Creutzfeldt-Jakob Disease
Medically reviewed by Drugs.com. Last updated on Aug 4, 2025.
What do I need to know about Creutzfeldt-Jakob disease (CJD)?
CJD damages your brain, spine, and nerves. CJD causes a loss of mental, emotional, and physical abilities. CJD is fatal over time.
What causes CJD?
CJD is caused by a kind of protein known as prion. Prions are normally found in your body. With CJD, an abnormal form of prion in your brain, nerves, and spine causes harm. Healthcare providers do not know how most people get an abnormal protein. In some cases, the abnormal protein may be inherited or transmitted during an organ or tissue transplant.
What are the signs and symptoms of CJD?
- Confusion or memory loss
- Sudden, jerking movements
- Moodiness, depression, or anxiety
- Trouble speaking or swallowing
- Trouble standing or walking
Related medications
How is CJD diagnosed?
- A biopsy is a procedure to remove small piece of tissue from your brain. Healthcare providers will send the sample to a lab to check for abnormal prions.
- An EEG, or electroencephalogram, is done to see activity in your brain. Healthcare providers will place small pads on your head and connect them to a machine to see how your brain is working.
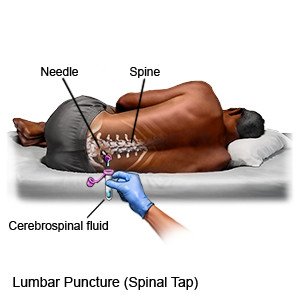
- A lumbar puncture, or spinal tap, is a procedure to remove fluid from around your spinal cord through a needle. It may show signs of inflammation or infection.
- An MRI may show areas of your brain that are damaged. You may be given contrast liquid to help your brain show up better in the pictures. Tell the healthcare provider if you have ever had an allergic reaction to contrast liquid. Do not enter the MRI room with anything metal. Metal can cause serious injury. Tell the healthcare provider if you have any metal in or on your body.
How is CJD treated?
There is no cure for CJD. The goal of treatment is to help reduce your symptoms. Medicines are given to decrease pain, anxiety, and muscle spasms. You may need treatment or therapy to help with speaking or swallowing.
Call your local emergency number (911 in the US) or have someone call if:
- You have trouble breathing.
- You have sudden changes in your vision.
When should I call my doctor?
- You have a fever.
- You are depressed and feel you cannot cope with your condition.
- You are anxious or nervous even after you take medicine.
- You have severe muscle twitching or pain even after you take medicine.
- You have questions or concerns about your condition or care.
Care Agreement
You have the right to help plan your care. Learn about your health condition and how it may be treated. Discuss treatment options with your healthcare providers to decide what care you want to receive. You always have the right to refuse treatment. The above information is an educational aid only. It is not intended as medical advice for individual conditions or treatments. Talk to your doctor, nurse or pharmacist before following any medical regimen to see if it is safe and effective for you.© Copyright Merative 2025 Information is for End User's use only and may not be sold, redistributed or otherwise used for commercial purposes.
Learn more about Creutzfeldt-Jakob Disease
Treatment options
Care guides
Further information
Always consult your healthcare provider to ensure the information displayed on this page applies to your personal circumstances.