Crofab: Package Insert / Prescribing Info
Package insert / product label
Generic name: ovine crotalidae venoms immune fab
Dosage form: injection, powder, lyophilized, for solution
Drug class: Antitoxins and antivenins
J Code (medical billing code): J0840 (Up to 1 gm, injection)
Medically reviewed by Drugs.com. Last updated on Nov 25, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
CROFAB® crotalidae polyvalent immune fab (ovine)
Lyophilized Powder for Solution for Intravenous Injection
Initial U.S. Approval: 2000
Recent Major Changes
Indications and Usage for Crofab
CROFAB is a sheep-derived antivenin indicated for the management of adult and pediatric patients with North American crotalid envenomation. (1)
Crofab Dosage and Administration
For intravenous use only
- Initiate administration as soon as possible after snake bite in patients who develop signs of envenomation (e.g. local injury, coagulation abnormality or systemic signs of envenomation)
- Dose: (2.1)
- Recommended initial dose is between 4 and 6 vials
- Observe patient for up to one hour after the initial dose and give an additional 4-6 vial dose as needed to gain initial control of envenomation
- After initial control is established, administer additional 2-vial doses every 6 hours for 18 hours (total of 3 doses)
- Preparation and Administration: (2.2)
- Reconstitute each vial of CROFAB with 18 mL of 0.9% Sodium Chloride and mix by continuous manual inversion at the rate of one to two inversions per second until no solid material is visible in the vial (contents will remain opalescent)
- Do not shake as this can cause foaming
- Further dilute the entire dose with 0.9% Sodium Chloride to a total volume of 250 mL
- Infuse each dose intravenously over at least 1 hour
Dosage Forms and Strengths
CROFAB is available as lyophilized powder. Each vial contains up to 1 gram of total protein and not less than the indicated number of mouse LD50 neutralizing units: (3)
| Snake Species Used for Antivenin Component | Minimum mouse LD50Units per vial |
| C. atrox
(Western Diamondback rattlesnake) | 1270 |
| C. adamanteus
(Eastern Diamondback rattlesnake) | 420 |
| C. scutulatus
(Mojave rattlesnake) | 5570 |
| A. piscivorus
(Cottonmouth or Water Moccasin) | 780 |
Contraindications
Do not administer CROFAB to patients with a known history of hypersensitivity to any of its components, or to papaya or papain unless the benefits outweigh the risks and appropriate management for anaphylactic reactions is readily available. (4)
Warnings and Precautions
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥5% of subjects): urticaria, rash, nausea and pruritus. Allergic reaction (severe hives and a severe rash and pruritus) has occurred following treatment.
Recurrent coagulopathy due to envenomation and requiring additional treatment may occur. (5.1)
To report SUSPECTED ADVERSE REACTIONS, contact 1-877-377-3784 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2018
Full Prescribing Information
1. Indications and Usage for Crofab
CROFAB is indicated for the management of adult and pediatric patients with North American crotalid envenomation. The term crotalid is used to describe the Crotalinae subfamily (formerly known as Crotalidae) of venomous snakes which includes rattlesnakes, copperheads and cottonmouths/water moccasins.
2. Crofab Dosage and Administration
For intravenous use only
2.1 Dose
- Administer CROFAB as soon as possible in patients who develop any signs of envenomation (e.g., local injury, coagulation abnormality or systemic signs of envenomation) to prevent clinical deterioration. CROFAB was shown in clinical studies to be effective when given within 6 hours of snakebite.
- Antivenin dosage requirements are contingent upon an individual patient’s response. Based on clinical experience with CROFAB, the recommended initial dose is 4 to 6 vials; however, the starting dose may vary from a minimum of 4 vials to a maximum of 12 vials based on clinical judgment and severity of envenomation [3].
- Observe the patient for up to 1 hour following completion of the first dose to determine if initial control of envenomation has been achieved. Initial control is achieved when local signs of envenomation are arrested (leading edge of local injury is not progressing), systemic symptoms are resolved and coagulation parameters have normalized or are trending toward normal. If initial control is not achieved by the first dose, an additional dose of 4 to 6 vials should be administered repeatedly until initial control of the envenomation syndrome has been achieved.
- Once initial control has been achieved, additional 2-vial doses of CROFAB every 6 hours for up to 18 hours (3 doses) are recommended. Optimal dosing following the 18-hour scheduled dose of CROFAB has not been determined. Additional 2-vial doses may be administered as deemed necessary by the treating physician, based on the patient’s clinical course.
- Infusion reactions, such as fever, low back pain, wheezing and nausea, may be related to the rate of infusion and can be controlled by decreasing the rate of administration of the solution [12]. Poison control centers are a helpful resource for individual treatment advice.
2.2 Preparation and Administration
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- Reconstitute each vial of CROFAB with 18 mL of 0.9% Sodium Chloride (diluent not included) and mix by continuous manual inversion at the rate of one to two inversions per second until no solid material is visible in the vial (the fully reconstituted product will still be opalescent). Do not shake as this can cause foaming. Further dilute the contents of all of the reconstituted vials to a total volume of 250 mL with 0.9% Sodium Chloride and mix by gently swirling.
- Use the reconstituted and diluted product within 4 hours.
- Infuse the dose intravenously over 60 minutes. However, the infusion should proceed slowly over the first 10 minutes at a 25- 50 mL/hour rate with careful observation for any allergic reaction. If no such reaction occurs, the infusion rate may be increased to the full 250 mL/hour rate until completion. Close patient monitoring is necessary.
3. Dosage Forms and Strengths
CROFAB is available as a sterile, nonpyrogenic, purified, lyophilized powder. Each vial contains up to 1 gram of total protein and not less than the indicated number of mouse LD50 neutralizing units*:
| * As of 2008, the potency assay has been optimized for a new strain of mice, which has resulted in changes to the minimum mouse LD50 neutralizing units. These changes do not reflect any change in product potency, but only a different biological response of the mouse strain to the venom. | |
| Snake Species Used for Antivenin Component | Minimum mouse LD50 Units per vial |
| C. atrox (Western Diamondback rattlesnake) | 1270 |
| C. adamanteus (Eastern Diamondback rattlesnake) | 420 |
| C. scutulatus (Mojave rattlesnake) | 5570 |
| A. piscivorus (Cottonmouth or Water Moccasin) | 780 |
4. Contraindications
Do not administer CROFAB to patients with a known history of hypersensitivity to papaya or papain unless the benefits outweigh the risks and appropriate management for anaphylactic reactions is readily available.
5. Warnings and Precautions
5.1 Coagulopathy
Coagulopathy is a complication noted in many victims of crotalid envenomation that arises due to the ability of the snake venom to interfere with the blood coagulation cascad [5, 9, 10], and is seen more frequently in severely envenomated patients.
In clinical trials with CROFAB, recurrent coagulopathy (the return of a coagulation abnormality after it has been successfully treated with antivenin), characterized by decreased fibrinogen, decreased platelets and elevated prothrombin time, occurred in approximately half of patients studied. The clinical significance of these recurrent abnormalities is not known; however, one systematic review of the literature showed that medically significant bleeding following treatment of Crotaline snake envenomation is uncommon, occurring with an estimated frequency of 0.5% [13]. Recurrent coagulation abnormalities were observed only in patients who experienced coagulation abnormalities during their initial hospitalization, although coagulopathy can initially appear at any time before, during or after treatment. Optimal dosing to completely prevent recurrent coagulopathy has not been determined. Because CROFAB has a shorter persistence in the blood than the snake venoms that can leak from depot sites over a prolonged period of time, repeat dosing to prevent or treat such recurrence may be necessary [see Dosage and Administration (2)].
Recurrent coagulopathy may persist for 1 to 2 weeks or more. Patients who experience coagulopathy due to snakebite during hospitalization for initial treatment should be monitored for signs and symptoms of recurrent coagulopathy for up to 1 week or longer at the physician’s discretion. During this period, the physician should carefully assess the need for re-treatment with CROFAB and use of any type of anticoagulant or anti-platelet drug.
Because snake envenomation can cause coagulation abnormalities, the following conditions, which are also associated with coagulation defects, should be considered: cancer, collagen disease, congestive heart failure, diarrhea, elevated temperature, hepatic disorders, hyperthyroidism, poor nutritional state, steatorrhea, vitamin K deficiency.
5.2 Hypersensitivity Reactions
Severe hypersensitivity reactions may occur with CROFAB. In case of acute hypersensitivity reactions, including anaphylaxis and anaphylactoid reactions, discontinue infusion and institute appropriate emergency treatment.
CROFAB contains purified immunoglobulin fragments from the blood of sheep that have been immunized with snake venoms [see Description (11)]. Injection of heterologous animal proteins can cause severe acute and delayed hypersensitivity reactions (late serum reaction or serum sickness) and a possible febrile response to immune complexes formed by animal antibodies and neutralized venom components [11].
Papain is used to cleave antibodies into fragments during the processing of CROFAB, and trace amounts of papain or inactivated papain residues may be present. Patients allergic to papain, chymopapain, other papaya extracts, or the pineapple enzyme bromelain may also have an allergic reaction to CROFAB. Some dust mite allergens and some latex allergens share antigenic structures with papain and patients with these allergies may be allergic to papain [7, 8].
Use the following precautions to manage hypersensitivity reactions:
- Emergency medical care (e.g., epinephrine, intravenous antihistamines and/or albuterol) should be readily available.
- Carefully monitor patients for signs and symptoms of an acute allergic reaction (e.g., urticaria, pruritus, erythema, angioedema, bronchospasm with wheezing or cough, stridor, laryngeal edema, hypotension, tachycardia).
- Follow-up all patients for signs and symptoms of delayed allergic reactions or serum sickness (e.g., rash, fever, myalgia, arthralgia).
Patients who receive a course of treatment with a foreign protein such as CROFAB may become sensitized to it. Therefore, caution should be used when administering a repeat course of treatment with CROFAB for a subsequent envenomation episode.
Skin testing has not been used in clinical trials of CROFAB and is not required.
6. Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥5% of subjects) were urticaria, rash, nausea, pruritus and back pain.
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of CROFAB was evaluated in 289 patients who received CROFAB for crotalid envenomation. Forty-two patients were from two prospective clinical trials in patients who experienced mild to moderate crotalid envenomation; 247 patients were from a retrospective study in patients who experienced mild, moderate or severe envenomation. There also were safety information extracted from literature review of publications on CROFAB that contained patient exposure data.
Premarketing Prospective Clinical Trials
A total 42 patients received CROFAB for treatment of mild to moderate crotalid envenomation in two clinical studies. The patients were aged 11 to 76 years, and 34 were male and 8 were female. Nineteen patients experienced an adverse reaction for a total of 26 adverse reactions. Adverse reactions involving the skin and appendages (primarily rash, urticaria, and pruritus) were reported in 12 of the 42 patients (see Table 1). One patient discontinued CROFAB therapy due to an allergic reaction.
Of the 19 patients who experienced adverse reactions, 3 patients experienced severe or serious adverse reactions.
- 1 patient who experienced a serious adverse reaction had recurrent coagulopathy due to envenomation, which required re-hospitalization and additional antivenin administration. This patient eventually made a complete recovery.
- 2 patients had severe adverse reactions that consisted of 1 patient who developed severe urticaria following treatment and 1 patient who developed a severe rash and pruritus several days following treatment. Both patients recovered following treatment with antihistamines and prednisone.
| † Allergic reaction consisted of urticaria, dyspnea and wheezing in 1 patient. | |
| Adverse Reaction | n=42 Number of Reactions |
| Body as a Whole | |
| Back pain | 2 |
| Allergic reaction† | 1 |
| Serum sickness | 1 |
| Skin and Appendages | |
| Urticaria | 7 |
| Rash | 3 |
| Pruritus | 2 |
| Subcutaneous nodule | 1 |
| Respiratory System | |
| Cough | 1 |
| Digestive System | |
| Nausea | 3 |
| Anorexia | 1 |
| Hematologic/Lymphatic | |
| Coagulation disorder | 1 |
| Ecchymosis | 1 |
| Musculoskeletal | |
| Myalgia | 1 |
| Nervous System | |
| Nervousness | 1 |
Six of 42 patients experienced an adverse reaction associated with an early serum reaction and 4 experienced an adverse reaction associated with a late serum reaction. Two additional patients were considered to have experienced a late serum reaction by the investigator, although no associated adverse reaction was reported. Serum reactions are defined as reactions associated with CROFAB infusion. They consisted mainly of urticaria and rash, and all patients recovered without sequelae. Table 2 lists the incidence of early and late serum reactions. There were 7 events classified as early serum reactions and 5 events classified as late serum reactions; none was serious.
| ** Allergic reaction consisted of urticaria, dyspnea and wheezing in 1 patient. | |||
| † Serum sickness consisted of severe rash and pruritus in 1 patient. | |||
| n=42 Number of Events |
|||
| Early Serum Reactions | |||
| Urticaria | 5 | ||
| Cough | 1 | ||
| Allergic reaction** | 1 | ||
| Late Serum Reactions | |||
| Rash | 2 | ||
| Pruritus | 1 | ||
| Urticaria | 1 | ||
| Serum sickness† | 1 | ||
Postmarketing Retrospective Study
This was a retrospective study of data collected from postmarketing use of CROFAB to compare treatment and outcome characteristics between patients with severe envenomation to those with mild to moderate envenomation. A total of 247 patients received CROFAB for treatment of mild, moderate or severe crotalid envenomation. The patients were aged 1 to 91 years, and 206 were male and 41 were female. Of the 247 patients, 209 were classified as mild/moderate (n=181) or severe (n=28), while 38 patients did not have enough data to calculate an initial severity score.
There were a total of 36 immediate adverse drug reactions reported in 15 patients (6.1%, n = 247).
- There were 11 immediate serious adverse events related to CROFAB administration reported in four patients. The events included two episodes each of hypotension and tongue swelling, and one episode each of chest discomfort, angioedema, bronchospasm, wheezing, tracheal edema, dyspnea, and lip swelling.
- There were 22 immediate non-serious adverse events related to CROFAB administration reported in 12 patients. The events included four episodes each of rash and pruritus, three episodes of urticaria and one episode each of tachycardia, tachypnea, erythema, swelling, hyperhidrosis, dizziness, headache, musculoskeletal chest pain, chills, feeling cold and nervousness.
Delayed hypersensitivity reactions were reported for two patients. In one patient, symptoms described as hives, itching and epigastric pressure occurred 6 days post-dosing and were not serious. In the second patient, symptoms were not described in the medical records and therefore were not captured in this study.
Recurrent coagulopathy developed in 5 severely envenomated patients and in 6 mild/moderate envenomated patients. In addition, 7 mild/moderate patients experienced delayed-onset coagulopathy. One severely envenomated patient with recurrent coagulopathy experienced medically significant bleeding.
Additional Published Clinical Studies Experience
From a literature review of nine publications on CROFAB that contained patient exposure data, 15 of 313 (4.8%) patients receiving CROFAB experienced acute hypersensitivity reactions. The most common signs and symptoms associated with these reactions were rash (10 patients) and wheezing (3 patients). Most reactions were mild, resolved after antihistamine therapy, and did not require discontinuation of antivenom therapy. No patient developed a life-threatening hypersensitivity reaction, required intubation, suffered lasting ill-effect or died as a result of CROFAB administration.
Follow up data (minimum of six days after treatment) were available in 94 of the 313 patients. Delayed hypersensitivity reactions were reported in 10 cases. The most common signs and symptoms of delayed hypersensitivity were rash (9 patients) and fever (3 patients). Most were mild and treated with antihistamines and steroids.
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during the post approval use of CROFAB. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to product exposure:
- Delayed allergic reaction manifested by fever, pruritis and/or rash
- Delayed or recurrent coagulopathy or thrombocytopenia
- Failure to achieve initial control
- Recurrent swelling refractory to treatment
- Thrombocytopenia refractory to treatment
- Prolonged hospitalization
- Bleeding
- Tremor
- Treatment failure resulting in death
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Animal reproduction studies have not been conducted with CROFAB. It is also not known whether CROFAB can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. CROFAB should be given to a pregnant woman only if clearly needed. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
It is not known whether CROFAB is excreted in human breast milk. Because
many drugs are excreted in human milk, caution should be exercised when
CROFAB is administered to a nursing woman.
8.4 Pediatric Use
Thirty-two percent (78/247) of patients studied in a post-marketing retrospective study of CROFAB were 16 years of age or younger (median 8.5 years [range 1 to 16 years]). Initial control of envenomation was achieved in 64/72 (89%) of pediatric patients, which was similar to the initial control rate in adults (103/128, 80%). Changes in severity score, recurrence and incidence adverse reactions were similar between pediatric and adult patients, indicating safety and efficacy in the pediatric population are comparable to that in adults. Limited published clinical experience has not shown that a dosage adjustment for age should be made [14, 15].
11. Crofab Description
CROFAB [Crotalidae Polyvalent Immune Fab (Ovine)] is a sterile, nonpyrogenic, purified, lyophilized preparation of ovine Fab (monovalent) immunoglobulin fragments obtained from the blood of healthy sheep flocks immunized with one of the following North American snake venoms: Crotalus atrox (Western Diamondback rattlesnake), Crotalus adamanteus (Eastern Diamondback rattlesnake), Crotalus scutulatus (Mojave rattlesnake), and Agkistrodon piscivorus (Cottonmouth or Water Moccasin). To obtain the final antivenin product, the four different monospecific antivenins are mixed. Each monospecific antivenin is prepared by fractionating the immunoglobulin from the ovine serum, digesting it with papain, and isolating the venom specific Fab fragments on ion exchange and affinity chromatography columns.
CROFAB is standardized by its ability to neutralize the lethal action of each of the four venom immunogens following intravenous injection in mice. The potency of the product will vary from batch to batch; however, a minimum number of mouse LD50 neutralizing units against each of the four venoms is included in every vial of final product, as shown in Table 3.
| 1 One neutralizing unit is determined as the amount of the mixed monospecific Fab proteins necessary to neutralize one LD50 of each of the four venoms, where the LD50 is the amount of venom that would be lethal in 50% of mice. | |||
| 2 As of 2008, the potency assay has been optimized for a new strain of mice, which has resulted in changes to the minimum mouse LD50 neutralizing units. These changes do not reflect any change in product potency, but only a different biological response of the mouse strain to the venom. | |||
| Venom | Minimum Potency per Vial of CROFAB2 | ||
| Crotalus atrox | ≥ 1270 | ||
| Crotalus adamanteus | ≥ 420 | ||
| Crotalus scutulatus | ≥ 5570 | ||
| Agkistrodon piscivorus | ≥ 780 | ||
Each vial of CROFAB contains up to 1 gram of total protein and sodium phosphate buffer consisting of dibasic sodium phosphate USP and sodium chloride USP. The product is intended for intravenous administration after reconstitution with 18 mL of 0.9% Sodium Chloride.
12. Crofab - Clinical Pharmacology
12.1 Mechanism of Action
CROFAB is a venom-specific Fab fragment of immunoglobulin G (IgG) that works by binding and neutralizing venom toxins, facilitating their redistribution away from target tissues and their elimination from the body.
12.3 Pharmacokinetics
A limited number of samples were collected from three patients in the pharmacokinetic study of CROFAB. Based on these data, estimates of elimination half-life were made. The elimination half life for total Fab ranged from approximately 12 to 23 hours. These limited pharmacokinetic estimates of half-life are augmented by data obtained with an analogous ovine Fab product produced by BTG International Inc. using a similar production process. In that study, 8 healthy subjects were given 1 mg of intravenous digoxin followed by an approximately equimolar neutralizing dose of 76 mg of digoxin immune Fab (ovine). Total Fab was shown to have a volume of distribution of 0.3 L/kg, a systemic clearance of 32 mL/min (approximately 0.4 mL/min/kg) and an elimination half-life of approximately 15 hours.
13. Nonclinical Toxicology
13.2 Animal Toxicology and/or Pharmacology
CROFAB was effective in neutralizing the venoms of 10 clinically important North American crotalid snakes in a murine lethality model (see Table 4) [1]. In addition, preliminary data from experiments in mice using whole IgG from the sheep immunized for CROFAB production suggest that CROFAB might possess antigenic cross-reactivity against the venoms of some Middle Eastern and North African snakes; however, there are no clinical data available to confirm these findings.
| Study Objective & Design | Endpoint Measured | Major Findings and Conclusions |
| To determine the cross-neutralizing ability of CROFAB to protect mice from the lethal effects of venom from clinically important species. Separate groups of mice were injected with increasing doses of CROFAB pre-mixed with two LD50 of each venom tested. | ED50 for each venom | (Note: Lower numbers represent increased
potency against venoms listed) Challenge Venom ED50 (mg antivenin/mg venom) C. atrox 3 C. adamanteus 18 C. scutulatus 8 A. piscivorus 4 C. h. atricaudatus 11 C. v. helleri 6 C. m. molossus 5 A. c. contortrix 8 S. m. barbouri 12 C. h. horridus 6 |
| Based on data from studies in mice, CROFAB has relatively good cross-protection against venoms not used in the immunization of flocks used to produce it. For C. v. helleri and C. m. molossus, higher doses may be required based on historical data. |
14. Clinical Studies
The safety and efficacy of CROFAB in crotalid envenomation were evaluated in two premarketing prospective and one postmarketing retrospective studies. The prospective studies evaluated patients suffering from minimal to moderate North American crotalid envenomation. The postmarketing study evaluated patients suffering from mild, moderate or severe envenomation. The definition of minimal, moderate, and severe envenomation in clinical studies of CROFAB is provided in Table 5.
| Envenomation Severity | Definition |
| Minimal | Swelling, pain, and ecchymosis limited to the immediate bite site;
Systemic signs and symptoms absent; Coagulation parameters normal with no clinical evidence of bleeding |
| Moderate | Swelling, pain, and ecchymosis involving less than a
full extremity or, if bite was sustained on the trunk,
head or neck, extending less than 50 cm;
Systemic signs and symptoms may be present but not life threatening, including but not limited to nausea, vomiting, oral paresthesia or unusual tastes, mild hypotension (systolic blood pressure >90 mmHg), mild tachycardia (heart rate <150), and tachypnea Coagulation parameters may be abnormal, but no clinical evidence of bleeding present. (Minor hematuria, gum bleeding and nosebleeds are allowed if they are not considered severe in the investigator’s judgment) |
| Severe | Swelling, pain, and ecchymosis involving more than an
entire extremity or threatening the airway
Systemic signs and symptoms are markedly abnormal, including severe alteration of mental status, severe hypotension, severe tachycardia, tachypnea, or respiratory insufficiency Coagulation parameters are abnormal, with serious bleeding or severe threat of bleeding |
Premarketing Prospective Studies
Two premarketing prospectively defined, open label, multi-center trials were conducted in otherwise healthy patients 11 years of age or older who had experienced minimal or moderate North American crotalid envenomation that showed evidence of progression. Progression was defined as the worsening of any evaluation parameter used in the grading of an envenomation: local injury, laboratory abnormality or symptoms and signs attributable to crotalid snake venom poisoning. Both clinical trials excluded patients with Copperhead envenomation.
Efficacy was determined using a Snakebite Severity Score (SSS) [2] (referred to as the efficacy score or ES) and an investigator’s clinical assessment (ICA) of efficacy. The SSS is a tool used to measure the severity of envenomation based on six body categories: local wound (e.g., pain, swelling and ecchymosis), pulmonary, cardiovascular, gastrointestinal, hematological and nervous system effects. A higher score indicates worse symptoms.. CROFAB was required to prevent an increase in the ES in order to demonstrate efficacy.
The ICA was based on the investigator’s clinical judgment as to whether the patient had a:
- Clinical response (pre-treatment signs and symptoms of envenomation were arrested or improved after treatment)
- Partial response (signs and symptoms of envenomation worsened, but at a slower rate than expected after treatment)
- Non-response (the patient’s condition was not favorably affected by the treatment)
Safety was assessed during CROFAB infusion by monitoring for early allergic events, such as anaphylaxis and early serum reactions, and late events, such as late serum reactions [see Adverse Reactions (6.1)]
In clinical study 1, 11 patients received an intravenous dose of 4 vials of CROFAB over 60 minutes. An additional 4-vial dose of CROFAB was administered after completion of the first CROFAB infusion, if deemed necessary by the investigator. At the 1-hour assessment, 10 out of 11 patients had no change or a decrease in their ES. Ten of 11 patients were also judged to have a clinical response by the ICA. After initial clinical response, two patients demonstrated a need for additional antivenom to stem progressive or recurrent symptoms and signs; one patient received an additional 4 vials of CROFAB. No patient in this study experienced an anaphylactic or anaphylactoid response or evidence of an early or late serum reaction as a result of administration of CROFAB.
Based on observations from the first study, the second clinical study compared two different CROFAB dosage schedules. Patients were given an initial intravenous dose of 6 vials of CROFAB with an option to retreat with an additional 6 vials, if needed, to achieve initial control of the envenomation syndrome. Initial control was defined as complete arrest of local manifestations and normalization of coagulation tests and systemic signs. Once initial control was achieved, patients were randomized to receive additional CROFAB either every 6 hours for 18 hours (Scheduled Group) or as needed (PRN Group).
In this study, CROFAB was administered to 31 patients with minimal or moderate crotalid envenomation. All 31 patients enrolled in the study achieved initial control of their envenomation with CROFAB, and 30, 25 and 26 of the 31 patients achieved a clinical response based on the ICA at 1, 6 and 12 hours respectively following initial control. Additionally, the mean ES was significantly decreased across the patient groups by the 12-hour evaluation time point (p=0.05 for the Scheduled Group; p=0.05 for the PRN Group) (see Table 6). There was no statistically significant difference between the Scheduled Group and the PRN Group with regard to the decrease in ES.
| * No change or a decline in the Efficacy Score was considered an indication of clinical response and a sign of efficacy. | |||
| ** For both the Scheduled and the PRN Groups, differences in the Efficacy Score at the four post-baseline assessment times were statistically decreased from baseline by Friedman’s test (p < 0.001) | |||
| Time Period | Scheduled Group (n=15) Efficacy Score* Mean ± SD | PRN Group (n=16) Efficacy Score* Mean ± SD |
|
| Baseline | 4.0 ± 1.3 | 4.7 ± 2.5 | |
| End of Initial Control Antivenin Infusion(s) | 3.2 ± 1.4 | 3.3 ± 1.3 | |
| 1 hour after Initial Control achieved | 3.1 ± 1.3 | 3.2 ± 0.9 | |
| 6 hours after Initial Control achieved | 2.6 ± 1.5 | 2.6 ± 1.3 | |
| 12 hours after Initial Control achieved | 2.4 ± 1.1** | 2.4 ± 1.2** | |
It has been noted in published literature accounts of rattlesnake bites, that a decrease in platelets can accompany moderately severe envenomation, that was not corrected by whole blood transfusions [3]. These reductions in platelet count have been observed to last from several hours and to several days following the venomous bite [3, 4, 5]. In clinical study 2, 6 patients had pre-dosing platelet counts below 100,000/mm3 (baseline average of 44,000/mm3). Platelet counts for all 6 patients increased to normal levels (average 209,000/ mm3) at 1 hour following initial control dosing with CROFAB (see Figure 1).
Figure 1 Platelet Counts from Baseline to 36 Hours for Patients with Counts <100,000/mm3 at Baseline (Clinical Study 2)
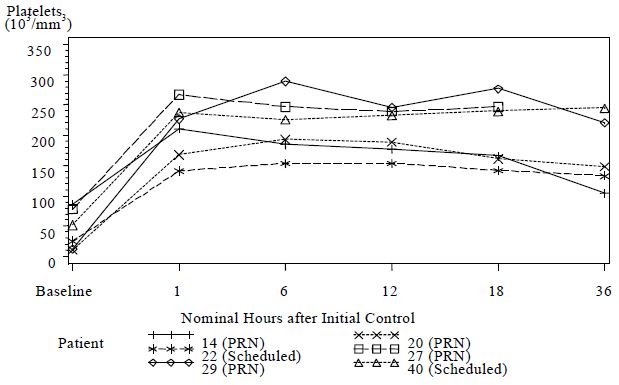
Although the difference in the decrease in ES between the two treatment groups was not significant, the data suggest that Scheduled dosing may provide better control of envenomation symptoms caused by continued leakage of venom from depot sites. Scheduled patients experienced a lower incidence of coagulation abnormalities at follow up compared with PRN patients (see Table 7) and more patients in the PRN Group showed a reduction in platelet count after discharge than in the Scheduled Group (see Figure 2). The need to administer additional CROFAB to patients in the PRN Group after initial control suggests there is continued need of antivenin for adequate treatment.
| ^ Numbers are expressed as percent of patients that had a follow-up
platelet count that was less than the count at hospital discharge, or a
fibrinogen level less than 50% of the level at hospital discharge * Follow-up data not available for one patient ** Statistically significant difference, p=0.04 by Fisher’s Exact test |
||
| Scheduled Group (n=14)*
(percent of patients with abnormal values)^ | PRN Group (n=16) (percent of patients with abnormal values)^ |
|
| Platelet | 2/14 (14%)** | 9/16 (56%)** |
| Fibrinogen | 2/14 (14%) | 7/16 (44%) |
Figure 2 Change in Platelet Counts in Individual Patients between Follow-Up Visits and Discharge
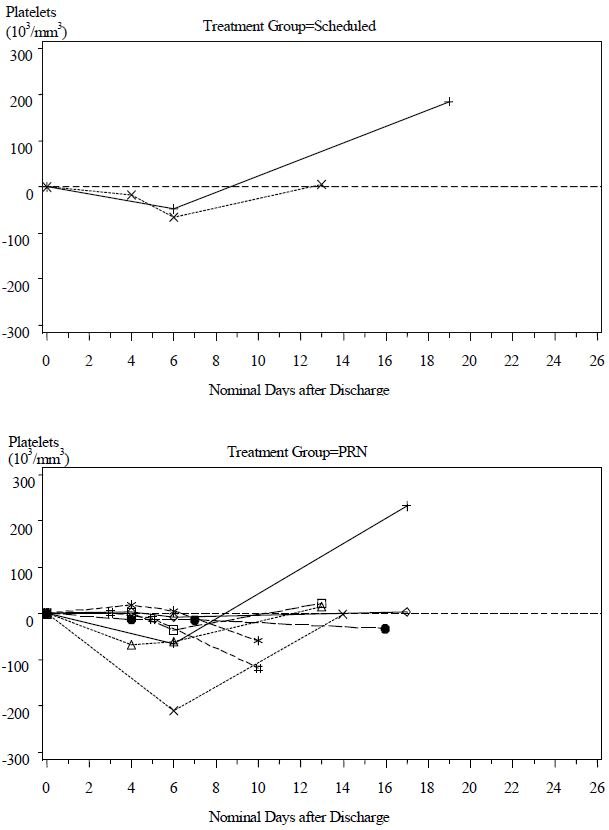
Patients in the Scheduled and PRN Groups are plotted separately
Only patients showing a reduced platelet count after discharge are shown
Postmarketing Retrospective Study
Following marketing approval of CROFAB, a retrospective study was conducted to assess the efficacy of CROFAB in mild, moderate and severe crotalid envenomation. This study was a multi-center, retrospective chart review of medical records of snakebite patients treated with CROFAB that compared treatment and outcomes of severe envenomation with those of mild and moderate envenomation. The primary efficacy variable was severity of envenomation as determined by a 7-point severity score. Patients were classified as having mild, moderate or severe envenomation based on their scores just prior to receiving antivenom. Those subjects with a severity score of 5 or 6 at the start of antivenom therapy were a priori defined as severe envenomations; those with a score of 3 or 4 were defined as moderate envenomations; and those with a score of 1 or 2 were defined as mild envenomations (see Table 5). A total of 247 patients of all severities were included in the study. Patients with enough data to determine baseline severity were included in the efficacy evaluation. This comprised a cohort of 209 patients, of whom 28 were classified as severe.
Improvement in severity score was observed in all 28 severely envenomated patients. Improvement was noted for each severe venom effect studied, including limb pain and swelling; cardiovascular, respiratory, gastrointestinal and neurologic effects; and coagulopathy/defibrination syndrome, thrombocytopenia and significant/spontaneous bleeding. The median dose of CROFAB administered to treat these severe venom effects was 9 vials (median of 2 doses). Initial control of envenomation was achieved in 57% (16/28) of severely envenomated patients and 87% (158/181) of mild/moderate envenomated patients. In both groups, failure to achieve initial control was most commonly attributable to persistent coagulopathy and/or thrombocytopenia, which occurred in 1 severe case. All 12 severe patients who did not achieve initial control received only one bolus dose of 4 to 6 vials to try to achieve initial control of envenomation. Of the 23 mild/moderate cases who did not achieve initial control, 19 did not follow recommended dosing for number of doses and vials. Whether initial control could have been achieved with larger initial doses of antivenom cannot be determined from this retrospective study. All patients, whether or not they achieved initial control, experienced significant improvement of venom effects and decreased severity scores after receiving CROFAB. Among patients with severe envenomation who did not achieve initial control, median severity score improved from 5 (range: 5 – 6) before CROFAB administration to 2 (range: 1 – 4) at the last loading dose. No patient in this analysis had a severity score greater than 3 at time of final clinical assessment.
15. References
- Consroe P, Egen NB, Russell FE, Gerrish K, Smith DC, Sidki A, et al. Comparison of a new ovine antigen binding fragment (Fab) antivenin for United States Crotalidae with the commercial antivenin for protection against venom induced lethality in mice. J Trop Med Hyg 1995; 53(5):507 510.
- Dart RC, Hurlbut KM, Garcia R, Boren J. Validation of a severity score for the assessment of Crotalid snakebite. Ann Emerg Med 1996; 27(3):321 326.
- Lavonas EJ, Ruha AM, Banner W, Bebarta V, Bernstein JN, Bush SP, Kerns WP, Richardson WH, Seifert SA, Tanen DA, Curry SC, Dart RC. Unified treatment algorithm for the management of crotaline snakebite in the United States: results of an evidence-informed consensus workshop. BMC Emerg Med February 3 2011;11:2 (http://www.biomedcentral.com/1471-227X/11/2).
- La Grange RG and Russell FE. Blood platelet studies in man and rabbits following Crotalus envenomation. Proc West Pharmacol Soc 1970;13:99-105.
- Lyons WJ. Profound thrombocytopenia associated with Crotalus ruber ruber envenomation: a clinical case. Toxicon 1971; 9:237 240.
- Tallon RW, Koch KL, Barnes SG, Ballard JO. Letter to Editor. N Engl J Med 1981;305:1347.
- Quarre JP, Lecomte J, Lauwers D, Gilbert P, Thiriaux J. Allergy to latex and papain. J Allergy Clin Immunol 1995; 95(4):922.
- Baur X, Chen Z, Rozynek P, Düser D, Raulf Heimsoth M. Cross reacting IgE antibodies recognizing latex allergens, including Hev b 1, as well as papain. Allergy 1995; 50(7):604 609.
- Furlow TG, Brennan LV. Purpura following timber rattlesnake (Crotalus horridus horridus) envenomation. Cutis 1985; 35:234 236.
- Budzynski AZ, Pandya BV, Rubin RN, Brizuela BS, Soszka T, Stewart GJ. Fibrinogenolytic afibrinogenemia after envenomation by western diamondback rattlesnake (Crotalus atrox). Blood 1984; 63(1):1 14.
- Kojis FG. Serum sickness and anaphylaxis. Am J Dis Child 1997;93 350.
- Kirkpatrick CH, The Digibind Study Advisory Panel. Allergic histories and reactions of patients treated with digoxin immune Fab (ovine) antibody. Am J Emerg Med 1991; 9(2 Suppl 1):7 10.
- Lavonas EJ, Khatri V, Daugherty C, Bucher-Bartelson B, King T, Dart RC. Medically significant late bleeding after treated Crotaline envenomation: A systematic review. Ann Emerg Med 2014;63(1):71-78.
- Pizon AF, Riley BD, LoVecchio F, and Gill R. Safety and Efficacy of Crotalidae Polyvalent Immune Fab in Pediatric Crotaline Envenomations. Acad Emerg Med 2007;14:373-376.
- Offerman SR, Bush SP, Moynihan JA, Clark RF. Crotaline Fab Antivenom for the Treatment of Children with Rattlesnake Envenomation. Pediatrics 2002; 110(5):968-971.
16. How is Crofab supplied
How Supplied
CROFAB is supplied as a carton that contains 2 vials of product (diluent not included) [NDC 50633-110-12]. Each vial of CROFAB contains up to 1 gram of lyophilized total protein and not less than the indicated number of mouse LD50 neutralizing units:
| Snake Species Used for Antivenin Component | Minimum mouse LD50 Units per vial |
| C. atrox (Western Diamondback rattlesnake) | 1270 |
| C. adamanteus (Eastern Diamondback rattlesnake) | 420 |
| C. scutulatus (Mojave rattlesnake) | 5570 |
| A. piscivorus (Cottonmouth or Water Moccasin) | 780 |
Storage and Handling
- Store vials at 2° to 8°C (36° to 46°F). A temperature excursion for no longer than 7 days within the range of -20° to 25°C (-4° to 77°F) is permitted.
- Do not freeze.
- Use within 4 hours after reconstitution.
17. Patient Counseling Information
- Advise patients to contact their physician immediately if they experience unusual bruising or bleeding (e.g., nosebleeds, excessive bleeding after brushing teeth, the appearance of blood in stools or urine, excessive menstrual bleeding, petechiae, excessive bruising or persistent oozing from superficial injuries) after hospital discharge.Such bruising or bleeding may occur for up to 1 week or longer following initial treatment.
- Advise patients to contact their physician immediately if they experience any signs and symptoms of delayed allergic reactions or serum sickness (e.g., rash, pruritus, urticaria) after hospital discharge.
Manufactured for
and distributed by:
BTG International Inc.
West Conshohocken, PA 19428
U.S. License No. 1861
CroFab® is a registered trademark of BTG International Inc.
BTG and the BTG roundel logo are registered trademarks of BTG International Ltd.
P11012F


| CROFAB
ovine crotalidae venoms immune fab injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - BTG International Inc. (617382395) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bora Pharmaceuticals Injectables Inc. | 119352788 | MANUFACTURE(50633-110) , LABEL(50633-110) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Protherics UK Limited | 536591589 | ANALYSIS(50633-110) , API MANUFACTURE(50633-110) | |
More about CroFab (antivenin (crotalidae) polyvalent)
- Check interactions
- Compare alternatives
- Reviews (2)
- Side effects
- Dosage information
- During pregnancy
- Drug class: antitoxins and antivenins