Xenpozyme: Package Insert / Prescribing Info
Package insert / product label
Generic name: olipudase alfa-rpcp
Dosage form: injection, powder, lyophilized, for solution
Drug class: Lysosomal enzymes
J Code (medical billing code): J0218 (1 mg, injection)
Medically reviewed by Drugs.com. Last updated on Mar 9, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
XENPOZYME® (olipudase alfa-rpcp) for injection, for intravenous use
Initial U.S. Approval: 2022
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
See full prescribing information for complete boxed warning.
Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available. If a severe hypersensitivity reaction occurs, discontinue XENPOZYME immediately and initiate appropriate medical treatment. (5.1)
Recent Major Changes
Indications and Usage for Xenpozyme
XENPOZYME is a hydrolytic lysosomal sphingomyelin-specific enzyme indicated for treatment of non–central nervous system manifestations of acid sphingomyelinase deficiency (ASMD) in adult and pediatric patients. (1)
Xenpozyme Dosage and Administration
- See Full Prescribing Information for important recommendations prior to XENPOZYME treatment initiation. (2.1)
- Adults: Recommended starting dose is 0.1 mg/kg administered as an intravenous infusion. (2.2)
- Pediatrics: Recommended starting dose is 0.03 mg/kg administered as an intravenous infusion. (2.3)
- See Full Prescribing Information for the recommended dose escalation and maintenance dosage, dosage modifications to reduce the risk of adverse reactions, and preparation and administration instructions. (2.2, 2.3, 2.5, 2.6, 2.7)
Dosage Forms and Strengths
For injection: 4 mg or 20 mg of olipudase alfa-rpcp as a lyophilized powder in a single-dose vial for reconstitution. (3)
Contraindications
None. (4)
Warnings and Precautions
- Infusion-Associated Reactions (IARs): If severe IARs occur, discontinue XENPOZYME and initiate appropriate medical treatment. (5.2)
- Elevated Transaminases: Assess ALT and AST within one month prior to initiation of XENPOZYME, within 72 hours prior to any infusion during dose escalation, or prior to the next scheduled XENPOZYME infusion upon resuming treatment following a missed dose. (5.3)
- Risk of Fetal Malformations During Dosage Initiation or Escalation in Pregnancy: XENPOZYME dosage initiation or escalation, at any time during pregnancy, is not recommended as it may lead to elevated sphingomyelin metabolite levels that may increase the risk of fetal malformations. Advise females of reproductive potential to use effective contraception during treatment and for 14 days after the last dose if XENPOZYME is discontinued. (5.4, 8.1, 8.3)
Adverse Reactions/Side Effects
- Most common adverse reactions in adult patients (incidence ≥10%) are headache, cough, diarrhea, hypotension, and ocular hyperemia. (6.1)
- Most common adverse reactions in pediatric patients (incidence ≥20%) are pyrexia, cough, diarrhea, rhinitis, abdominal pain, vomiting, headache, urticaria, nausea, rash, arthralgia, pruritus, fatigue, and pharyngitis. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genzyme Corporation at 1-800-745-4447 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2024
Full Prescribing Information
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
Patients treated with XENPOZYME have experienced life-threatening hypersensitivity reactions, including anaphylaxis. Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available during XENPOZYME administration. If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue XENPOZYME immediately and initiate appropriate medical treatment. In patients with severe hypersensitivity reactions, a desensitization procedure to XENPOZYME may be considered [see Warnings and Precautions (5.1)].
1. Indications and Usage for Xenpozyme
XENPOZYME is indicated for treatment of non–central nervous system manifestations of acid sphingomyelinase deficiency (ASMD) in adult and pediatric patients.
2. Xenpozyme Dosage and Administration
2.1 Important Recommendations Prior to XENPOZYME Treatment Initiation
Therapy with XENPOZYME should be directed in consultation with physicians knowledgeable in the management of ASMD. In order to avoid dosing errors including overdosage [see Overdosage (10)], follow all instructions for dosage and administration.
Laboratory Testing
Before initiating XENPOZYME:
- Obtain baseline transaminase (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]) levels in all patients within 1 month prior to treatment initiation [see Warnings and Precautions (5.3)].
- Verify pregnancy status in females of reproductive potential [see Use in Specific Populations (8.1, 8.3)].
Premedication
Prior to XENPOZYME administration, consider premedicating with antihistamines, antipyretics, and/or corticosteroids [see Warnings and Precautions (5.1, 5.2)].
Medical Support
Appropriate medical support measures including cardiopulmonary resuscitation equipment should be readily available during XENPOZYME administration [see Warnings and Precautions (5.1)].
Weight-Based Dosing Information
The recommended adult and pediatric dosages of XENPOZYME for the dose escalation and maintenance phases [see Dosage and Administration (2.2, 2.3)] are based on body weight as follows for patients with a body mass index (BMI):
- Less than or equal to 30, the dosage is based on actual body weight (kg)
- Greater than 30, the dosage is based on adjusted body weight (kg). Calculate an adjusted body weight (kg) based on height in meters as described below:
- Adjusted body weight (kg) = (actual height in m)2 × 30
2.2 Recommended Dosage in Adult Patients
Dose Escalation Phase
The recommended starting dose of XENPOZYME in adults is 0.1 mg/kg.
In order to reduce the risk of infusion-associated reactions or elevated transaminase levels, follow the dose escalation regimen in Table 1 [see Warnings and Precautions (5.1, 5.2, 5.3)].
Administer XENPOZYME via intravenous infusion every 2 weeks.
| Adult Patients (18 years and older) | |
|---|---|
|
|
| First dose (Day 1/Week 0) | 0.1 mg/kg |
| Second dose (Week 2) | 0.3 mg/kg |
| Third dose (Week 4) | 0.3 mg/kg |
| Fourth dose (Week 6) | 0.6 mg/kg |
| Fifth dose (Week 8) | 0.6 mg/kg |
| Sixth dose (Week 10) | 1 mg/kg |
| Seventh dose (Week 12) | 2 mg/kg |
| Eighth dose (Week 14)† | 3 mg/kg (recommended maintenance dose) |
2.3 Recommended Dosage in Pediatric Patients
Dose Escalation Phase
The recommended starting dose of XENPOZYME in pediatric patients is 0.03 mg/kg.
In order to reduce the risk of hypersensitivity and infusion-associated reactions or elevated liver enzyme elevations, follow the dose escalation regimen in Table 2 [see Warnings and Precautions (5.1, 5.2, 5.3)].
Administer XENPOZYME via intravenous infusion every 2 weeks.
| Pediatric Patients (0 to 17 years) | |
|---|---|
|
|
| First dose (Day 1/Week 0) | 0.03 mg/kg |
| Second dose (Week 2) | 0.1 mg/kg |
| Third dose (Week 4) | 0.3 mg/kg |
| Fourth dose (Week 6) | 0.3 mg/kg |
| Fifth dose (Week 8) | 0.6 mg/kg |
| Sixth dose (Week 10) | 0.6 mg/kg |
| Seventh dose (Week 12) | 1 mg/kg |
| Eighth dose (Week 14) | 2 mg/kg |
| Ninth dose (Week 16)† | 3 mg/kg (recommended maintenance dose) |
2.4 Missed Doses
A dose is considered missed when it is not administered within 3 days of the scheduled date. When a dose of XENPOZYME is missed, refer to Table 3. Follow the instructions in the "Escalation Phase" or "Maintenance Phase" depending on which phase the patient misses the dose.
| Consecutive Missed Doses In: | Escalation Phase | Maintenance Phase |
|---|---|---|
|
||
| 1 missed dose |
| First and subsequent doses after missed dose: Administer maintenance dose |
| 2 consecutive missed doses |
|
|
| 3 or more consecutive missed doses |
| First and subsequent doses after missed doses: Restart dosing at 0.3 mg/kg and follow Table 1 for adult patients or Table 2 for pediatric patients.
|
2.5 Dosage and Administration Modifications and Monitoring
- In the event of a severe hypersensitivity reaction (e.g., anaphylaxis) or a severe infusion-associated reaction (IAR), immediately discontinue XENPOZYME administration and initiate appropriate medical treatment [see Warnings and Precautions (5.1, 5.2)].
- In the event of a mild to moderate hypersensitivity reaction or a mild to moderate IAR, consider temporarily holding or slowing the infusion rate, and/or reducing the XENPOZYME dose. If dose is reduced, re-escalate following dose escalation described in Tables 1 and 2 for adult and pediatric patients, respectively [see Warnings and Precautions (5.1, 5.2)].
- If transaminase levels are elevated above baseline and >2 times the ULN prior to the next scheduled administration, the XENPOZYME dose can be adjusted (prior dose repeated or reduced) or treatment can be temporarily withheld until the liver transaminases return to the patient's baseline value [see Warnings and Precautions (5.3)].
2.6 Preparation Instructions
Use aseptic technique during preparation. Reconstitute and dilute XENPOZYME in the following manner:
Reconstitution and Dilution Instructions
- 1.
- Determine the number of XENPOZYME vials to be reconstituted based on the calculated dose [see Dosage and Administration (2.2, 2.3)].
- 2.
- Remove XENPOZYME vials from refrigeration and set aside for approximately 20 to 30 minutes to allow vials to reach room temperature.
- 3.
- Reconstitute each vial with:
- 1.1 mL of Sterile Water for Injection, USP into the 4 mg vial
- 5.1 mL of Sterile Water for Injection, USP into the 20 mg vial
- by directing the diluent flow to the inside wall of the vial to avoid foaming.
- 4.
- Gently roll and tilt vial(s) to reconstitute XENPOZYME and avoid foaming. Each reconstituted vial will yield a 4 mg/mL clear, colorless solution.
- 5.
- Visually inspect the reconstituted solution in the vials for particulate matter and discoloration. The solution should be clear and colorless. Discard if the solution is discolored or if visible particulate matter is present.
- 6.
- Withdraw the required volume of XENPOZYME from the vial(s) and dilute the XENPOZYME solution for infusion with 0.9% Sodium Chloride Injection, USP in a syringe or infusion bag depending on the volume of infusion (see Table 4).
- For patients who weigh less than 10 kg receiving 0.03 mg/kg and 0.1 mg/kg and patients who weigh between 10 to 20 kg receiving 0.03 mg/kg dose, the volume of infusion will vary to achieve a fixed final concentration of 0.1 mg/mL (see Table 4). Prepare the required dose diluted to a final concentration of 0.1 mg/mL in a syringe for infusion.
- For all other patient weights and doses, the final concentration will vary to achieve a fixed total volume (see Table 4).
- -
- For total volume less than or equal to 20 mL prepare a syringe for infusion:
- Inject the required volume of the reconstituted XENPOZYME solution (4 mg/mL) from step 3 slowly down the inside wall of the syringe.
- Add slowly the quantity sufficient of 0.9% Sodium Chloride Injection, USP to obtain the required total infusion volume (avoid foaming within the syringe).
- -
- For a total volume of greater than or equal to 50 mL prepare an infusion bag:
- Add slowly the required volume of the reconstituted XENPOZYME solution (4 mg/mL) from step 3 into the appropriate size 0.9% Sodium Chloride Injection, USP infusion bag (avoid foaming within the bag) to achieve a fixed total volume per Table 4.
- 7.
- Gently invert the syringe or the infusion bag to mix. Do not shake. Because this is a protein solution, slight flocculation (described as thin translucent fibers) occurs occasionally after dilution.
- 8.
- Vials are for single dose only. Discard any unused solution.
Storage and Handling of the Reconstituted and Diluted Solutions
- If the reconstituted XENPOZYME vials are not used immediately, store refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours or at controlled room temperature at 20°C to 25°C (68°F to 77°F) for up to 6 hours. Discard the unused XENPOZYME reconstituted solution after 24 hours if stored refrigerated or 6 hours if stored at controlled room temperature.
- If the diluted solution is not used immediately, refrigerate the diluted solution at 2°C to 8°C (36°F to 46°F) for up to 24 hours or store at room temperature at 20°C to 25°C (68°F to 77°F) for up to 12 hours (inclusive of infusion time), or discard.
- Do not freeze.
| Pediatric Patients (0 to 17 years) | Adult patients (18 years and older) | |||
|---|---|---|---|---|
| Body Weight ≥2 kg and <10 kg | Body Weight ≥10 kg and <20 kg | Body Weight ≥20 kg | Body Weight ≥20 kg |
|
| XENPOZYME Dose | Total Infusion Volume | |||
|
||||
| 0.03 mg/kg | Actual volume will vary†
(0.6 mL to 3 mL) | Actual volume will vary†
(3 mL to 6 mL) | 5 mL | NA |
| 0.1 mg/kg | Actual volume will vary†
(2 mL to 10 mL) | 5 mL | 10 mL | 20 mL |
| 0.3 mg/kg | 5 mL | 10 mL | 20 mL | 100 mL |
| 0.6 mg/kg | 10 mL | 20 mL | 50 mL | 100 mL |
| 1 mg/kg | 20 mL | 50 mL | 100 mL | 100 mL |
| 2 mg/kg | 50 mL | 75 mL | 200 mL | 100 mL |
| 3 mg/kg | 50 mL | 100 mL | 250 mL | 100 mL |
2.7 Administration Instructions
- Prior to administration, inspect the syringe or infusion bag for foaming. If foaming is present, let foam dissipate before administering XENPOZYME.
- Use a low-protein binding, 0.2 micron, in-line filter during administration. The following materials can be used: polyolefin or polyvinylchloride (PVC) with DEHP for infusion bags, polypropylene for syringes, polyurethane or PVC DEHP-free for infusion sets and polyethersulfone or polytetrafluoroethylene for in-line filters.
- Infuse XENPOZYME using the infusion rates described in Table 5 and Table 6. In absence of infusion-associated reactions, increase infusion rate per the steps of infusion as indicated (+/- 5 minutes). Each step of infusion will last for 20 minutes with the exception of the final step which should last until completion of the infusion volume.
- At the end of the infusion, flush the infusion line with 0.9% Sodium Chloride Injection, USP using the same infusion rate as the one used for the last part of the infusion.
- Do not infuse XENPOZYME in the same intravenous line with other products.
| Dose | Infusion Rate | |||
|---|---|---|---|---|
| step 1 | step 2 | step 3 | step 4 | |
| NA: Not applicable. | ||||
| Start infusion at step 1 and in absence of infusion-associated reaction increase infusion rate sequentially per the steps of infusion. | ||||
| 0.1 mg/kg | 20 mL/hour | 60 mL/hour | NA | NA |
| 0.3 to 3 mg/kg | 3.33 mL/hour | 10 mL/hour | 20 mL/hour | 33.33 mL/hour |
| Dose | Infusion rate | |||
|---|---|---|---|---|
| step 1 | step 2 | step 3 | step 4 | |
| NA: Not applicable. | ||||
| Start infusion at step 1 and in absence of infusion-associated reactions increase infusion rate sequentially per the steps of infusion. | ||||
| 0.03 mg/kg | 0.1 mg/kg/hour for the full length of the infusion | NA | NA | NA |
| 0.1 mg/kg | 0.1 mg/kg/hour | 0.3 mg/kg/hour | NA | NA |
| 0.3 mg/kg | 0.1 mg/kg/hour | 0.3 mg/kg/hour | 0.6 mg/kg/hour | NA |
| 0.6 mg/kg | 0.1 mg/kg/hour | 0.3 mg/kg/hour | 0.6 mg/kg/hour | 1 mg/kg/hour |
| 1 mg/kg | ||||
| 2 mg/kg | ||||
| 3 mg/kg | ||||
Home Infusion
Home administration under the supervision of a healthcare provider may be considered for patients on maintenance dose [see Dosage and Administration (2.2, 2.3)] and who are tolerating their infusion well. The decision to have patients moved to home infusion should be made after evaluation and recommendation by a physician.
The dose and infusion rate used in the home setting should remain the same as were used in the supervised clinical setting and should not be changed without supervision of a physician. In case of missed dose(s) or delayed infusion, contact a physician as subsequent infusions may occur in a supervised clinical setting.
3. Dosage Forms and Strengths
For injection: 4 mg or 20 mg of olipudase alfa-rpcp as a sterile, white to off white lyophilized powder in a single-dose vial for reconstitution.
5. Warnings and Precautions
5.1 Hypersensitivity Reactions Including Anaphylaxis
Life-threatening hypersensitivity reactions, including anaphylaxis, have been reported in olipudase alfa-treated patients. One 18-month-old XENPOZYME-treated patient experienced an anaphylactic reaction during the sixth infusion in the dose escalation period in Trial 2 [see Adverse Reactions (6.1)]. Additionally, a 16-month-old patient with ASMD type A, treated with a version of olipudase alfa manufactured from a different process, experienced two anaphylactic reactions during the fifth and sixth infusions in the dose escalation period; the patient received an immune tolerance induction therapy prior to treatment. In both of these pediatric patients with anaphylaxis, anti-olipudase alfa-rpcp IgE (IgE ADA) and IgG (IgG ADA) antibodies were detected [see Adverse Reactions (6.1) and Clinical Pharmacology (12.6)].
Hypersensitivity reactions that were mild to moderate in severity occurred in 10 (33%) XENPOZYME-treated adult patients and 4 (50%) XENPOZYME-treated pediatric patients in clinical trials. Hypersensitivity reactions in adults included urticaria, pruritus, erythema, rash, rash erythematous, eczema, angioedema, and erythema nodosum. Hypersensitivity reactions in pediatric patients included urticaria, pruritus, rash, erythema, and localized edema [see Adverse Reactions (6)].
Prior to XENPOZYME administration, consider premedicating with antihistamines, antipyretics, and/or corticosteroids. Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available during XENPOZYME administration.
- If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue XENPOZYME immediately and initiate appropriate medical treatment. Consider the risks and benefits of re-administering XENPOZYME following a severe hypersensitivity reaction (including anaphylaxis). One patient has been rechallenged using slower infusion rates at a dosage lower than the recommended dosage. In patients with a severe hypersensitivity reaction, a tailored desensitization procedure to XENPOZYME may be considered. If the decision is made to readminister XENPOZYME, ensure the patient tolerates the infusion. If the patient tolerates the infusion, the dosage (dose and/or the rate) may be increased to reach the recommended dosage.
- Consider testing for IgE ADA in XENPOZYME-treated patients who experienced severe hypersensitivity reactions, including anaphylaxis [see Adverse Reactions (6.1)]. Testing for antibodies against olipudase alfa-rpcp are available through Genzyme Corporation (at 1-800-745-4447). Consider other clinical laboratory testing such as serum tryptase and complement activation in patients who experience anaphylaxis.
- If a mild or moderate hypersensitivity reaction occurs, consider temporarily holding the infusion, slowing the infusion rate, and/or reducing the XENPOZYME dose [see Dosage and Administration (2.5)].
5.2 Infusion-Associated Reactions
IARs occurred in approximately 75% of pediatric and 50% of adult XENPOZYME-treated patients in the clinical trials; a severe IAR occurred in one (12.5%) of the pediatric patients. The most frequent IARs in:
- ≥10% of adult patients were headache, pruritus, vomiting, and urticaria
- >20% of pediatric patients were urticaria, erythema, headache, nausea, pyrexia, and vomiting
Acute phase reaction (APR), an acute inflammatory response accompanied by elevations in inflammatory serum protein concentrations, was observed in one XENPOZYME-treated adult and one XENPOZYME-treated pediatric patient. Most of the APRs occurred at 48 hours post infusion during the dose escalation period. Elevations of C-reactive protein, calcitonin, and IL-6, and reduction of serum iron were observed. The most common clinical symptoms associated with APRs were pyrexia, vomiting, and diarrhea. Acute phase reactions were managed similar to other IARs. In the postmarketing setting, 24 hours after receiving XENPOZYME at a higher than recommended initial dose, a 2-year-old male patient with ASMD, experienced fever, respiratory distress, hypotension, and death [see Overdosage (10)].
Prior to XENPOZYME administration, consider pre-medicating with antihistamines, antipyretics, and/or corticosteroids to reduce the risk of infusion-associated reactions (IARs). However, IARs may still occur in patients after receiving pre-treatment. Follow the dose escalation regimen to minimize IARs [see Dosage & Administration (2.2 & 2.3)].
- If a severe IAR occurs, discontinue XENPOZYME immediately and initiate appropriate medical treatment. Consider the risks and benefits of re-administering XENPOZYME following a severe IAR. One patient has been rechallenged using slower infusion rates at a dosage lower than the recommended dosage. If the patient tolerates the infusion, the dosage (dose and/or the rate) may be increased to reach the recommended dosage.
- If a mild or moderate IAR occurs, consider temporarily holding the infusion, slowing the infusion rate, and/or reducing the XENPOZYME dosage [see Dosage and Administration (2.5)].
5.3 Elevated Transaminase Levels
XENPOZYME may be associated with elevated transaminases (ALT, AST, or both) within 24 to 48 hours after infusion. Elevated transaminase levels were reported in 4 (13%) XENPOZYME-treated adults and 1 (13%) XENPOZYME-treated pediatric patient during the dose escalation phase in clinical trials. At the time of the next scheduled infusion, these elevated transaminase levels generally returned to levels observed prior to the XENPOZYME infusion [see Adverse Reactions (6.1)].
To manage the risk of elevated transaminase levels, assess ALT and AST within one month prior to initiation of XENPOZYME, within 72 hours prior to any infusion during dose escalation, which includes the first 3 mg/kg dose outlined in Tables 1 and 2, or prior to the next scheduled XENPOZYME infusion upon resuming treatment following a missed dose.
If either the baseline or pre-infusion transaminase level (during the dose escalation phase) is >2 times the ULN, repeat transaminase levels within 72 hours after the end of the infusion. If the pre-infusion transaminase levels are elevated above baseline and >2 times the ULN prior to the next scheduled administration, the XENPOZYME dose can be reduced (repeat prior lower dose or reduce the dose) or XENPOZYME can be temporarily withheld until the liver transaminases return to the patient's baseline value.
Upon reaching the recommended maintenance dose, transaminase testing is recommended to be continued as part of routine clinical management of ASMD.
5.4 Risk of Fetal Malformations During Dosage Initiation or Escalation in Pregnancy
There is no evidence that olipudase alfa-rpcp crosses the human placenta. However, published literature reports that early embryonic exposure to a metabolite of sphingomyelin (ceramide) or the S1P receptor modulator fingolimod can produce exencephaly in chicks and mice, respectively. In animal reproduction studies, exencephaly, a neural tube defect occurring in the first trimester of pregnancy, was observed in mouse fetuses at exposures less than the exposure at the maximum recommended human dose of olipudase alfa-rpcp.
XENPOZYME dosage initiation or escalation, at any time during pregnancy, is not recommended as it may lead to elevated sphingomyelin metabolite levels that may increase the risk of fetal malformations [see Use in Specific Population (8.1), Clinical Pharmacology (12.2)]. The decision to continue or discontinue XENPOZYME maintenance dosing in pregnancy should consider the female's need for XENPOZYME, the potential drug-related risks to the fetus, and the potential adverse outcomes from untreated maternal ASMD disease.
Verify the pregnancy status in females of reproductive potential prior to initiating XENPOZYME treatment. Advise females of reproductive potential to use effective contraception during treatment and for 14 days after the last dose if XENPOZYME is discontinued [see Use in Specific Populations (8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hypersensitivity Reactions Including Anaphylaxis [see Warnings and Precautions (5.1)]
- Infusion-Associated Reactions (IARs) [see Warnings and Precautions (5.2)]
- Elevated Transaminase Levels [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety analysis from 3 clinical trials included a total of 38 XENPOZYME-treated patients (30 adult and 8 pediatric patients) with age range from 1.5 to 59 years old receiving intravenous doses up to 3 mg/kg every 2 weeks [see Clinical Studies (14)]. The median exposure duration was 2.5 years (range: 0.4 to 3.7 years) in adult patients and 2.7 years (range: 2.5 to 3.2 years) in pediatric patients.
Serious adverse reactions of anaphylactic reaction were reported in 2 (25%) XENPOZYME-treated pediatric patients.
Most frequently reported adverse drug reactions in adults (incidence ≥10%) were headache, cough, diarrhea, hypotension, and ocular hyperemia.
Most frequently reported adverse drug reactions in pediatric patients (incidence ≥20%) were pyrexia, cough, diarrhea, rhinitis, abdominal pain, vomiting, headache, urticaria, nausea, rash, arthralgia, pruritus, fatigue, and pharyngitis.
Adult patients with ASMD type B and type A/B (Trial 1)
In Trial 1, 13 adult patients received XENPOZYME once every 2 weeks for 52 weeks (primary analysis period (PAP)) at dosages escalating from 0.1 mg/kg to a target dose of 3 mg/kg [see Clinical Studies (14.2)].
Adverse reactions that occurred in at least 7% of XENPOZYME-treated adult patients during the PAP are described in Table 7.
| Adverse Reaction | XENPOZYME N=13 | Placebo N=18 |
|---|---|---|
| Headache | 7 (54%) | 8 (44%) |
| Cough | 4 (31%) | 2 (11%) |
| Diarrhea | 2 (15%) | 2 (11%) |
| Hypotension | 2 (15%) | 2 (11%) |
| Ocular hyperemia | 2 (15%) | 1 (6%) |
| Erythema | 1 (8%) | 1 (6%) |
| Asthenia | 1 (8%) | 1 (6%) |
| Pharyngitis | 1 (8%) | 1 (6%) |
| Dyspnea | 1 (8%) | 0 |
| Urticaria | 1 (8%) | 0 |
| Papule | 1 (8%) | 0 |
| Myalgia | 1 (8%) | 0 |
| Throat irritation | 1 (8%) | 0 |
| C-reactive protein abnormal | 1 (8%) | 0 |
Pediatric Patients with ASMD type B and type A/B (Trial 2 and Trial 3)
In Trial 2, 8 pediatric patients less than or equal to 17 years of age received XENPOZYME intravenously once every 2 weeks for 64 weeks [see Clinical Studies (14.3)]. After 64 weeks, all pediatric patients entered into Trial 3.
Adverse reactions that occurred in at least 13% of pediatric patients are described in Table 8.
| Adverse Reactions | XENPOZYME N=8 |
|---|---|
| Abdominal pain includes abdominal pain and abdominal pain upper | |
| Fatigue includes fatigue and asthenia | |
| Rash includes rash and erythema | |
|
|
| Pyrexia | 8 (100%) |
| Cough | 6 (75%) |
| Diarrhea | 6 (75%) |
| Rhinitis | 6 (75%) |
| Abdominal pain | 5 (63%) |
| Vomiting | 4 (50%) |
| Headache | 4 (50%) |
| Urticaria | 4 (50%) |
| Nausea | 3 (38%) |
| Rash | 3 (38%) |
| Arthralgia | 3 (38%) |
| Pruritus | 2 (25%) |
| Fatigue | 2 (25%) |
| Pharyngitis | 2 (25%) |
| C-reactive protein increased | 1 (13%) |
| Hypotension | 1 (13%) |
| Anaphylactic reaction | 1 (13%) |
| Hypersensitivity | 1 (13%) |
| Infusion site swelling | 1 (13%) |
| Tachycardia | 1 (13%) |
| Pharyngeal swelling | 1 (13%) |
Treatment related serious adverse reactions, hypersensitivity reactions including anaphylaxis, and IARs occurred within 24 hours of infusion and were observed in a higher percentage of pediatric patients than in adult patients.
Laboratory Adverse Reaction
Elevated transaminase levels ranging from 3 times to 14 times the upper limit of normal (ULN) were reported in 4 (13%) adults and 1 (13%) pediatric patient during the XENPOZYME dose escalation phase in clinical trials.
Immunogenicity: Antidrug Antibody-Associated Adverse Reactions
In Trial 1, infusion-associated reactions (including hypersensitivity reactions) occurred in a higher percentage in XENPOZYME-treated patients who developed IgG ADA compared to those who did not develop IgG ADA (73% versus 44%) [see Clinical Pharmacology (12.6) and Clinical Studies (14.2)].
In Trial 2, one XENPOZYME-treated pediatric patient (18-months old) experienced an anaphylactic reaction during the sixth infusion and developed IgE ADA and the highest IgG ADA titers (ADA peak titer 1,600) of the patients in this trial. After treatment discontinuation, XENPOZYME was resumed four months later using a diluted drug solution and a desensitization procedure.
One pediatric patient (16-months old) with ASMD type A, treated with a version of olipudase alfa manufactured from a different process, experienced anaphylactic reactions (both during the fifth and sixth infusions) and developed IgG ADA (highest titer 1,600) and IgE ADA [see Warnings and Precautions (5.1)].
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies, XENPOZYME may cause embryo-fetal harm when administered to a pregnant female. XENPOZYME dosage initiation or escalation, at any time during pregnancy, is not recommended as it may lead to elevated sphingomyelin metabolite levels that may increase the risk of fetal malformations (see Data), [see Clinical Pharmacology (12.2)]. However, the decision to continue or discontinue XENPOZYME maintenance dosing in pregnancy should consider the female's need for XENPOZYME, the potential drug-related risks to the fetus, and the potential adverse outcomes from untreated maternal ASMD disease.
In an embryo-fetal toxicity study in pregnant mice, a rare malformation (exencephaly) was observed in offspring at an exposure less than the exposure at the maximum recommended human dose (MRHD) of olipudase alfa-rpcp (see Data).
There are no available data on XENPOZYME use in pregnant females to evaluate for a drug associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Advise the pregnant female of the potential risk to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, miscarriage, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in pregnant mice, olipudase alfa-rpcp was administered intravenously at doses of 3, 10, or 30 mg/kg daily from gestation day (GD) 6 through GD 15. Exencephaly was observed in 1 litter at each of the 10 and 30 mg/kg dose groups (2 and 3 fetuses, respectively). These data are consistent with published literature reports that brief embryonic exposures to sphingomyelin metabolites or a sphingosine-1-phosphate (S1P) receptor modulator produced neural tube defects, including exencephaly, in chicks and mice.
The developmental No Observed Adverse Effect Level (NOAEL) is 3 mg/kg. The AUC associated with this dose is 0.14-fold the clinical exposure at the MRHD. The developmental Lowest-Observed-Adverse-Effect Level (LOAEL), 10 mg/kg, is also associated with an exposure that is less than the clinical exposure at the MRHD.
In an embryo-fetal development study in pregnant rabbits, olipudase alfa-rpcp was administered intravenously at doses of 3, 10, or 30 mg/kg daily from GD 6 through GD 19. There was no maternal or developmental toxicity. The developmental NOAEL was 30 mg/kg; the AUC0–24 at this dose is approximately 10.5-fold the exposure at the MRHD.
In a study of pre- and postnatal development in mice, olipudase alfa-rpcp was administered intravenously every other day from GD 6 through GD 18; then resumed every other day after parturition, from Lactation Day (LD) 1 through LD 19. Olipudase alfa-rpcp did not induce any effect on maternal reproductive function or on developmental and reproductive parameters of male and female offspring. Therefore, the maternal and developmental NOAELs are 30 mg/kg. Exposures at this dose, based on the embryo-fetal development study, were estimated to be approximately 1.5-fold the MRHD of olipudase alfa-rpcp.
8.2 Lactation
Risk Summary
There are no data on the presence of olipudase alfa-rpcp in human milk, the effects on the breastfed infant, or the effects on milk production. Olipudase alfa-rpcp is present in animal milk. (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for XENPOZYME and any potential adverse effects on the breastfed infant from XENPOZYME or from the underlying maternal condition.
Data
Olipudase alfa-rpcp was administered as a single intravenous dose (3 mg/kg) to lactating CD1 mice on post-partum day 7. Milk was not evaluated until post-partum day 9, at which time concentrations of olipudase alfa-rpcp detected were approximately 1.3% the estimated maximal maternal plasma concentration.
8.3 Females and Males of Reproductive Potential
XENPOZYME may cause embryo-fetal harm when administered during the first trimester of pregnancy [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of XENPOZYME for the treatment of non-central nervous system manifestations of acid sphingomyelinase deficiency (ASMD) have been established in pediatric patients down to birth.
Use of XENPOZYME for this indication is supported by evidence from an adequate, and well-controlled trial (Trial 1) in adults with supportive efficacy, safety, and tolerability data in pediatric patients (Trial 2 and Trial 3) [see Adverse Reactions (6.1) and Clinical Studies (14.2, 14.3, 14.4)].
Compared to adults, a higher percentage of pediatric patients experienced treatment related serious adverse reactions, anaphylaxis, hypersensitivity reactions, and IARs that occurred within 24 hours of infusion [see Adverse Reactions (6.1)]. Two pediatric patients, an 18 month old receiving XENPOZYME and a 16 month old with ASMD type A that received a version of olipudase alfa manufactured from a different process developed anaphylaxis [see Warnings and Precautions (5.1)].
8.5 Geriatric Use
Of the total number of XENPOZYME-treated adult patients in these trials, 1 (3%) was 65 to 74 years of age, and none were 75 years of age and older [see Clinical Studies (14)].
Clinical trials of XENPOZYME did not include sufficient numbers of patients 65 years of age and older to determine whether they respond differently from younger adult patients.
10. Overdosage
Cases of overdosage with XENPOZYME have been reported in pediatric patients during dose escalation. Some patients experienced serious adverse reactions including death within 24 hours of initial dose [see Warnings and Precautions (5.2)]. The clinical findings included fever, hypotension, gastrointestinal bleeding, marked elevation in liver tests, metabolic acidosis, respiratory failure, and vomiting.
There is no known specific antidote for XENPOZYME overdosage. In the event of overdosage, immediately stop the infusion, and monitor the patient closely in a hospital setting for the development of hypersensitivity reactions and IARs including acute phase reactions. For the management of adverse reactions, see Warnings and Precautions (5.1, 5.2, 5.3) and Adverse Reactions (6.1).
11. Xenpozyme Description
Olipudase alfa-rpcp is a hydrolytic lysosomal sphingomyelin-specific enzyme consisting of 570 amino acids produced in a Chinese hamster ovary cell line by recombinant DNA technology. The molecular weight of olipudase alfa-rpcp is approximately 76 kDa.
XENPOZYME (olipudase alfa-rpcp) for injection is supplied as a sterile, preservative-free, white to off-white lyophilized powder for reconstitution and dilution to be administered via intravenous infusion. XENPOZYME is supplied in single-dose vials.
Each 4 mg vial contains 4 mg olipudase alfa-rpcp, dibasic sodium phosphate (0.89 mg), methionine (14.92 mg), monobasic sodium phosphate (1.63 mg), and sucrose (50 mg). After reconstitution with 1.1 mL of Sterile Water for Injection, USP, the final concentration is 4 mg/mL [see Dosage and Administration (2.6)].
Each 20 mg vial contains 20 mg olipudase alfa-rpcp, dibasic sodium phosphate (4.47 mg), methionine (74.6 mg), monobasic sodium phosphate (8.17 mg), and sucrose (250 mg). After reconstitution with 5.1 mL of Sterile Water for Injection, USP, the final concentration is 4 mg/mL [see Dosage and Administration (2.6)].
The pH is 6.5 after reconstitution.
12. Xenpozyme - Clinical Pharmacology
12.1 Mechanism of Action
ASMD is a lysosomal storage disease that results from reduced activity of the enzyme acid sphingomyelinase (ASM), caused by pathogenic variants in the sphingomyelin phosphodiesterase 1 gene. ASM degrades sphingomyelin to ceramide and phosphocholine. The deficiency of ASM causes an intra-lysosomal accumulation of sphingomyelin (as well as cholesterol and other cell membrane lipids) in various tissues. XENPOZYME provides an exogenous source of ASM.
XENPOZYME is not expected to cross the blood-brain barrier or modulate the CNS manifestations of ASMD.
12.2 Pharmacodynamics
Plasma Ceramide Levels
Ceramide is elevated in plasma of adult and pediatric patients with ASMD. Plasma ceramide levels showed a transient increase after each administration (post infusion) of XENPOZYME. In the dose escalation phase, plasma ceramide levels were substantially increased compared to the baseline level. Plasma ceramide levels gradually decreased following repeated administration of XENPOZYME and the pre-infusion levels were generally lower than the baseline level during the maintenance phase of treatment.
- In adult patients with ASMD in Trial 1 [see Clinical Studies (14.2)], the mean (standard deviation, SD) pre-infusion plasma ceramide concentration was 3.7 (1.4) mg/L at baseline and decreased to 2.2 (0.6) mg/L at Week 52 following treatment with XENPOZYME.
- In pediatric patients with ASMD in Trial 2 [see Clinical Studies (14.3)], the mean (SD) pre-infusion plasma ceramide concentration was 4.7 (0.9) mg/L at baseline and decreased to 1.8 (0.3) mg/L at Week 52 following treatment with XENPOZYME.
Plasma Lysosphingomyelin Levels
Lysosphingomyelin is substantially elevated in plasma of adult and pediatric patients with ASMD. Plasma lysosphingomyelin levels decreased after repeated administration of XENPOZYME.
- In adult patients with ASMD in Trial 1 [see Clinical Studies (14.2)], the mean (SD) pre-infusion plasma lysosphingomyelin concentration was 379 (204) mcg/L at baseline and decreased to 99 (118) mcg/L at Week 52 following treatment with XENPOZYME.
- In pediatric patients with ASMD in Trial 2 [see Clinical Studies (14.3)], the mean (SD) pre-infusion plasma lysosphingomyelin concentration was 625 (339) mcg/L at baseline and decreased to 80 (47) mcg/L at Week 52 following treatment with XENPOZYME.
12.3 Pharmacokinetics
In adult patients with ASMD, the mean (SD) maximum plasma olipudase alfa-rpcp concentration (Cmax) and area under the concentration-time curve (AUC) at steady state were 30 (5) mcg/mL and 607 (120) mcg∙h/mL, respectively, at the recommended maintenance dose of 3 mg/kg administered once every 2 weeks. Olipudase alfa-rpcp Cmax and AUC increase proportionally over a dose range of 0.1 to 3 mg/kg (0.03 to 1 times the approved recommended maintenance dose).
Distribution
The mean (SD) volume of distribution of olipudase alfa-rpcp was 13 (2) L in adult patients with ASMD.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of olipudase alfa-rpcp or of other olipudase alfa products.
Following 0.4 to 3.7 years of XENPOZYME treatment in Trial 1 [see Clinical Studies (14.2)], 9 out of 30 (30%) XENPOZYME-treated adult patients with ASMD developed anti-olipudase alfa-rpcp IgG antibodies (referred to as IgG ADA). The median time to seroconversion from first XENPOZYME infusion was approximately 8 weeks. One out of these 9 (11%) adult patients had neutralizing antibodies (NAb) that inhibited the olipudase alfa-rpcp enzyme activity. None of these 9 patients had NAb that inhibit the cellular uptake of olipudase alfa-rpcp.
Following 2.5 to 3.2 years of XENPOZYME treatment in Trial 2 and 3 [see Clinical Studies (14.3)], 6 out of 8 (75%) XENPOZYME-treated pediatric patients with ASMD developed IgG ADA. The median time to seroconversion from first XENPOZYME infusion was 10 weeks. One out of the 6 (17%) pediatric patients developed NAb that inhibited olipudase alfa-rpcp enzyme activity. None of these 6 patients had NAb that inhibited the cellular uptake of olipudase alfa-rpcp.
Infusion-associated reactions (including hypersensitivity reactions) occurred in a higher percentage in XENPOZYME-treated patients who developed ADA compared to those who did not develop ADA [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
There was no identified clinically significant effect of ADA on pharmacokinetics of XENPOZYME.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Studies to evaluate the carcinogenic potential of olipudase alfa-rpcp have not been conducted.
Mutagenesis
Studies to evaluate the mutagenic potential of olipudase alfa-rpcp have not been conducted.
Impairment of Fertility
Intravenous administration of olipudase alfa-rpcp every other day at doses up to 30 mg/kg had no adverse effects in a combined study of fertility in male and female mice. Exposures at this dose, based on the embryo-fetal development study, were estimated to be approximately 1.5-fold those of the MRHD of olipudase alfa-rpcp.
13.2 Animal Toxicology and/or Pharmacology
In acid sphingomyelinase knockout (ASMKO) mice (a disease model of ASMD), mortality was observed after a single dose ≥10 mg/kg administered as an IV bolus injection. Observations (lethargy, coolness to touch, and unwillingness to move), combined with the adrenal hemorrhage, suggested that hypotensive shock may be the cause of death. These findings were accompanied by necrosis and apoptosis in the liver and adrenal gland, elevations of ceramide, sphingosine and sphingosine 1-phosphate in the serum, catabolites of accumulated sphingomyelin as well as elevations in the serum concentrations of inflammatory mediators, such as cytokines and acute phase proteins.
In ASMKO mice, a dose-dependent reduction in heart rate accompanied by a decrease in motor activity and followed by a slow decline in blood pressure was noted after a single IV administration at 3, 10, and 20 mg/kg. After 2 doses of olipudase alfa-rpcp at 3 and 10 mg/kg to ASMKO mice, a slight decline in heart rate was noted following the second administration.
Repeated dose studies in adult ASMKO mice show that administration of olipudase alfa-rpcp via a dose escalation regimen, (3 mg/kg administered IV every other day, followed by a single IV dose of 20 mg/kg 3 days later) did not result in toxicity. The lack of adverse findings in BALB/c, C57BL/6 mice, rats, dogs, and monkeys at comparable olipudase alfa-rpcp doses suggested that the dose-related toxicity observed in ASMKO mice may be due to the rate and amount of substrate degradation.
14. Clinical Studies
14.1 Overview of Clinical Trials
The efficacy of XENPOZYME for the treatment of non–central nervous system manifestations of acid sphingomyelinase deficiency (ASMD) has been evaluated in 3 clinical trials involving a total of 61 patients with ASMD:
- Trial 1 in adult patients (NCT02004691),
- Trial 2 in pediatric patients (NCT02292654), and
- Trial 3 a long-term trial in pediatric patients (NCT02004704).
14.2 Clinical Trial in Adult Patients with ASMD
Trial 1 was a multicenter, randomized, double-blinded, placebo-controlled, repeat-dose phase II/III trial in adult patients with ASMD (clinical diagnosis consistent with ASMD type B and A/B). In this trial, patients received either XENPOZYME or placebo. Treatment was administered in both groups as an intravenous infusion once every 2 weeks. XENPOZYME was dosed as follows: 0.1 mg/kg (Day 1, Week 0), 0.3 mg/kg (Weeks 2 and 4), 0.6 mg/kg (Weeks 6 and 8), 1 mg/kg (Week 10), 2 mg/kg (Week 12), and then a maintenance dose of 3 mg/kg (Week 14 onwards). The trial was divided into 2 consecutive periods: a randomized placebo-controlled, double-blinded primary analysis period (PAP) which lasted to Week 52, followed by an extension treatment period (ETP) for up to 4 years. Patients randomized to the placebo arm in the PAP crossed over to receive XENPOZYME treatment in the ETP to reach the targeted dose of 3 mg/kg, while patients in the original XENPOZYME arm continued treatment.
Patients enrolled in the trial had a diffusion capacity of the lungs for carbon monoxide (DLco) ≤70% of the predicted normal value and a spleen volume ≥6 multiples of normal (MN) measured by magnetic resonance imaging (MRI). The trial population included 87% White, 7% Asian, and 7% other; for ethnicity, 32% identified as Hispanic/Latino, 65% as non-Hispanic/Latino, and 3% were not reported.
Five males and 13 females with a median age of 34 years (range: 18 to 66) were included in the placebo arm and 8 males and 5 females with a median age of 34 years (range: 20 to 59) were included in the XENPOZYME arm. The XENPOZYME and placebo groups included 1 patient (8%) and 2 patients (11%) with mild renal impairment (60 mL/minute ≤ creatinine clearance <90 mL/minute), respectively. There were no patients with moderate or severe renal impairment.
Key efficacy endpoints included assessment of % predicted DLco, spleen volume, liver volume, and platelet count.
At Week 52 during the PAP, an increase of 21% in the mean percent change in % predicted DLco was observed in the XENPOZYME-treated patients compared to the placebo-treated patients (Table 9). A reduction in spleen volume of 39% was observed in the XENPOZYME-treated patients compared to the placebo-treated patients. The changes in % predicted DLco and spleen volume were noted at Week 26 of treatment, the first post-dose endpoint assessment (Figures 1 and 2).
A decrease in mean liver volume and an increase in mean platelet count were noted in the XENPOZYME-treated patients compared to the placebo-treated patients at Week 52 (Table 9).
| Placebo | XENPOZYME | Difference [95% CI] |
|
|---|---|---|---|
| Nominal p value: | |||
| DLco | |||
| n | 18 | 13 | |
| Mean % predicted DLco at baseline (SD) | 48.5 (10.8) | 49.1 (9.7) | NA |
| n | 17 | 12 | |
| Mean % predicted DLco at Week 52 (SD) | 49.9 (11.1) | 59.4 (9.6) | NA |
| n | 17 | 12 | |
| LS Mean Percent change in % predicted DLco at Week 52 (SE) | 3.0 (3.3) | 23.9 (3.8) | 20.9 (5.0)*
[10.6, 31.2] |
| Spleen volume | |||
| n | 18 | 13 | |
| Mean Spleen Volume (MN) at baseline (SD) | 11.2 (3.8) | 11.5 (4.7) | NA |
| n | 17 | 13 | |
| Mean Spleen Volume (MN) at Week 52 (SD) | 11.2 (4.2) | 7.2 (3.9) | NA |
| n | 17 | 13 | |
| LS Mean Percent change in Spleen Volume (in MN) at Week 52 (SE) | 0.5 (2.62) | -38.9 (3.0) | -39.4 (4.0)†
[-47.6, -31.2] |
| Liver volume | |||
| n | 18 | 13 | |
| Mean Liver Volume (MN) at baseline (SD) | 1.6 (0.5) | 1.4 (0.3) | NA |
| n | 17 | 12 | |
| Mean Liver Volume (MN) at Week 52 (SD) | 1.6 (0.5) | 1.0 (0.2) | NA |
| n | 17 | 12 | |
| LS Mean Percent change in Liver Volume from baseline to Week 52 (SE) | -1.8 (2.7) | -26.5 (3.2) | -24.7 (4.2)†
[-33.4, -16.1] |
| Platelet count | |||
| n | 18 | 13 | |
| Mean Platelet Count (109/L) at baseline (SD) | 115.6 (36.3) | 109.3 (30.6) | NA |
| n | 16 | 13 | |
| Mean Platelet Count (109/L) at Week 52 (SD) | 120.2 (43.2) | 126.4 (29.0) | NA |
| n | 16 | 13 | |
| LS Mean Percent change in Platelet Count from baseline to Week 52 (SE) | 2.7 (4.5) | 18.3 (5.0) | +15.6 (6.7) ‡
[1.8, 29.4] |
Seventeen of 18 patients previously receiving placebo and 13 of 13 patients previously treated with XENPOZYME for 52 weeks (in the PAP) started or continued treatment with XENPOZYME, respectively, for up to 4 years. At Week 104, patients initially randomized to placebo had received XENPOZYME for 52 weeks and demonstrated the following LS mean (SE) percent changes in clinical parameters from baseline (before first administration of XENPOZYME): increase in % predicted DLco was 26.8% (6.2) (Figure 1); reduction in spleen volume (MN) was 36.5% (2.5) (Figure 2); reduction in liver volume (MN) was 29.5 (2.6); and increase in platelet count was 19.5 (6.7).
Patients in the previous XENPOZYME group demonstrated improvement from baseline to Week 104 in the following parameters: LS mean (SE) percent increase in % predicted DLco was 34.1% (7.9) (Figure 1); LS mean (SE) percent reduction in spleen volume (MN) was 48.3 (2.9) (Figure 2); LS mean (SE) percent reduction in liver volume (MN) was 31.7 (2.9); LS mean (SE) percent increase in platelet count was 24.0 (8.2).
Figure 1: Plot of the LS Means (95% CI) of the Percentage Change in DLco (% predicted) from Baseline to Week 104 in Adult Patients with ASMD (Trial 1)
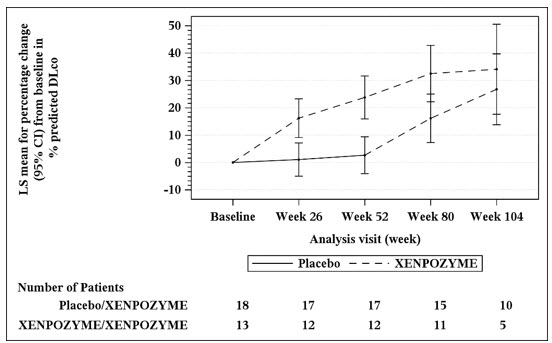
The vertical bars represent the 95% CIs for the LS means.
The LS means and 95% CIs are based on a mixed model for repeated measures approach, using data up to Week 104. Patients in placebo/XENPOZYME group received placebo by Week 52 and switched to XENPOZYME thereafter.
Figure 2: Plot of the LS Means (95% CI) of the Percentage Change in Spleen Volume (MN) from Baseline to Week 104 in Patients with ASMD (Trial 1)
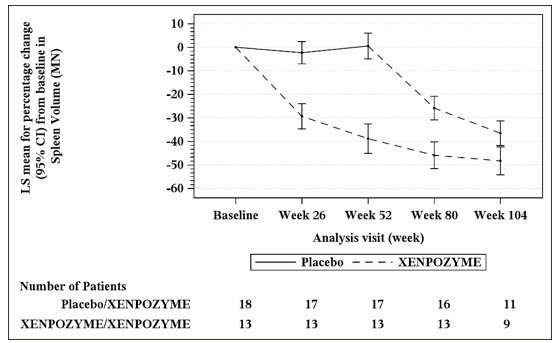
The vertical bars represent the 95% CIs for the LS means.
The LS means and 95% CIs are based on a mixed model for repeated measures approach, using data up to Week 104. Patients in placebo/XENPOZYME group received placebo by Week 52 and switched to XENPOZYME thereafter.
14.3 Clinical Trial in Pediatric Patients with ASMD
Trial 2 was a multi-center, open-label, repeated-dose trial of XENPOZYME administered intravenously once every 2 weeks (via infusion) for 64 weeks in pediatric patients aged <18 years with a clinical diagnosis consistent with ASMD type B and A/B. Exploratory efficacy endpoints related to organomegaly, pulmonary and liver functions, and linear growth were evaluated at Week 52. XENPOZYME was dosed as follows: 0.03 mg/kg (Day 1, Week 0), 0.1 mg/kg (Weeks 2), 0.3 mg/kg (Weeks 4 and 6), 0.6 mg/kg (Week 8 and 10), 1 mg/kg (Week 12), 2 mg/kg (Week 14), and then a maintenance dose of 3 mg/kg (Week 16 onwards).
In Trial 2, 8 patients (7 patients from 2 to <12 years old, and 1 patient <2 years old) received an initial dose of 0.03 mg/kg XENPOZYME and all but one completed the dose escalation up to the maintenance dose of 3 mg/kg within 22 weeks. All patients were White and of non-Hispanic/Latino ethnicity.
Patients enrolled in the trial had a spleen volume ≥5 MN measured by MRI. Age of patients treated with XENPOZYME ranged from 1 to 10 years old, with both sexes equally represented.
Treatment with XENPOZYME resulted in improvements in mean percent change in % predicted DLco, spleen and liver volumes, platelet counts, and linear growth progression (as measured by height Z-scores) at Week 52 as compared to baseline (Table 10).
| Baseline Values | Week 52 Values | |
|---|---|---|
| (n=3) | (n=3) | |
| Mean % predicted DLco (SD) | 48.5 (8.1) | 70.9 (13.7) |
| LS Mean Percent change in % predicted DLco* (SE) | 45.9 (22.7) | |
| 95% CI | -12.5, 104.3 | |
| (n=8) | (n=8) | |
| Mean Spleen Volume (MN) (SD) | 18.3 (5.6) | 9.50 (2.4) |
| LS Mean Percent change in Spleen Volume (in MN) (SE) | -46.7 (3.6) | |
| 95% CI | -55.5, -37.9 | |
| (n=8) | (n=8) | |
| Mean Liver Volume (MN) (SD) | 2.5 (0.5) | 1.6 (0.3) |
| LS Mean Percent change in Liver Volume (in MN) (SE) | -38.1 (2.9) | |
| 95% CI | -44.1, -32.0 | |
| (n=8) | (n=7) | |
| Mean Platelet Count (109/L) (SD) | 136.7 (33.2) | 184.5 (54.2) |
| LS Mean Percent change in Platelet Count (SE) | 37.6 (13.7) | |
| 95% CI | 8.5, 66.7 | |
| (n=8) | (n=7) | |
| Mean height Z-scores (SD) | -1.9 (0.8) | -1.5 (1.0) |
| LS Mean Change in height Z-scores (SE) | 0.5 (0.1) | |
| 95% CI | 0.2, 0.8 |
14.4 Extension Trial in ASMD Pediatric Patients
The 8 pediatric patients 2 to <12 years of age from Trial 2 continued treatment in an open label long term trial (Trial 3) and were treated with XENPOZYME for 2.5 to 3.2 years.
Efficacy analyses showed continued improvements in the 3 patients evaluated for % predicted DLco, 6 patients evaluated for platelet counts, and all 8 patients evaluated for spleen and liver volumes, compared to baseline, during the additional 6 months extension. In addition, the height Z-score increased by 1.3 from baseline when evaluated through 24 months of XENPOZYME treatment. Bone age, as assessed by hand x-ray, was delayed by a mean of 26.4 months at baseline in the 7 pediatric patients enrolled in Trial 2 with a bone age measured at Month 24 in Trial 3. The bone age improved to within a mean of 12 months of the chronological age when assessed at Month 24 in these 7 patients.
16. How is Xenpozyme supplied
How Supplied
XENPOZYME (olipudase alfa-rpcp) for injection is supplied as a sterile white to off-white lyophilized powder for reconstitution in a single-dose vial. XENPOZYME does not contain any preservatives.
XENPOZYME is available supplied as:
- Carton containing one 20 mg single-dose vial (NDC 58468-0050-1)
- Carton containing one 4 mg single-dose vial (NDC 58468-0051-1)
Storage and Handling
Store refrigerated at 2°C to 8°C (36°F to 46°F). Do not freeze.
For storage of reconstituted and diluted solution [see Dosage and Administration (2.6)].
17. Patient Counseling Information
Hypersensitivity Reactions (Including Anaphylaxis) and Infusion-Associated Reactions (IARs)
Advise the patient and caregiver that reactions related to the infusion may occur during and after XENPOZYME treatment, including life-threatening hypersensitivity reactions, anaphylaxis, and IARs. Inform the patient and/or caregiver of the signs and symptoms of hypersensitivity reactions and IARs and to seek immediate medical care should signs and symptoms occur [see Warnings and Precautions (5.1, 5.2)].
Embryo-Fetal Toxicity
XENPOZYME may cause embryo-fetal harm. Advise the pregnant female and females of reproductive potential of the potential risk to the fetus. Advise a female patient and caregiver to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Advise a female of reproductive potential to use effective contraception during treatment and for 14 days after the last dose if XENPOZYME is discontinued [see Use in Specific Populations (8.1, 8.3)].
Manufactured by:
Genzyme Corporation
450 Water Street
Cambridge, MA 02141
U.S. License Number: 1596
©2024 Sanofi. All rights reserved. XENPOZYME is a registered trademark of Genzyme Corporation.

| XENPOZYME
olipudase alfa-rpcp injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| XENPOZYME
olipudase alfa-rpcp injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Genzyme Corporation (025322157) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Corporation | 968278916 | ANALYSIS(58468-0050, 58468-0051) , API MANUFACTURE(58468-0050, 58468-0051) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bionique Testing Laboratories, Inc. | 108684523 | ANALYSIS(58468-0050, 58468-0051) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bioreliance Corporation | 147227730 | ANALYSIS(58468-0050, 58468-0051) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Ireland Limited | 985127419 | ANALYSIS(58468-0050, 58468-0051) , MANUFACTURE(58468-0050, 58468-0051) , PACK(58468-0050, 58468-0051) , LABEL(58468-0050, 58468-0051) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Corporation | 050424395 | PACK(58468-0050, 58468-0051) , LABEL(58468-0050, 58468-0051) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Eurofins Biopharma Product Testing Ireland Limited | 238239933 | ANALYSIS(58468-0050, 58468-0051) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Patheon Biologics LLC | 965750420 | ANALYSIS(58468-0050, 58468-0051) , API MANUFACTURE(58468-0050, 58468-0051) | |
Biological Products Related to Xenpozyme
Find detailed information on biosimilars for this medication.
Frequently asked questions
- How is Tay-Sachs different from Niemann-Pick disease?
- How effective is Xenpozyme?
- How does Xenpozyme work?
More about Xenpozyme (olipudase alfa)
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: lysosomal enzymes
- Breastfeeding
- En español