to 18 hours in a refrigerator (2°C to 8°C; 36°F to 46°F) and for up to 6 hours at room temperature (up to 30°C). The allowed time at room temperature (up to 30°C) includes the time required for preparation of the diluted solution, equilibration, if needed, and the 2 hours needed to administer to the patient. PROTECT FROM LIGHT and DO NOT FREEZE.
Administration
- •
- Use an in-line 0.2 micron polyethersulfone (PES) filter for infusion of MYLOTARG.
- •
- Protect the intravenous bag from light using a light-blocking cover during infusion. The infusion line does not need to be protected from light.
- •
- Infuse the diluted solution over 2 hours using an infusion set made of polyvinyl chloride (PVC) with DEHP, PVC non-DEHP, polyethylene, or polyurethane. The infusion must be completed prior to the end of the allowed 6-hour storage of the diluted solution at room temperature (up to 30°C).
- •
- Do not mix MYLOTARG with, or administer as an infusion with, other medicinal products.
3. Dosage Forms and Strengths
For injection: 4.5 mg as a white to off-white lyophilized cake or powder in a single-dose vial for reconstitution and further dilution.
4. Contraindications
MYLOTARG is contraindicated in patients with a history of hypersensitivity to the active substance in MYLOTARG or any of its components or to any of the excipients. Reactions have included anaphylaxis [see Warnings and Precautions (5.2), Adverse Reactions (6)].
5. Warnings and Precautions
5.1 Hepatotoxicity, Including Veno-occlusive Liver Disease (VOD)
Hepatotoxicity, including life-threatening and sometimes fatal hepatic VOD events, have been reported in patients receiving MYLOTARG as a single agent or as part of a combination chemotherapy regimen [see Adverse Reactions (6)].
In ALFA-0701, VOD events were reported in 6/131 (5%) adult patients during or following treatment with MYLOTARG, or following later hematopoietic stem cell transplantation (HSCT). The median time from the MYLOTARG dose to onset of VOD was 9 days (range: 2–298 days), with 5 events occurring within 28 days of any dose of MYLOTARG and 1 event occurring greater than 28 days after the last dose of MYLOTARG. Three of the 6 VOD events were fatal. VOD was also reported in 2 patients in the control arm of ALFA-0701 after receiving MYLOTARG as a therapy for relapsed AML.
In MyloFrance-1 (MYLOTARG 3 mg/m2 on Days 1, 4 and 7), VOD events were reported in none of the 57 patients during or following treatment, or following HSCT after completion of MYLOTARG treatment.
In AAML0531, VOD events were reported in 25/520 (5%) pediatric patients in the MYLOTARG arm. VOD was fatal in 2 patients. Among 187 pediatric patients who underwent HSCT in the MYLOTARG arm, VOD occurred within 30 days post-HSCT in 20 (11%) patients.
Based on an analysis across trials, the risk of VOD was higher in adult patients who received higher doses of MYLOTARG as monotherapy, in patients with moderate or severe hepatic impairment prior to receiving MYLOTARG, in patients treated with MYLOTARG after HSCT, and in patients who underwent HSCT after treatment with MYLOTARG. Patients who had moderate/severe hepatic impairment prior to treatment with MYLOTARG were 8.7 times more likely to develop VOD compared to patients without moderate/severe hepatic impairment at baseline. Patients treated with MYLOTARG for relapse after HSCT were 2.6 times more likely to develop VOD compared to patients without prior HSCT. Patients who underwent HSCT following MYLOTARG treatment were 2.9 times more likely to develop VOD after HSCT compared to patients without HSCT following MYLOTARG treatment. Although no relationship was found between VOD and time of HSCT relative to higher MYLOTARG monotherapy doses, the ALFA-0701 study recommended an interval of 2 months between the last dose of MYLOTARG and HSCT. In MyloFrance-1, no patients underwent HSCT within 3.5 months of MYLOTARG therapy.
Assess ALT, AST, total bilirubin, and alkaline phosphatase prior to each dose of MYLOTARG. After treatment with MYLOTARG, monitor frequently for signs and symptoms of VOD; these may include elevations in ALT, AST, total bilirubin, hepatomegaly (which may be painful), rapid weight gain, and ascites. Monitoring only total bilirubin may not identify all patients at risk of VOD. For patients who develop abnormal liver tests, more frequent monitoring of liver tests and clinical signs and symptoms of hepatotoxicity is recommended. For patients who proceed to HSCT, monitor liver tests frequently during the post-HSCT period, as appropriate.
Manage signs or symptoms of hepatic toxicity by dose interruption or discontinuation of MYLOTARG [see Dosage and Administration (2.3)]. In patients who experience VOD, discontinue MYLOTARG and treat according to standard medical practice.
5.2 Infusion-Related Reactions (Including Anaphylaxis)
Life-threatening or fatal infusion-related-reactions can occur during or within 24 hours following infusion of MYLOTARG [see Adverse Reactions (6)]. Signs and symptoms of infusion-related reactions may include fever, chills, hypotension, tachycardia, hypoxia and respiratory failure.
Premedicate prior to MYLOTARG infusion [see Dosage and Administration (2.1)]. Monitor vital signs frequently during infusion. Interrupt infusion immediately for patients who develop evidence of infusion reaction, especially dyspnea, bronchospasm, or hypotension. Monitor patients during and for at least 1 hour after the end of the infusion or until signs and symptoms completely resolve. Discontinue use of MYLOTARG in patients who develop signs or symptoms of anaphylaxis, including severe respiratory symptoms or clinically significant hypotension [see Dosage and Administration (2.2)].
5.3 Hemorrhage
MYLOTARG is myelosuppressive and can cause fatal or life-threatening hemorrhage due to prolonged thrombocytopenia. In ALFA-0701, (MYLOTARG in combination with chemotherapy), all grades and Grade 3–4 bleeding events were reported in 118/131 (90%) and 27/131 (21%) patients, respectively. Fatal bleeding events (including cerebral hematoma, intracranial hematoma, and subdural hematoma) occurred in 4/131 (3%) patients. Thrombocytopenia with platelet counts less than 50 Gi/L persisting more than 42 days occurred in 19 (19%) patients in the induction phase [see Adverse Reactions (6)]. The proportion of patients with persistent thrombocytopenia increased with progressive treatment phases and was higher in patients treated with MYLOTARG plus chemotherapy than with chemotherapy alone [see Adverse Reactions (6)].
In AAML0531, fatal bleeding occurred in 3/520 (<1%) of the pediatric patients. Grade 3 or 4 bleeding was reported in 66/520 (13%) of the pediatric patients in the MYLOTARG arm.
In AML-19 (MYLOTARG monotherapy at 6 mg/m2 Day 1 and 3 mg/m2 Day 8), all grades and Grade 3 or higher bleeding were reported in 28/111 (25%) and 14/111 (13%) patients, respectively. Fatal bleeding occurred in 1/111 (1%). In MyloFrance-1 (MYLOTARG 3 mg/m2 as monotherapy), Grade 3 bleeding was reported in 4/57 (7%) patients, but no patient experienced Grade 4 hemorrhage.
Assess blood counts prior to each dose of MYLOTARG and monitor blood counts frequently after treatment with MYLOTARG until resolution of cytopenias. Monitor patients for signs and symptoms of bleeding during treatment with MYLOTARG. Manage severe bleeding, hemorrhage or persistent thrombocytopenia using dose delay or permanent discontinuation of MYLOTARG [see Dosage and Administration (2.2)], and provide supportive care per standard practice.
5.4 QT Interval Prolongation
QT interval prolongation has been observed in patients treated with other drugs containing calicheamicin. When administering MYLOTARG to patients who have a history of or predisposition for QTc prolongation, who are taking medicinal products that are known to prolong QT interval, and in patients with electrolyte disturbances, obtain electrocardiograms (ECGs) and electrolytes prior to the start of treatment and as needed during administration.
5.5 Use in AML with Adverse-Risk Cytogenetics
In subgroup analyses in ALFA-0701, the addition of MYLOTARG to standard combination chemotherapy did not improve event-free survival in the subgroup of patients having adverse-risk cytogenetics (HR 1.11; 95% CI: 0.63, 1.95). For patients being treated with MYLOTARG in combination with daunorubicin and cytarabine for newly-diagnosed de novo AML, when cytogenetics testing results become available consider whether the potential benefit of continuing treatment with MYLOTARG outweighs the risks for the individual patient.
5.6 Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal studies, MYLOTARG can cause embryo-fetal harm when administered to a pregnant woman. In animal studies, gemtuzumab ozogamicin caused embryo-fetal toxicity, starting at a dose that was approximately 0.4 times the exposure in patients at the maximum recommended dose, based on the area under the concentration-time curve (AUC).
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with MYLOTARG and for at least 6 months after the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with MYLOTARG and for at least 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- •
- Hepatotoxicity, including VOD [see Warnings and Precautions (5.1)]
- •
- Infusion-related reactions [see Warnings and Precautions (5.2)]
- •
- Hemorrhage [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Combination Therapy in Newly-Diagnosed De Novo CD33-positive AML
The safety of MYLOTARG in first-line combination therapy was evaluated in two prospective clinical trials, Study ALFA-0701 in adults and Study AAML0531 in pediatric patients.
Study ALFA-0701
The safety evaluation of MYLOTARG (3 mg/m2 Day 1, 4 and 7 in combination with daunorubicin and cytarabine [DA]) in adults is based on data from ALFA-0701 for 131 patients treated with MYLOTARG plus DA and in 137 patients treated with DA alone [see Clinical Studies (14.1)]. In this study, 123 patients received all 3 fractionated doses of MYLOTARG and 7 patients missed at least 1 dose, with a mean total dose administered during induction of 14.51 mg (range: 4.6–18.0). MYLOTARG was received by 91 (70%) patients in the MYLOTARG arm during Consolidation 1 and 64 (49%) patients in the MYLOTARG arm during Consolidation 2.
Safety data consisting of selected TEAEs considered most important for understanding the safety profile of MYLOTARG as well as all adverse events (AEs) that led to the permanent discontinuation of treatment were retrospectively collected. The selected TEAEs consisted of all grades hemorrhages, all grades VOD, and severe infections.
Discontinuation due to any adverse reaction occurred in 31% of patients in the MYLOTARG arm versus 7% in the DA arm. The most frequent (greater than or equal to 1%) adverse reactions for patients treated with MYLOTARG that led to permanent discontinuation were thrombocytopenia (15%), VOD (3%), and septic shock (2%).
Fatal adverse reactions occurred in 8 patients (6%) in the MYLOTARG arm versus 3 patients (2%) in the DA arm. In the MYLOTARG arm, 3 patients died of VOD, 4 patients died of hemorrhage-related events (CNS hemorrhage, hemorrhagic shock), and 1 patient died of suspected cardiac cause. In the DA arm, 3 patients died of sepsis.
| MYLOTARG + Daunorubicin + Cytarabine
(n, %) | Daunorubicin + Cytarabine
(n, %) |
|
|---|---|---|
| Abbreviations: AML=acute myeloid leukemia; N=number of patients; PT=preferred term. | ||
|
Induction |
N = 131 |
N = 137 |
|
Infection* |
61 (47%) |
53 (39%) |
|
Hemorrhage† |
24 (18%) |
12 (9%) |
|
Veno-occlusive liver disease‡ |
3 (2%) |
0 |
|
Consolidation 1 |
N = 91 |
N = 103 |
|
Infection* |
50 (55%) |
43 (42%) |
|
Hemorrhage† |
5 (5%) |
0 |
|
Veno-occlusive liver disease‡ |
0 |
0 |
|
Consolidation 2 |
N = 64 |
N = 107 |
|
Infection* |
32 (50%) |
54 (50%) |
|
Hemorrhage† |
4 (6%) |
0 |
|
Veno-occlusive liver disease‡ |
0 |
0 |
All patients in ALFA-0701 developed severe neutropenia, thrombocytopenia and anemia. The incidence of Grade 3–4 thrombocytopenia that was prolonged in the absence of active leukemia was higher in patients treated with MYLOTARG (Table 3).
| MYLOTARG + Daunorubicin + Cytarabine
(n/N, %) | Daunorubicin + Cytarabine
(n/N, %) |
|
|---|---|---|
|
||
|
Induction |
||
|
Prolonged thrombocytopenia |
19/101 (19%) |
7/97 (7%) |
|
Prolonged neutropenia |
3/106 (3%) |
0/101 (0%) |
|
Consolidation 1 |
||
|
Prolonged thrombocytopenia |
21/87 (24%) |
6/91 (7%) |
|
Prolonged neutropenia |
3/88 (3%) |
1/97 (1%) |
|
Consolidation 2 |
||
|
Prolonged thrombocytopenia |
22/62 (35%) |
25/103 (24%) |
|
Prolonged neutropenia |
1/62 (2%) |
2/105 (2%) |
Table 4 summarizes shifts in selected chemistry abnormalities by treatment arm for patients treated in ALFA-0701.
| MYLOTARG + Daunorubicin + Cytarabine | Daunorubicin + Cytarabine | |||
|---|---|---|---|---|
| Laboratory Abnormality | Subjects (n) with baseline Grade less than or equal to 2 | Progressed to Grade greater than or equal to 3 (n, %) | Subjects (n) with baseline Grade less than or equal to 2 | Progressed to Grade greater than or equal to 3 (n, %) |
|
Hypophosphatemia |
117 |
75 (64%) |
127 |
52 (41%) |
|
Hypokalemia |
127 |
73 (57%) |
133 |
41 (31%) |
|
Hyponatremia |
129 |
57 (44%) |
134 |
36 (27%) |
|
Alkaline phosphatase increased |
120 |
16 (13%) |
128 |
7 (5%) |
|
Aspartate aminotransferase increased |
126 |
18 (14%) |
132 |
11 (8%) |
|
Alanine aminotransferase increased |
124 |
13 (10%) |
132 |
20 (15%) |
|
Blood bilirubin increased |
119 |
9 (8%) |
126 |
5 (4%) |
Study AAML0531
The safety evaluation of MYLOTARG in combination with chemotherapy in pediatric patients is based on data from AAML0531 [see Clinical Studies (14.1)] in randomized and treated patients (N = 520 MYLOTARG and chemotherapy and N = 517 chemotherapy alone). In the MYLOTARG arm of this study, 520 patients received Induction 1 and 326 patients received Intensification 2.
Safety data collected included only Grade 3 and 4 nonhematologic adverse events, deaths, VOD/SOS, and prolongation of neutropenia and thrombocytopenia.
Table 5 shows the Grade 3 or 4 adverse reactions (≥5%) in the MYLOTARG + chemotherapy or chemotherapy alone arms in patients with newly-diagnosed de novo AML in AAML0531.
In the MYLOTARG + chemotherapy arm, fatal adverse reactions (by grouped terms) were infection (14 [3%]), multi-organ failure (5 [1%]), anemia (1 [<1%]), and hemorrhage (3 [<1%]). In the chemotherapy arm, fatal adverse reactions included infection (7 [1%]), multi-organ failure (6 [1%]), hepatic failure (1 [<1%]), hypotension (3 [<1%]), and hemorrhage (3 [<1%]).
| Induction 1 | Intensification 2 | |||
|---|---|---|---|---|
| MYLOTARG + Chemotherapy
N = 520 n (%) | Chemotherapy alone
N = 517 n (%) | MYLOTARG + Chemotherapy
N = 326 n (%) | Chemotherapy alone
N = 304 n (%) |
|
|
||||
|
Infection* |
186 (36%) |
181 (35%) |
220 (67%) |
211 (69%) |
|
Febrile neutropenia |
167 (32%) |
157 (30%) |
79 (24%) |
68 (22%) |
|
Decreased appetite |
78 (15%) |
79 (15%) |
61 (19%) |
36 (12%) |
|
Hyperglycemia |
59 (11%) |
55 (11%) |
36 (11%) |
28 (9%) |
|
Mucositis* |
55 (11%) |
64 (12%) |
25 (8%) |
15 (5%) |
|
Hypoxia |
35 (7%) |
26 (5%) |
19 (6%) |
22 (7%) |
|
Hemorrhage* |
36 (7%) |
19 (4%) |
19 (6%) |
9 (3%) |
|
Transaminase Increased* |
33 (6%) |
24 (5%) |
23 (7%) |
13 (4%) |
|
Diarrhea |
21 (4%) |
36 (7%) |
15 (5%) |
10 (3%) |
|
Nausea |
21 (4%) |
18 (4%) |
23 (7%) |
10 (3%) |
|
Hypotension |
16 (3%) |
26 (5%) |
28 (9%) |
23 (8%) |
The addition of MYLOTARG to chemotherapy was associated with a higher incidence of prolonged thrombocytopenia and neutropenia particularly when used in Intensification 2. During Intensification 2, prolonged thrombocytopenia (platelets <50 Gi/L lasting past cycle Day 42 in the absence of active leukemia) was reported in 64% (190/297) of patients in the MYLOTARG + chemotherapy arm compared with 55% (146/264) in the chemotherapy alone arm. Prolonged neutropenia (neutrophils <0.5 Gi/L lasting past cycle Day 42 in the absence of active leukemia) occurred in 47% (142/300) versus 43% (118/275) of patients, respectively. The prolonged cytopenias were associated with more deaths in remission in the MYLOTARG + chemotherapy arm (29 [5%]) compared to the chemotherapy alone arm (15 [3%]).
VOD events were reported in 25 (5%) patients in the MYLOTARG + chemotherapy arm as well as 25 (5%) of the chemotherapy alone arm. VOD was fatal in 2 (<1%) and 7 (1%) patients in the MYLOTARG + chemotherapy arm and chemotherapy alone arm, respectively.
Monotherapy for Newly-Diagnosed CD33-positive AML
The safety evaluation of MYLOTARG (6 mg/m2 then 3 mg/m2, with 7 days between the doses) as monotherapy is based on a randomized, open-label, Phase 3 trial of MYLOTARG (N=118) versus best supportive care (BSC) (N=119) in patients with previously untreated AML who were considered ineligible for intensive chemotherapy in Study AML-19 [see Clinical Studies (14.1)].
The overall incidence of any Grade adverse reactions reported in AML-19 was 87% in the MYLOTARG arm and 90% in the BSC arm. The incidence of Grade greater than or equal to 3 adverse reactions was 61% in the MYLOTARG arm and 68% in the BSC arm. Death due to any Adverse Event was reported in the MYLOTARG arm of 19 (17%) compared to the BSC arm of 23 (20%).
| MYLOTARG
n=111 | Best Supportive Care
n=114 |
|||
|---|---|---|---|---|
| Any Grade | Grade ≥3 | Any Grade | Grade ≥3 | |
|
Liver |
57 (51%) |
8 (7%) |
52 (46%) |
7 (6%) |
|
Fatigue |
51 (46%) |
13 (12%) |
69 (61%) |
24 (21%) |
|
Infection |
49 (44%) |
39 (35%) |
48 (42%) |
39 (34%) |
|
Cardiac |
31 (28%) |
7 (6%) |
37 (33%) |
16 (14%) |
|
Bleeding |
28 (25%) |
14 (13%) |
34 (30%) |
14 (12%) |
|
Febrile neutropenia |
20 (18%) |
20 (18%) |
27 (24%) |
27 (24%) |
|
Metabolic |
18 (16%) |
4 (4%) |
17 (15%) |
7 (6%) |
|
Renal |
7 (6%) |
4 (4%) |
9 (8%) |
5 (4%) |
Monotherapy for Relapsed or Refractory CD33-positive AML
The adverse reactions described in this section reflect exposure to MYLOTARG 3 mg/m2 on Days 1, 4 and 7 as monotherapy in 57 patients with relapsed AML treated on MyloFrance-1 [see Clinical Studies (14.1)]. All 57 (100%) patients received the 3 planned doses of MYLOTARG.
During the treatment period, Grade 3 treatment-emergent adverse events (TEAEs) that occurred in greater than 1% patients included sepsis (32%), fever (16%), rash (11%), pneumonia (7%), bleeding (7%), mucositis (4%), pain (4%), diarrhea (2%), headaches (2%), tachycardia (2%), and lung edema (2%). No Grade 4 toxicity was observed. All grade TEAEs that occurred in greater than 15% of patients included fever (79%), infection (42%), increased AST (40%), bleeding (23%), nausea and vomiting (21%), constipation (21%), mucositis (21%), headache (19%), increased ALT (16%), and rash (16%). No infectious deaths occurred. Grade 1 or 2 hyperbilirubinemia developed in 4 (7%) patients. No episodes of VOD occurred. Seven patients received HSCT after MYLOTARG treatment. Three patients received an allogeneic BMT and 4 patients were treated with autologous BMT. No patients developed VOD following HSCT.
6.2 Postmarketing Experience
The following adverse drug reactions have been identified during post-approval use of MYLOTARG. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal Disorders: Neutropenic colitis1
Infections and Infestations: fungal lung infections including Pulmonary mycosis and Pneumocystis jirovecii pneumonia1; and bacterial infections including Stenotrophomonas infection
Renal and Urinary Disorders: Hemorrhagic cystitis1
Respiratory, Thoracic and Mediastinal Disorders: Interstitial pneumonia1
- 1
- including fatal events
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and findings from animal studies [see Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)], MYLOTARG can cause embryo-fetal harm when administered to a pregnant woman. There are no available data on MYLOTARG use in pregnant women to evaluate for a drug-associated risk. In animal reproduction studies, gemtuzumab ozogamicin caused embryo-fetal toxicity, including structural abnormalities and alterations to growth, at maternal systemic exposures that were greater than or equal to 0.4 times the exposure in patients at the maximum recommended dose based on AUC (see Data). Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Animal Data
In an embryo-fetal development study in rats, pregnant animals received daily intravenous doses up to 1.2 mg/m2/day gemtuzumab ozogamicin during the period of organogenesis. Embryo-fetal toxicities including fetal growth retardation as evidenced by decreased live fetal weights, incidence of fetal wavy ribs and delayed skeletal ossification were observed at greater than or equal to 0.15 mg/m2/day. Increased embryo-fetal lethality and fetal morphological anomalies (digital malformations, absence of the aortic arch, anomalies in the long bones in the forelimbs, misshapen scapula, absence of a vertebral centrum, and fused sternebrae) were observed at greater than or equal to 0.36 mg/m2/day. All doses with embryo-fetal effects were observed in the presence of maternal toxicity that included decreases in gestational body weight gain, food consumption, and gravid uterine weight. The lowest dose at which embryo-fetal effects were observed in rats (0.15 mg/m2/day) was 0.4 times the exposure in patients at the maximum recommended human dose, based on AUC.
8.2 Lactation
Risk Summary
There are no data on the presence of gemtuzumab ozogamicin or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed infant, advise women not to breastfeed during treatment with MYLOTARG and for at least 1 month after the final dose.
8.3 Females and Males of Reproductive Potential
MYLOTARG can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status in females of reproductive potential prior to initiating MYLOTARG.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with MYLOTARG and for at least 6 months after the last dose [see Nonclinical Toxicology (13.1)].
Males
Advise males with female partners of reproductive potential to use effective contraception during treatment with MYLOTARG and for at least 3 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Females
Based on findings in animals, MYLOTARG may impair female fertility [see Nonclinical Toxicology (13.1)].
Males
Based on findings in animals, MYLOTARG may impair male fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of MYLOTARG in combination with standard chemotherapy have been established in pediatric patients 1 month and older with newly-diagnosed de novo AML. The use of MYLOTARG for this indication is supported by evidence of effectiveness from adequate and well-controlled studies in adults with supportive data on safety and effectiveness in Study AAML0531 (NCT00372593) [see Adverse Reactions (6.1), Clinical Studies (14.1)]. AAML0531 included patients in the following age groups: 2 patients less than 27 days old, 94 patients 28 days to less than 2 years old, 225 patients 2 years to less than 12 years old, 175 patients 12 years old to less than 18 years old, and 36 patients 18 years or older in the MYLOTARG plus chemotherapy arm. The safety and effectiveness of MYLOTARG with standard chemotherapy in pediatric patients less than 1 month of age with newly-diagnosed de novo AML have not been established.
The safety and effectiveness of MYLOTARG as a single agent in pediatric patients with newly-diagnosed AML have not been established.
The safety and effectiveness of MYLOTARG as a single agent in the pediatric patients with relapsed or refractory AML is supported by a single-arm trial in 29 patients in the following age groups: 1 patient 1 month to less than 2 years old, 13 patients 2 years to less than 12 years old, and 15 patients 12 years to 18 years old. A literature review included an additional 96 patients with ages ranging from 0.2 to 21 years. No differences in efficacy and safety were observed by age. The information on this use is discussed throughout the labeling. The safety and effectiveness of MYLOTARG as a single agent in pediatric patients less than 2 years of age with relapsed or refractory AML have not been established.
8.5 Geriatric Use
Use of MYLOTARG in combination with daunorubicin and cytarabine in newly-diagnosed adult patients with de novo AML is supported by a randomized, controlled trial that included 50 patients greater than or equal to 65 years old. No overall differences in safety or effectiveness were observed between these subjects and younger subjects. Use of MYLOTARG monotherapy in newly-diagnosed adult patients with AML is supported by a randomized controlled trial with 118 patients treated with MYLOTARG. All patients were over the age of 60 years and 65% of patients were above 75 years. No overall differences in effectiveness were observed by age.
Use of MYLOTARG as single-agent treatment of relapsed or refractory AML is supported by a single-arm trial that included 27 patients 65 years or older. No overall differences in effectiveness were observed between these patients and younger patients. Elderly patients experienced a higher rate of fever and severe or greater infections.
11. Mylotarg Description
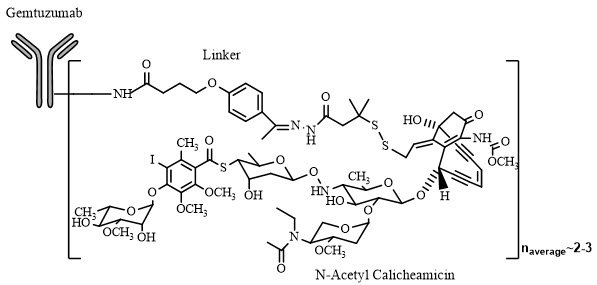
Gemtuzumab ozogamicin is an antibody-drug conjugate (ADC) composed of the CD33-directed monoclonal antibody (hP67.6; recombinant humanized immunoglobulin [Ig] G4, kappa antibody produced by mammalian cell culture in NS0 cells) that is covalently linked to the cytotoxic agent N-acetyl gamma calicheamicin. Gemtuzumab ozogamicin consists of conjugated and unconjugated gemtuzumab. The conjugated molecules differ in the number of activated calicheamicin derivative moieties attached to gemtuzumab. The number of conjugated calicheamicin derivatives per gemtuzumab molecule ranges from predominantly zero to 6, with an average of 2 to 3 moles of calicheamicin derivative per mole of gemtuzumab.
MYLOTARG (gemtuzumab ozogamicin) for injection is supplied as a sterile, white to off-white, preservative-free lyophilized cake or powder for intravenous administration. Each single-dose vial delivers 4.5 mg gemtuzumab ozogamicin. Inactive ingredients are dextran 40 (41.0 mg), sodium chloride (26.1 mg), sodium phosphate dibasic anhydrous (2.7 mg), sodium phosphate monobasic monohydrate (0.45 mg), and sucrose (69.8 mg). After reconstitution with 5 mL of Sterile Water for Injection USP, the concentration is 1 mg/mL of gemtuzumab ozogamicin with a deliverable volume of 4.5 mL (4.5 mg).
12. Mylotarg - Clinical Pharmacology
12.1 Mechanism of Action
Gemtuzumab ozogamicin is a CD33-directed antibody-drug conjugate (ADC). The antibody portion (hP67.6) recognizes human CD33 antigen. The small molecule, N-acetyl gamma calicheamicin, is a cytotoxic agent that is covalently attached to the antibody via a linker. Nonclinical data suggest that the anticancer activity of gemtuzumab ozogamicin is due to the binding of the ADC to CD33-expressing tumor cells, followed by internalization of the ADC-CD33 complex, and the intracellular release of N-acetyl gamma calicheamicin dimethyl hydrazide via hydrolytic cleavage of the linker. Activation of N-acetyl gamma calicheamicin dimethyl hydrazide induces double-strand DNA breaks, subsequently inducing cell cycle arrest and apoptotic cell death.
12.2 Pharmacodynamics
Saturation of a high percentage of CD33 antigenic sites is presumed to be required for maximum delivery of calicheamicin to leukemic blast cells. Near maximal peripheral CD33 saturation was observed across studies after gemtuzumab ozogamicin dosing at dose levels of 2 mg/m2 and above.
At 9 mg/m2 gemtuzumab ozogamicin (2 doses, 14 days apart), the risk for VOD increases as the Cmax of the first dose of gemtuzumab ozogamicin increases. The increase in VOD is more prominent in patients with prior stem cell transplantation.
12.3 Pharmacokinetics
There are no clinical PK data for the fractionated regimen. When gemtuzumab ozogamicin is administered at 9 mg/m2 (2 doses, 14 days apart), the Cmax following the first dose for patients who received 9 mg/m2 gemtuzumab ozogamicin was 3.0 mg/L and increased to 3.6 mg/L after the second dose.
Distribution
N-acetyl gamma calicheamicin dimethyl hydrazide is approximately 97% bound to human plasma proteins in vitro. Population PK analyses found the total volume of distribution of hP67.6 antibody (sum of V1 [6.31 L] and V2 [15.1 L]) to be approximately 21.4 L in patients.
Elimination
The clearance (CL) value of hP67.6 from plasma was 0.35 L/h after the first dose and 0.15 L/h after the second dose, a decrease of roughly 60%. The terminal plasma half-life (t½) for hP67.6 was 62 hours after the first dose and 90 hours after the second dose.
Metabolism
In vitro studies demonstrated that N-acetyl gamma calicheamicin dimethyl hydrazide is extensively metabolized, primarily via nonenzymatic reduction of the disulfide moiety.
Specific Populations
Age, race, sex, mild or moderate renal impairment (creatinine clearance [CLcr] 30–89 mL/min calculated by the Cockcroft-Gault equation) or mild hepatic impairment had no clinically significant effect on the pharmacokinetics of gemtuzumab ozogamicin. The pharmacokinetics of gemtuzumab ozogamicin in patients with severe renal impairment (CLcr 15–29 mL/min) or moderate (total bilirubin greater than 1.5× to 3.0× ULN) and severe hepatic impairment (total bilirubin greater than 3× ULN) is unknown.
In Vitro Studies
At clinically relevant concentrations, gemtuzumab ozogamicin had a low potential to:
- •
- Inhibit CYP450 Enzymes: CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5.
At clinically relevant concentrations, N-acetyl gamma calicheamicin dimethyl hydrazide had a low potential to:
- •
- Inhibit CYP450 Enzymes: CYP1A2, CYP2B6 , CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5.
- •
- Induce CYP450 Enzymes: CYP1A2, CYP2B6, and CYP3A4.
- •
- Inhibit UGT Enzymes: UGT1A1, UGT1A4, UGT1A6, UGT1A9, and UGT2B7.
- •
- Inhibit Drug Transporters: P-gp (P-glycoprotein), breast cancer resistance protein (BCRP), organic anion transporter (OAT)1 and OAT3, organic cation transporter (OCT)2, and organic anion transporting polypeptide (OATP)1B1 and OATP1B3.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Formal carcinogenicity studies have not been conducted with gemtuzumab ozogamicin. In toxicity studies, rats were dosed weekly for 6 weeks with gemtuzumab ozogamicin at doses up to 7.2 mg/m2/week. After 6 weeks of dosing, rats developed oval cell hyperplasia in the liver, which is considered a potentially preneoplastic finding, at 7.2 mg/m2/week (approximately 16 times the exposure in patients at the maximum recommended dose, based on AUC). Other preneoplastic or neoplastic changes observed with other antibody-calicheamicin conjugates in rats included basophilic and/or eosinophilic altered cell foci and hepatocellular adenomas. The relevance of these animal findings to humans is uncertain.
Gemtuzumab ozogamicin was clastogenic in vivo in the bone marrow of mice that received single doses greater than or equal to 22.1 mg/m2. This is consistent with the known induction of DNA breaks by calicheamicin. N-acetyl gamma calicheamicin dimethyl hydrazide (the released cytotoxic agent) was mutagenic in the bacterial reverse mutation assay and clastogenic in the in vitro micronucleus assay in human TK6 cells.
In a female fertility study, female rats were administered daily intravenous doses of gemtuzumab ozogamicin up to 1.08 mg/m2 for 14 days before mating with untreated male rats. Significant decreases in the numbers of corpora lutea and implants were observed at 1.08 mg/m2, and dose-related decreases and increases in the number of live and dead embryos were observed at doses tested (approximately 0.4 times the exposure in patients at the maximum recommended dose, based on AUC). Increased embryofetal lethality at ≥0.36 mg/m2 was observed in the presence of maternal toxicity that included decreases in gestational body weight and food consumption. Additional findings in female reproductive organs (ovarian atrophy and decreased numbers of follicles associated with atrophy of the uterus, vagina and mammary glands) occurred in rats and monkeys after dosing with other antibody-calicheamicin conjugates.
Fertility was assessed in male rats administered daily intravenous doses of gemtuzumab ozogamicin from 0.12 to 1.08 mg/m2 for 28 days, followed by mating with untreated females, either at the end of the dosing period or after a 9-week drug-free period. Male fertility index was decreased at doses ≥0.12 mg/m2 (approximately 1.2 times the exposure in patients at the maximum recommended dose, based on AUC). Effects on testes and epididymides occurred at ≥0.12 mg/m2, including smaller size and lower weights in addition to adverse effects on sperm. Partial recovery was noted for some effects. Additional effects in male reproductive organs occurred in repeat-dose toxicology studies and included effects on mammary gland, testes, and epididymides in rats at ≥2.4 mg/m2/week and effects on testes and epididymides in monkeys at 21.6 mg/m2/week. Testicular effects in male monkeys with other antibody-calicheamicin conjugates included degeneration of seminiferous tubules and decreased epididymidal sperm, which did not reverse following a 6-week drug-free period.
14. Clinical Studies
14.1 Newly-Diagnosed CD33-positive AML
Study ALFA-0701
MYLOTARG in combination with chemotherapy was evaluated in ALFA-0701 (NCT00927498), a multicenter, randomized, open-label Phase 3 study of 271 patients with newly-diagnosed de novo AML ages 50 to 70 years. Patients were randomized (1:1) to receive induction therapy consisting of daunorubicin (60 mg/m2 on Days 1 to 3) and cytarabine (200 mg/m2 on Days 1 to 7) (DA) with (n=135) or without (n=136) MYLOTARG 3 mg/m2 (up to maximum of one vial) on Days 1, 4, and 7. Patients who did not achieve a response after first induction could receive a second induction with daunorubicin and cytarabine alone (daunorubicin 35 mg/m2/day on Days 1 and 2, and cytarabine 1 g/m2 every 12 hours, on Day 1 to Day 3 without MYLOTARG). Patients with response received consolidation therapy with 2 courses of treatment including daunorubicin (60 mg/m2 on Day 1 of consolidation course 1; 60 mg/m2 on Days 1 and 2 of consolidation course 2) and cytarabine (1 g/m2 every 12 hours on Days 1 to 4) with or without MYLOTARG 3 mg/m2 (up to a maximum of one vial) on Day 1 according to their initial randomization. Patients who experienced remission were also eligible for allogeneic transplantation. An interval of at least 2 months between the last dose of MYLOTARG and transplantation was recommended.
The median age of the patients was 62 years (range, 50–70), 137 female and 134 male, and 88% had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 to 1 at baseline. Baseline characteristics were balanced between treatment arms with the exception of gender as a higher percentage of males were enrolled in the MYLOTARG arm (55%) than in the DA alone arm (44%). Overall, 59%, 65%, and 70% of patients had documented favorable/intermediate risk and 33%, 27%, and 21% had poor/adverse disease by the National Comprehensive Cancer Network (NCCN), European LeukemiaNet (ELN), and cytogenetic risk classifications, respectively. CD33 expression on AML blasts by flow cytometry harmonized from local laboratory results was determined in 194/271 (72%) patients overall. Few patients (14%) had low CD33 expression (less than 30% of blasts), and none had no expression of CD33.
Efficacy was established on the basis of event-free survival (EFS), measured from the date of randomization until induction failure, relapse, or death by any cause. Per protocol, induction failure was defined as failure to achieve CR or CRp in induction, and date of induction failure was defined as date of marrow evaluation after the last course of induction. Median EFS was 17.3 months in the MYLOTARG arm versus 9.5 months in the control arm; hazard ratio (HR) 0.56 (95% CI: 0.42–0.76); 2-sided p less than 0.001 by log-rank test.
In an exploratory analysis of EFS defined as failure to achieve CR in induction, relapse, or death from any cause and using the date of randomization as the date of induction failure, median EFS was 13.6 months for MYLOTARG + DA and 8.8 months for DA with HR 0.68 (95% CI: 0.51–0.91).
The Kaplan-Meier plot for per-protocol EFS is shown in Figure 1. There was no statistically significant difference between treatment arms in overall survival.
Figure 1. Kaplan-Meier Plot of Event-Free Survival (mITT Population) ALFA-0701 Trial
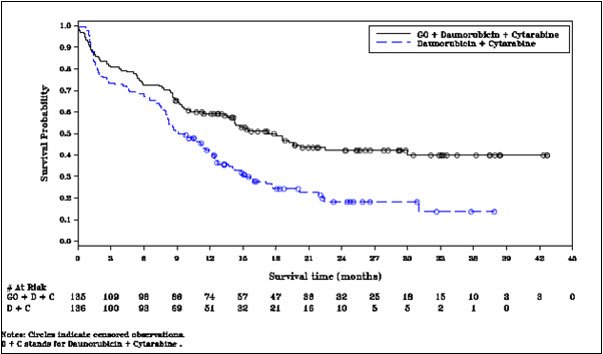
Abbreviations: C=cytarabine; D=daunorubicin; GO=gemtuzumab ozogamicin; mITT=modified intent-to-treat.
Study AAML0531
MYLOTARG in combination with chemotherapy was evaluated in AAML0531 (NCT00372593), a multicenter, randomized study of 1,063 patients with newly-diagnosed AML ages 0 to 29 years. Patients were randomized to 5-cycle chemotherapy alone or with a single dose of MYLOTARG (3 mg/m2/dose) administered once on Day 6 in Induction 1 and once on Day 7 in Intensification 2. All patients proceeded to Induction 2 regardless of remission status after Induction 1. In the absence of active disease, a neutrophil count (ANC) >1 Gi/L and a platelet count >75 Gi/L was recommended before proceeding with subsequent cycles of therapy. Patients not in remission after Induction 2 discontinued protocol therapy permanently. All other patients proceeded to Intensification 1. Patients with high- and intermediate-risk disease with 5/6 or 6/6 matched family donors (MFD) proceeded to HSCT following Intensification 1. Patients with high-risk disease proceeded to HSCT with an alternative donor if no MFD was available. All patients with low-risk disease and any high- and intermediate-risk patients without appropriate donors proceeded with Intensification 2 with or without MYLOTARG according to their initial randomization, followed by Intensification 3. All patients in remission were to proceed on to Intensification 2 or allogeneic HSCT. In Intensification 2, patients received MYLOTARG according to the initial randomization. Patients in remission after Intensification 2 proceeded to Intensification 3.
There were 532 patients randomized to treatment with MYLOTARG + chemotherapy and 531 to chemotherapy alone. Overall, 94% of patients were <18 years of age, and 6% were adults; median age was 9.0 years (range: 0–29 years). The patients were 49% male, 51% female, 73% White, 11% Black, 5% Asian, 11% other or missing race, and 18% Hispanic. The proportion of patients in each disease risk group: low risk (23% vs 23%), intermediate risk (57% vs 57%), and high risk (15% vs 17%).
Supportive evidence of efficacy was provided by event-free survival (EFS), measured from the date of study entry until induction failure, relapse, or death by any cause. Induction failure was defined as failure to achieve CR by the end of Induction 2 period, and date of induction failure was defined as Day 1 on study. The EFS hazard ratio was 0.84 (95% CI: 0.71–0.99). The estimated percentage of patients free of induction failure, relapse, or death at five years was 48% (95% CI: 43%–52%) in the MYLOTARG + chemotherapy arm versus 40% (95% CI: 36%–45%) in the chemotherapy alone arm.
The Kaplan-Meier plot for EFS is shown in Figure 2. No difference between treatment arms in overall survival was demonstrated.
Figure 2. Kaplan-Meier Plot of Event-Free Survival (Full Analysis Set) Study AAML0531 Trial
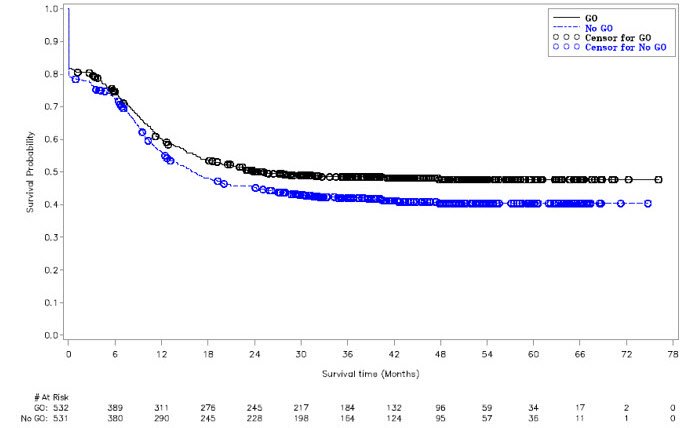
Abbreviations: GO=gemtuzumab ozogamicin.
Study AML-19
MYLOTARG single-agent therapy was evaluated in Study AML-19 (NCT00091234), a multicenter, randomized, open-label Phase 3 study comparing MYLOTARG to best supportive care (BSC) for patients with newly-diagnosed AML who were a) greater than 75 years of age or b) 61 to 75 years of age with a World Health Organization performance status (WHO PS) greater than 2 or were unwilling to receive intensive chemotherapy. Patients were randomized 1:1 and stratified by age (61–75 vs 76–80 years vs ≥81 years), CD33 positivity of bone marrow blasts (less than 20 % vs 20–80% vs greater than 80% vs unknown), initial white blood cell count (less than 30 vs greater than or equal to 30 × 109/L), WHO PS (0–1 vs 2 vs 3–4), and institution.
During induction, MYLOTARG 6 mg/m2 was given on Day 1 and MYLOTARG 3 mg/m2 was given on Day 8. Patients with no evidence of disease progression or significant toxicities after MYLOTARG induction received continuation therapy as outpatients with up to 8 courses of treatment including MYLOTARG 2 mg/m2 on Day 1 every 4 weeks. Patients continued therapy if they did not experience significant toxicities, relapse, or disease progression. BSC included standard supportive care measures and hydroxyurea or other anti-metabolites for palliative purposes.
In total, 118 patients were randomized to treatment with MYLOTARG and 119 patients to BSC. Overall, the median age of patients was 77 years (range, 62–88 years), and most patients (65%) had a WHO PS of 0 to 1 at baseline. Baseline characteristics were balanced between treatment arms with the exception of gender and cytogenetics. Compared to the BSC arm, the MYLOTARG arm had a higher percentage of females (52% vs 39%) and patients with favorable/intermediate risk cytogenetics (50% vs 38%). The proportion with adverse cytogenetics was similar between arms (28% vs 27%). Fewer patients on the MYLOTARG arm had missing cytogenetics data (22% vs 35%). CD33 expression on AML blasts by flow cytometry at a centralized location was determined in 235/237 (99%) patients; 10% had CD33 expression less than 20%.
The efficacy of MYLOTARG was established on the basis of improvement in overall survival (OS). The hazard ratio (HR) for OS was 0.69 (95% CI: 0.53–0.90) (2-sided p=0.005 by log-rank test). Median OS was 4.9 months in the MYLOTARG arm versus 3.6 months in the control arm.
14.2 Relapsed or refractory CD33-positive AML
Study MyloFrance-1
The efficacy of MYLOTARG as a single agent was evaluated in MyloFrance-1 a phase 2, single-arm, open-label study in adults with CD33-positive AML in first relapse. Patients with secondary leukemia or a prior autologous or allogeneic stem cell transplantation were excluded. Study treatment included a single course of MYLOTARG 3 mg/m2 on Days 1, 4, and 7. Consolidation therapy consisted of cytarabine intravenously every 12 hours for 3 days. The cytarabine dose was 3 g/m2 for patients less than 55 years old and 1 g/m2 for patients 55 years or older and/or patients with a creatinine clearance below 50 mL/minute. Hematopoietic stem cell transplantation (HSCT) was allowed after treatment with MYLOTARG, but it was recommended to delay HSCT by at least 90 days following MYLOTARG.
There were 57 patients treated with MYLOTARG. Overall, the median age of patients was 64 years (range 22–80 years). The median duration of first remission was 10 months. Forty-four (78%) patients had intermediate-risk and 12 (22%) poor-risk cytogenetics.
The efficacy of MYLOTARG was established on the basis of complete remission (CR) rate and duration of remission. Fifteen (26%; 95% CI 16% – 40%) patients achieved CR following a single course of MYLOTARG. Median relapse-free survival, measured from the first documentation of CR to the date of relapse or death, was 11.6 months.
16. How is Mylotarg supplied
MYLOTARG (gemtuzumab ozogamicin) for injection is a white to off-white lyophilized cake or powder supplied in a carton (NDC 0008-4510-01) containing one 4.5 mg single-dose vial [see Dosage and Administration (2)].
17. Patient Counseling Information
Hepatotoxicity, Including Veno-occlusive Liver Disease (VOD)
Inform patients that liver problems, including severe, life-threatening, or fatal VOD may develop during MYLOTARG treatment. Prior to receiving MYLOTARG, inform patients who previously received, or will receive an HSCT that they may be at increased risk for developing VOD. Inform patients that the risk of developing VOD after an allogeneic HSCT is increased after receiving treatment with MYLOTARG. Inform patients that signs or symptoms of liver toxicity, including rapid weight gain, right upper quadrant pain and tenderness, hepatomegaly, and ascites should be monitored regularly during treatment, but these symptoms may not identify all patients at risk or prevent the complications of liver toxicity. Inform patients that liver problems may require dosing interruption or permanent discontinuation of MYLOTARG [see Warnings and Precautions (5.1)].
Hemorrhage
Inform patients that decreased platelet counts, which may be life-threatening, may develop during MYLOTARG treatment and that complications associated with decreased platelet counts may include bleeding/hemorrhage events, which may be life-threatening or fatal. Inform patients to report signs and symptoms of bleeding/hemorrhage during treatment with MYLOTARG. Inform patients that severe bleeding/hemorrhage may require dosing interruption or permanent discontinuation of MYLOTARG [see Warnings and Precautions (5.3)].
Infusion-Related Reactions
Advise patients to contact their health care provider if they experience signs and symptoms of infusion-related reactions, including symptoms such as fever, chills, rash, or breathing problems [see Warnings and Precautions (5.2)].
Embryo-Fetal Toxicity
Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.6), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with MYLOTARG and for at least 6 months after the last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with MYLOTARG and for at least 3 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with MYLOTARG and for at least 1 month after the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise males and females of reproductive potential that MYLOTARG may impair fertility [see Use in Specific Populations (8.3)].
This product's labeling may have been updated. For the most recent prescribing information, please visit www.pfizer.com.

US License No. 003
LAB-0868-8.0


| MYLOTARG
gemtuzumab ozogamicin injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Wyeth Pharmaceuticals LLC, a subsidiary of Pfizer Inc. (113008515) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pharmacia & Upjohn Company LLC | 618054084 | PACK(0008-4510) , LABEL(0008-4510) , ANALYSIS(0008-4510) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Wyeth Pharmaceutical Division of Wyeth Holdings LLC | 054065909 | ANALYSIS(0008-4510) , MANUFACTURE(0008-4510) , API MANUFACTURE(0008-4510) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC | 174350868 | API MANUFACTURE(0008-4510) , ANALYSIS(0008-4510) | |
Biological Products Related to Mylotarg
Find detailed information on biosimilars for this medication.
More about Mylotarg (gemtuzumab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (2)
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: CD33 monoclonal antibodies
- Breastfeeding
- En español