Pombiliti: Package Insert / Prescribing Info
Package insert / product label
Generic name: cipaglucosidase alfa
Dosage form: injection, powder, lyophilized, for solution
Drug class: Lysosomal enzymes
J Code (medical billing code): J1203 (5 mg, injection)
Medically reviewed by Drugs.com. Last updated on Dec 10, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
POMBILITI (cipaglucosidase alfa-atga) for injection, for intravenous use
Initial U.S. Approval: 2023
WARNING: SEVERE HYPERSENSITIVITY REACTIONS, INFUSION-ASSOCIATED REACTIONS, and RISK OF ACUTE CARDIORESPIRATORY FAILURE IN SUSCEPTIBLE PATIENTS
See full prescribing information for complete boxed warning
-
Hypersensitivity Reactions Including Anaphylaxis
Appropriate medical support measures, including cardiopulmonary resuscitation equipment, should be readily available. If a severe hypersensitivity reaction occurs, POMBILITI should be discontinued immediately and appropriate medical treatment should be initiated. (5.1) -
Infusion-Associated Reactions (IARs)
If severe IARs occur, immediately discontinue POMBILITI and initiate appropriate medical treatment. (5.2) -
Risk of Acute Cardiorespiratory Failure in Susceptible Patients
Patients susceptible to fluid volume overload, or those with acute underlying respiratory illness or compromised cardiac or respiratory function, may be at risk of serious exacerbation of their cardiac or respiratory status during POMBILITI infusion. (5.3)
Indications and Usage for Pombiliti
POMBILITI is a hydrolytic lysosomal glycogen-specific enzyme indicated, in combination with Opfolda, an enzyme stabilizer, for the treatment of adult patients with late-onset Pompe disease (lysosomal acid alpha-glucosidase [GAA] deficiency) weighing ≥40 kg and who are not improving on their current enzyme replacement therapy (ERT). (1)
Pombiliti Dosage and Administration
- Verify pregnancy status in females of reproductive potential prior to initiating treatment. (2.1)
- Administer POMBILITI in combination with Opfolda. (2.2)
- Consider administering antihistamines, antipyretics, and/or corticosteroids prior to POMBILITI administration. (2.2)
- Recommended POMBILITI dosage is 20 mg/kg (of actual body weight) administered every other week as an intravenous infusion over approximately 4 hours. (2.2)
- Start POMBILITI in combination with Opfolda 2 weeks after the last ERT dose. (2.2)
- Initiate the POMBILITI infusion approximately 1 hour after oral administration of Opfolda. If the POMBILITI infusion cannot be started within 3 hours of oral administration of Opfolda, reschedule POMBILITI in combination with Opfolda at least 24 hours after Opfolda was last taken. If POMBILITI in combination with Opfolda are both missed, re-start treatment as soon as possible. (2.2)
- See the full prescribing information for dosage modifications due to hypersensitivity reactions or IARs. (2.3)
- Must be reconstituted and diluted prior to use. (2.4)
- See the full prescribing information for administration instructions. (2.6)
Dosage Forms and Strengths
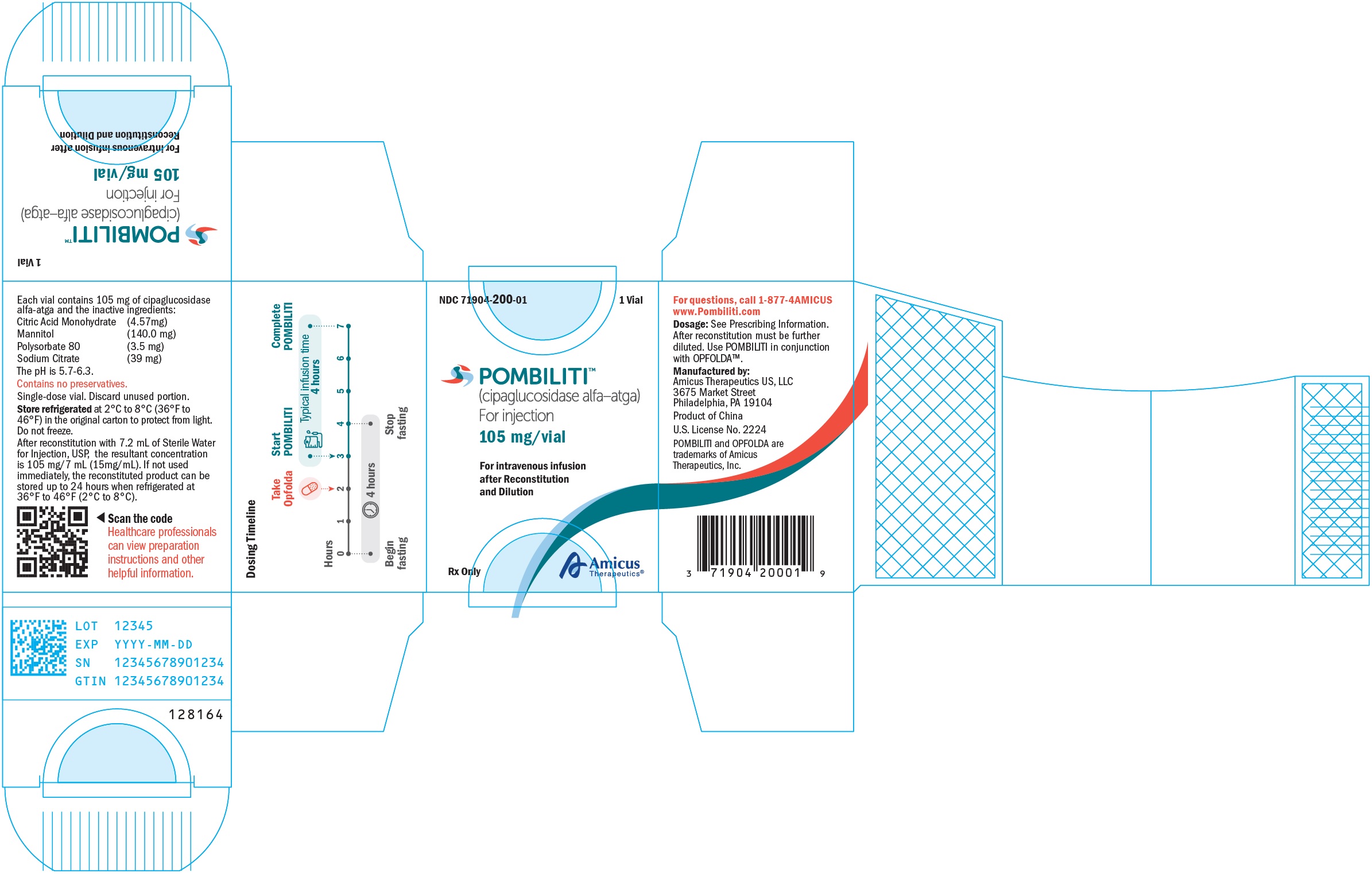
For injection: 105 mg of cipaglucosidase alfa-atga as a lyophilized powder in a single-dose vial for reconstitution. (3)
Warnings and Precautions
- Embryo-Fetal Toxicity: May cause embryo-fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment and for at least 60 days after the last dose. (4, 5.4, 8.1, 8.3)
- Risks Associated with Opfolda: Refer to the Opfolda Prescribing Information for a description of additional risks for Opfolda. (5.5)
Adverse Reactions/Side Effects
Most common adverse reactions ≥ 5% are headache, diarrhea, fatigue, nausea, abdominal pain and pyrexia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amicus Therapeutics at 1-877-4AMICUS or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
Lactation: Breastfeeding not recommended. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2024
Full Prescribing Information
WARNING: SEVERE HYPERSENSITIVITY REACTIONS, INFUSION-ASSOCIATED REACTIONS, and RISK OF ACUTE CARDIORESPIRATORY FAILURE IN SUSCEPTIBLE PATIENTS
Hypersensitivity Reactions Including Anaphylaxis
Patients treated with POMBILITI have experienced life-threatening hypersensitivity reactions, including anaphylaxis. Appropriate medical support measures, including cardiopulmonary resuscitation equipment, should be readily available during POMBILITI administration. If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, POMBILITI should be discontinued immediately, and appropriate medical treatment should be initiated. In patients with severe hypersensitivity reaction, desensitization measures to POMBILITI may be considered [see Warnings and Precautions (5.1)].
Infusion-Associated Reactions (IARs)
Patients treated with POMBILITI have experienced severe IARs. If severe IARs occur, immediately discontinue the POMBILITI infusion, initiate appropriate medical treatment, and assess the benefits and risks of readministering POMBILITI following severe IARs. Patients with an acute underlying illness at the time of POMBILITI infusion may be at greater risk for IARs. Patients with advanced Pompe disease may have compromised cardiac and respiratory function, which may predispose them to a higher risk of severe complications from IARs [see Warnings and Precautions (5.2)].
Risk of Acute Cardiorespiratory Failure in Susceptible Patients
Patients susceptible to fluid volume overload, or those with acute underlying respiratory illness or compromised cardiac or respiratory function for whom fluid restriction is indicated may be at risk of serious exacerbation of their cardiac or respiratory status during POMBILITI infusion. More frequent monitoring of vitals should be performed during POMBILITI infusion in such patients [see Warnings and Precautions (5.3)].
1. Indications and Usage for Pombiliti
POMBILITI is indicated, in combination with Opfolda, for the treatment of adult patients with late-onset Pompe disease (lysosomal acid alpha-glucosidase [GAA] deficiency) weighing ≥40 kg and who are not improving on their current enzyme replacement therapy (ERT).
2. Pombiliti Dosage and Administration
2.1 Pregnancy Evaluation Prior to Initiating Treatment
Verify the pregnancy status of females of reproductive potential prior to initiating POMBILITI in combination with Opfolda [see Use in Specific Populations (8.1, 8.3)].
2.2 Recommended Dosage and Administration
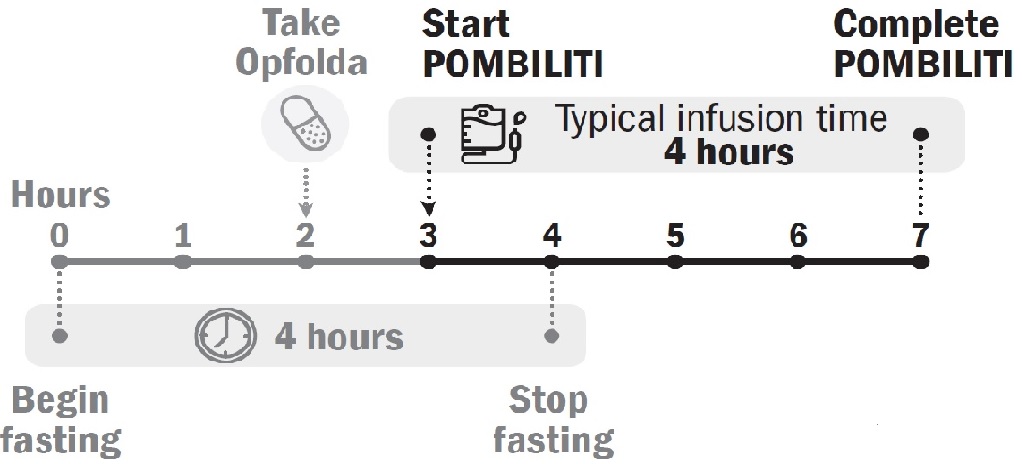
POMBILITI must be administered in combination with Opfolda (see Figure 1 for the dosing timeline). If the Opfolda dose is missed, POMBILITI should not be administered. Refer to the Opfolda Prescribing Information for Opfolda dosage and administration recommendations.
Prior to POMBILITI administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids [see Warnings and Precautions (5.1, 5.2)]. If premedication was used with previous enzyme replacement therapy (ERT), prior to POMBILITI administration, pretreat with antihistamines, antipyretics, and/or corticosteroids.
The recommended dosage of POMBILITI is 20 mg/kg (of actual body weight) administered every other week as an intravenous infusion over approximately 4 hours (see Table 1 for the recommended total infusion volume based on the patient’s weight).
Start POMBILITI in combination with Opfolda 2 weeks after the last ERT dose.
Initiate the POMBILITI infusion approximately 1 hour after oral administration of Opfolda. If the POMBILITI infusion cannot be started within 3 hours of oral administration of Opfolda, reschedule POMBILITI in combination with Opfolda at least 24 hours after Opfolda was last taken. If POMBILITI in combination with Opfolda are both missed, re-start treatment as soon as possible.
2.3 Dosage and Administration Modifications Due to Hypersensitivity Reactions and/or Infusion-Associated Reactions
In the event of a severe hypersensitivity reaction (including anaphylaxis) or a severe infusion-associated reaction (IAR), immediately discontinue the POMBILITI infusion, and initiate appropriate medical treatment. For additional recommendations in the event of a severe hypersensitivity reaction [see Warnings and Precautions (5.1, 5.2)].
In the event of a mild to moderate hypersensitivity reaction or moderate IAR, consider temporarily holding or slowing the infusion rate and initiating appropriate medical treatment [see Warnings and Precautions (5.1, 5.2)]. If symptoms:
- Persist despite temporarily holding or slowing the infusion, stop the infusion for 30 to 60 minutes, monitor the patient, and consider resuming the infusion at a reduced rate if symptoms have improved. If symptoms continue to persist, discontinue the infusion, and consider re-initiating the infusion within 7 to 14 days with appropriate premedication.
- Subside following holding or slowing the infusion, increase the infusion rate to the rate at which the reaction occurred and consider continuing to increase the rate (every 30 minutes) in a stepwise manner up to the target infusion rate. Closely monitor the patient.
2.4 Reconstitution and Dilution Instructions
Use aseptic technique during preparation. Reconstitute and dilute POMBILITI in the following manner:
Reconstitute the Lyophilized Powder
- Determine the number of POMBILITI vials to be reconstituted based on the calculated dose (based on patient’s actual body weight in kg) [see (Dosage and Administration 2.2)].
- Remove vials from the refrigerator and set aside for approximately 30 minutes to allow vials to come to room temperature.
- Reconstitute each vial by slowly injecting 7.2 mL of Sterile Water for Injection, down the inside wall of each vial to avoid foaming. Avoid forceful impact of Sterile Water for Injection on the lyophilized powder and avoid foaming.
- Roll and tilt each vial to allow the lyophilized powder to dissolve completely which typically takes 2 minutes. Each vial will yield a concentration of 15 mg/mL. Do not invert, swirl, or shake.
- Visually inspect the reconstituted solution for particulate matter and discoloration. The reconstituted solution appears as a clear to opalescent, colorless to yellowish solution essentially particle free. Discard if foreign matter is observed or the solution is discolored.
Dilute the Reconstituted Solution
- Remove airspace within a bag of 0.9% Sodium Chloride Injection. Remove an equal volume of 0.9% Sodium Chloride Injection from the bag that will be replaced by the total volume (mL) of reconstituted POMBILITI (see Table 1 for the recommended total infusion volume based on the patient’s weight).
- Slowly withdraw 7 mL of reconstituted solution from each of the vials until the patient’s dose is obtained. Discard any remaining reconstituted solution in the last vial.
- Add the reconstituted solution slowly and directly into the infusion bag.
- To prevent foaming, gently invert infusion bag to mix the solution and avoid vigorous shaking or agitation. After dilution, the solution will have a final concentration of 0.5 to 4 mg/mL of cipaglucosidase alfa-atga. Do not use a pneumatic tube to transport the infusion bag.
- Administer the diluted solution at room temperature without delay [see (Dosage and Administration 2.6)].
2.5 Storage Instructions for the Reconstituted and Diluted Product
Storage of the Reconstituted Solution
If the reconstituted POMBILITI vials are not used immediately, store refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours. Do not freeze.
Storage of the Diluted Solution
If the diluted solution is not administered immediately, store refrigerated at 2°C to 8°C (36°F to 46°F) for up to 16 hours. Storage at room temperature is not recommended. Do not freeze.
After removal of the diluted solution from the refrigerator:
- Completely infuse within 6 hours.
- Do not restore in the refrigerator.
Discard the diluted solution if refrigerated more than 16 hours or if the diluted solution is not able to be completely infused within 6 hours after removal from the refrigerator.
2.6 Administration Instructions
Prior to administration, inspect the infusion bag for foaming. If foaming is present, let foam dissipate before administering POMBILITI. If the diluted solution has been refrigerated, allow solution to equilibrate to room temperature for 30 minutes prior to infusion.
Use an administration set with an inline low protein binding 0.2‑micron filter to administer POMBILITI. Change filter if the filter becomes blocked.
Initiate the POMBILITI infusion approximately 1 hour after oral administration of Opfolda. In the event of POMBILITI infusion delay, the starting infusion time should not exceed 3 hours from the oral administration of Opfolda [see (Dosage and Administration 2.2)].
The initial recommended infusion rate is 1 mg/kg/hour (see Table 1). Gradually increase the infusion rate by 2 mg/kg/hour every 30 minutes if there are no signs of hypersensitivity or infusion-associated reactions (IARs) until a maximum rate of 7 mg/kg/hour is reached; then, maintain the infusion rate at 7 mg/kg/hour until the infusion is complete. The approximate total infusion duration is 4 hours.
See Table 1 for the rate of infusion at each step, expressed as mL/hour based on the recommended infusion volume by patient weight.
Do not infuse POMBILITI in the same intravenous line with other products.
Table 1. Recommended POMBILITI Infusion Volumes and Rates by Patient Weight
| Patient Weight Range | Total Infusion Volume | Step 1 1 mg/kg/hour | Step 2 3 mg/kg/hour | Step 3 5 mg/kg/hour | Step 4 7 mg/kg/hour |
| Infusion Rate in mL/hour | |||||
| 40–50 kg | 250 mL | 13 | 38 | 63 | 88 |
| 50.1–60 kg | 300 mL | 15 | 45 | 75 | 105 |
| 60.1-100 kg | 500 mL | 25 | 75 | 125 | 175 |
| 100.1–120 kg | 600 mL | 30 | 90 | 150 | 210 |
| 120.1–140 kg | 700 mL | 35 | 105 | 175 | 245 |
3. Dosage Forms and Strengths
For injection: 105 mg of cipaglucosidase alfa-atga as a white to slightly yellowish lyophilized powder with a cake-like appearance in a single-dose vial for reconstitution
4. Contraindications
POMBILITI in combination with Opfolda is contraindicated in pregnancy [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1)].
5. Warnings and Precautions
5.1 Hypersensitivity Reactions Including Anaphylaxis
Life-threatening hypersensitivity reactions, including anaphylaxis, have been reported in POMBILITI-treated patients. In clinical trials, 43 (27%) POMBILITI-treated patients experienced hypersensitivity reactions, including 4 (3%) patients who reported severe hypersensitivity reactions and 4 (3%) additional patients who experienced anaphylaxis (fulfilling at least one of the Sampson criteria). Three of the 4 (2%) patients experiencing anaphylaxis discontinued from the trial [see Clinical Studies (14)]. Two of the 4 patients who experienced anaphylaxis developed high anti-cipaglucosidase alfa-atga antibody titers [see Clinical Pharmacology (12.6)]. Anaphylaxis signs and symptoms included dyspnea, rash, hypotension, bronchospasm, edema, pharyngeal edema, and tongue swelling. Symptoms of severe hypersensitivity reactions included urticaria, pruritus, and flushing.
Prior to POMBILITI administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids. Appropriate medical support measures, including cardiopulmonary resuscitation equipment, should be readily available during POMBILITI administration.
- If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, POMBILITI should be discontinued immediately, and appropriate medical treatment should be initiated. The risks and benefits of readministering POMBILITI following severe hypersensitivity reaction (including anaphylaxis) should be considered. Patients may be rechallenged using slower infusion rates. In patients with severe hypersensitivity reaction, desensitization measures to POMBILITI may be considered. If the decision is made to readminister POMBILITI, ensure the patient tolerates the infusion. If the patient tolerates the infusion, the dosage (dose and/or the rate) may be increased to reach the approved recommended dosage.
- If a mild or moderate hypersensitivity reaction occurs, the infusion rate may be slowed or temporarily stopped [see Dosage and Administration (2.3)].
5.2 Infusion-Associated Reactions
In clinical trials, IARs were reported to occur at any time during and/or within a few hours after the POMBILITI infusion and were more likely to occur with higher infusion rates. IARs were reported in 48 (32%) POMBILITI-treated patients in clinical trials. In these trials, 4 (3%) POMBILITI-treated patients reported 11 severe IARs including symptoms of pharyngeal edema, anaphylactic reaction, urticaria, pruritus, chills, dyspnea, and flushing. The majority of IARs were assessed as mild to moderate. IARs that led to treatment discontinuation were urticaria, anaphylactic reaction, chills, and hypotension.
Antihistamines, antipyretics, and/or corticosteroids can be given prior to POMBILITI administration to reduce the risk of infusion-associated reactions (IARs). However, IARs may still occur in patients after receiving pretreatment.
- If severe IARs occur, immediately discontinue the POMBILITI infusion, initiate appropriate medical treatment, and assess the benefits and risks of readministering POMBILITI following severe IARs. Patients may be rechallenged using slower infusion rates. Once a patient tolerates the infusion, the infusion rate may be increased to reach the recommended infusion rate.
- If mild or moderate IARs occur regardless of pretreatment, decreasing the infusion rate or temporarily stopping the infusion may ameliorate the symptoms [see Dosage and Administration (2.3)].
Patients with an acute underlying illness at the time of POMBILITI infusion may be at greater risk for IARs. Patients with advanced Pompe disease may have compromised cardiac and respiratory function, which may predispose them to a higher risk of severe complications from IARs.
5.3 Risk of Acute Cardiorespiratory Failure in Susceptible Patients
Patients susceptible to fluid volume overload, or those with acute underlying respiratory illness or compromised cardiac or respiratory function for whom fluid restriction is indicated may be at risk of serious exacerbation of their cardiac or respiratory status during the POMBILITI infusion. More frequent monitoring of vitals should be performed during POMBILITI infusion in these patients. Some patients may require prolonged observation times.
5.4 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies, POMBILITI in combination with Opfolda may cause embryo-fetal harm when administered to a pregnant female and is contraindicated during pregnancy. In a rabbit embryo-fetal development study, great vessel and cardiac malformations were increased in offspring of pregnant rabbits treated with cipaglucosidase alfa‑atga in combination with oral miglustat at 16‑fold and 3‑fold, respectively, the maximum recommended human dose (MRHD) based on plasma AUC exposure.
Verify the pregnancy status in females of reproductive potential prior to initiating treatment with POMBILITI in combination with Opfolda. Advise females of reproductive potential to use effective contraception during treatment with POMBILITI in combination with Opfolda and for at least 60 days after the last dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hypersensitivity Reactions Including Anaphylaxis [see Warnings and Precautions (5.1)]
- Infusion-Associated Reactions [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions from the Pooled Clinical Trials Including Trial 1
The pooled safety analysis from 3 clinical trials included 151 adult patients with late-onset Pompe disease (LOPD) treated with POMBILITI in combination with Opfolda including:
- 85 patients in the randomized, double-blind, active-controlled trial in adults (Trial 1) [see Clinical Studies (14)],
- 37 patients in the open-label extension trial where patients switched from a non‑U.S.‑approved alglucosidase alfa product [see Clinical Studies (14)] to POMBILITI in combination with Opfolda,
- 29 patients in an open-label trial.
The total median duration of exposure in these trials was 21 months, with 120 patients having at least 12 months exposure to POMBILITI in combination with Opfolda. In these trials, 78% (n=117) of the patients received previous ERT (ERT‑experienced) with a mean treatment duration of 7.7 years.
In these trials, serious adverse reactions reported in 2 or more patients treated with POMBILITI in combination with Opfolda were anaphylaxis and urticaria. A total of 5 patients treated with POMBILITI in combination with Opfolda in these trials permanently discontinued POMBILITI due to adverse reactions, including 4 of these patients who discontinued the treatment because of a serious adverse reaction.
The most common adverse reactions (≥5%) reported in the pooled safety population of patients treated with POMBILITI in combination with Opfolda in the 3 clinical trials were headache, diarrhea, fatigue, nausea, abdominal pain and pyrexia.
In these trials, IARs were reported in 48 (32%) patients treated with POMBILITI in combination with Opfolda. IARs reported in more than 1 patient included headache, myalgia, diarrhea, nausea, fatigue, muscle spasms, pyrexia, dizziness, cough, chills, rash, vomiting, dyspnea, pain, abdominal distension, tachycardia, urticaria, flatulence, pruritus, abdominal pain, chest discomfort, flushing, hyperhidrosis, dysgeusia, hypotension, and hypertension [see Warnings and Precautions (5.2)].
Adverse Reactions from Trial 1
Trial 1 (a randomized, double‑blind, active‑controlled trial) included 123 adult patients with LOPD who were randomized in a 2:1 ratio to receive treatment with POMBILITI in combination with Opfolda or a non-U.S.-approved alglucosidase alfa product with placebo [see Clinical Studies (14)].
The duration of exposure was similar for both treatment groups (overall mean exposure of 12 months). Most patients (77%) were ERT‑experienced, and a majority of patients in both treatment groups had >5 years of prior treatment with ERT (69% and 63% of patients in the POMBILITI in combination with Opfolda group and the non-U.S.-approved alglucosidase alfa product with placebo group, respectively).
The most common adverse reactions (≥5%) reported in the patients who received POMBILITI in combination with Opfolda in Trial 1 were headache and diarrhea.
Table 2 summarizes frequent adverse reactions that occurred in patients treated with POMBILITI in combination with Opfolda in Trial 1. Trial 1 was not designed to demonstrate a statistically significant difference in the incidence of adverse reactions in the POMBILITI in combination with Opfolda and the non-U.S.-approved alglucosidase alfa product with placebo groups.
Table 2. Adverse Reactions that Occurred in Adults with LOPD at an Incidence of ≥2% in Trial 1
| LOPD: late-onset Pompe disease | ||
| ∗ Headache included migraine and migraine with aura. | ||
| † Rash included erythematous rash and macular rash. | ||
| ‡ Abdominal pain included upper and lower abdominal pain. | ||
| § Tachycardia included sinus tachycardia. | ||
| ¶ Urticaria included mechanical urticaria and urticarial rash. | ||
| Adverse Reaction | POMBILITI in Combination with Opfolda (n=85) N (%) | A Non-U.S.-Approved Alglucosidase alfa Product with Placebo (n=38) N (%) |
| Headache∗ | 7 (8.2) | 3 (7.9) |
| Diarrhea | 5 (5.9) | 2 (5.3) |
| Dizziness | 4 (4.7) | 2 (5.3) |
| Dyspnea | 3 (3.5) | 0 |
| Abdominal distention | 3 (3.5) | 2 (5.3) |
| Pyrexia | 3 (3.5) | 1 (2.6) |
| Rash† | 3 (3.5) | 0 |
| Abdominal pain‡ | 2 (2.4) | 4 (10.5) |
| Nausea | 2 (2.4) | 5 (13.2) |
| Chills | 2 (2.4) | 0 |
| Dysgeusia | 2 (2.4) | 0 |
| Flushing | 2 (2.4) | 0 |
| Muscle spasms | 2 (2.4) | 0 |
| Pruritus | 2 (2.4) | 2 (5.3) |
| Tachycardia§ | 2 (2.4) | 0 |
| Urticaria¶ | 2 (2.4) | 0 |
Additional adverse reactions reported in at least 2% of patients treated with POMBILITI in combination with Opfolda across the 3 clinical trials include: myalgia, arthralgia, increased blood pressure, pain, tremor, dyspepsia, asthenia, constipation, infusion site swelling, flank pain, malaise, paresthesia, somnolence, and decreased platelet count.
Immunogenicity: Anti-Drug Antibody-Associated Adverse Reactions in Trial 1
Anaphylaxis or serious infusion-associated reactions (IARs) occurred in 1 (1.5%) POMBILITI‑treated patient (who previously received U.S.-approved alglucosidase alfa or a non-U.S.-approved alglucosidase alfa product) who had peak anti-cipaglucosidase alfa-atga antibody titer of 6,553,600 during the 52-week treatment period [see Clinical Pharmacology (12.6)].
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies, POMBILITI in combination with Opfolda may cause embryo-fetal harm when administered to a pregnant female and is contraindicated during pregnancy. In a rabbit embryo-fetal development study, great vessel and cardiac malformations were increased in offspring of pregnant rabbits treated with cipaglucosidase alfa-atga in combination with miglustat at 16-fold and 3-fold, respectively, the MRHD of POMBILITI and Opfolda based on plasma AUC exposure. A No Observed Adverse Effect Level (NOAEL) was not identified for the combination. In a pre- and post-natal development study in rats, increases in pup mortality were seen following maternal treatment with cipaglucosidase alfa-atga (400 mg/kg) in combination with miglustat, or with cipaglucosidase alfa-atga (400 mg/kg) alone. The NOAEL for cipaglucosidase alfa-atga alone is 150 mg/kg (5-fold the POMBILITI MRHD margin). A NOAEL for the combination was not identified. Margins at the lowest observed adverse effect level (LOAEL), relative to exposures at the MRHD of POMBILITI and Opfolda were 20-fold and 4-fold, respectively, based on plasma AUC exposure (see Data).
There are no available human data on POMBILITI in combination with Opfolda use in pregnant females to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes.
Animal Data
Reproductive toxicity studies of cipaglucosidase alfa-atga in rats and rabbits included pretreatment with diphenhydramine (DPH) to prevent or minimize hypersensitivity reactions.
In a rabbit embryo-fetal development study, cipaglucosidase alfa-atga (30, 70, or 175 mg/kg) was administered intravenously every other day to pregnant females during organogenesis (Gestation Day [GD] 7 through GD 19). Additional experimental groups received 25 mg/kg oral miglustat alone, or in combination with intravenous cipaglucosidase alfa-atga 175 mg/kg, with the same dosing frequency during organogenesis. Clusters of great vessel and cardiac malformations were increased in offspring of pregnant rabbits treated with the combination of cipaglucosidase alfa-atga and miglustat at 16-fold and 3-fold the MRHD of POMBILITI and Opfolda, respectively, based on plasma AUC exposure. A NOAEL for the combination was not identified. One fetus treated with cipaglucosidase alfa-atga alone (175 mg/kg) and one fetus treated with miglustat alone (25 mg/kg), each showed a similar cluster of these great vessel and cardiac malformations.
In a rat embryo-fetal development study, cipaglucosidase alfa-atga (75, 150, or 400 mg/kg) was administered intravenously every other day to pregnant rats during organogenesis (GD 6 through GD 18). Additional experimental groups received 60 mg/kg oral miglustat alone, or in combination with intravenous cipaglucosidase alfa-atga 400 mg/kg, with the same dosing frequency during organogenesis. No evidence of adverse effects was noted in pregnant rats or their offspring in any experimental group. The margin at the NOAEL for cipaglucosidase alfa‑atga (400 mg/kg) was 20-fold the POMBILITI MRHD based on plasma AUC exposure. The margin at the NOAEL for miglustat (60 mg/kg) was 4-fold the Opfolda MRHD based on plasma AUC exposure.
In a pre-and post-natal development study in rats, cipaglucosidase alfa-atga (75, 150, or 400 mg/kg) was administered intravenously every other day to pregnant females from GD 6 through GD 18, and from Lactation Day (LD) 1 through LD 19. Additional experimental groups received 60 mg/kg oral miglustat alone, or in combination with intravenous cipaglucosidase alfa‑atga 400 mg/kg, with the same dosing frequency during pregnancy and lactation. Maternal and pup mortality were increased with the combination, and pup mortality was also increased with cipaglucosidase alfa-atga 400 mg/kg alone. The NOAEL for cipaglucosidase alfa-atga alone is 150 mg/kg (5-fold the POMBILITI MRHD margin). A NOAEL was not identified for the combination, for which LOAEL margins at the MRHD of POMBILITI and Opfolda were 20-fold and 4-fold, respectively, based on plasma AUC exposure.
8.2 Lactation
Risk Summary
There are no data on the presence of cipaglucosidase alfa-atga, alone or in combination with miglustat, in human milk, the effects on the breastfed infant, or the effects on milk production. Cipaglucosidase alfa‑atga is present in animal milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Based on findings in animal studies, the use of POMBILITI in combination with Opfolda may lead to serious adverse reactions in breastfed infants. Advise females that breastfeeding is not recommended while on treatment with POMBILITI in combination with Opfolda.
Evaluation of milk in rats from the pre- and post-natal development study of cipaglucosidase alfa atga in combination with miglustat (400 mg/kg and 60 mg/kg, respectively) showed excretion of cipaglucosidase alfa-atga and secretion of miglustat in rat milk. In this study, the ratio of cipaglucosidase alfa-atga exposure in rat milk to cipaglucosidase alfa-atga exposure in rat plasma was <4%, and the ratio of miglustat exposure in rat milk to the miglustat exposure in rat plasma was 1.7.
The concentration of drug in animal milk does not necessarily predict the concentration of drug in human milk.
8.3 Females and Males of Reproductive Potential
POMBILITI in combination with Opfolda may cause embryo-fetal harm when administered to a pregnant female [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status in females of reproductive potential prior to initiating treatment with POMBILITI in combination with Opfolda.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with POMBILITI in combination with Opfolda and for at least 60 days after the last dose.
Infertility
Females
Based on preimplantation loss observed in female rats treated with intravenous cipaglucosidase alfa-atga (400 mg/kg) in combination with oral miglustat (60 mg/kg) every other day for 14 days prior to mating, and continuing through GD 7, POMBILITI in combination with Opfolda may impair human female fertility. A NOAEL for the combination was not identified. The LOAEL margins are 27-fold and 4-fold the MRHD for POMBILITI and Opfolda, respectively. It is not known whether this preimplantation loss in female rats would be sustained if dosing with the combination were discontinued prior to mating [see Nonclinical Toxicology (13.1)].
Males
Based on reversible increases in preimplantation loss in male rats treated with the combination every other day for 28 days prior to mating, POMBILITI in combination with Opfolda may impair human male fertility. A NOAEL for the combination was not identified. The LOAEL margins are 27-fold and 4-fold the MRHD for POMBILITI and Opfolda, respectively [see Nonclinical Toxicology (13.1)].
For additional information about male fertility with the use of Opfolda, see the Opfolda Prescribing Information.
8.4 Pediatric Use
Safety and effectiveness of POMBILITI in combination with Opfolda have not been established in pediatric patients with late-onset Pompe disease.
8.5 Geriatric Use
Of the total number of patients treated with POMBILITI in combination with Opfolda in clinical trials for LOPD, 17 (11%) were 65 to 74 years of age, and none were 75 years of age and older [see Clinical Studies (14)].
Clinical trials of POMBILITI in combination with Opfolda did not include sufficient numbers of patients 65 years of age and older to determine whether they respond differently from younger adult patients.
11. Pombiliti Description
Cipaglucosidase alfa-atga is a hydrolytic lysosomal glycogen‑specific recombinant human α‑glucosidase (rhGAA) enzyme derived from a Chinese Hamster Ovary (CHO) cell line using perfusion methodology, resulting in cellularly (CHO)‑derived N‑glycans. Cipaglucosidase alfa‑atga is a glycoprotein with 1.3 mols of bis‑mannose‑6‑phosphate (bis‑M6P) per mol of enzyme. Cipaglucosidase alfa-atga has a molecular weight of approximately 110 kDa.
POMBILITI (cipaglucosidase alfa-atga) for injection is a sterile, white to slightly yellowish lyophilized powder with a cake-like appearance for intravenous use after reconstitution and dilution. Each single-dose vial contains 105 mg of cipaglucosidase alfa-atga and the inactive ingredients citric acid monohydrate (4.57 mg), mannitol (140 mg), polysorbate 80 (3.5 mg), and sodium citrate (39 mg). After reconstitution with Sterile Water for Injection, USP, the resultant concentration is 15 mg/mL with a pH of between 5.7 to 6.3.
12. Pombiliti - Clinical Pharmacology
12.1 Mechanism of Action
Pompe disease (also known as glycogen storage disease type II, acid maltase deficiency, and glycogenosis type II) is an inherited disorder of glycogen metabolism caused by a deficiency of lysosomal acid alpha‑glucosidase (GAA) that degrades glycogen to glucose in the lysosome. GAA deficiency results in intra-lysosomal accumulation of glycogen in various tissues.
Cipaglucosidase alfa-atga provides an exogenous source of GAA. The bis-M6P on cipaglucosidase alfa-atga mediates binding to M6P receptors on the cell surface with high affinity. After binding, it is internalized and transported into lysosomes where it undergoes proteolytic cleavage and N‑glycans trimming which are both required to yield the most mature and active form of GAA. Cipaglucosidase alfa-atga then exerts enzymatic activity in cleaving glycogen.
Miglustat binds with, stabilizes, and reduces inactivation of cipaglucosidase alfa-atga in the blood after infusion.
12.2 Pharmacodynamics
In patients with LOPD, excess of glycogen is degraded to hexose tetrasaccharide (Hex4) which is then excreted in urine. The urinary Hex4 assay measures its major component, glucose tetrasaccharide (Glc4). Treatment with POMBILITI in combination with Opfolda resulted in reductions of urinary Glc4 concentrations (normalized by urine creatinine and reported as mmol Glc4/mol creatinine) in adult patients with LOPD.
In ERT‑experienced patients in Trial 1 [see Clinical Studies (14)], the baseline mean urinary Glc4 concentration was 4.6 mmol/mol and 7.2 mmol/mol in the POMBILITI in combination with Opfolda and the non-U.S.-approved alglucosidase alfa product with placebo groups, respectively. The mean percentage (SD) change in urinary Glc4 concentration from baseline to Week 52 was ‑29% (32) and +20% (32) in the POMBILITI in combination with Opfolda and the non-U.S.-approved alglucosidase alfa product with placebo groups, respectively.
12.3 Pharmacokinetics
The maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) of cipaglucosidase alfa-atga following administration with 20 mg/kg of cipaglucosidase alfa-atga in combination with a single oral dose of 260 mg of Opfolda in adult patients with LOPD are summarized in Table 3.
Table 3. Geometric Mean (CV%) Pharmacokinetic Parameters of Cipaglucosidase alfa‑atga in ERT‑experienced Adult Patients with LOPD
| CV: coefficient of variation; ERT: enzyme replacement therapy; LOPD: late-onset Pompe disease; PK: pharmacokinetic parameters; Cmax: maximum plasma concentration; AUC: plasma concentration-time curve | ||
| † POMBILITI in combination with Opfolda is not approved for use without Opfolda [see Indications and Usage (1)]. | ||
| PK Parameter | Cipaglucosidase alfa-atga 20 mg/kg in Combination with Opfolda 260 mg | Cipaglucosidase alfa-atga 20 mg/kg† |
| Cmax (mcg/mL) | 345 (18.5) | 325 (13.5) |
| AUC0-inf (mcg*h/mL) | 1812 (20.8) | 1410 (15.9) |
Distribution
The volume of distribution of cipaglucosidase alfa-atga ranged from 2.0 to 4.7 L.
Elimination
When cipaglucosidase alfa-atga was administered with Opfolda, the mean total body clearance (CL) of cipaglucosidase alfa-atga was 0.8 L/hour and the mean half-life of cipaglucosidase alfa-atga was 2.1 hours in ERT‑experienced patients.
Metabolism
The metabolic pathway of cipaglucosidase alfa-atga has not been characterized. Cipaglucosidase alfa-atga is expected to be metabolized into small peptides and amino acid via catabolic pathways.
Specific Populations
Age (18 to 74 years) and sex did not have clinically meaningful effects on the pharmacokinetics of cipaglucosidase alfa-atga.
12.6 Immunogenicity
The observed incidence of antidrug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of antidrug antibodies (ADA) in the trials described below with the incidence of antidrug antibodies in other trials, including those of cipaglucosidase alfa-atga or of other cipaglucosidase alfa products.
Table 4 presents the incidence of ADA and neutralizing antibodies against cipaglucosidase alfa-atga in adult patients with LOPD treated with POMBILITI in combination with Opfolda (who previously received U.S.-approved alglucosidase alfa or a non-U.S.-approved alglucosidase alfa product) during the 52-week treatment period in Trial 1 [see Clinical Studies (14)].
Antibodies against cipaglucosidase alfa-atga (anti-cipaglucosidase alfa-atga antibodies) were cross-reactive to alglucosidase alfa.
Patients who had an IAR post‑treatment were tested for anti-cipaglucosidase alfa-atga IgE antibodies after the occurrence of the IAR. For ERT‑experienced patients, 25% of POMBILITI‑treated patients had IARs, with 1 patient having positive results for anti‑cipaglucosidase alfa-atga IgE antibodies [see Adverse Reactions (6.1)]. Overall, there was no clear trend in IAR occurrence with the development of anti-cipaglucosidase alfa-atga IgE antibodies.
There was no identified clinically significant effect of ADA on pharmacokinetics or pharmacodynamics of POMBILITI in combination with Opfolda over the treatment duration of 52 weeks. Because of the small number of patients with negative ADA, the effect of ADA on the effectiveness of POMBILITI in combination with Opfolda is unknown.
Table 4. Incidence of Anti-Cipaglucosidase alfa-atga IgG Antibodies in ERT-experienced POMBILITI-treated Adult Patients with LOPD (Trial 1)*
| ERT: enzyme replacement therapy; LOPD: late-onset Pompe disease | ||
| ∗ In this table, ADAs (antidrug antibodies) are anti-cipaglucosidase alfa-atga IgG antibodies, and neutralizing IgG antibodies are against cipaglucosidase alfa-atga. | ||
| † Enzyme activity measured as 4-methylumbelliferone-α-D-glucopyranoside (4-MU-α-Glc) hydrolysis | ||
| ‡ Enzyme activity measured as glycogen hydrolysis | ||
| § Inhibition of cation independent mannose-6-phosphate receptor (CI-MPR) binding | ||
| ¶ ERT‑experienced patients received U.S.-approved alglucosidase alfa or a non-U.S.-approved alglucosidase alfa product before they received POMBILITI. | ||
| Antidrug Antibodies | ERT-experienced Patients¶ (N=65) |
|
| ADA at Baseline | n=55 (85%) | |
| ADA at Week 52 | n=58 (89%) | |
| Inhibition of enzyme activity† | n=26 (40%) | |
| Inhibition of enzyme activity‡ | n=24 (37%) | |
| Inhibition of CI-MPR binding§ | n=53 (82%) | |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Studies in animals to evaluate the carcinogenic potential of cipaglucosidase alfa-atga have not been conducted. For information on an evaluation of carcinogenicity of miglustat in animals, see the Opfolda Prescribing Information.
Mutagenesis
Studies to evaluate the mutagenic or genotoxic potential of cipaglucosidase alfa-atga have not been conducted. For information regarding the mutagenesis of miglustat, see the Opfolda Prescribing Information.
Impairment of Fertility
In a fertility study in rats, cipaglucosidase alfa-atga (75, 150, or 400 mg/kg) was administered intravenously to male and female rats every other day. Dosing in males was initiated 28 days prior to cohabitation with untreated females. Dosing in females was initiated 14 days prior to cohabitation with untreated males and continued through GD 7. Additional experimental groups received 60 mg/kg oral miglustat alone, or in combination with intravenous cipaglucosidase alfa‑atga 400 mg/kg, with the same frequency over the same pre‑mating interval (males) or pre-mating and pregnancy interval (females).
There was no effect on male or female rat fertility in any experimental group. Treatment of male rats with the combination was associated with increased preimplantation loss that was reversible. Treatment of female rats with the combination, or with miglustat alone, resulted in preimplantation loss; whether this would be reversible if treatment were discontinued prior to cohabitation is unknown. NOAELs were not identified for the combination in either male or female rats. The LOAEL margins for these doses represent 27-fold and 4-fold the MRHD of POMBILITI and Opfolda, respectively, based on plasma AUC exposure.
14. Clinical Studies
Trial 1 was a randomized, double‑blind, active‑controlled, international, multi‑center clinical trial (NCT#03729362) in patients ≥18 years old diagnosed with LOPD. Patients were randomized 2:1 to receive POMBILITI (20 mg/kg by intravenous infusion) in combination with Opfolda (260 mg orally for those ≥50 kg or 195 mg orally for those ≥40 kg to <50 kg) or a non-U.S.-approved alglucosidase alfa product with placebo every other week for 52 weeks. The efficacy population included a total of 123 patients of whom 95 (77%) had received prior treatment with U.S.-approved alglucosidase alfa or a non-U.S.-approved alglucosidase alfa product (ERT‑experienced) and 28 (23%) were ERT‑naïve. More than two thirds (n=64, 67%) of ERT‑experienced patients had been on ERT treatment for more than 5 years prior to entering Trial 1 (mean of 7.4 years).
Demographics, baseline sitting forced vital capacity (FVC) (% predicted), and 6-minute walk distance (6MWD) were generally similar between the 2 treatment groups (see Table 5 for baseline sitting FVC [percent predicted] values). Of the 123 randomized patients, 56 were males, baseline mean age was 47 years old (range from 19 to 74 years old), and mean age at diagnosis was 39 years old (range from 1 to 66). The racial groups for the patients consisted of 104 White (85%), 6 Japanese (5%), 6 Other racial group (5%), 4 Asian (3%), 1 Native Hawaiian or other Pacific Islander (1%), 1 American Indian or Alaska Native (1%), and 1 Black or African American (1%).
Key efficacy endpoints included assessment of sitting FVC (% predicted) and 6MWD (Table 5 and Table 6).
Sitting FVC (Percent‑predicted) at 52 Weeks
Patients treated with POMBILITI in combination with Opfolda showed a mean change in sitting FVC from baseline at Week 52 of -1.1% as compared with patients treated with a non-U.S.-approved alglucosidase alfa product with placebo of -3.3%; the estimated treatment difference was 2.3% (95% CI: 0.02, 4.62).
The ERT‑experienced patients treated with POMBILITI in combination with Opfolda showed a numerically favorable change in sitting FVC from baseline at Week 52 (Table 5 and Figure 2).
Table 5. Summary of Sitting FVC in Adults with LOPD by ERT Status at 52 Weeks in Trial 1
| FVC: forced vital capacity; LOPD: late-onset Pompe disease; ERT: enzyme replacement therapy; SD: standard deviation; Diff.: difference; SE: standard error; CI: confidence interval | ||||
| ∗ POMBILITI in combination with Opfolda is not approved for use in ERT-naïve patients with LOPD [see Indications and Usage (1)]. The ERT-naïve patient subgroup enrolled too few patients to conclusively interpret the data. For the ERT-naïve group, the treatment difference was estimated using a 2-sample t-test. | ||||
| † A U.S.-approved alglucosidase alfa product was not used in this clinical trial. Conclusions cannot be drawn from this clinical trial regarding comparative effectiveness between a U.S.-approved alglucosidase alfa product and POMBILITI in combination with Opfolda for the treatment of adult patients with LOPD weighing ≥40 kg and who are not improving on their current ERT. | ||||
| ‡ For the ERT‑experienced group, the treatment difference of the mean was estimated by analysis of covariance which included treatment, gender, baseline FVC, age, weight, and height in the model. Nominal p=0.006. Missing data at Week 52 was imputed using last observed values. | ||||
| Efficacy Endpoint | ERT-experienced | ERT-naïve* | ||
| Sitting FVC (% predicted) | POMBILITI in Combination with Opfolda | A Non-U.S.-Approved Alglucosidase alfa Product† with Placebo | POMBILITI in Combination with Opfolda | A Non-U.S.-Approved Alglucosidase alfa Product† with Placebo |
| Baseline n Mean (SD) Median |
n=65 67.9 (19.1) 68.0 |
n=30 67.5 (21.0) 69.0 |
n=20 80.2 (18.7) 82.3 |
n=8 79.6 (21.0) 88.5 |
| Change from baseline at Week 52 n Mean (SD) Median |
n=55 0.1 (5.9) 0.5 |
n=26 -3.5 (4.7) -2.5 |
n=19 -4.7 (6.2) -4.5 |
n=7 -2.4 (6.3) -3.0 |
| Change to Week 52 Diff. of means (SE) (95% CI) |
3.5 (1.3) (1.0, 6.0)‡ |
-1.9 (2.7) (-7.3, 3.6) |
||
Figure 2. Mean Change (± SE) in Sitting FVC (% predicted) from Baseline to Week 52 in ERT-experienced Adults with LOPD in Trial 1*
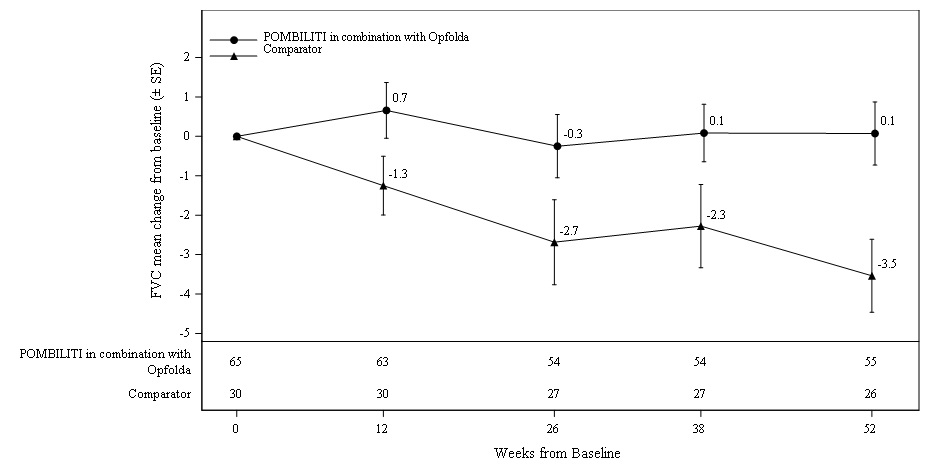
SE: standard error; FVC: forced vital capacity; ERT: enzyme replacement therapy; LOPD: late-onset Pompe disease
∗ A U.S.-approved alglucosidase alfa product was not used in this clinical trial. Conclusions cannot be drawn from this clinical trial regarding comparative effectiveness between a U.S.-approved alglucosidase alfa product and POMBILITI in combination with Opfolda for the treatment of adult patients with LOPD weighing ≥40 kg and who are not improving on their current ERT.
6 Minute Walk Distance (6MWD) at 52 Weeks
Patients treated with POMBILITI in combination with Opfolda walked on average 21 meters farther from baseline as compared to those treated with a non-U.S.-approved alglucosidase alfa product with placebo who walked 8 meters farther from baseline; the estimated treatment difference was 14 meters (95% CI: -1, 28).
The ERT‑experienced patients treated with POMBILITI in combination with Opfolda showed a numerically favorable change in 6MWD from baseline at Week 52 (Table 6 and Figure 3).
Table 6. Summary of 6MWD in Adults with LOPD by ERT Status at 52 Weeks in Trial 1
| 6MWD: 6-minute walk distance; LOPD: late-onset Pompe disease; ERT: enzyme replacement therapy; SD: standard deviation; Diff.: difference; SE: standard error; CI: confidence interval | ||||
| ∗ POMBILITI in combination with Opfolda is not approved for use in ERT‑naïve patients with LOPD [see Indications and Usage (1)]. The ERT-naïve patient subgroup enrolled too few patients to conclusively interpret the data. For the ERT‑naïve group, the treatment difference was estimated using a 2-sample t-test. One ERT‑naïve subject in the control arm was excluded from this table because their change of 355 meters in 6MWD from baseline at Week 52 was a statistical outlier and not considered clinically plausible. | ||||
| † A U.S.-approved alglucosidase alfa product was not used in this clinical trial. Conclusions cannot be drawn from this clinical trial regarding comparative effectiveness between a U.S.-approved alglucosidase alfa product and POMBILITI in combination with Opfolda for the treatment of adult patients with LOPD weighing ≥40 kg and who are not improving on their current ERT. | ||||
| ‡ For the ERT‑experienced group, the treatment difference of the mean was estimated by nonparametric analysis of covariance which included treatment, gender, baseline 6MWD, age, weight, and height in the model. Nominal p=0.047. Missing data at Week 52 was imputed using last observed values. | ||||
| Efficacy Endpoint | ERT‑experienced | ERT‑naïve* | ||
| 6MWD | POMBILITI in Combination with Opfolda | A Non-U.S.-Approved Alglucosidase alfa Product† with Placebo | POMBILITI in Combination with Opfolda | A Non-U.S.-Approved Alglucosidase alfa Product† with Placebo |
| Baseline n Mean (SD) Median |
n=65 347 (110) 353 |
n=30 335 (114) 344 |
n=20 394 (112) 375 |
n=7 421 (136) 386 |
| Change from baseline at Week 52 n Mean (SD) Median |
n=61 16 (39) 10 |
n=29 1 (40) -9 |
n=20 33 (49) 24 |
n=7 38 (29) 34 |
| Change to Week 52 Diff. of means (SE) (95% CI) |
17 (8) (0.2, 33)‡ |
-5 (20) (-45, 36) |
||
Figure 3. Mean Change (± SE) of 6MWD from Baseline to Week 52 in ERT‑experienced Adults with LOPD in Trial 1*
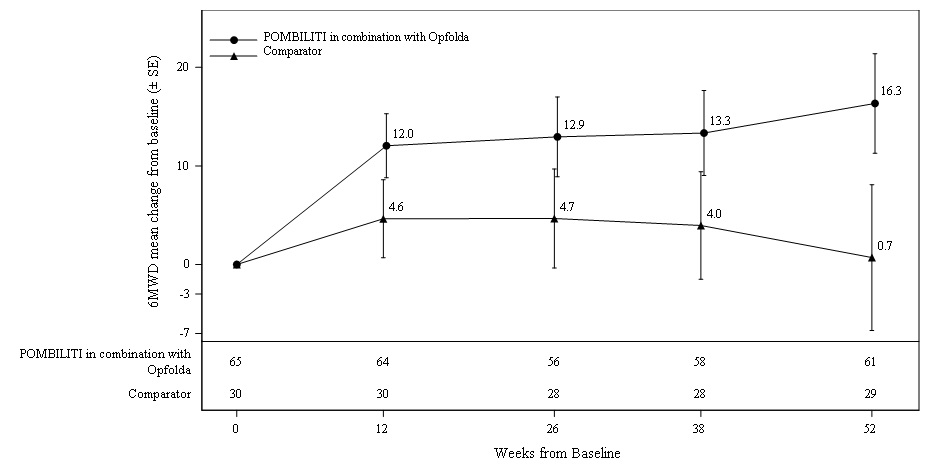
SE: standard error; 6MWD: 6-minute walk distance; ERT: enzyme replacement therapy; LOPD: late-onset Pompe disease
∗ A U.S.-approved alglucosidase alfa product was not used in this clinical trial. Conclusions cannot be drawn from this clinical trial regarding comparative effectiveness between a U.S.-approved alglucosidase alfa product and POMBILITI in combination with Opfolda for the treatment of adult patients with LOPD weighing ≥40 kg and who are not improving on their current ERT.
16. How is Pombiliti supplied
How Supplied
POMBILITI (cipaglucosidase alfa-atga) for injection is supplied as a sterile, white to slightly yellowish lyophilized powder with a cake-like appearance in a single‑dose vial. POMBILITI does not contain any preservatives. See Table 7 for the available POMBILITI packages.
| Carton | NDC |
| Contains one (1) 105 mg single-dose vial | 71904‑200‑01 |
| Contains ten (10) 105 mg single-dose vials | 71904‑200‑02 |
| Contains twenty-five (25) 105 mg single-dose vials | 71904‑200‑03 |
Storage and Handling
Store refrigerated at 2℃ to 8℃ (36℉ to 46℉) in the original carton to protect from light. Do not freeze.
For storage of the reconstituted solution and diluted solution [see Dosage and Administration (2.5)].
17. Patient Counseling Information
POMBILITI must be administered in combination with Opfolda. Refer to the Opfolda Prescribing Information for Opfolda patient counseling information.
Administration
Advise the patient and caregiver to follow the timeline recommendations for taking Opfolda prior to the intravenous infusion with POMBILITI [see Dosage and Administration (2.2)].
Hypersensitivity Reactions (Including Anaphylaxis) and Infusion-Associated Reactions (IARs)
Advise the patient and caregiver that reactions related to the infusion may occur during and after POMBILITI in combination with Opfolda treatment, including life-threatening hypersensitivity reactions, including anaphylaxis, and IARs. Inform the patient and caregiver of the signs and symptoms of hypersensitivity reactions and IARs and to seek medical care should signs and symptoms occur [see Warnings and Precaution (5.1, 5.2)].
Risk of Acute Cardiorespiratory Failure
Advise the patient and caregiver that a patient with underlying respiratory illness or compromised cardiac or respiratory function may be at risk of acute cardiorespiratory failure from volume overload during POMBILITI infusion and to seek medical care should signs and symptoms occur [see Warnings and Precaution (5.3)].
Embryo-Fetal Toxicity
POMBILITI in combination with Opfolda may cause embryo-fetal harm. Advise a female patient and caregiver to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1)].
Advise a female of reproductive potential to use effective contraception during treatment with POMBILITI in combination with Opfolda and for at least 60 days after the last dose [see Use in Specific Populations (8.1, 8.3)].
Lactation
Advise a lactating female not to breastfeed during treatment with POMBILITI in combination with Opfolda [see Use in Specific Populations (8.2)].
Infertility
Advise the male or female of reproductive potential that POMBILITI in combination with Opfolda may impair fertility [see Use in Specific Populations (8.3)].
Manufactured by:
Amicus Therapeutics US, LLC
3675 Market Street
Philadelphia, PA 19104
U.S. License No. 2224
POMBILITI and Opfolda are registered trademarks of Amicus Therapeutics, Inc.
For more information, go to pombilitiopfoldahcp.com.



| POMBILITI
ATGA
cipaglucosidase alfa-atga injection, powder, lyophilized, for solution |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - AMICUS THERAPEUTICS US, LLC (080932337) |
Biological Products Related to Pombiliti
Find detailed information on biosimilars for this medication.
More about Pombiliti (cipaglucosidase alfa)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: lysosomal enzymes
- Breastfeeding
- En español