Depen: Package Insert / Prescribing Info
Package insert / product label
Generic name: penicillamine
Dosage form: tablet
Drug class: Antirheumatics
Medically reviewed by Drugs.com. Last updated on Nov 5, 2023.
On This Page
Physicians planning to use penicillamine should thoroughly familiarize themselves with its toxicity, special dosage considerations, and therapeutic benefits. Penicillamine should never be used casually. Each patient should remain constantly under the close supervision of the physician. Patients should be warned to report promptly any symptoms suggesting toxicity.
Depen Description
Penicillamine is 3-mercapto-D-valine, a disease modifying antirheumatic drug. It is a white or practically white, crystalline powder, freely soluble in water, slightly soluble in alcohol, and insoluble in ether, acetone, benzene, and carbon tetrachloride. Although its configuration is D, it is levorotatory as usually measured:
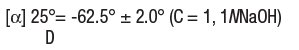
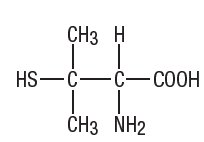
The empirical formula is C5H11NO2S, giving it a molecular weight of 149.21. The structural formula is:
It reacts readily with formaldehyde or acetone to form a thiazolidine-carboxylic acid.
Depen® (penicillamine tablets, USP) Titratable Tablets for oral administration contain 250 mg of penicillamine.
Other ingredients (inactive): edetate disodium, hypromellose, lactose, magnesium stearate, magnesium trisilicate, polyethylene glycol, povidone, starch, and stearic acid.
Depen - Clinical Pharmacology
Penicillamine is a chelating agent recommended for the removal of excess copper in patients with Wilson’s disease. From in vitro studies which indicate that one atom of copper combines with two molecules of penicillamine, it would appear that one gram of penicillamine should be followed by the excretion of about 200 milligrams of copper; however, the actual amount excreted is about one percent of this.
Penicillamine also reduces excess cystine excretion in cystinuria. This is done, at least in part, by disulfide interchange between penicillamine and cystine, resulting in formation of penicillamine-cysteine disulfide, a substance that is much more soluble than cystine and is excreted readily.
Penicillamine interferes with the formation of cross-links between tropocollagen molecules and cleaves them when newly formed.
The mechanism of action of penicillamine in rheumatoid arthritis is unknown, although it appears to suppress disease activity. Unlike cytotoxic immunosuppressants, penicillamine markedly lowers IgM rheumatoid factor but produces no significant depression in absolute levels of serum immunoglobulins. Also unlike cytotoxic immunosuppressants, which act on both, penicillamine in vitro depresses T-cell activity but not B-cell activity.
In vitro, penicillamine dissociates macroglobulins (rheumatoid factor) although the relationship of the activity to its effect in rheumatoid arthritis is not known.
In rheumatoid arthritis, the onset of therapeutic response to DEPEN may not be seen for two or three months. In those patients who respond, however, the first evidence of suppression of symptoms such as pain, tenderness, and swelling usually is generally apparent within three months. The optimum duration of therapy has not been determined. If remissions occur, they may last from months to years but usually require continued treatment (see DOSAGE AND ADMINISTRATION).
In all patients receiving penicillamine, it is important that DEPEN be given on an empty stomach, at least one hour before meals or two hours after meals, and at least one hour apart from any other drug, food or milk. This permits maximum absorption and reduces the likelihood of inactivation by metal binding in the gastrointestinal tract.
Methodology for determining the bioavailability of penicillamine is not available; however, penicillamine is known to be a very soluble substance.
Indications and Usage for Depen
DEPEN is indicated in the treatment of Wilson’s disease, cystinuria, and in patients with severe, active rheumatoid arthritis who have failed to respond to an adequate trial of conventional therapy. Available evidence suggests that DEPEN is not of value in ankylosing spondylitis.
Wilson’s Disease - Wilson’s disease (hepatolenticular degeneration) results from the interaction of an inherited defect and an abnormality in copper metabolism. The metabolic defect, which is the consequence of the autosomal inheritance of one abnormal gene from each parent, manifests itself in a greater positive copper balance than normal. As a result, copper is deposited in several organs and appears eventually to produce pathologic effects most prominently seen in the brain, where degeneration is widespread; in the liver, where fatty infiltration, inflammation, and hepatocellular damage progress to postnecrotic cirrhosis; in the kidney, where tubular and glomerular dysfunction results; and in the eye, where characteristic corneal copper deposits are known as Kayser-Fleischer rings.
Two types of patients require treatment for Wilson’s disease: (1) the symptomatic, and (2) the asymptomatic in whom it can be assumed the disease will develop in the future if the patient is not treated.
Diagnosis, suspected on the basis of family or individual history, physical examination, or a low serum concentration of ceruloplasmin*, is confirmed by the demonstration of Kayser-Fleischer rings or, particularly in the asymptomatic patient, by the quantitative demonstration in a liver biopsy specimen of a concentration of copper in excess of 250 mcg/g dry weight.
Treatment has two objectives:
- (1)
- to minimize dietary intake and absorption of copper.
- (2)
- to promote excretion of copper deposited in tissues.
The first objective is attained by a daily diet that contains no more than one or two milligrams of copper. Such a diet should exclude, most importantly, chocolate, nuts, shellfish, mushrooms, liver, molasses, broccoli, and cereals enriched with copper, and be composed to as great an extent as possible of foods with a low copper content. Distilled or demineralized water should be used if the patient’s drinking water contains more than 0.1 mg of copper per liter.
For the second objective, a copper chelating agent is used.
In symptomatic patients, this treatment usually produces marked neurologic improvement, fading of Kayser-Fleischer rings, and gradual amelioration of hepatic dysfunction and psychic disturbances.
Clinical experience to date suggests that life is prolonged with the above regimen.
Noticeable improvement may not occur for one to three months. Occasionally, neurologic symptoms become worse during initiation of therapy with DEPEN. Despite this, the drug should not be discontinued permanently. Although temporary interruption may result in clinical improvement of the neurological symptoms, it carries an increased risk of developing a sensitivity reaction upon resumption of therapy (See WARNINGS).
* For quantitative test for serum ceruloplasmin see: Morell, A.G.; Windsor, J.; Sternlieb, I; Scheinberg, I.H.: Measurement of the concentration of ceruloplasmin in serum by determination of its oxidase activity, in “Laboratory Diagnosis of Liver Disease,” F.W. Sunderman; F.W. Sunderman, Jr., (eds.), St. Louis, Warren H. Green, Inc., 1968, pp. 193-195.
Treatment of asymptomatic patients has been carried out for over ten years. Symptoms and signs of the disease appear to be prevented indefinitely if daily treatment with DEPEN can be continued.
Cystinuria - Cystinuria is characterized by excessive urinary excretion of the dibasic amino acids, arginine, lysine, ornithine, and cystine, and the mixed disulfide of cysteine and homocysteine. The metabolic defect that leads to cystinuria is inherited as an autosomal, recessive trait. Metabolism of the affected amino acids is influenced by at least two abnormal factors: (1) defective gastrointestinal absorption and (2) renal tubular dysfunction.
Arginine, lysine, ornithine, and cysteine are soluble substances, readily excreted. There is no apparent pathology connected with their excretion in excessive quantities.
Cystine, however, is so slightly soluble at the usual range of urinary pH that it is not excreted readily, and so crystallizes and forms stones in the urinary tract. Stone formation is the only known pathology in cystinuria. Normal daily output of cystine is 40 to 80 mg. In cystinuria, output is greatly increased and may exceed 1 g/day. At 500 to 600 mg/day, stone formation is almost certain. When it is more than 300 mg/day, treatment is indicated.
Conventional treatment is directed at keeping urinary cystine diluted enough to prevent stone formation, keeping the urine alkaline enough to dissolve as much cystine as possible, and minimizing cystine production by a diet low in methionine (the major dietary precursor of cystine). Patients must drink enough fluid to keep urine specific gravity below 1.010, take enough alkali to keep urinary pH at 7.5 to 8, and maintain a diet low in methionine. This diet is not recommended in growing children and probably is contraindicated in pregnancy because of its low protein content (see PRECAUTIONS).
When these measures are inadequate to control recurrent stone formation, DEPEN may be used as additional therapy. When patients refuse to adhere to conventional treatment, DEPEN may be a useful substitute. It is capable of keeping cystine excretion to near normal values, thereby hindering stone formation and the serious consequences of pyelonephritis and impaired renal function that develop in some patients.
Bartter and colleagues depict the process by which penicillamine interacts with cystine to form penicillamine-cysteine mixed disulfide as:
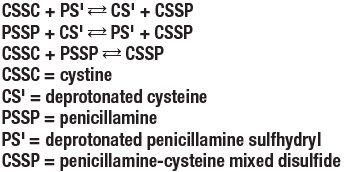
In this process, it is assumed that the deprotonated form of penicillamine, PS', is the active factor in bringing about the disulfide interchange.
Rheumatoid Arthritis - Because DEPEN can cause severe adverse reactions, its use in rheumatoid arthritis should be restricted to patients who have severe, active disease and who have failed to respond to an adequate trial of conventional therapy. Even then, benefit-to-risk ratio should be carefully considered. Other measures, such as rest, physiotherapy, salicylates, and corticosteroids should be used, when indicated, in conjunction with DEPEN (see PRECAUTIONS).
Contraindications
Except for treatment of Wilson’s disease or certain cases of cystinuria, use of penicillamine during pregnancy is contraindicated (See WARNINGS).
Although breast milk studies have not been reported in animals or humans, mothers on therapy with penicillamine should not nurse their infants.
Patients with a history of penicillamine-related aplastic anemia or agranulocytosis should not be restarted on penicillamine (see WARNINGS and ADVERSE REACTIONS). Because of its potential for causing renal damage, penicillamine should not be administered to rheumatoid arthritis patients with a history or other evidence of renal insufficiency.
Warnings
The use of penicillamine has been associated with fatalities due to certain diseases, such as aplastic anemia, agranulocytosis, thrombocytopenia, Goodpasture’s syndrome, and myasthenia gravis.
Because of the potential for serious hematological and renal adverse reactions to occur at any time, routine urinalysis, white and differential blood cell count, hemoglobin determination, and direct platelet count must be done every two weeks for at least the first six months of penicillamine therapy and monthly thereafter. Patients should be instructed to report promptly the development of signs and symptoms of granulocytopenia and/or thrombocytopenia such as fever, sore throat, chills, bruising, or bleeding. The above laboratory studies should then be promptly repeated.
Leukopenia and thrombocytopenia have been reported to occur in up to five percent of patients during penicillamine therapy. Leukopenia is of the granulocytic series and may or may not be associated with an increase in eosinophils. A confirmed reduction in WBC below 3500 per cubic mL mandates discontinuance of penicillamine therapy. Thrombocytopenia may be on an idiosyncratic basis with decreased or absent megakaryocytes in the marrow, when it is part of an aplastic anemia. In other cases the thrombocytopenia is presumably on an immune basis since the number of megakaryocytes in the marrow has been reported to be normal or sometimes increased. The development of a platelet count below 100,000 per cubic mL, even in the absence of clinical bleeding, requires at least temporary cessation of penicillamine therapy. A progressive fall in either platelet count or WBC in three successive determinations, even though values are still within the normal range, likewise requires at least temporary cessation.
Proteinuria and/or hematuria may develop during therapy and may be warning signs of membranous glomerulopathy which can progress to a nephrotic syndrome. Close observation of these patients is essential. In some patients the proteinuria disappears with continued therapy; in others penicillamine must be discontinued. When a patient develops proteinuria or hematuria the physician must ascertain whether it is a sign of drug-induced glomerulopathy or is unrelated to penicillamine.
Rheumatoid arthritis patients who develop moderate degrees of proteinuria may be continued cautiously on penicillamine therapy, provided that quantitative 24-hour urinary protein determinations are obtained at intervals of one to two weeks. Penicillamine dosage should not be increased under these circumstances. Proteinuria which exceeds 1 g/24 hours, or proteinuria which is progressively increasing requires either discontinuance of the drug or a reduction in the dosage. In some patients, proteinuria has been reported to clear following reduction in dosage.
In rheumatoid arthritis patients, penicillamine should be discontinued if unexplained gross hematuria or persistent microscopic hematuria develops.
In patients with Wilson’s disease or cystinuria the risks of continued penicillamine therapy in patients manifesting potentially serious urinary abnormalities must be weighed against the expected therapeutic benefits.
When penicillamine is used in cystinuria, an annual x-ray for renal stones is advised. Cystine stones form rapidly, sometimes in six months.
Up to one year or more may be required for any urinary abnormalities to disappear after penicillamine has been discontinued.
Because of rare reports of intrahepatic cholestasis and toxic hepatitis, liver function tests are recommended every six months for the duration of therapy.
Goodpasture’s syndrome has occurred rarely. The development of abnormal urinary findings associated with hemoptysis and pulmonary infiltrates on x-ray requires immediate cessation of penicillamine.
Obliterative bronchiolitis has been reported rarely. The patient should be cautioned to report immediately pulmonary symptoms such as exertional dyspnea, unexplained cough, or wheezing. Pulmonary function studies should be considered at that time.
Myasthenic syndrome sometimes progressing to myasthenia gravis has been reported. Ptosis and diplopia, with weakness of the extraocular muscles, are often early signs of myasthenia. In the majority of cases, symptoms of myasthenia have receded after withdrawal of penicillamine.
Most of the various forms of pemphigus have occurred during treatment with penicillamine. Pemphigus vulgaris and pemphigus foliaceus are reported most frequently, usually as a late complication of therapy. The seborrhea-like characteristics of pemphigus foliaceus may obscure an early diagnosis. When pemphigus is suspected, DEPEN should be discontinued. Treatment has consisted of high doses of corticosteroids alone or, in some cases, concomitantly with an immunosuppressant. Treatment may be required for only a few weeks or months, but may need to be continued for more than a year.
Once instituted for Wilson’s disease or cystinuria, treatment with penicillamine should, as a rule, be continued on a daily basis. Interruptions for even a few days have been followed by sensitivity reactions after reinstitution of therapy.
Use In Pregnancy
Penicillamine has been shown to be teratogenic in rats when given in doses 6 times higher than the highest dose recommended for human use (based on a standard weight of 50 kg). Skeletal defects, cleft palates, and fetal toxicity (resorptions) have been reported.
There are no controlled studies on the use of penicillamine in pregnant women. Although normal outcomes have been reported, characteristic congenital cutis laxa and associated birth defects have been reported in infants born of mothers who received therapy with penicillamine during pregnancy. Penicillamine should be used in women of childbearing potential only when the expected benefits outweigh the possible hazards. Women on therapy with penicillamine who are of childbearing potential should be apprised of this risk, advised to report promptly any missed menstrual periods or other indications of possible pregnancy, and followed closely for early recognition of pregnancy.
Wilson’s Disease - Reported experience* shows that continued treatment with penicillamine throughout pregnancy protects the mother against relapse of the Wilson’s disease, and that discontinuation of penicillamine has deleterious effects on the mother.
If penicillamine is administered during pregnancy to patients with Wilson’s disease, it is recommended that the daily dosage be limited to 1 g. If cesarean section is planned, the daily dosage should be limited to 250 mg during the last six weeks of pregnancy and post-operatively until wound healing is complete.
Cystinuria - If possible, penicillamine should not be given during pregnancy to women with cystinuria (see CONTRAINDICATIONS). There are reports of women with cystinuria on therapy with penicillamine who gave birth to infants with generalized connective tissue defects who died following abdominal surgery. If stones continue to form in these patients, the benefits of therapy to the mother must be evaluated against the risk to the fetus.
Rheumatoid Arthritis - Penicillamine should not be administered to rheumatoid arthritis patients who are pregnant (see CONTRAINDICATIONS) and should be discontinued promptly in patients in whom pregnancy is suspected or diagnosed.
There is a report that a woman with rheumatoid arthritis treated with less than one gram a day of penicillamine during pregnancy gave birth (cesarean delivery) to an infant with growth retardation, flattened face with broad nasal bridge, low set ears, short neck with loose skin folds, and unusually lax body skin.
Precautions
Some patients may experience drug fever, a marked febrile response to penicillamine, usually in the second or third week following initiation of therapy. Drug fever may sometimes be accompanied by a macular cutaneous eruption.
In the case of drug fever in patients with Wilson’s disease or cystinuria, penicillamine should be temporarily discontinued until the reaction subsides. Then penicillamine should be reinstituted with a small dose that is gradually increased until the desired dosage is attained. Systemic steroid therapy may be necessary, and is usually helpful, in such patients in whom toxic reactions develop a second or third time.
In the case of drug fever in rheumatoid arthritis patients, because other treatments are available, penicillamine should be discontinued and another therapeutic alternative tried, since experience indicates that the febrile reaction will recur in a very high percentage of patients upon readministration of penicillamine.
The skin and mucous membranes should be observed for allergic reactions. Early and late rashes have occurred. Early rash occurs during the first few months of treatment and is more common. It is usually a generalized pruritic, erythematous, maculopapular, or morbilliform rash and resembles the allergic rash seen with other drugs. Early rash usually disappears within days after stopping penicillamine and seldom recurs when the drug is restarted at a lower dosage. Pruritus and early rash may often be controlled by the concomitant administration of antihistamines. Less commonly, a late rash may be seen, usually after six months or more of treatment, and requires discontinuation of penicillamine. It is usually on the trunk, is accompanied by intense pruritus, and is usually unresponsive to topical corticosteroid therapy. Late rash may take weeks to disappear after penicillamine is stopped and usually recurs if the drug is restarted.
The appearance of a drug eruption accompanied by fever, arthralgia, lymphadenopathy, or other allergic manifestations usually requires discontinuation of penicillamine. Certain patients will develop a positive anti-nuclear antibody (ANA) test and some of these may show a lupus erythematosus-like syndrome similar to drug-induced lupus associated with other drugs. The lupus erythematosus-like syndrome is not associated with hypocomplementemia and may be present without nephropathy. The development of a positive ANA test does not mandate discontinuance of the drug; however, the physician should be alerted to the possibility that a lupus erythematosus-like syndrome may develop in the future.
Some patients may develop oral ulcerations which in some cases have the appearance of aphthous stomatitis. The stomatitis usually recurs on rechallenge but often clears on a lower dosage. Although rare, cheilosis, glossitis, and gingivostomatitis have also been reported. These oral lesions are frequently dose-related and may preclude further increase in penicillamine dosage or require discontinuation of the drug.
Hypogeusia (a blunting or diminution in taste perception) has occurred in some patients. This may last two to three months or more and may develop into a total loss of taste; however, it is usually self-limited, despite continued penicillamine treatment. Such taste impairment is rare in patients with Wilson’s disease.
* Scheinberg, I.H., Sternlieb, I.: N Engl J Med 293: 1300-1302, December 18, 1975.
Penicillamine should not be used in patients who are receiving concurrently gold therapy, antimalarial or cytotoxic drugs, oxyphenbutazone, or phenylbutazone because these drugs are also associated with similar serious hematologic and renal adverse reactions. Patients who have had gold salt therapy discontinued due to a major toxic reaction may be at greater risk of serious adverse reactions with penicillamine, but not necessarily of the same type.
Patients who are allergic to penicillin may theoretically have cross-sensitivity to penicillamine. The possibility of reactions from contamination of penicillamine by trace amounts of penicillin has been eliminated now that penicillamine is being produced synthetically rather than as a degradation product of penicillin.
Because of their dietary restrictions, patients with Wilson’s disease and cystinuria should be given 25 mg/day of pyridoxine during therapy, since penicillamine increases the requirement for this vitamin. Patients also may receive benefit from a multivitamin preparation, although there is no evidence that deficiency of any vitamin other than pyridoxine is associated with penicillamine. In Wilson’s disease, multivitamin preparations must be copper-free.
Rheumatoid arthritis patients whose nutrition is impaired should also be given a daily supplement of pyridoxine. Mineral supplements should not be given, since they may block the response to penicillamine.
Iron deficiency may develop, especially in children and in menstruating women. In Wilson’s disease, this may be a result of adding the effects of the low copper diet, which is probably also low in iron, and the penicillamine to the effects of blood loss or growth. In cystinuria, a low methionine diet may contribute to iron deficiency, since it is necessarily low in protein. If necessary, iron may be given in short courses, but a period of two hours should elapse between administration of penicillamine and iron, since orally administered iron has been shown to reduce the effects of penicillamine.
Penicillamine causes an increase in the amount of soluble collagen. In the rat this results in inhibition of normal healing and also a decrease in tensile strength of intact skin. In man this may be the cause of increased skin friability at sites especially subject to pressure or trauma, such as shoulders, elbows, knees, toes, and buttocks. Extravasations of blood may occur and may appear as purpuric areas, with external bleeding if the skin is broken, or as vesicles containing dark blood. Neither type is progressive. There is no apparent association with bleeding elsewhere in the body and no associated coagulation defect has been found. Therapy with penicillamine may be continued in the presence of these lesions. They may not recur if dosage is reduced. Other reported effects probably due to the action of penicillamine on collagen are excessive wrinkling of the skin and development of small, white papules at venipuncture and surgical sites.
The effects of penicillamine on collagen and elastin make it advisable to consider a reduction in dosage to 250 mg/day when surgery is contemplated. Reinstitution of full therapy should be delayed until wound healing is complete.
Adverse Reactions/Side Effects
To report SUSPECTED ADVERSE REACTIONS, contact Meda Pharmaceuticals Inc. at 1-800-526-3840 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Penicillamine is a drug with a high incidence of untoward reactions, some of which are potentially fatal. Therefore, it is mandatory that patients receiving penicillamine therapy remain under close medical supervision throughout the period of drug administration (see WARNINGS and PRECAUTIONS).
Reported incidences (%) for the most commonly occurring adverse reactions in rheumatoid arthritis patients are noted, based on 17 representative clinical trials reported in the literature (1270 patients).
Allergic - Generalized pruritus, early and late rashes (5%), pemphigus (see WARNINGS), and drug eruptions which may be accompanied by fever, arthralgia, or lymphadenopathy have occurred (see WARNINGS and PRECAUTIONS). Some patients may show a lupus erythematosus-like syndrome similar to drug-induced lupus produced by other pharmacological agents (see PRECAUTIONS).
Urticaria and exfoliative dermatitis have occurred.
Thyroiditis has been reported; hypoglycemia in association with anti-insulin antibodies has been reported. These reactions are extremely rare.
Some patients may develop a migratory polyarthralgia, often with objective synovitis (see DOSAGE AND ADMINISTRATION).
Gastrointestinal - Anorexia, epigastric pain, nausea, vomiting, or occasional diarrhea may occur (17%).
Isolated cases of reactivated peptic ulcer have occurred, as have hepatic dysfunction and pancreatitis. Intrahepatic cholestasis and toxic hepatitis have been reported rarely. There have been a few reports of increased serum alkaline phosphatase, lactic dehydrogenase, and positive cephalin flocculation and thymol turbidity tests.
Some patients may report a blunting, diminution, or total loss of taste perception (12%); or may develop oral ulcerations. Although rare, cheilosis, glossitis, and gingivo-stomatitis have been reported (see PRECAUTIONS).
Gastrointestinal side effects are usually reversible following cessation of therapy.
Hematological - Penicillamine can cause bone marrow depression (see WARNINGS). Leukopenia (2%) and thrombocytopenia (4%) have occurred. Fatalities have been reported as a result of thrombocytopenia, agranulocytosis, aplastic anemia, and sideroblastic anemia.
Thrombotic thrombocytopenic purpura, hemolytic anemia, red cell aplasia, monocytosis, leukocytosis, eosinophilia, and thrombocytosis have also been reported.
Renal - Patients on penicillamine therapy may develop proteinuria (6%) and/or hematuria which, in some, may progress to the development of the nephrotic syndrome as a result of an immune complex membranous glomerulopathy (see WARNINGS).
Central Nervous System - Tinnitus, optic neuritis, and peripheral sensory and motor neuropathies (including polyradiculoneuropathy, i.e., Guillain-Barre Syndrome) have been reported. Muscular weakness may or may not occur with the peripheral neuropathies.
Neuromuscular - Myasthenia gravis (see WARNINGS).
Other - Adverse reactions that have been reported rarely include thrombophlebitis; hyperpyrexia (see PRECAUTIONS); falling hair or alopecia; lichen planus; polymyositis; dermatomyositis; mammary hyperplasia; elastosis perforans serpiginosa; toxic epidermal necrolysis; anetoderma (cutaneous macular atrophy); and Goodpasture’s syndrome, a severe and ultimately fatal glomerulonephritis associated with intra-alveolar hemorrhage (see WARNINGS). Fatal renal vasculitis has also been reported. Allergic alveolitis, obliterative bronchiolitis, interstitial pneumonitis, and pulmonary fibrosis have been reported in patients with severe rheumatoid arthritis, some of whom were receiving penicillamine. Bronchial asthma also has been reported.
Increased skin friability, excessive wrinkling of skin, and development of small, white papules at venipuncture and surgical sites have been reported (see PRECAUTIONS).
The chelating action of the drug may cause increased excretion of other heavy metals such as zinc, mercury, and lead.
There have been reports associating penicillamine with leukemia. However, circumstances involved in these reports are such that a cause and effect relationship to the drug has not been established.
Related/similar drugs
Depen Dosage and Administration
In all patients receiving penicillamine, it is important that DEPEN be given on an empty stomach, at least one hour before meals or two hours after meals, and at least one hour apart from any other drug, food, or milk. Because penicillamine increases the requirement for pyridoxine, patients may require a daily supplement of pyridoxine (see PRECAUTIONS).
Wilson’s Disease - Optimal dosage can be determined by measurement of urinary copper excretion and the determination of free copper in the serum. The urine must be collected in copper-free glassware, and should be quantitatively analyzed for copper before and soon after initiation of therapy with DEPEN.
Determination of 24-hour urinary copper excretions is of greatest value in the first week of therapy with penicillamine. In the absence of any drug reaction, a dose between 0.75 and 1.5 g that results in an initial 24-hour cupriuresis of over 2 mg should be continued for about three months, by which time the most reliable method of monitoring maintenance treatment is the determination of free copper in the serum. This equals the difference between quantitatively determined total copper and ceruloplasmin-copper. Adequately treated patients will usually have less than 10 mcg free copper/dL of serum. It is seldom necessary to exceed a dosage of 2 g/day. If the patient is intolerant to therapy with DEPEN, alternative treatment is trientine hydrochloride.
In patients who cannot tolerate as much as 1 g/day initially, initiating dosage with 250 mg/day, and increasing gradually to the requisite amount, gives closer control of the effects of the drug and may help to reduce the incidence of adverse reactions.
Cystinuria - It is recommended that DEPEN be used along with conventional therapy. By reducing urinary cystine, it decreases crystalluria and stone formation. In some instances, it has been reported to decrease the size of, and even to dissolve, stones already formed.
The usual dosage of DEPEN in the treatment of cystinuria is 2 g/day for adults, with a range of 1 to 4 g/day. For pediatric patients, dosage can be based on 30 mg/kg/day. The total daily amount should be divided into four doses. If four equal doses are not feasible, give the larger portion at bedtime. If adverse reactions necessitate a reduction in dosage, it is important to retain the bedtime dose.
Initiating dosage with 250 mg/day, and increasing gradually to the requisite amount, gives closer control of the effects of the drug and may help to reduce the incidence of adverse reactions.
In addition to taking DEPEN, patients should drink copiously. It is especially important to drink about a pint of fluid at bedtime and another pint once during the night when urine is more concentrated and more acid than during the day. The greater the fluid intake, the lower the required dosage of DEPEN.
Dosage must be individualized to an amount that limits cystine excretion to 100-200 mg/day in those with no history of stones, and below 100 mg/day in those who have had stone formation and/or pain. Thus, in deter-mining dosage, the inherent tubular defect, the patient’s size, age, and rate of growth, and his diet and water intake all must be taken into consideration.
The standard nitroprusside cyanide test has been reported useful as a qualitative measure of the effective dose*:
* Lotz, M., Potts, J.T. and Bartter, F.C.: BritMed J 2: 521, August 28, 1965 (in Medical Memoranda).
Add 2 mL of freshly prepared 5 percent sodium cyanide to 5 mL of a 24-hour aliquot of protein-free urine and let stand ten minutes. Add 5 drops of freshly prepared 5 percent sodium nitroprusside and mix. Cystine will turn the mixture magenta. If the result is negative, it can be assumed that cystine excretion is less than 100 mg/g creatinine.
Although penicillamine is rarely excreted unchanged, it also will turn the mixture magenta. If there is any question as to which substance is causing the reaction, a ferric chloride test can be done to eliminate doubt: Add 3 percent ferric chloride dropwise to the urine. Penicillamine will turn the urine an immediate and quickly fading blue. Cystine will not produce any change in appearance.
Rheumatoid Arthritis - The principal rule of treatment with DEPEN in rheumatoid arthritis is patience. The onset of therapeutic response is typically delayed. Two or three months may be required before the first evidence of a clinical response is noted (see CLINICAL PHARMACOLOGY).
When treatment with DEPEN has been interrupted because of adverse reactions or other reasons, the drug should be reintroduced cautiously by starting with a lower dosage and increasing slowly.
Initial Therapy - The currently recommended dosage regimen in rheumatoid arthritis begins with a single daily dose of 125 mg or 250 mg which is thereafter increased at one to three month intervals, by 125 mg or 250 mg/day, as patient response and tolerance indicate. If a satisfactory remission of symptoms is achieved, the dose associated with the remission should be continued (see Maintenance Therapy). If there is no improvement and there are no signs of potentially serious toxicity after two to three months of treatment with doses of 500-750 mg/day, increases of 250 mg/day at two to three month intervals may be continued until a satisfactory remission occurs (see Maintenance Therapy) or signs of toxicity develop (see WARNINGS and PRECAUTIONS). If there is no discernible improvement after three to four months of treatment with 1000 to 1500 mg of penicillamine/day, it may be assumed the patient will not respond and DEPEN should be discontinued.
Maintenance Therapy - The maintenance dosage of DEPEN must be individualized, and may require adjustment during the course of treatment. Many patients respond satisfactorily to a dosage within the 500-750 mg/day range. Some need less.
Changes in maintenance dosage levels may not be reflected clinically or in the erythrocyte sedimentation rate for two to three months after each dosage adjustment.
Some patients will subsequently require an increase in the maintenance dosage to achieve maximal disease suppression. In those patients who do respond, but who evidence incomplete suppression of their disease after the first six to nine months of treatment, the daily dosage of DEPEN may be increased by 125 mg or 250 mg/day at three-month intervals. It is unusual in current practice to employ a dosage in excess of 1 g/day, but up to 1.5 g/day has sometimes been required.
Management of Exacerbations - During the course of treatment some patients may experience an exacerbation of disease activity following an initial good response. These may be self-limited and can subside within twelve weeks. They are usually controlled by the addition of nonsteroidal anti-inflammatory drugs, and only if the patient has demonstrated a true “escape” phenomenon (as evidenced by failure of the flare to subside within this time period) should an increase in the maintenance dose ordinarily be considered.
In the rheumatoid patient, migratory polyarthralgia due to penicillamine is extremely difficult to differentiate from an exacerbation of the rheumatoid arthritis. Discontinuance or a substantial reduction in the dosage of DEPEN for up to several weeks will usually determine which of these processes is responsible for the arthralgia.
Duration of Therapy - The optimum duration of DEPEN therapy in rheumatoid arthritis has not been determined. If the patient has been in remission for six months or more, a gradual, stepwise dosage reduction in decrements of 125 mg or 250 mg/day at approximately three month intervals may be attempted.
Concomitant Drug Therapy - DEPEN should not be used in patients who are receiving gold therapy, anti-malarial or cytotoxic drugs, oxyphenbutazone, or phenylbutazone (see PRECAUTIONS). Other measures, such as salicylates, other nonsteroidal anti-inflammatory drugs or systemic corticosteroids may be continued when DEPEN is initiated. After improvement commences, analgesic and anti-inflammatory drugs may be slowly discontinued as symptoms permit. Steroid withdrawal must be done gradually, and many months of DEPEN treatment may be required before steroids can be completely eliminated.
Dosage Frequency - Based on clinical experience, dosages up to 500 mg/day can be given as a single daily dose. Dosages in excess of 500 mg/day should be administered in divided doses.
How is Depen supplied
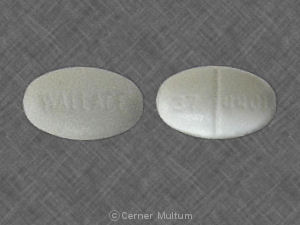
Depen® (penicillamine tablets, USP) Titratable Tablets: 250 mg functionally scored, oval, white tablets coded with 37-4401; available in bottles of 100 (NDC 0037-4401-01).
Storage: Store at controlled room temperature 20°–25°C (68°–77°F). Protect from moisture.
Dispense in a tight container.
To report SUSPECTED ADVERSE REACTIONS, contact Meda Pharmaceuticals Inc. at 1-800-526-3840 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Manufactured for:
Meda Pharmaceuticals Inc.
1000 Mylan Blvd
Canonsburg, PA 15317
©2023 Viatris Inc.
DEPEN is a registered trademark of Meda Pharmaceuticals Inc., a Viatris company.
500493-06 Rev. 7/2023
IN-4401-04
Package Label - Principal Display Panel – 100 Tablet Bottle, Depen Titratable Tablets
NDC 0037-4401-01
100 Tablets
Depen®
(penicillamine
tablets, USP)
Titratable Tablets
250 mg
Rx Only
Meda
Pharmaceuticals®
Usual Dosage: See attached package
circular for full prescribing information.
Store at controlled room temperature
20°-25°C (68°-77°F). Protect from moisture.
Dispense in a tight container.
© 2023 Viatris Inc.
Manufactured for:
Meda Pharmaceuticals Inc.
Canonsburg, PA 15317 U.S.A.
LB-4401A-02 Rev. 7/2023
400942-03

| DEPEN
penicillamine tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Meda Pharmaceuticals Inc. (051229602) |
More about Depen (penicillamine)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- Drug class: antirheumatics
- Breastfeeding
- En español