Enspryng: Package Insert / Prescribing Info
Package insert / product label
Generic name: satralizumab
Dosage form: injection, solution
Drug class: Interleukin inhibitors
Medically reviewed by Drugs.com. Last updated on May 19, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
ENSPRYNG® (satralizumab-mwge) injection, for subcutaneous use
Initial U.S. Approval: 2020
Indications and Usage for Enspryng
ENSPRYNG is an interleukin-6 (IL-6) receptor antagonist indicated for the treatment of neuromyelitis optica spectrum disorder (NMOSD) in adult patients who are anti-aquaporin-4 (AQP4) antibody positive. (1)
Enspryng Dosage and Administration
- Hepatitis B virus, tuberculosis, and liver transaminase screening is required before the first dose. (2.1)
- Prior to every use, determine if there is an active infection. (2.2)
- The recommended loading dosage of ENSPRYNG for the first three administrations is 120 mg by subcutaneous injection at Weeks 0, 2, and 4, followed by a maintenance dosage of 120 mg every 4 weeks. (2.2)
- See Full Prescribing Information for important preparation and administration instructions. (2.3)
Dosage Forms and Strengths
Injection: 120 mg/mL in a single-dose prefilled syringe (3)
Contraindications
Warnings and Precautions
- Infections: Delay ENSPRYNG administration in patients with an active infection until the infection is resolved. Vaccination with live or live-attenuated vaccines is not recommended during treatment. (5.1)
- Elevated Liver Enzymes: Monitor ALT and AST levels during treatment; interruption of ENSPRYNG may be required. (5.2)
- Decreased Neutrophil Counts: Monitor neutrophils during treatment. (5.3)
Adverse Reactions/Side Effects
The most common adverse reactions (incidence at least 15%) are nasopharyngitis, headache, upper respiratory tract infection, gastritis, rash, arthralgia, extremity pain, fatigue, and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2022
Full Prescribing Information
1. Indications and Usage for Enspryng
ENSPRYNG is indicated for the treatment of neuromyelitis optica spectrum disorder (NMOSD) in adult patients who are anti-aquaporin-4 (AQP4) antibody positive.
2. Enspryng Dosage and Administration
2.1 Assessments Prior to the First Dose of ENSPRYNG
Hepatitis B Virus Screening
Prior to initiating ENSPRYNG, perform Hepatitis B virus (HBV) screening. ENSPRYNG is contraindicated in patients with active HBV confirmed by positive results for surface antigen [HBsAg] and anti-HBV tests. For patients who are negative for HBsAg and positive for HB core antibody [HBcAb+] or are carriers of HBV [HBsAg+], consult liver disease experts before starting and during treatment with ENSPRYNG [see Contraindications (4) and Warnings and Precautions (5.1)].
Tuberculosis Screening
Prior to initiating ENSPRYNG, evaluate for active tuberculosis and test for latent infection. For patients with active tuberculosis or positive tuberculosis screening without a history of appropriate treatment, consult infectious disease experts before initiating treatment with ENSPRYNG [see Contraindications (4) and Warnings and Precautions (5.1)].
Liver Transaminase Screening
Liver transaminases and serum bilirubin should be assessed prior to initiation of treatment with ENSPRYNG [see Warnings and Precautions (5.2)].
Caution should be exercised when considering initiation of ENSPRYNG treatment in patients whose aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels are greater than 1.5 times the upper limit of normal (ULN).
Vaccinations
Because vaccination with live-attenuated or live vaccines is not recommended during treatment with ENSPRYNG, administer all immunizations according to immunization guidelines at least 4 weeks prior to initiation of ENSPRYNG for live or live-attenuated vaccines and, whenever possible, at least 2 weeks prior to initiation of ENSPRYNG for non-live vaccines [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage
For subcutaneous use only.
Prior to every use of ENSPRYNG, advise patients to consult with their healthcare professional (HCP) if they suspect an active infection, including localized infections. In case of active infection, delay use of ENSPRYNG until the infection is resolved [see Warnings and Precautions (5.1)].
The recommended loading dosage of ENSPRYNG for the first three administrations is 120 mg by subcutaneous injection at Weeks 0, 2, and 4, followed by a maintenance dosage of 120 mg every 4 weeks.
Missed Dose
If a dose of ENSPRYNG is missed for any reason other than increases in liver enzymes [see Dosage and Administration (2.4)], administer as described in Table 1.
| Last Dose Administered | Recommended Dosage for Delayed or Missed Doses |
|---|---|
|
|
| Less than 8 weeks during the maintenance period or missed a loading dose | Administer 120 mg by subcutaneous injection as soon as possible, and do not wait until the next planned dose. Maintenance period After the delayed or missed dose is administered, reset the dose schedule to every 4 weeks. Loading period If the second loading dose is delayed or missed, administer as soon as possible and administer the 3rd and final loading dose 2 weeks later. If the third loading dose is delayed or missed, administer as soon as possible and administer the 1st maintenance dose 4 weeks later. |
| 8 weeks to less than 12 weeks | 120 mg by subcutaneous injection at 0* and 2 weeks, followed by 120 mg every 4 weeks. |
| 12 weeks or longer | 120 mg by subcutaneous injection at 0*, 2, and 4 weeks followed by 120 mg every 4 weeks. |
2.3 Important Administration Instructions
- ENSPRYNG is intended for patient self-administration by subcutaneous injection under the guidance of a health care professional (HCP). After proper training in subcutaneous injection technique, a patient may self-inject ENSPRYNG or the patient's caregiver may administer ENSPRYNG, if the HCP determines that it is appropriate. See ENSPRYNG "Instructions for Use" (IFU) for more detailed instructions on the preparation and administration of ENSPRYNG.
- Patients or caregivers should seek immediate medical attention if the patient develops symptoms of a serious allergic reaction and should not administer further doses until evaluated by a HCP [see Contraindications (4) and Warning and Precautions (5.4)].
- Prior to use, remove the prefilled syringe from the refrigerator and allow to sit at room temperature outside of the carton for 30 minutes. Do not warm ENSPRYNG in any other way.
- Inspect visually for particulate matter and discoloration prior to administration. ENSPRYNG solution should be clear and colorless to slightly yellow. Do not use ENSPRYNG if the solution is cloudy, discolored, or contains particles, or if any part of the prefilled syringe appears to be damaged.
- Instruct patients to inject the full amount in the syringe (1 mL), which provides 120 mg of ENSPRYNG, according to the directions provided in the IFU.
- Administer ENSPRYNG by subcutaneous injection in the abdomen or thigh. Rotate injection sites with each administration. Do not give injection into moles, scars, or areas where the skin is tender, bruised, red, hard, or not intact.
2.4 Safety Monitoring During Treatment
Liver Transaminases
Monitor ALT and AST levels every 4 weeks for the first 3 months of treatment with ENSPRYNG, followed by every 3 months for one year, and thereafter as clinically necessary [see Warnings and Precautions (5.2)].
If an ALT or AST elevation of greater than 5 times the ULN occurs, discontinue ENSPRYNG as follows:
- If associated with any bilirubin elevation, discontinue ENSPRYNG, and reinitiation is not recommended.
- If not associated with any bilirubin elevation above the ULN, when the ALT or AST level has returned to the normal range and following a benefit-risk assessment of the patient, treatment with ENSPRYNG can be restarted per the schedule in Table 2.
If treatment is restarted, the liver parameters must be closely monitored, and if any subsequent increase in ALT/AST and/or bilirubin above the ULN is observed, ENSPRYNG should be discontinued, and another reinitiation is not recommended.
Table 2 Recommended Dosage for Restart of Treatment After Liver Transaminase Elevation Last Dose Administered Recommended Dosage for Restart of Treatment - *
- "0 weeks" refers to time of the first administration after the missed dose.
Less than 12 weeks Restart at a dosage of 120 mg by subcutaneous injection every 4 weeks. 12 weeks or longer Restart at a dose of 120 mg by subcutaneous injection at Weeks 0*, 2, and 4, followed by a dosage of 120 mg every 4 weeks.
Neutrophil Counts
Monitor neutrophils 4 to 8 weeks after initiation of therapy and thereafter at regular clinically determined intervals. If the neutrophil count is below 1.0 × 109/L and confirmed by repeat testing, ENSPRYNG should be interrupted until the neutrophil count is > 1.0 × 109/L [see Warnings and Precautions (5.3)].
3. Dosage Forms and Strengths
Injection: 120 mg/mL clear, and colorless to slightly yellow solution in single-dose prefilled syringe.
4. Contraindications
ENSPRYNG is contraindicated in patients with:
- A known hypersensitivity to satralizumab or any of the inactive ingredients [see Warnings and Precautions (5.4)]
- Active Hepatitis B infection [see Warnings and Precautions (5.1)]
- Active or untreated latent tuberculosis [see Warnings and Precautions (5.1)]
5. Warnings and Precautions
5.1 Infections
An increased risk of infections, including serious and potentially fatal infections, has been observed in patients treated with IL-6 receptor antagonists, including ENSPRYNG.
The most common infections reported in a randomized clinical trial of patients treated with ENSPRYNG who were not on other chronic immunosuppressant therapies (Study 1), and that occurred more often than in patients receiving placebo, were nasopharyngitis (12%) and cellulitis (10%). The most common infections in patients who were on an additional concurrent immunosuppressant, and that occurred more often than in patients receiving placebo, were nasopharyngitis (31%), upper respiratory infection (19%), and pharyngitis (12%).
Delay ENSPRYNG administration in patients with an active infection, including localized infections, until the infection is resolved.
Hepatitis B Virus (HBV) Reactivation
Risk of HBV reactivation has been observed with other immunosuppressant therapies. Patients with chronic HBV infection were excluded from clinical trials. Perform HBV screening in all patients before initiation of treatment with ENSPRYNG. Do not administer ENSPRYNG to patients with active hepatitis. For patients who are chronic carriers of HBV [HBsAg+] or are negative for HBsAg and positive for HB core antibody [HBcAb+], consult liver disease experts before starting and during treatment with ENSPRYNG.
Tuberculosis
Tuberculosis has occurred in patients treated with other interleukin-6 receptor antagonists. Patients should be evaluated for tuberculosis risk factors and tested for latent infection prior to initiating ENSPRYNG. Consider anti-tuberculosis therapy prior to initiation of ENSPRYNG in patients with a history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent tuberculosis but having risk factors for tuberculosis infection. Consult infectious disease experts regarding whether initiating anti-tuberculosis therapy is appropriate before starting treatment. Patients should be monitored for the development of symptoms and signs of tuberculosis with ENSPRYNG, even if initial tuberculosis testing is negative.
Vaccinations
Live or live-attenuated vaccines should not be given concurrently with ENSPRYNG because clinical safety has not been established. Administer all immunizations according to immunization guidelines at least 4 weeks prior to initiation of ENSPRYNG for live or live-attenuated vaccines and, whenever possible, at least 2 weeks prior to initiation of ENSPRYNG for non-live vaccines.
5.2 Elevated Liver Enzymes
Mild and moderate elevations of liver enzymes have been observed in patients treated with ENSPRYNG at a higher incidence than in patients receiving placebo [see Adverse Reactions (6.1)].
ALT and AST levels should be monitored every 4 weeks for the first 3 months of treatment, followed by every 3 months for one year, and thereafter, as clinically indicated [see Dosage and Administration (2.4)].
5.3 Decreased Neutrophil Counts
Decreases in neutrophil counts were observed in patients treated with ENSPRYNG at a higher incidence than placebo [see Adverse Reactions (6.1)].
Neutrophil counts should be monitored 4 to 8 weeks after initiation of therapy, and thereafter at regular clinically determined intervals [see Dosage and Administration (2.4)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Infections [see Warnings and Precautions (5.1)]
- Elevated Liver Enzymes [see Warnings and Precautions (5.2)]
- Decreased Neutrophil Counts [see Warnings and Precautions (5.3)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice.
The safety of ENSPRYNG was evaluated in two randomized, placebo-controlled clinical trials [Study 1 evaluated ENSPRYNG without concurrent immunosuppressive therapy (IST) and Study 2 evaluated ENSPRYNG with concurrent IST], which included 41 anti-AQP4 seropositive patients treated with ENSPRYNG in Study 1 and 26 anti-AQP4 seropositive patients treated with ENSPRYNG in Study 2 [see Clinical Studies (14)]. In the double-blind, controlled period, the median exposure time on ENSPRYNG treatment was approximately 2 years in Study 1 and approximately 3 years in Study 2. The median exposure time on placebo treatment was approximately 1 year in both Study 1 and Study 2.
Adverse reactions that occurred in Study 1 and Study 2 in more than 5% of patients treated with ENSPRYNG, and at a greater incidence than in patients who received placebo, are shown in Table 3 and Table 4, respectively. The most common adverse reactions (15% or greater with ENSPRYNG in either) were nasopharyngitis, headache, upper respiratory tract infection, gastritis, rash, arthralgia, extremity pain, fatigue, and nausea.
| Adverse Reaction | ENSPRYNG (N = 41) % | PLACEBO (N = 23) % |
|---|---|---|
| Rash | 17 | 0 |
| Arthralgia | 17 | 0 |
| Pain in extremity | 15 | 9 |
| Fatigue | 15 | 4 |
| Nausea | 15 | 9 |
| Nasopharyngitis | 12 | 4 |
| Pruritus | 10 | 0 |
| Depression | 10 | 0 |
| Cellulitis | 10 | 0 |
| Neutropenia | 10 | 4 |
| Blood creatine phosphokinase increased | 10 | 4 |
| Fall | 10 | 4 |
| Adverse Reaction | ENSPRYNG + IST (N = 26) % | PLACEBO + IST (N = 26) % |
|---|---|---|
| Nasopharyngitis | 31 | 15 |
| Headache | 27 | 12 |
| Upper respiratory tract infection | 19 | 12 |
| Gastritis | 15 | 0 |
| Arthralgia | 12 | 0 |
| Pharyngitis | 12 | 8 |
Injection-Related Reactions
In Study 1 and Study 2, injection-related reactions were reported in 9% of patients treated with ENSPRYNG compared with 8% in patients receiving placebo. These reactions in the ENSPRYNG-treated patients were predominantly mild to moderate in severity, and most occurred within 24 hours after the injection. The most commonly reported systemic symptom was diarrhea. The reported local injection site reactions were pruritus, injection site reaction, and skin mass.
Infections
In Study 1, the rate of patients with infections was 51 patients/100 patient-years (95% CI: 32, 78) in patients treated with ENSPRYNG compared with 108 patients/100 patient-years (95% CI: 52, 199) in patients receiving placebo. The rate of patients with serious infections was 5 patients/100 patient-years (95% CI: 1, 14) in patients treated with ENSPRYNG compared with 4 patients/100 patient-years (95% CI: 0, 21) in patients receiving placebo.
In Study 2, the rate of patients with infections was 168 patients/100 patient-years (95% CI: 100, 265) in patients treated with ENSPRYNG compared with 143 patients/100 patient-years (95% CI: 83, 229) in patients treated with placebo. The rate of patients with serious infections was 4 patients/100 patient-years (95% CI: 1, 15) in patients treated with ENSPRYNG compared with 10 patients/100 patient-years (95% CI: 2, 28) in patients receiving placebo.
Laboratory Abnormalities
Decreased Neutrophil Count
Of the patients treated with ENSPRYNG, 10% had neutrophils below 1 × 109/L compared to 9% in placebo in Study 1. In Study 2, 15% patients had neutrophils below 1 × 109/L compared to 4% in placebo. There was one patient in Study 1 treated with ENSPRYNG with neutrophil counts < 0.5 × 109/L, and one patient in Study 2 discontinued ENSPRYNG because of neutropenia.
Decreased Platelet Count
In Study 1, a shift in platelet count decreases from normal at baseline to below the lower limit of normal (LLN) occurred in 26% of patients treated with ENSPRYNG compared to 5% of patients receiving placebo. In Study 2, decreases in platelet counts from normal at baseline to below the LLN occurred in 35% of patients treated with ENSPRYNG and in 17% of patients receiving placebo. None of the patients had a decrease in platelet count to less than 50 × 109/L.
Elevated Liver Enzymes
In Study 1, increases from normal at baseline to above ULN in ALT or AST occurred in 43% and 25% of patients treated with ENSPRYNG, respectively, compared to 13% and 9% of patients receiving placebo. In Study 2, increases from normal at baseline to above the ULN in ALT or AST occurred in 8% and 8% of patients treated with ENSPRYNG, respectively, compared to 12% and 19% of patients receiving placebo.
In Study 1 and Study 2 combined, elevations of ALT or AST greater than 3 times the ULN occurred in 3% of patients treated with ENSPRYNG, compared to no patients treated with placebo. These elevations were not associated with increases in total bilirubin. One patient receiving ENSPRYNG in Study 2 had an elevation of ALT above 5 times the ULN, which was observed 4 weeks after initiation of therapy, normalizing 78 days after discontinuation of ENSPRYNG.
Lipid Abnormalities
In Study 1 and Study 2, elevations in total cholesterol above 7.75 mmol/L (300 mg/dl) occurred in 12% and 15% of patients treated with ENSPRYNG, respectively, compared to no patients receiving placebo.
Elevations in triglycerides above 3.42 mmol/L (300 mg/dl) occurred in 27% and 12% of patients treated with ENSPRYNG in Study 1 and Study 2, respectively, compared to 13% and 8% of patients receiving placebo.
Fibrinogen Levels
In Study 1, the median percent reduction in fibrinogen was 38% in patients treated with ENSPRYNG compared to 5% in patients receiving placebo. In Study 2, the median percent reduction in fibrinogen level was 33% in patients treated with ENSPRYNG compared to 0% in patients receiving placebo.
Complement Levels
In Study 1, the median percent reduction in the C3 and C4 components of complement was 23% and 50% in patients treated with ENSPRYNG, respectively, compared to 0% and 1% patients receiving placebo. In Study 2, the median percent reduction in the C3 and C4 components of complement was 20% and 53% in patients treated with ENSPRYNG, respectively, compared to 0% and 1% in patients receiving placebo.
Body Weight
In the pool of Studies 1 and 2, body weight increases of at least 7% from baseline occurred in 28% of patients treated with ENSPRYNG compared to 8% of patients receiving placebo.
Body weight increases of at least 15% from baseline occurred in 4% of patients treated with ENSPRYNG compared to 4% of patients receiving placebo.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of anti-satralizumab-mwge antibodies in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In Study 1 and Study 2, anti-drug-antibodies (ADAs) were observed in 73% and 38% of patients receiving ENSPRYNG in the double-blind period, respectively. The ability of these ADAs to neutralize satralizumab-mwge binding is unknown. Patients with higher body weight and lower exposure were more likely to develop ADAs (irrespective of treatment with IST). Exposure was lower in ADA positive patients. Although anti-satralizumab-mwge antibody development was not found to affect the efficacy of ENSPRYNG in these patients, the available data are too limited to make definitive conclusions. Immunogenicity does not have a clinically-relevant impact on safety. Based on the available information, neither dose interruption nor modification is warranted in those patients who develop ADAs.
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ENSPRYNG during pregnancy. Healthcare providers are encouraged to register patients and pregnant women are encouraged to register themselves by calling 1-833-277-9338.
Risk Summary
There are no adequate data on the developmental risk associated with the use of ENSPRYNG in pregnant women. In an animal reproduction study, no adverse effects on maternal animals or fetal development were observed in pregnant monkeys and their offspring, with administration of satralizumab-mwge at doses up to 50 mg/kg/week (see Data).
In the U.S. general population, the estimated background risk of major birth defect and miscarriage in clinically recognized pregnancies is 2 – 4% and 15 – 20%, respectively. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown.
Clinical Considerations
Fetal/neonatal adverse reactions
Monoclonal antibodies are increasingly transported across the placenta as pregnancy progresses, with the largest amount transferred during the third trimester. Risks and benefits should be considered prior to administering live or live-attenuated vaccines to infants exposed to ENSPRYNG in utero [see Warnings and Precautions (5.1)].
Data
Animal Data
Weekly subcutaneous administration of satralizumab-mwge (0, 2, or 50 mg/kg) to monkeys throughout pregnancy resulted in no adverse effects on postnatal development of the offspring; however, immune function was impaired in offspring at both doses. Plasma exposures (Cave) in dams at the low and high doses were approximately 3 and 100 times, respectively, that in humans at the recommended monthly maintenance dose of 120 mg.
8.2 Lactation
Risk Summary
No information is available on the presence of satralizumab-mwge in human milk, the effects of the satralizumab-mwge on the breastfed infant, or the effects of the satralizumab-mwge on milk production. Satralizumab-mwge was excreted in the milk of lactating monkeys administered satralizumab-mwge throughout pregnancy. Human IgG is excreted in human milk and the potential for absorption in the infant is unknown. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ENSPRYNG and any potential adverse effects on the breastfed infant from ENSPRYNG or from the underlying maternal condition.
8.5 Geriatric Use
Clinical studies of ENSPRYNG did not include sufficient numbers of patients aged 65 years and older to determine whether they respond differently from younger patients. However, population pharmacokinetic analyses in patients with NMOSD did not show that age affected the pharmacokinetics of satralizumab-mwge [see Clinical Pharmacology (12.3)]. In general, caution should be used when dosing the elderly, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant diseases or other drug therapy.
11. Enspryng Description
Satralizumab-mwge is a recombinant humanized anti-human interleukin 6 (IL-6) receptor monoclonal antibody based on a human IgG2 framework. Each light chain and heavy chain consists of 214 and 443 amino acids, respectively. Satralizumab-mwge is a glycoprotein with an approximate molecular weight of 143 kDa and is produced by recombinant DNA technology in Chinese hamster ovary cells. The binding of satralizumab-mwge to the IL-6 receptor is pH-sensitive.
ENSPRYNG (satralizumab-mwge) injection for subcutaneous administration is supplied as a sterile, clear, colorless to slightly yellow solution with no preservative with an approximate pH of 6. ENSPRYNG is supplied in a single-dose prefilled syringe. Each syringe delivers 1 mL of solution containing 120 mg of satralizumab-mwge, L-arginine (26.1 mg), L-histidine (3.1 mg), poloxamer 188 (0.5 mg), L-aspartic acid (pH adjustment), and Water for Injection, USP.
12. Enspryng - Clinical Pharmacology
12.1 Mechanism of Action
The precise mechanism by which satralizumab-mwge exerts therapeutic effects in NMOSD is unknown but is presumed to involve inhibition of IL-6-mediated signaling through binding to soluble and membrane-bound IL-6 receptors.
12.2 Pharmacodynamics
The relationship between any of the pharmacodynamic effects of ENSPRYNG and clinical outcomes in NMOSD is unknown.
12.3 Pharmacokinetics
The pharmacokinetics of ENSPRYNG have been characterized both in Japanese and Caucasian healthy volunteers, and in NMOSD patients. The pharmacokinetics in NMOSD patients using the recommended dose were characterized using population pharmacokinetic analysis methods based on a database of 154 patients.
The concentration-time course of ENSPRYNG in patients with NMOSD was accurately described by a two-compartment population pharmacokinetic model with parallel linear and target-mediated (Michaelis-Menten) elimination and first-order subcutaneous absorption. ENSPRYNG clearance and volume parameters allometrically scaled by body weight (through power function with the fixed power coefficient of 0.75 and 1 for clearance and volume parameters, respectively). Body weight was shown to be a significant covariate, with clearance and Vc for patients weighing 123 kg (97.5th percentile of the weight distribution) increased by 71.3% and 105%, respectively, compared to a patient weighing 60 kg.
Steady state pharmacokinetics were achieved after the loading period (8 weeks) as follows [mean (±SD)]: Cmin: 19.7 (12.2) mcg/mL, Cmax: 31.5 (14.9) mcg/mL, and AUC: 737 (386) mcg.mL/day.
Distribution
Satralizumab-mwge undergoes biphasic distribution. The central volume of distribution was 3.46 L and the peripheral volume of distribution was 2.07 L. The inter-compartmental clearance was 0.336 L/day.
Elimination
The total clearance of satralizumab-mwge is concentration-dependent. Linear clearance (accounting for approximately half of the total clearance at steady state using the recommended dose in NMOSD patients) is estimated to be 0.0601 L/day. The associated terminal t1/2 is approximately 30 days (range 22 – 37 days) based on data pooled from Study 1 and Study 2.
Metabolism
The metabolism of satralizumab-mwge has not been directly studied, as antibodies are cleared principally by catabolism.
Excretion
Monoclonal antibodies, including satralizumab-mwge, are not eliminated via renal or hepatic pathways.
Specific Populations
Population pharmacokinetic analyses in patients with NMOSD showed that age, gender, and race did not meaningfully influence the pharmacokinetics of satralizumab-mwge.
Drug Interaction Studies
No formal drug-drug interaction studies have been performed with ENSPRYNG.
Based on population pharmacokinetic analyses of the available data, the impact of commonly used small molecule drugs on the pharmacokinetics of satralizumab-mwge remains inconclusive.
Suppression of IL-6 signaling by treatment with ENSPRYNG, from the low baseline levels seen in Study 1 and Study 2, is expected to have a minor impact on exposure of concomitant medications metabolized by CYP450 enzymes. The clinical significance of this is unknown.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Mutagenesis
Genetic toxicology studies of satralizumab-mwge were not conducted. As an antibody, satralizumab-mwge is not expected to interact directly with DNA.
Impairment of Fertility
In monkeys administered satralizumab-mwge (0, 2, 10, or 50 mg/kg) weekly by subcutaneous injection for 26 weeks, no effects on sperm, estrus cycle, or male and female reproductive organs were observed. At the high dose, plasma exposures (Cave) were approximately 100 times that in humans at the recommended monthly maintenance dose of 120 mg.
14. Clinical Studies
The efficacy of ENSPRYNG for the treatment of NMOSD in adult patients was established in two studies. Study 1 was a randomized (2:1), placebo-controlled trial in 95 patients without concurrent IST (Study 1, NCT02073279) in which 64 patients were anti-AQP4 antibody positive and 31 patients were anti-AQP4 antibody negative.
Study 2 was a randomized (1:1), placebo-controlled trial in 76 adult patients with concurrent IST (Study 2, NCT02028884). Of these, 52 adult patients were anti-AQP4 antibody positive and 24 adult patients were anti-AQP4 antibody negative.
Patients met the following eligibility criteria:
- Study 1: Clinical evidence of 1 relapse in the previous 12 months
- Study 2: Clinical evidence of at least 2 relapses in the previous 2 years, at least one of which must have occurred in the previous year
- EDSS score of 0 to 6.5 (both studies)
- Study 1: Patients were excluded if previously treated with IST within an interval specified for each such therapy
- Study 2: One of the following baseline treatments at a stable dose as a monotherapy for 8 weeks prior to baseline: azathioprine, mycophenolate mofetil, oral corticosteroids
In Study 1, 41 anti-AQP4 antibody positive adult patients were randomized to and received ENSPRYNG and 23 received placebo. Females accounted for 76% of the ENSPRYNG group and 96% of the placebo group. The remaining baseline demographic characteristics were balanced between the treatment groups. The mean age was 44 years. Fifty percent were White, 22% were Black or African-American, and 20% were Asian. The mean EDSS score was 3.8.
In Study 2, 26 anti-AQP4 antibody positive adult patients were randomized to and received ENSPRYNG and 26 received placebo. All patients were receiving either concurrent azathioprine (42%), oral corticosteroids (52%), or mycophenolate mofetil (6%) during the trial. The baseline demographic and disease characteristics were balanced between the treatment groups. Females accounted for 100% of the study population. Forty-six percent of patients were White and 52% were Asian. The mean age was 46 years. The mean EDSS score was 4.0.
All potential relapses were adjudicated by a blinded Clinical Endpoint Committee (CEC). The primary efficacy endpoint for both studies was the time to the first CEC-confirmed relapse.
In Study 1, the time to the first CEC-confirmed relapse was significantly longer in ENSPRYNG-treated patients compared to patients who received placebo (risk reduction 55%; hazard ratio 0.45; p = 0.0184). In the anti-AQP4 antibody positive population, there was a 74% risk reduction; hazard ratio 0.26; p = 0.0014 (Table 5; Figure 1). There was no evidence of a benefit in the anti-AQP4 antibody negative patients.
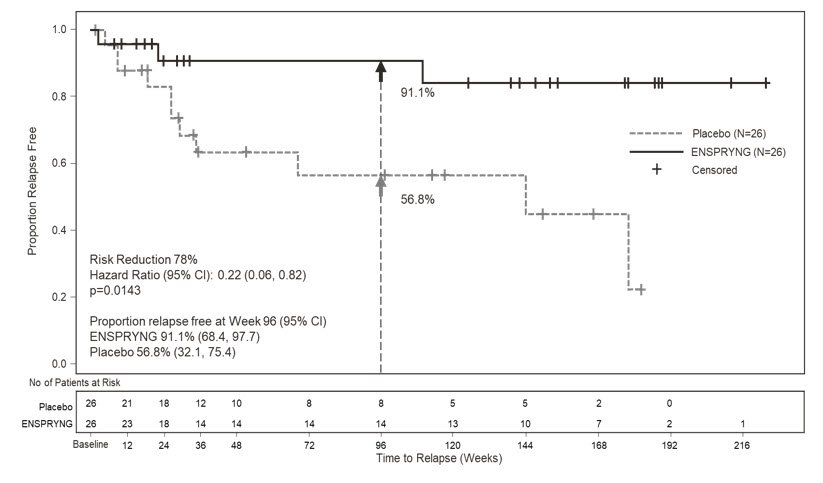
In Study 2, the time to the first CEC-confirmed relapse was significantly longer in patients treated with ENSPRYNG compared to patients who received placebo (risk reduction 62%; hazard ratio 0.38; p = 0.0184). In the anti-AQP4 antibody positive population, there was a 78% risk reduction; hazard ratio 0.22; p = 0.0143 (Table 5; Figure 2). There was no evidence of a benefit in the anti-AQP4 antibody negative patients.
| Study 1 | Study 2 | |||
|---|---|---|---|---|
| ENSPRYNG N=41 | Placebo N=23 | ENSPRYNG + IST*
N= 26 | Placebo + IST N=26 |
|
| Time to Clinical Endpoint Committee (CEC)-Determined Relapse (Primary Efficacy Endpoint) | ||||
|
||||
| Number (%) of Patients with Relapse | 9 (22) | 13 (56.5) | 3 (11.5) | 11 (42.3) |
| Hazard Ratio (95% CI) | 0.26 (0.11, 0.63) | 0.22 (0.06, 0.82) |
||
| p-value | 0.0014 | 0.0143 | ||
| Risk Reduction | 74% | 78% | ||
| Proportion of Protocol Defined Relapse-Free Patients at 96 Weeks | 76.5% | 41.1% | 91.1% | 56.8% |
Figure 1 Study 1: Time to First CEC-Determined NMOSD Relapse in the Randomized Controlled Period in the ITT Population Anti-AQP4 Antibody Positive Patients
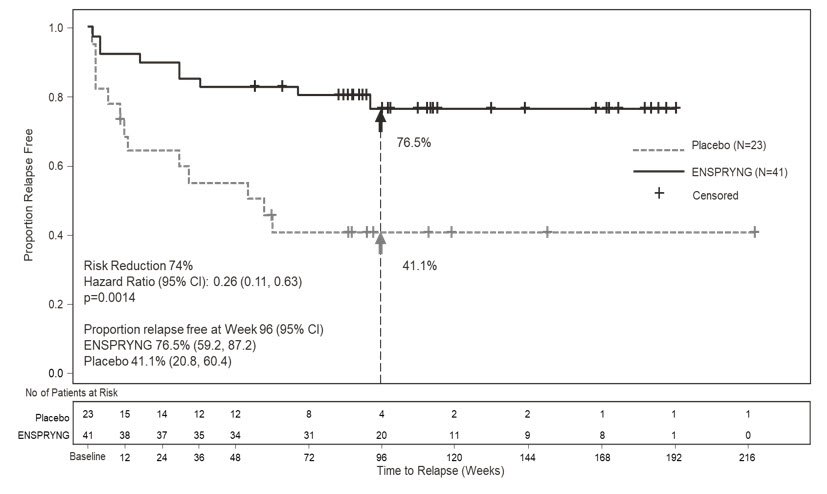
Figure 2 Study 2: Time to First CEC-Determined NMOSD Relapse in the Randomized Controlled Period in the ITT Population Anti-AQP4 Antibody Positive Patients
16. How is Enspryng supplied
16.1 How Supplied
ENSPRYNG (satralizumab-mwge) injection is available as a sterile, preservative-free, clear, colorless to slightly yellow solution in single-dose prefilled syringe (PFS) with needle safety device.
ENSPRYNG PFS is not made with natural rubber latex. Each ENSPRYNG carton contains one single-dose 120 mg/mL prefilled syringe (NDC 50242-007-01).
16.2 Storage and Handling
- Refrigerate at 2°C to 8°C (36°F to 46°F) in original carton to protect from light. Do not freeze. Do not shake.
- Prior to administration, ENSPRYNG, if unopened, can be removed from and returned to the refrigerator, if necessary. The total combined time out of refrigeration should not exceed 8 days at a temperature that does not exceed 30°C (86°F).
17. Patient Counseling Information
Advise the patients to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Infections
Inform patients that an increased risk of infections, including serious and potentially fatal infections, has been observed in patients treated with IL-6 receptor antagonists, including ENSPRYNG. Instruct patients to contact their healthcare provider immediately when symptoms suggesting infection (e.g., fever, chills, constant cough, or sore throat) appear during treatment [see Warning and Precautions (5.1)].
Vaccinations
Advise patients to complete any required vaccinations at least 4 weeks prior to initiation of ENSPRYNG for live or live-attenuated vaccines and, whenever possible, at least 2 weeks prior to initiation of ENSPRYNG for non-live vaccines [see Warnings and Precautions 5.1].
Elevated Liver Enzymes
Inform patients on the importance of liver enzyme testing [see Warnings and Precautions (5.2)].
Decreased Neutrophil Counts
Inform patients on the importance of neutrophil count testing [see Warnings and Precautions (5.3)].
Hypersensitivity Reactions
Inform patients about the signs and symptoms of hypersensitivity reactions and anaphylaxis and advise them to contact their healthcare provider immediately if these symptoms occur [see Warnings and Precautions (5.4)].
Instruction on Injection Technique
Instruct patients and caregivers to read the Instructions for Use before the patient starts using ENSPRYNG, and each time the patient gets a refill as there may be new information they need to know.
Perform the first injection under the guidance of a qualified healthcare professional. If a patient or caregiver is to administer subcutaneous ENSPRYNG, instruct him/her in injection techniques and assess his/her ability to inject subcutaneously to ensure proper administration of subcutaneous ENSPRYNG and the suitability for home use [see Dosage and Administration (2.3) and Instructions for Use].
Instruct patients to remove the prefilled syringe from the refrigerator prior to use and allow to sit at room temperature outside of the carton for 30 minutes. Do not warm ENSPRYNG in any other way.
Advise patients to consult their healthcare provider if the full dose is not received.
A puncture-resistant container for disposal of syringes should be used and should be kept out of the reach of children. Instruct patients or caregivers in the technique as well as proper prefilled syringe disposal, and caution against reuse of these items.
Pregnancy Exposure Registry
Encourage patients to enroll in the ENSPRYNG Pregnancy Registry if they become pregnant while taking ENSPRYNG. The Registry monitors fetal outcomes of pregnant women exposed to ENSPRYNG [see Use in Specific Populations (8.1)].
ENSPRYNG® [satralizumab-mwge]
Manufactured by:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990
ENSPRYNG® is a registered trademark of
Chugai Pharmaceutical Co., Ltd., Tokyo, Japan
©2022 Genentech, Inc. All rights reserved.
U.S. License No.: 1048
| This Medication Guide has been approved by the U.S. Food and Drug Administration | Issued: 3/2022 | |
| MEDICATION GUIDE
ENSPRYNG® (en-spryng) (satralizumab-mwge) injection, for subcutaneous use |
||
| What is the most important information I should know about ENSPRYNG? ENSPRYNG may cause serious side effects including:
|
||
| What is ENSPRYNG?
ENSPRYNG is a prescription medicine used to treat neuromyelitis optica spectrum disorder (NMOSD) in adults who are aquaporin-4 (AQP4) antibody positive. It is not known if ENSPRYNG is safe and effective in children. |
||
Do not take ENSPRYNG if you:
|
||
Before you take ENSPRYNG, tell your healthcare provider about all of your medical conditions, including if you:
|
||
How should I take ENSPRYNG?
|
||
| What are the possible side effects of ENSPRYNG? ENSPRYNG may cause serious side effects, including:
|
||
|
|
|
|
||
|
|
|
| These are not all the possible side effects of ENSPRYNG. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Genentech at 1-888-835-2555. |
||
How should I store ENSPRYNG?
|
||
| General information about the safe and effective use of ENSPRYNG.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ENSPRYNG for a condition for which it was not prescribed. Do not give ENSPRYNG to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ENSPRYNG that is written for health professionals. |
||
| What are the ingredients in ENSPRYNG?
Active ingredient: satralizumab-mwge Inactive ingredients: L-arginine, L-histidine, poloxamer 188, L-aspartic acid, and Water for Injection. Manufactured by: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990 ENSPRYNG® is a registered trademark of Chugai Pharmaceutical Co., Ltd., Tokyo, Japan U.S. License No.: 1048 For more information, go to www.ENSPRYNG.com or call 1-844-NSPRYNG. |
||
Instructions for Use ENSPRYNG® (en-spryng) (satralizumab-mwge) Injection
Read this Instructions for Use before you start using your prefilled syringe and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. Your healthcare provider will decide if you or a caregiver can give you injections of ENSPRYNG at home. They will also show you or a caregiver the correct and safe way to use the syringe before you use it for the first time.
Talk to your healthcare provider if you have any questions.
Important Information
- Each syringe is prefilled with a medicine called ENSPRYNG.
- Each carton of ENSPRYNG contains only 1 prefilled syringe.
- Each prefilled syringe can be used only 1 time.
Do not share your ENSPRYNG syringe with other people. You may give them a serious infection or get a serious infection from them.
Do not:
- take the needle cap off until you are ready to inject ENSPRYNG.
- use the syringe if it has been dropped or damaged.
- try to take the syringe apart at any time.
- leave the syringe unattended.
- re-use the same syringe.
How should I store the ENSPRYNG prefilled syringe?
- Keep the unused syringe in the refrigerator between 36°F to 46°F (2°C to 8°C) until ready to use.
- Before giving an injection, if the ENSPRYNG is not opened, it can be removed from and placed back in the refrigerator if needed. The total combined time out of the refrigerator should not be more than 8 days at a temperature that does not go above 86°F (30°C).
- Keep the syringe in its original carton away from direct sunlight.
- Always keep the syringe dry.
Keep the ENSPRYNG syringe and all medicines out of the reach of children.
Do not:
- freeze the syringe.
- use the syringe if it has been frozen.
- shake.
Supplies needed to give your injection
Each ENSPRYNG carton contains:
- 1 prefilled syringe for 1-time use only.
Not included in the carton:
- 1 alcohol pad
- 1 sterile cotton ball or gauze
- 1 small bandage
- 1 FDA-cleared puncture-resistant sharps container for safe disposal of the needle cap and used syringe. See Step 21 "Disposing of ENSPRYNG" at the end of this Instructions for Use.
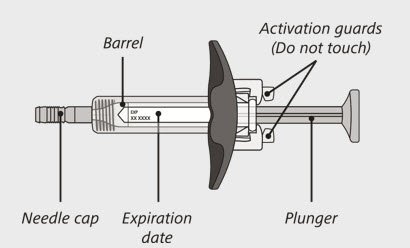
ENSPRYNG prefilled syringe
(See Figure A and Figure B)
Before use:
After use:
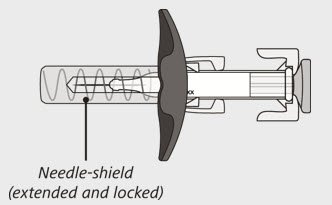
The syringe has a needle-shield that automatically covers the needle when the injection is complete.
Prepare to use ENSPRYNG
- 1.
- Take the carton containing the syringe out of the refrigerator and place it on a clean, flat work surface (like a table).
- 2.
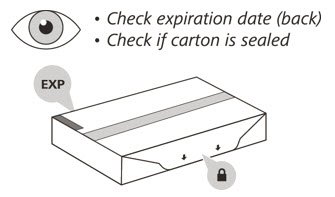
- Check the expiration (EXP) date on the back of the carton (See Figure C). Do not use if the carton has expired.
- 3.
- Check the front of the carton to make sure it is sealed (Figure C). Do not use if the seal has been broken.
If the expiration date has passed or the seal is broken, do not use. Then go to Step 21 "Disposing of ENSPRYNG" and contact your healthcare provider.
- 4.
- Open the sealed carton (See Figure D).
- 5.
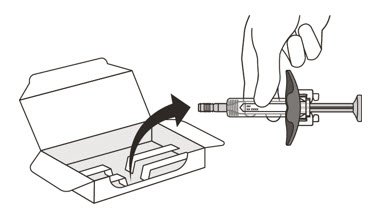
- Carefully lift the syringe out of the carton by holding the barrel (See Figure E).
Do not:- turn the carton upside down to remove the syringe.
- touch the activation guards because this may damage the syringe.
- hold the plunger or needle cap.
Check the syringe
(See Figure F)
- 6.
- Check the expiration date on the syringe. Do not use the syringe if it has expired.
- 7.
- Check the syringe for any damage. Do not use if it is cracked or broken.
- 8.
- Check that the liquid through the viewing window is clear and colorless to slightly yellow. Do not inject the medicine if the liquid is cloudy, discolored, or has particles in it.
- There may be some small air bubbles in the syringe. This is normal and you should not try to remove them.
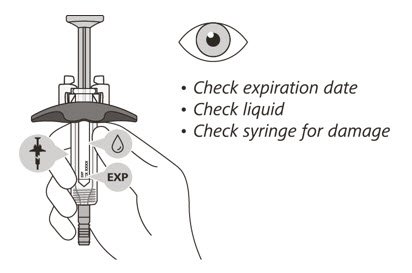
If the expiration (EXP) date has passed, the syringe is damaged or the liquid is cloudy, discolored or has particles in it, do not use. Then go to Step 21 "Disposing of ENSPRYNG" and contact your healthcare provider.
Let your syringe warm up
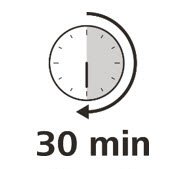
- 9.
- After you have checked the syringe, place it on a clean, flat work surface (like a table) for 30 minutes. This will allow it to reach room temperature. (See Figure G).
It is important to let the syringe gently warm up as injecting cold medicine may feel uncomfortable and make it harder to push.
Do not:- speed up the warming process in any way, such as using a microwave or placing the syringe in warm water.
- remove the needle cover while the syringe is reaching room temperature.
Wash your hands
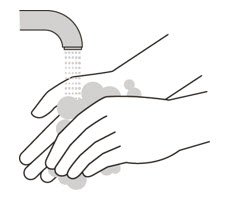
- 10.
- Wash your hands with soap and water. (See Figure H).
Choose the injection site
- 11.
- Choose your injection site in either:
- the lower part of your stomach (abdomen) or
- the front and middle of your thighs. (See Figure I).
- Do not inject into the 2-inch (5 cm) area around your belly button.
- Do not inject into moles, scars, bruises, or areas where the skin is tender, red, hard or broken.
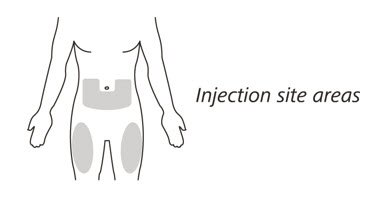
Choose a different injection site for each new injection. Choose a different place to inject which is at least 1 inch (2.5 cm) away from the place where you last injected.
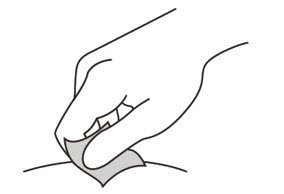
Clean the injection site
- 12.
- Wipe the injection site with an alcohol pad and let it air dry.
Do not:- fan or blow on the area which you have cleaned.
- touch the injection site again before you give the injection.
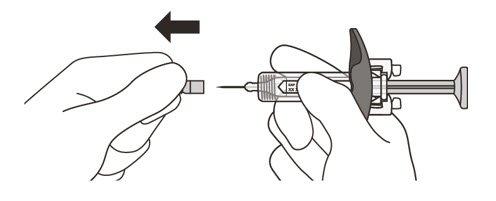
Inject ENSPRYNG
- 13.
- Hold the barrel of the syringe between your thumb and index finger. With your other hand, pull the needle cap straight off. You may see a drop of liquid at the end of the needle. This is normal and will not affect your dose (See Figure K).
-
Use the syringe within 5 minutes of removing the cap or the needle may clog.
Do not: - take the needle cap off until you are ready to inject ENSPRYNG.
- put the needle cap back on after it has been removed as this may damage the needle.
- touch the needle or let it touch any surfaces after removing the needle cap.
-
Use the syringe within 5 minutes of removing the cap or the needle may clog.
- 14.
- Throw away the needle cap in a puncture-resistant sharps container immediately. See Step 21 "Disposing of ENSPRYNG".
- 15.
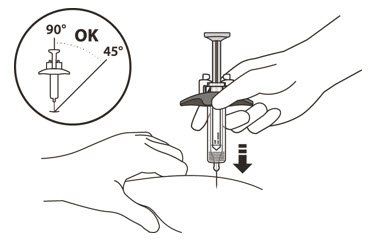
- Hold the barrel of the syringe using your thumb and index finger. With your other hand, pinch the area of skin you have cleaned (See Figure L).
- 16.
- Use a quick, dart-like motion to insert the needle at an angle between 45° to 90°(See Figure L).
Do not:- insert the needle through clothing.
- change the angle of the injection.
- insert the needle again.
- 17.
- After the needle is inserted, let go of the pinched skin.
- 18.
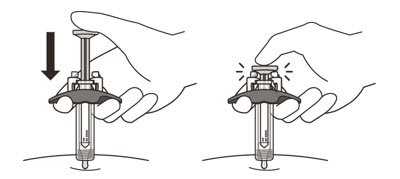
- Slowly inject all of the medicine by gently pushing the plunger all the way down until it touches the activation guards (See Figure M).
- 19.
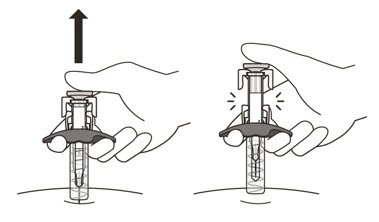
- Gently release the plunger and allow the needle to come out of the skin at the same angle it was inserted (See Figure N).
- The needle will now be covered by the needle-shield. If the needle is not covered, carefully place the syringe into a puncture-resistant sharps container to avoid injury. See Step 21 "Disposing of ENSPRYNG".
Taking care of the injection site
- 20.
- There may be a little bleeding at the injection site. You can press a cotton ball or gauze over the injection site but do not rub it. If needed, you may also cover the area you injected with a small bandage. If the medicine gets into contact with your skin, wash the area with water.
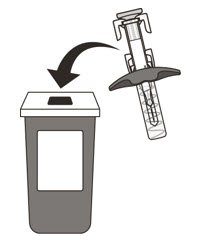
- 21.
- Put your used syringe in an FDA-cleared sharps disposal container immediately after use (See Figure O). Do not throw away (dispose of) the syringe in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out
- upright and stable during use
- leak-resistant
- properly labeled to warn of hazardous waste inside the container
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about the safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in trash unless your community guidelines permit this.
- Do not recycle your used sharps disposal container.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990
U.S. License No.: 1048
Approved: 8/2020
ENSPRYNG® is a registered trademark of Chugai Pharmaceutical Co., Ltd., Tokyo, Japan
© 2020 Genentech, Inc. All rights reserved.
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
PRINCIPAL DISPLAY PANEL - 120 mg/mL Syringe Carton
NDC 50242-007-01
Rx only
Enspryng®
(satralizumab-mwge)
Injection
120 mg/mL
For Subcutaneous Use.
Single-Dose Prefilled Syringe.
Refrigerate at 2°C to 8°C (36°F to 46°F) in original carton to protect from light.
Do not freeze. Do not shake.
ATTENTION PHARMACIST: Each patient is required to receive the enclosed
Medication Guide.
1 prefilled syringe
Genentech
10240933

| ENSPRYNG
satralizumab injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Genentech Inc. (080129000) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| F. Hoffmann-La Roche Ltd. | 485244961 | ANALYSIS(50242-007) , LABEL(50242-007) , PACK(50242-007) | |
Biological Products Related to Enspryng
Find detailed information on biosimilars for this medication.
Frequently asked questions
More about Enspryng (satralizumab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: interleukin inhibitors
- Breastfeeding
- En español