Benlysta: Package Insert / Prescribing Info
Package insert / product label
Generic name: belimumab
Dosage form: injection, powder, lyophilized, for solution
Drug class: Selective immunosuppressants
J Code (medical billing code): J0490 (10 mg, injection)
Medically reviewed by Drugs.com. Last updated on Jul 8, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
BENLYSTA (belimumab) for injection, for intravenous use
BENLYSTA (belimumab) injection, for subcutaneous use
Initial U.S. Approval: 2011
Indications and Usage for Benlysta
BENLYSTA is a B-lymphocyte stimulator (BLyS)-specific inhibitor indicated for the treatment of patients 5 years of age and older with:
- •
- Active systemic lupus erythematosus (SLE) who are receiving standard therapy; (1)
- •
- Active lupus nephritis who are receiving standard therapy. (1)
Limitations of Use:
The efficacy of BENLYSTA has not been evaluated in patients with severe active central nervous system lupus. Use of BENLYSTA is not recommended in this situation. (1)
Benlysta Dosage and Administration
- •
- See Full Prescribing Information for complete preparation and administration information. (2.1, 2.2, 2.3)
- •
- Intravenous dosage for active SLE or lupus nephritis:
- •
- Subcutaneous dosage for active SLE or lupus nephritis: (2.3)
| a Prefilled syringe has not been studied in children less than 18 years of age. | ||
| b The 400-mg dose requires administration of two 200‑mg injections. | ||
|
Indication |
Adults (Autoinjector or Prefilled Syringe) |
Pediatric Patients 5 Years of Age and Older (Weight‑Based Dosing) (Autoinjector Only)a |
|
Active SLE |
200 mg once weekly |
|
|
Active Lupus Nephritis |
400 mgb once weekly for 4 doses, followed by 200 mg once weekly |
|
Dosage Forms and Strengths
- •
- Intravenous Infusion (for injection): 120 mg or 400 mg of belimumab lyophilized powder in single‑dose vial for reconstitution and dilution prior to intravenous infusion. (3)
- •
- Subcutaneous Injection (injection): 200 mg/mL of belimumab in single‑dose prefilled autoinjector or single‑dose prefilled syringe. (3)
Contraindications
Previous anaphylaxis to belimumab. (4)
Warnings and Precautions
- •
- Serious Infections: Serious and sometimes fatal infections have occurred in patients receiving immunosuppressive agents, including BENLYSTA. Use with caution in patients with severe or chronic infections. Consider interrupting therapy with BENLYSTA if patients develop a new infection during treatment with BENLYSTA. (5.1)
- •
- Progressive Multifocal Leukoencephalopathy (PML): Evaluate patients with new-onset or deteriorating neurological signs and symptoms for PML. If PML is suspected, immunosuppressant therapy, including BENLYSTA, must be suspended until PML has been excluded. If PML is confirmed, immunosuppressant therapy, including BENLYSTA, must be discontinued. (5.1)
- •
- Hypersensitivity Reactions, including Anaphylaxis: Serious and fatal reactions have been reported. (5.2)
- •
- Depression and Suicidality: Depression and suicidality were reported in trials with BENLYSTA. Assess for depression and risk of suicide before treatment with BENLYSTA and monitor during treatment. Instruct patients to contact their healthcare provider if new or worsening depression, suicidal thoughts, or other mood changes occur. (5.3)
- •
- Immunization: Live vaccines should not be given concurrently with BENLYSTA. (5.5)
Adverse Reactions/Side Effects
Common adverse reactions (≥5%): nausea, diarrhea, pyrexia, nasopharyngitis, bronchitis, insomnia, pain in extremity, depression, migraine, pharyngitis, and injection site reactions (subcutaneous administration). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact GlaxoSmithKline at 1-877-423-6597 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2025
Full Prescribing Information
1. Indications and Usage for Benlysta
BENLYSTA is indicated for the treatment of patients 5 years of age and older with:
- •
- Active systemic lupus erythematosus (SLE) who are receiving standard therapy, and
- •
- Active lupus nephritis who are receiving standard therapy.
Limitations of Use
The efficacy of BENLYSTA has not been evaluated in patients with severe active central nervous system (CNS) lupus. Use of BENLYSTA is not recommended in this situation.
2. Benlysta Dosage and Administration
2.1 Important Administration Instructions
BENLYSTA may be administered intravenously or subcutaneously [see Dosage and Administration (2.2, 2.3)]. Vials are intended for intravenous use only (not for subcutaneous use) and autoinjectors and prefilled syringes are intended for subcutaneous use only (not for intravenous use).
Precautions Prior to Intravenous Use
BENLYSTA should be administered by healthcare providers prepared to manage anaphylaxis [see Warnings and Precautions (5.2)].
Prior to intravenous dosing with BENLYSTA, consider administering premedication for prophylaxis against infusion reactions and hypersensitivity reactions [see Warnings and Precautions (5.2), Adverse Reactions (6.1)].
2.2 Recommended Intravenous Dosage, and Preparation and Administration Instructions
BENLYSTA for intravenous use must be reconstituted and diluted prior to administration. Do not administer as an intravenous push or bolus.
Recommended Dosage and Administration
The recommended intravenous BENLYSTA dosage in patients 5 years of age and older with active SLE or lupus nephritis is 10 mg/kg at 2‑week intervals for the first 3 doses and at 4‑week intervals thereafter.
Reconstitute, dilute, and administer as an intravenous infusion over a period of 1 hour. Do not concomitantly infuse BENLYSTA in the same intravenous line with other agents. No physical or biochemical compatibility studies have been conducted to evaluate the coadministration of BENLYSTA with other agents.
The infusion rate may be slowed or interrupted if the patient develops an infusion reaction. The infusion must be discontinued immediately if the patient experiences a serious hypersensitivity reaction [see Contraindications (4), Warnings and Precautions (5.2)].
Preparation of the Intravenous Solution
BENLYSTA for intravenous use is provided as a lyophilized powder in a single‑dose vial and should be reconstituted and diluted by a healthcare professional using aseptic technique as follows. Use of a 21- to 25-gauge needle is recommended when piercing the vial stopper for reconstitution and dilution.
Reconstitution Instructions for Intravenous Use:
- 1.
- Remove the vial of BENLYSTA from the refrigerator and allow to stand for 10 to 15 minutes for the vial to reach room temperature.
- 2.
- Reconstitute the BENLYSTA powder with Sterile Water for Injection, USP, as follows. The reconstituted solution will contain a concentration of 80 mg/mL belimumab.
- •
- Reconstitute the 120-mg vial with 1.5 mL Sterile Water for Injection, USP.
- •
- Reconstitute the 400-mg vial with 4.8 mL Sterile Water for Injection, USP.
- 3.
- Direct the stream of sterile water towards the side of the vial to minimize foaming. Gently swirl the vial for 60 seconds. Allow the vial to sit at room temperature during reconstitution, gently swirling the vial for 60 seconds every 5 minutes until the powder is dissolved. Do not shake. Reconstitution is typically complete within 10 to 15 minutes after the sterile water has been added, but it may take up to 30 minutes. Protect the reconstituted solution from sunlight.
- 4.
- If a mechanical reconstitution device (swirler) is used to reconstitute BENLYSTA, do not exceed 500 rpm or swirl the vial for more than 30 minutes.
- 5.
- Once reconstitution is complete, the solution should be opalescent and colorless to pale yellow, and without particles. Small air bubbles, however, are expected and acceptable.
Dilution Instructions for Intravenous Use:
- 1.
- Dextrose intravenous solutions are incompatible with BENLYSTA. BENLYSTA should only be diluted in 0.9% Sodium Chloride Injection, USP (normal saline), 0.45% Sodium Chloride Injection, USP (half-normal saline), or Lactated Ringer’s Injection, USP to a volume of 250 mL for intravenous infusion. To prepare the intravenous infusion solution for patients whose body weight is less than or equal to 40 kg, a 100 mL bag or bottle of normal saline, half-normal saline, or Lactated Ringer’s Injection may be used such that the resulting belimumab concentration in the infusion bag does not exceed 4 mg/mL. From a 250‑mL (or 100‑mL) infusion bag or bottle of normal saline, half-normal saline, or Lactated Ringer’s Injection, withdraw and discard a volume equal to the volume of the reconstituted solution of BENLYSTA required for the patient’s dose. Then add the required volume of the reconstituted solution of BENLYSTA into the intravenous infusion solution in the infusion bag or bottle. Gently invert the bag or bottle to mix the intravenous infusion solution. Any unused solution in the vials must be discarded.
- 2.
- Visually inspect parenteral drug products for particulate matter and discoloration prior to administration, whenever solution and container permit. Discard the solution if any particulate matter or discoloration is observed.
- 3.
- If the reconstituted solution of BENLYSTA is not used immediately, store, protect from direct sunlight and refrigerate at 36°F to 46°F (2°C to 8°C). Store solutions of BENLYSTA diluted in normal saline, half‑normal saline, or Lactated Ringer’s Injection at 36°F to 46°F (2°C to 8°C) or room temperature. The total time from reconstitution of BENLYSTA to completion of infusion should not exceed 8 hours.
- 4.
- No incompatibilities between BENLYSTA and polyvinylchloride or polyolefin bags have been observed.
2.3 Recommended Subcutaneous Dosage, and Preparation and Administration Instructions
The recommended subcutaneous BENLYSTA dosage in patients 5 years of age and older with active SLE or lupus nephritis is provided in Table 1. Administer BENLYSTA subcutaneously in the abdomen or thigh. For patients less than 10 years of age, BENLYSTA must be administered by a healthcare professional or trained caregiver.
| a The prefilled syringe has not been studied in pediatric patients less than 18 years of age. | ||
| b The 400-mg dose requires administration of two 200-mg injections. | ||
|
Indication |
Adults (Autoinjector or Prefilled Syringe) |
Pediatric Patients 5 Years of Age and Older (Weight‑Based Dosing) (Autoinjector Only)a |
|
Active SLE |
200 mg once weekly |
|
|
Active Lupus Nephritis |
400 mgb once weekly for 4 doses, followed by 200 mg once weekly |
|
Administration Instructions for Subcutaneous Injection
- 1.
- Administer the first subcutaneous injection of BENLYSTA under the supervision of a healthcare provider. Provide patients or caregivers with proper training on subcutaneous administration and education about signs and symptoms of hypersensitivity reactions [see Warnings and Precautions (5.2)]. For adults and pediatric patients 10 years of age and older, subsequent subcutaneous BENLYSTA administrations may be performed by the patient or trained caregiver, if determined to be appropriate. For pediatric patients less than 10 years of age, subsequent subcutaneous BENLYSTA administrations must be performed by a healthcare provider or trained caregiver.
- 2.
- Instruct the patient or caregiver to follow the directions for administration provided in the Instructions for Use.
- 3.
- Instruct the patient or caregiver to remove the autoinjector or prefilled syringe from the refrigerator and allow it to sit at room temperature for 30 minutes prior to the subcutaneous injection. Do not warm BENLYSTA in any other way.
- 4.
- Prior to administration, instruct the patient or caregiver to visually inspect the window of the autoinjector or the prefilled syringe for particulate matter or discoloration. BENLYSTA should be clear to opalescent and colorless to pale yellow. Do not use BENLYSTA if the product exhibits discoloration or particulate matter. Instruct the patient or caregiver not to use the BENLYSTA autoinjector or prefilled syringe if dropped on a hard surface.
- 5.
- When injecting in the same body region, advise the patient or caregiver to use a different injection site for each injection; never give injections into areas where the skin is tender, bruised, red, or hard. When administering a 400‑mg dose, inject each 200‑mg injection at least 5 cm (approximately 2 inches) apart.
- 6.
- Instruct the patient or caregiver to administer BENLYSTA, preferably on the same day each week or the same day of alternate weeks, as appropriate.
- 7.
- If a dose is missed, instruct the patient or caregiver to administer a dose as soon as the patient remembers. Thereafter, the patient can resume dosing on their usual day of administration or start a new schedule from the day that the missed dose was administered.
2.4 Switching from Intravenous to Subcutaneous BENLYSTA Use
Active SLE
Administer the first subcutaneous BENLYSTA dose 1 to 4 weeks after the last intravenous dose. To switch:
- •
- Adults and pediatric patients greater than or equal to 40 kg, administer the first 200 mg subcutaneous dose 1 to 4 weeks after the last intravenous dose, then continue 200 mg subcutaneous dosing once weekly.
- •
- Pediatric patients 15 kg to less than 40 kg, administer the first 200 mg subcutaneous dose 1 to 4 weeks after the last intravenous dose, then continue 200 mg subcutaneous dosing every 2 weeks.
Active Lupus Nephritis
Patients may be switched from intravenous BENLYSTA treatment to subcutaneous BENLYSTA treatment after completing at least 2 intravenous doses. To switch:
- •
- Adults and pediatric patients greater than or equal to 40 kg, administer the first 200 mg subcutaneous dose 1 to 2 weeks after the last intravenous dose, then continue 200 mg subcutaneous dosing once weekly.
- •
- Pediatric patients 15 kg to less than 40 kg, administer the first 200 mg subcutaneous dose 1 to 2 weeks after the last intravenous dose, then continue 200 mg subcutaneous dosing every 2 weeks.
3. Dosage Forms and Strengths
Intravenous Infusion
For injection: 120 mg or 400 mg of belimumab as a lyophilized powder in single‑dose vials for reconstitution and dilution prior to intravenous infusion.
Subcutaneous Injection
Injection: 200 mg/mL of belimumab as a clear to opalescent and colorless to pale yellow solution in a single-dose prefilled autoinjector or a single-dose prefilled glass syringe.
4. Contraindications
BENLYSTA is contraindicated in patients who have had anaphylaxis with belimumab.
5. Warnings and Precautions
5.1 Serious Infections
Serious and sometimes fatal infections have been reported in patients receiving immunosuppressive agents, including BENLYSTA. Overall, the incidence of serious infections in controlled trials was similar in subjects receiving BENLYSTA compared with placebo, whereas fatal infections occurred more frequently in subjects receiving BENLYSTA [see Adverse Reactions (6.1)].
Consider the risk and benefit before initiating treatment with BENLYSTA in patients with severe or chronic infections. Consider interrupting therapy with BENLYSTA in patients who develop a new infection while receiving it and monitor these patients closely.
Progressive Multifocal Leukoencephalopathy (PML)
Cases of JC virus-associated PML resulting in neurological deficits, including fatal cases, have been reported in patients with SLE receiving immunosuppressants, including BENLYSTA. Risk factors for PML include treatment with immunosuppressant therapies and impairment of immune function. Consider the diagnosis of PML in any patient presenting with new-onset or deteriorating neurological signs and symptoms and consult with a neurologist or other appropriate specialist as clinically indicated. In patients with suspected PML, immunosuppressant therapy, including BENLYSTA, must be suspended until PML has been excluded. If PML is confirmed, immunosuppressant therapy, including BENLYSTA, must be discontinued.
5.2 Hypersensitivity Reactions, including Anaphylaxis
Acute hypersensitivity reactions, including anaphylaxis and death, and infusion-related reactions have been reported in association with BENLYSTA [see Adverse Reactions (6.1)]. These events generally occurred within hours of the infusion; however, they may occur later. Non-acute hypersensitivity reactions including rash, nausea, fatigue, myalgia, headache, and facial edema have been reported and typically occurred up to a week following the most recent infusion. Hypersensitivity, including serious reactions, has occurred in patients who have previously tolerated infusions of BENLYSTA. Limited data suggest that patients with a history of multiple drug allergies or significant hypersensitivity may be at increased risk.
Due to overlap in signs and symptoms, it was not possible to distinguish between hypersensitivity reactions and infusion-related reactions in all cases. In the controlled clinical trials of BENLYSTA administered intravenously in adults with SLE, some subjects (13%) received premedication, which may have mitigated or masked a hypersensitivity response or infusion-related reaction; however, there is insufficient evidence to determine whether premedication diminishes the frequency or severity of hypersensitivity reactions or infusion-related reaction.
BENLYSTA for intravenous use should be administered by healthcare providers prepared to manage anaphylaxis and infusion-related reactions. Healthcare providers should be aware of the risk of hypersensitivity reactions, which may present as infusion-related reactions. In the event of a serious reaction, discontinue BENLYSTA immediately and administer appropriate medical therapy. With intravenous administration, the infusion rate may be slowed or interrupted if the patient develops an infusion reaction. Monitor patients during infusion and for an appropriate period of time after intravenous administration of BENLYSTA. Consider administering premedication as prophylaxis prior to intravenous dosing [see Dosage and Administration (2.1, 2.2)].
Inform patients receiving BENLYSTA of the signs and symptoms of hypersensitivity reactions and instruct them to seek immediate medical care should a reaction occur.
5.3 Depression and Suicidality
In controlled clinical trials, depression and suicidality were reported in subjects receiving BENLYSTA [see Adverse Reactions (6.1)]. Assess the risk of depression and suicide considering the patient’s medical history and current psychiatric status before treatment with BENLYSTA and continue to monitor patients during treatment. Instruct patients receiving BENLYSTA (and caregivers, if applicable) to contact their healthcare provider if they experience new or worsening depression, suicidal thoughts or behavior, or other mood changes. Consider the risk and benefit of continued treatment with BENLYSTA for patients who develop such symptoms.
5.4 Malignancy
There is an increased risk of malignancies with the use of immunosuppressants. The impact of treatment with BENLYSTA on the development of malignancies is not known [see Adverse Reactions (6.1)].
Consider the individual benefit-risk in patients with known risk factors for the development or reoccurrence of malignancy prior to prescribing BENLYSTA. In patients who develop malignancies, consider the risk and benefit of continued treatment with BENLYSTA.
5.5 Immunization
Because of its mechanism of action, BENLYSTA may interfere with the response to immunizations. Live vaccines should not be given for 30 days before or concurrently with BENLYSTA as clinical safety has not been established. No data are available on the secondary transmission of infection from persons receiving live vaccines to patients receiving BENLYSTA or the effect of BENLYSTA on new immunizations.
5.6 Concomitant Use with Other Biologic Therapies
Available data do not support the safety and efficacy of concomitant use of BENLYSTA with rituximab in patients with SLE. An increased incidence of serious infections and post-injection systemic reactions in subjects receiving BENLYSTA concomitantly with rituximab compared to subjects receiving BENLYSTA alone has been observed [see Adverse Reactions (6.1)]. The safety and efficacy of BENLYSTA concomitantly with other biologic therapies, including B-cell-targeted therapies, have not been established. Caution should be exercised if BENLYSTA is administered in combination with other biologic therapies [see Warnings and Precautions (5)].
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described below and in the Warnings and Precautions section:
- •
- Serious Infections [see Warnings and Precautions (5.1)]
- •
- Hypersensitivity Reactions, including Anaphylaxis [see Warnings and Precautions (5.2)]
- •
- Depression and Suicidality [see Warnings and Precautions (5.3)]
- •
- Malignancy [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Intravenous Administration in Adult Subjects with Active SLE
The data described in Table 2 reflect exposure to BENLYSTA administered intravenously plus standard therapy compared with placebo plus standard therapy in 2,133 adult subjects with active SLE in 3 controlled trials (Trials 1, 2, and 3). Subjects received BENLYSTA plus standard therapy at doses of 1 mg/kg (n = 673), 4 mg/kg (n = 111; Trial 1 only), or 10 mg/kg (n = 674), or placebo plus standard therapy (n = 675) intravenously over a 1‑hour period on Days 0, 14, 28, and then every 28 days. In 2 of the trials (Trial 1 and Trial 3), treatment was given for 48 weeks, while in the other trial (Trial 2) treatment was given for 72 weeks [see Clinical Studies (14.2)]. Because there was no apparent dose‑related increase in the majority of adverse events observed with BENLYSTA, the safety data summarized below are presented for the 3 intravenous doses pooled, unless otherwise indicated; the adverse reaction table displays the results for the recommended intravenous dose of 10 mg/kg compared with placebo.
In Trials 1, 2, and 3, 93% of subjects treated with BENLYSTA plus standard therapy reported an adverse event compared with 92% treated with placebo plus standard therapy.
The most common serious adverse events were serious infections (6% and 5.2% in the groups receiving BENLYSTA and placebo plus standard therapy, respectively), some of which were fatal.
The most commonly reported adverse events, occurring in ≥5% of subjects in Trials 1, 2, and 3 were nausea, diarrhea, pyrexia, nasopharyngitis, bronchitis, insomnia, pain in extremity, depression, migraine, and pharyngitis.
The proportion of subjects who discontinued treatment due to any adverse reaction during Trials 1, 2, and 3 was 6.2% for subjects receiving BENLYSTA plus standard therapy and 7.1% for subjects receiving placebo plus standard therapy. The most common adverse reactions resulting in discontinuation of treatment (≥1% of subjects receiving BENLYSTA or placebo) were infusion reactions (1.6% BENLYSTA and 0.9% placebo), lupus nephritis (0.7% BENLYSTA and 1.2% placebo), and infections (0.7% BENLYSTA and 1% placebo).
Table 2 lists adverse reactions, regardless of causality, occurring in at least 3% of subjects with active SLE who received BENLYSTA 10 mg/kg plus standard therapy and at an incidence at least 1% greater than that observed with placebo plus standard therapy in 3 controlled trials (Trials 1, 2, and 3).
|
Adverse Reactions |
BENLYSTA 10 mg/kg + Standard Therapy (n = 674) % |
Placebo + Standard Therapy (n = 675) % |
|
Nausea |
15 |
12 |
|
Diarrhea |
12 |
9 |
|
Pyrexia |
10 |
8 |
|
Nasopharyngitis |
9 |
7 |
|
Bronchitis |
9 |
5 |
|
Insomnia |
7 |
5 |
|
Pain in extremity |
6 |
4 |
|
Depression |
5 |
4 |
|
Migraine |
5 |
4 |
|
Pharyngitis |
5 |
3 |
|
Cystitis |
4 |
3 |
|
Leukopenia |
4 |
2 |
|
Gastroenteritis viral |
3 |
1 |
Specific Adverse Reactions in Adult Subjects with Active SLE (Intravenous Administration)
Infections: In Trials 1, 2, and 3, the overall incidence of infections was 71% in subjects receiving BENLYSTA compared with 67% in subjects receiving placebo. The most frequent infections (>5% of subjects receiving BENLYSTA) were upper respiratory tract infection, urinary tract infection, nasopharyngitis, sinusitis, bronchitis, and influenza. Infections leading to discontinuation of treatment occurred in 0.7% of subjects receiving BENLYSTA and 1.0% of subjects receiving placebo.
Serious Infections: In Trials 1, 2, and 3, the incidence of serious infections was 6.0% in subjects receiving BENLYSTA and 5.2% in subjects receiving placebo. The most frequent serious infections included pneumonia, urinary tract infections, cellulitis, and bronchitis. Fatal infections occurred in 0.3% (4/1,458) of subjects receiving BENLYSTA and in 0.1% (1/675) of subjects receiving placebo.
In a randomized, double‑blind, placebo‑controlled, 52‑week, postmarketing safety trial of BENLYSTA administered intravenously in adults with active SLE (N = 4,003), the incidence of serious infections was 3.7% in subjects receiving BENLYSTA compared with 4.1% in subjects receiving placebo. Serious infections leading to discontinuation of treatment occurred in 1.0% of subjects receiving BENLYSTA and in 0.9% of subjects receiving placebo. Fatal infections occurred in 0.45% (9/2,002) of subjects receiving BENLYSTA and in 0.15% (3/2,001) of subjects receiving placebo, where the incidence of all‑cause mortality was 0.50% (10/2,002) in subjects receiving BENLYSTA and 0.40% (8/2,001) in subjects receiving placebo.
Hypersensitivity Reactions, including Anaphylaxis: In Trials 1, 2, and 3, hypersensitivity reactions (occurring on the same day of infusion) were reported in 13% (191/1,458) of subjects receiving BENLYSTA and 11% (76/675) of subjects receiving placebo. Anaphylaxis was observed in 0.6% (9/1,458) of subjects receiving BENLYSTA and 0.4% (3/675) of subjects receiving placebo. Manifestations included hypotension, angioedema, urticaria or other rash, pruritus, and dyspnea.
Infusion‑Related Reactions: In Trials 1, 2, and 3, adverse events associated with the infusion (occurring on the same day of the infusion) were reported in 17% (251/1,458) of subjects receiving BENLYSTA and 15% (99/675) of subjects receiving placebo. Serious infusion reactions (excluding hypersensitivity reactions) were reported in 0.5% of subjects receiving BENLYSTA and 0.4% of subjects receiving placebo and included bradycardia, myalgia, headache, rash, urticaria, and hypotension. The most common infusion reactions (≥3% of subjects receiving BENLYSTA) were headache, nausea, and skin reactions.
Depression and Suicidality: In Trials 1, 2, and 3, psychiatric events were reported more frequently with BENLYSTA (16%) than with placebo (12%), primarily related to depression‑related events (6.3% BENLYSTA; 4.7% placebo), insomnia (6% BENLYSTA; 5.3% placebo), and anxiety (3.9% BENLYSTA; 2.8% placebo). Serious psychiatric events were reported in 0.8% (12/1,458) of subjects receiving BENLYSTA and 0.4% (3/675) of subjects receiving placebo. Serious depression was reported in 0.4% (6/1,458) of subjects receiving BENLYSTA and 0.1% (1/675) of subjects receiving placebo. Two suicides (0.1%) were reported in subjects receiving BENLYSTA (one with 10 mg/kg and one with 1 mg/kg).
In a 52‑week postmarketing safety trial of BENLYSTA (N = 4,003), serious psychiatric events were reported in 1% (20/2,002) of subjects receiving BENLYSTA and 0.3% (6/2,001) of subjects receiving placebo. Serious depression was reported in 0.3% (7/2,002) of subjects receiving BENLYSTA and in <0.1% (1/2,001) receiving placebo. The overall incidence of serious suicidal ideation or behavior or self‑injury without suicidal intent was 0.7% (15/2,002) of subjects receiving BENLYSTA and 0.2% (5/2,001) of subjects receiving placebo. On the Columbia‑Suicide Severity Rating Scale (C‑SSRS), 2.4% (48/1,974) of subjects receiving BENLYSTA reported suicidal ideation or behavior compared with 2% (39/1,988) of subjects receiving placebo. No suicide was reported in either group.
The intravenous trials above did not exclude subjects with a history of psychiatric disorders.
Malignancy: In Trials 1, 2, and 3, malignancies (including non‑melanoma skin cancers) were reported in 0.4% of subjects receiving BENLYSTA and 0.4% of subjects receiving placebo. In the intravenous controlled clinical trials, malignancies, excluding non‑melanoma skin cancers, were observed in 0.2% (3/1,458) and 0.3% (2/675) of subjects receiving BENLYSTA and placebo, respectively.
Intravenous Administration in Black/African‑American Subjects with Active SLE
The safety of BENLYSTA 10 mg/kg administered intravenously in adults plus standard therapy (n = 331) compared with placebo plus standard therapy (n = 165) in Black subjects with active SLE (Trial 4) was consistent with the known safety profile of BENLYSTA administered intravenously plus standard therapy in the overall population [see Clinical Studies (14.2)].
Intravenous Administration in Adult Subjects with Active Lupus Nephritis
The safety of BENLYSTA 10 mg/kg administered intravenously plus standard therapy (n = 224) compared with placebo plus standard therapy (n = 224) was evaluated in adults with active lupus nephritis for up to 104 weeks (Trial 5) [see Clinical Studies (14.3)]. The adverse reactions observed were consistent with the known safety profile of BENLYSTA administered intravenously plus standard therapy in patients with active SLE. Cases of myelosuppression, including febrile neutropenia, leukopenia, and pancytopenia, were observed in subjects who received induction therapy with cyclophosphamide followed by maintenance therapy with azathioprine, or mycophenolate.
Specific Adverse Reactions in Adult Subjects with Active Lupus Nephritis (Intravenous Administration)
Infections: In Trial 5, the overall incidence of infections was 82% in subjects receiving BENLYSTA compared with 76% in subjects receiving placebo.
Serious Infections: In Trial 5, serious infections occurred in 14% of subjects receiving BENLYSTA and in 17% of subjects receiving placebo. Fatal infections occurred in 0.9% (2/224) of subjects receiving BENLYSTA and in 0.9% (2/224) of subjects receiving placebo.
Intravenous Administration in Pediatric Subjects 5 Years of Age and Older with Active SLE
The safety of BENLYSTA administered intravenously plus standard therapy (n = 53) compared with placebo plus standard therapy (n = 40) was evaluated in 93 pediatric subjects with active SLE (Trial 6). The adverse reactions observed were consistent with those observed in adults with SLE [see Clinical Studies (14.4)].
Subcutaneous Administration in Adult Subjects with Active SLE
The data described below reflect exposure to BENLYSTA administered subcutaneously plus standard therapy compared with placebo plus standard therapy in 836 adult subjects with active SLE in a controlled trial (Trial 7). In addition to standard therapy, subjects received BENLYSTA 200 mg (n = 556) or placebo (n = 280) (2:1 randomization) once weekly for up to 52 weeks [see Clinical Studies (14.5)].
In the trial, 81% of subjects treated with BENLYSTA plus standard therapy reported an adverse event compared with 84% treated with placebo plus standard therapy. The proportion of subjects who discontinued treatment due to any adverse reaction during the controlled clinical trial was 7.2% of subjects receiving BENLYSTA plus standard therapy and 8.9% of subjects receiving placebo plus standard therapy.
The safety profile observed for BENLYSTA administered subcutaneously plus standard therapy was consistent with the known safety profile of BENLYSTA administered intravenously plus standard therapy, with the exception of local injection site reactions.
Infections: In Trial 7, the overall incidence of infections was 55% in subjects receiving BENLYSTA compared with 57% in subjects receiving placebo. The most commonly reported infections with BENLYSTA administered subcutaneously were similar to those reported with BENLYSTA administered intravenously.
Serious Infections: In Trial 7, the incidence of serious infections was 4.1% in subjects receiving BENLYSTA and 5.4% in subjects receiving placebo. Fatal infections occurred in 0.5% (3/556) of subjects receiving BENLYSTA and in none of the subjects receiving placebo (0/280).
Depression and Suicidality: In Trial 7, which excluded subjects with a history of psychiatric disorders, psychiatric events were reported in 6% of subjects receiving BENLYSTA and 11% of subjects receiving placebo. Depression‑related events were reported in 2.7% (15/556) of subjects receiving BENLYSTA and 3.6% (10/280) of subjects receiving placebo. Serious psychiatric events were reported in 0.2% (1/556) of subjects receiving BENLYSTA and in no subjects receiving placebo. There were no serious depression‑related events or suicides reported in either group. On the C‑SSRS, 1.3% (7/554) of subjects receiving BENLYSTA reported suicidal ideation or behavior compared with 0.7% (2/277) of subjects receiving placebo.
Malignancy: In Trial 7, the reports of malignancies were similar to those reported with BENLYSTA administered intravenously.
Injection Site Reactions: In Trial 7, the frequency of injection site reactions was 6.1% (34/556) for subjects receiving BENLYSTA plus standard therapy and 2.5% (7/280) for subjects receiving placebo plus standard therapy. These injection site reactions (most commonly pain, erythema, hematoma, pruritus, and induration) were mild to moderate in severity. The majority (94%) did not necessitate discontinuation of treatment.
Concomitant Use of Subcutaneous BENLYSTA and Intravenous Rituximab in Adult Subjects with Active SLE
BENLYSTA administered subcutaneously in combination with intravenous rituximab was studied in a Phase 3, randomized, double‑blind, placebo‑controlled, 104‑week trial in adult subjects with active SLE. Subjects were randomized to 1 of the 3 treatment arms: BENLYSTA with a single cycle of rituximab (n = 144); BENLYSTA with placebo (n = 72); BENLYSTA plus standard therapy (n = 76). In general, adverse reactions were consistent with the known safety profile of BENLYSTA and rituximab. When compared with BENLYSTA and placebo or BENLYSTA plus standard therapy, BENLYSTA in combination with rituximab was associated with higher frequency of serious adverse events (13.9%, 19.7%, 22.2%, respectively), serious infections (2.8%, 5.3%, 9.0%, respectively), and post‑injection systemic reactions (9.7%, 5.3%, 13.2%, respectively).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of BENLYSTA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- •
- Fatal anaphylaxis [see Warnings and Precautions (5.2)].
Related/similar drugs
7. Drug Interactions
Formal drug interaction studies have not been performed with BENLYSTA. In clinical trials, BENLYSTA was administered concomitantly with other drugs, including corticosteroids, antimalarials, immunomodulatory and immunosuppressive agents (including azathioprine, cyclophosphamide, methotrexate, and mycophenolate), angiotensin pathway antihypertensives, HMG‑CoA reductase inhibitors (statins), and/or non-steroidal anti-inflammatory drugs (NSAIDs) without evidence of a clinically meaningful effect of these concomitant medications on belimumab pharmacokinetics. The effect of belimumab on the pharmacokinetics of other drugs has not been evaluated [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that evaluates pregnancy outcomes in women with lupus exposed to BENLYSTA during pregnancy. Healthcare professionals are encouraged to refer patients and pregnant women are encouraged to enroll themselves by calling 1-877-311-8972 or visiting https://mothertobaby.org/ongoing-study/benlysta-belimumab/.
Risk Summary
Available data on use of BENLYSTA in pregnant women, from observational studies, published case reports, and postmarketing surveillance, are insufficient to determine whether there is a drug-associated risk for major birth defects or miscarriage. There are risks to the mother and fetus associated with SLE (see Clinical Considerations). Monoclonal antibodies, such as belimumab, are actively transported across the placenta during the third trimester of pregnancy and may affect immune response in the in utero-exposed infant (see Clinical Considerations). In an animal combined embryo-fetal and pre- and post-natal development study with monkeys that received belimumab by intravenous administration, there was no evidence of fetal harm with exposures approximately 9 times (based on intravenous administration) and 20 times (based on subcutaneous administration) the exposure at the maximum recommended human dose (MRHD). Belimumab-related findings in monkey fetuses and/or infants included reductions of B-cell counts, reductions in the density of lymphoid tissue B-lymphocytes in the spleen and lymph nodes, and altered IgG and IgM titers. The no-adverse-effect-level (NOAEL) was not identified for these findings; however, they were reversible within 3 to 12 months after the drug was discontinued (see Data). Based on animal data and the mechanism of action of belimumab, the immune system in infants of treated mothers may be adversely affected. It is unknown, based on available data, whether immune effects, if identified, are reversible [see Clinical Pharmacology (12.1)].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Disease-Associated Maternal and/or Embryo/Fetal Risk: Pregnant women with SLE are at increased risk of adverse pregnancy outcomes, including worsening of the underlying disease, premature birth, miscarriage, and intrauterine growth restriction. Maternal lupus nephritis increases the risk of hypertension and preeclampsia/eclampsia. Passage of maternal autoantibodies across the placenta may result in adverse neonatal outcomes, including neonatal lupus and congenital heart block.
Fetal/Neonatal Adverse Reactions: Monoclonal antibodies are increasingly transported across the placenta as pregnancy progresses, with the largest amount transferred during the third trimester. Risks and benefits should be considered prior to administering live or live-attenuated vaccines to infants exposed to BENLYSTA in utero. Monitor an infant of a treated mother for B-cell reduction and other immune dysfunction [see Warnings and Precautions (5.5)].
Animal Data: In a combined embryo-fetal and pre- and post-natal development study, pregnant cynomolgus monkeys received belimumab at intravenous doses of 0, 5, or 150 mg/kg every 2 weeks from confirmation of pregnancy at Gestation Days (GD) 20 to 22, throughout the period of organogenesis (up to approximately GD 50), and continuing to either the day of scheduled cesarean section (GD 150 [late third trimester]) or the day of parturition. There was no evidence of maternal toxicity, effects on embryofetal and infant survival, or structural abnormalities at exposure approximately 9 times the MRHD of 10 mg/kg intravenously or 20 times the MRHD of 200 mg subcutaneously (on an area under the curve [AUC] basis with maternal animal intravenous doses up to 150 mg/kg). Belimumab-related findings in mothers included reductions of immature and mature B-cell counts and in fetuses and/or infants included reductions of immature and mature B-cell counts, reductions in the density of lymphoid tissue B-lymphocytes in the spleen and lymph nodes, reduced spleen weights, increased IgG titers, and reduced IgM titers. B-cell counts in infant monkeys exposed to belimumab in utero recovered by 3 months of age and in mothers after 1 year. Immunoglobulin G (IgG) and IgM levels in infant monkeys recovered by 6 months of age and the reductions in B-lymphocytes in the lymph nodes and spleen were reversed by 1 year of age. Belimumab crossed the placenta, as it was detected in fetal cord blood and amniotic fluid on GD 150.
8.2 Lactation
Risk Summary
No information is available on the presence of belimumab in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. Belimumab was detected in the milk of cynomolgus monkeys; however, due to species-specific differences in lactation physiology, animal data may not predict drug levels in human milk. Maternal IgG is known to be present in human milk. If belimumab is transferred into human milk, the effects of local exposure in the gastrointestinal tract and potential limited systemic exposure in the infant to belimumab are unknown. The lack of clinical data during lactation precludes clear determination of the risk of BENLYSTA to an infant during lactation; therefore, the developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for BENLYSTA, and any potential adverse effects on the breastfed child from BENLYSTA or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Following an assessment of benefit versus risk, if prevention of pregnancy is warranted, females of reproductive potential should use effective contraception during treatment and for at least 4 months after the final treatment.
8.4 Pediatric Use
The safety and effectiveness of BENLYSTA have been established for the treatment of active SLE and active lupus nephritis in pediatric patients 5 years of age and older who are receiving standard therapy.
The safety and effectiveness of BENLYSTA have not been established in pediatric patients less than 5 years of age.
Intravenous Use
Use of BENLYSTA intravenously in pediatric patients 5 years of age and older with active SLE is supported by evidence from pharmacokinetic (PK), safety, and efficacy results from a pediatric trial (Trial 6), as well as PK exposure and extrapolation of the established efficacy of BENLYSTA plus standard therapy from the Phase 3 intravenous studies in adults with active SLE (Trials 2 and 3). In Trial 6, the proportion of pediatric subjects achieving an SRI‑4 response was higher in subjects receiving BENLYSTA plus standard therapy compared with placebo plus standard therapy. Pediatric subjects receiving BENLYSTA plus standard therapy also had a lower risk of experiencing a severe flare compared with placebo plus standard therapy [see Clinical Studies (14.4)]. Pharmacokinetics were evaluated in a total of 53 pediatric subjects (Trial 6) and were consistent with the adult population with active SLE [see Clinical Pharmacology (12.3)].
Use of BENLYSTA intravenously in pediatric patients 5 years of age and older with active lupus nephritis is based on the extrapolation of efficacy from the intravenous trial (Trial 5) in adults (n = 224) with active lupus nephritis, and supported by pharmacokinetic data from intravenous studies in adults (n = 224) with active lupus nephritis and from pediatric subjects (n = 53) with active SLE (Trial 6). Estimated belimumab exposures for pediatric patients were comparable to adults with active lupus nephritis [see Clinical Pharmacology (12.3)].
Subcutaneous Use
Use of BENLYSTA, administered subcutaneously in pediatric patients 5 years of age and older who weigh at least 15 kg with active SLE, is supported by evidence from an open‑label pharmacokinetic trial (subcutaneous administration of BENLYSTA in pediatric subjects with active SLE) and Trial 6 (a pharmacokinetic, efficacy, and safety trial of intravenous dosing in pediatric subjects with active SLE). The pharmacokinetics of belimumab, following subcutaneous administration in pediatric patients, are estimated to be comparable to adults who receive BENLYSTA subcutaneously and pediatric patients who receive BENLYSTA intravenously [see Clinical Pharmacology (12.3)].
Use of BENLYSTA, administered subcutaneously in pediatric patients 5 years of age and older who weigh at least 15 kg with active lupus nephritis, is based on the extrapolation of efficacy from the intravenous trial in adults (n = 224) with active lupus nephritis (Trial 5), and is supported by pharmacokinetic data derived from the same adult trial (Trial 5), a pharmacokinetic, efficacy, and safety intravenous trial in pediatric subjects with active SLE (n = 53) (Trial 6), and an open‑label subcutaneous trial in pediatric subjects with active SLE (n = 25). Belimumab exposures for pediatric patients with active lupus nephritis following subcutaneous administration are estimated to be comparable to adults with active lupus nephritis who receive BENLYSTA intravenously [see Clinical Pharmacology (12.3)].
8.5 Geriatric Use
Clinical studies of BENLYSTA did not include sufficient numbers of subjects, 65 years of age or older, to determine whether they respond differently from younger subjects. Use with caution in geriatric patients.
8.6 Renal Impairment
The safety and efficacy of BENLYSTA were evaluated in studies that included subjects with SLE who had mild (creatinine clearance [CrCl] ≥60 and <90 mL/min), moderate (CrCl ≥30 and <60 mL/min), or severe (CrCl ≥15 and <30 mL/min) renal impairment. No dosage adjustment is recommended in patients with renal impairment.
8.7 Hepatic Impairment
No formal trials were conducted to examine the effects of hepatic impairment on the pharmacokinetics of belimumab. No dosage adjustment is recommended in patients with hepatic impairment.
8.8 Racial Groups
In Trial 2 and Trial 3 (intravenous dosing), SLE Responder Index‑4 (SRI‑4) response rates were lower for Black subjects receiving BENLYSTA plus standard therapy relative to Black subjects receiving placebo plus standard therapy [see Clinical Studies (14.2)].
In Trial 4 (intravenous dosing), a 2:1 randomized, placebo‑controlled trial in Black subjects, SLE Responder Index (SRI‑S2K) response rates were higher for Black subjects receiving BENLYSTA plus standard therapy (49%) relative to Black subjects receiving placebo plus standard therapy (42%). However, the treatment difference was not statistically significant [see Clinical Studies (14.2)].
In Trial 7 (subcutaneous dosing), SRI‑4 response was 45% (26/58) in Black subjects receiving BENLYSTA plus standard therapy compared with 39% (13/33) in Black subjects receiving placebo plus standard therapy [see Clinical Studies (14.5)].
The safety profile of BENLYSTA in Black subjects was consistent with the known safety profile of BENLYSTA administered in the overall population [see Adverse Reactions (6.1)].
10. Overdosage
There is limited experience with overdosage of belimumab.
Two doses of up to 20 mg/kg have been given intravenously to humans with no increase in incidence or severity of adverse reactions compared with doses of 1, 4, or 10 mg/kg.
11. Benlysta Description
Belimumab is a human IgG1λ monoclonal antibody specific for soluble human B lymphocyte stimulator protein (BLyS, also referred to as BAFF and TNFSF13B). Belimumab has a molecular weight of approximately 147 kDa. Belimumab is produced by recombinant DNA technology in a murine cell (NS0) expression system.
Intravenous Infusion
BENLYSTA (belimumab) for injection is a sterile, white to off-white, preservative‑free, lyophilized powder in a single-dose vial for reconstitution and dilution prior to intravenous infusion. BENLYSTA for injection is supplied as 120 mg per vial and 400 mg per vial and requires reconstitution with Sterile Water for Injection, USP (1.5 mL and 4.8 mL, respectively) to obtain a concentration of 80 mg/mL [see Dosage and Administration (2.2)]. After reconstitution, each vial allows for withdrawal of 1.5 mL (120 mg) or 5 mL (400 mg). Each mL delivers 80 mg belimumab, citric acid (0.16 mg), polysorbate 80 (0.4 mg), sodium citrate (2.7 mg), and sucrose (80 mg), with a pH of 6.5.
The vial stoppers are not made with natural rubber latex.
Subcutaneous Injection
BENLYSTA (belimumab) injection is a sterile, preservative-free, clear to opalescent, and colorless to pale yellow solution for subcutaneous use. It is supplied in a 1-mL single-dose prefilled autoinjector with a fixed 27-gauge, half-inch needle or in a 1-mL single-dose prefilled syringe with a fixed 27-gauge, half-inch needle with a needle guard. Each 1 mL delivers 200 mg belimumab, L-arginine hydrochloride (5.3 mg), L-histidine (0.65 mg), L-histidine monohydrochloride (1.2 mg), polysorbate 80 (0.1 mg), and sodium chloride (6.7 mg), with a pH of 6.0.
The autoinjectors and prefilled syringes are not made with natural rubber latex.
12. Benlysta - Clinical Pharmacology
12.1 Mechanism of Action
BENLYSTA is a BLyS-specific inhibitor that blocks the binding of soluble BLyS, a B-cell survival factor, to its receptors on B cells. BENLYSTA does not bind B cells directly, but by binding BLyS, BENLYSTA inhibits the survival of B cells, including autoreactive B cells, and reduces the differentiation of B cells into immunoglobulin-producing plasma cells.
12.2 Pharmacodynamics
Treatment with BENLYSTA in adult subjects significantly reduced circulating CD19+, CD20+, naïve, and activated B cells, and the SLE B‑cell subset at Week 52. Reductions in naïve and the SLE B‑cell subset were observed as early as Week 8 and sustained to Week 52. Memory cells increased initially and slowly declined toward baseline levels by Week 52.
Treatment with BENLYSTA in adult subjects led to reductions in IgG and anti‑double‑stranded DNA antibodies (anti‑dsDNA) which were observed as early as Week 8 and sustained through Week 52. In subjects with low complement levels at baseline, treatment led to increases in complement C3 and C4 as early as Week 12 and were sustained through Week 52.
The pharmacodynamic response observed in Black subjects (Trial 4) was consistent with the previous studies.
In subjects with active lupus nephritis (Trial 5), following treatment with BENLYSTA, there was a decrease in serum IgG as early as Week 4, and subsequently there was an increase in serum IgG levels which was associated with decreased proteinuria. Reductions in autoantibodies, increases in complement, and reductions in circulating total B cells and B‑cell subsets observed were consistent with the SLE studies.
In Trial 6 (pediatric dosing), the pharmacodynamic response was consistent with the adult data.
The clinical relevance of above mentioned pharmacodynamic biomarkers has not been established.
12.3 Pharmacokinetics
Intravenous Infusion in Adults
Systemic Lupus Erythematosus: The pharmacokinetic parameters displayed in Table 3 are based on population parameter estimates from 563 adult subjects with active SLE who received BENLYSTA 10 mg/kg.
| a Intravenous infusions were administered at 2‑week intervals for the first 3 doses and at 4‑week intervals thereafter. | |
|
Pharmacokinetic Parameter |
Population Estimates (n = 563) |
|
Peak concentration at steady state (Cmax,ss, mcg/mL) |
313 |
|
Area under the curve (AUC0-∞, day•mcg/mL) |
3,083 |
|
Distribution half-life (t½, days) |
1.8 |
|
Terminal half-life (t½, days) |
19.4 |
|
Systemic clearance (CL, mL/day) |
215 |
|
Volume of distribution at steady state (Vss, L) |
5 |
Lupus Nephritis: A population pharmacokinetic analysis was conducted in 224 adult subjects with active lupus nephritis who received belimumab 10 mg/kg intravenously (Days 0, 14, 28, and then every 28 days up to 104 weeks) plus standard therapy in Trial 5 [see Clinical Studies (14.3)]. In these subjects, due to additional clearance associated with proteinuria, belimumab exposure was initially lower than observed in SLE studies and lower belimumab exposure was observed in subjects with higher proteinuria. When the proteinuria was decreased to approximately ≤1 g/g after treatment, belimumab clearance and exposure were similar to that observed in subjects with active SLE who received belimumab 10 mg/kg intravenously. The available data do not support a dose adjustment in patients with high proteinuria.
Subcutaneous Injection in Adults
Systemic Lupus Erythematosus: The pharmacokinetic parameters displayed in Table 4 are based on population parameter estimates from 661 adult subjects with active SLE after subcutaneous administration of belimumab 200 mg once weekly. The time to reach maximum serum concentration (Cmax) was 2.6 days (Tmax) after administration at steady state. The bioavailability of belimumab was approximately 74%. With weekly subcutaneous administration there were minor fluctuations around the steady‑state average concentration (Cavg,ss 104 mcg/mL), with the steady‑state trough concentration (Cmin,ss) (97 mcg/mL) being only slightly below Cavg,ss.
|
Pharmacokinetic Parameter |
Population Estimates (n = 661) |
|
Peak concentration at steady state (Cmax,ss, mcg/mL) |
108 |
|
Area under the curve (AUC0-∞, day•mcg/mL) |
726 |
|
Distribution half-life (t½, days) |
1.1 |
|
Terminal half-life (t½, days) |
18.3 |
|
Systemic clearance (CL, mL/day) |
204 |
|
Volume of distribution at steady state (Vss, L) |
5 |
Lupus Nephritis: Based on population pharmacokinetic modeling and simulation of the subcutaneous 400-mg weekly loading dose, the average belimumab concentration during the first 12 weeks was predicted to be 78 mcg/mL, which is similar to the estimated concentration of 89 mcg/mL for intravenous administration. The loading dose of 400 mg weekly provides steady-state concentrations from Week 2 of dosing. The steady-state average concentrations of subcutaneous administration of belimumab 200 mg once weekly in adults with active lupus nephritis are predicted to be similar to those observed in adults with active lupus nephritis receiving belimumab 10 mg/kg intravenously every 4 weeks.
Specific Populations
The following information is based on the population pharmacokinetic analyses of intravenous administration and subcutaneous administration of BENLYSTA.
Age: Age did not significantly influence the pharmacokinetics of belimumab, where the majority of subjects were between 18 and 45 years of age (70% with intravenous dosing; 74% with subcutaneous dosing).
Geriatric Patients: Limited pharmacokinetic data are available for elderly patients as less than 2% of the subjects included in the pharmacokinetic analysis were 65 years of age or older [see Use in Specific Populations (8.5)].
Pediatric Patients:
- •
- Intravenous Administration in Pediatric Patients with Active SLE: The pharmacokinetic parameters of belimumab after intravenous administration in pediatric subjects with active SLE are based on individual parameter estimates from a population pharmacokinetic analysis of 53 pediatric subjects with active SLE (Trial 6). Following intravenous administration of 10 mg/kg on Days 0, 14, and 28, and at 4‑week intervals thereafter, belimumab exposures were similar between pediatric and adult subjects with active SLE. Steady‑state geometric mean Cmax, Cmin, Cavg, and AUC values were 305, 42, 92 mcg/mL, and 2,569 day•mcg/mL in the 5‑year to 11‑year‑old group, and 317, 52, 112 mcg/mL and 3,126 day•mcg/mL in the 12‑year to 17‑year‑old group [see Use in Specific Populations (8.4)].
- •
- Subcutaneous Administration in Pediatric Patients with Active SLE: The pharmacokinetic parameters of belimumab, following subcutaneous administration in pediatric subjects with active SLE, are based on a population pharmacokinetic analysis derived from 25 pediatric subjects with active SLE who received BENLYSTA subcutaneously and Trial 6 (a Phase 2 trial in pediatric subjects with active SLE receiving BENLYSTA intravenously). Following subcutaneous administration of 200 mg of BENLYSTA in pediatric subjects 5 years to less than 18 years of age, either weekly (subjects weighing greater than or equal to 40 kg) or every 2 weeks (subjects weighing 15 to less than 40 kg), the steady state average belimumab concentration is estimated to be similar to that of adult subjects with active SLE following subcutaneous administration of BENLYSTA 200 mg weekly and similar to that of pediatric subjects with active SLE following intravenous administration of 10 mg/kg BENLYSTA on Days 0, 14, and 28, and at 4‑week intervals thereafter. Simulated steady‑state geometric mean Cmax and AUC are estimated to be 110 mcg/mL and 1,328 day•mcg/mL for pediatric subjects (weighing 15 to less than 40 kg) receiving BENLYSTA 200 mg every 2 weeks, and 134 mcg/mL and 899 day•mcg/mL for pediatric subjects (weighing greater than or equal to 40 kg) receiving BENLYSTA 200 mg once weekly [see Use in Specific Populations (8.4)].
- •
- Intravenous Administration in Pediatric Patients with Active Lupus Nephritis: The pharmacokinetics of belimumab, following intravenous administration in pediatric patients with active lupus nephritis, were estimated based on a population pharmacokinetic model developed from intravenous pharmacokinetics data from 224 adults with active lupus nephritis in Trial 5 and validated using intravenous pharmacokinetics data from 53 pediatric subjects with active SLE in Trial 6. Based on modeling and simulation results, with intravenous administration of 10 mg/kg on Days 0, 14, and 28 and at 4‑week intervals thereafter, the simulated belimumab exposures for both the 5‑year to 11‑year‑old group and the 12‑year to 17‑year‑old group of pediatric subjects were estimated to be comparable to adults with active lupus nephritis [see Use in Specific Populations (8.4)].
- •
- Subcutaneous Administration in Pediatric Patients with Active Lupus Nephritis: The pharmacokinetics of belimumab, following subcutaneous administration in pediatric patients with active lupus nephritis, were extrapolated from previously developed pediatric and adult pharmacokinetic models (Trials 5 and 6) and validated using data from pediatric subjects with active SLE who received BENLYSTA intravenously (Trial 6) and subcutaneously. Based on the modeling and simulation results, following subcutaneous administration of BENLYSTA in pediatric subjects with active lupus nephritis aged 5 years to less than 18 years, receiving either 400 mg once weekly for 4 doses, then 200 mg every week thereafter (subjects weighing greater than or equal to 40 kg) or 200 mg once weekly for 4 doses then 200 mg once every 2 weeks (subjects weighing 15 to less than 40 kg), the simulated belimumab exposures were estimated to be comparable to that of adults with active lupus nephritis [see Use in Specific Populations (8.4)].
Male and Female Patients: Gender did not significantly influence belimumab pharmacokinetics in the largely female trial population (94% with intravenous dosing; 85% with subcutaneous dosing).
Racial Groups: Race did not significantly influence belimumab pharmacokinetics. The racial distribution with intravenous administration was 53% White, 16% Asian, 16% Alaska native/American Indian, and 14% Black in Trials 1, 2, and 3. Trial 4 enrolled only Black subjects. The racial distribution with subcutaneous administration (Trial 7) was 61% White, 20% Asian, 11% Black, and 6% Alaska native/American Indian.
Weight: Body weight and body mass index (BMI) had no clinically relevant effect on the pharmacokinetics of belimumab administered subcutaneously in adults. No dose adjustment is recommended in adults based on weight or BMI for subcutaneous administration.
The effects of body weight on belimumab exposure, after subcutaneous administration in pediatric patients, have been determined using a population pharmacokinetic model. Pediatric patients with lower body weight have lower belimumab clearance and volume of distribution. To ensure belimumab exposures remain within acceptable limits and are consistent across the pediatric weight range, patients with lower body weight are given a lower dose of BENLYSTA or administered BENLYSTA less frequently [see Dosage and Administration (2.3)].
Patients with Renal Impairment: No formal trials were conducted to examine the effects of renal impairment on the pharmacokinetics of belimumab. BENLYSTA was studied in a limited number of adult subjects with SLE who had mild (CrCl ≥60 and <90 mL/min), moderate (CrCl ≥30 and <60 mL/min), or severe (CrCl ≥15 and <30 mL/min) renal impairment: 770 subjects with mild renal impairment, 261 subjects with moderate renal impairment, and 14 subjects with severe renal impairment received belimumab intravenously; 121 subjects with mild renal impairment and 30 subjects with moderate renal impairment received belimumab subcutaneously [see Use in Specific Populations (8.6)].
Patients with Hepatic Impairment: No formal trials were conducted to examine the effects of hepatic impairment on the pharmacokinetics of belimumab. Baseline alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels did not significantly influence belimumab pharmacokinetics [see Use in Specific Populations (8.7)].
Drug Interaction Studies
No formal drug interaction studies have been conducted with BENLYSTA. Concomitant use of mycophenolate, cyclophosphamide, azathioprine, methotrexate, antimalarials, NSAIDs, aspirin, and/or HMG‑CoA reductase inhibitors did not significantly influence belimumab pharmacokinetics. Coadministration of steroids and angiotensin‑converting enzyme (ACE) inhibitors resulted in an increase of systemic clearance of belimumab that was not clinically significant because the magnitude was well within the range of normal variability of clearance. The effect of belimumab on the pharmacokinetics of other drugs has not been evaluated.
12.6 Immunogenicity
The observed incidence of anti‑drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti‑drug antibodies in other studies, including those of BENLYSTA or of other belimumab products.
In Trials 2 and 3 (intravenous dosing in adults with active SLE), anti‑belimumab antibodies were assessed during the respective 52‑week and 76‑week, placebo‑controlled periods and detected in 4 of 563 (0.7%) subjects receiving BENLYSTA 10 mg/kg and in 27 of 559 (4.8%) subjects receiving BENLYSTA 1 mg/kg. The reported frequency for the group receiving 10 mg/kg may underestimate the actual frequency due to lower assay sensitivity in the presence of high drug concentrations. Neutralizing antibodies were detected in 3 subjects receiving BENLYSTA 1 mg/kg. Three subjects with anti‑belimumab antibodies experienced mild infusion reactions of nausea, erythematous rash, pruritus, eyelid edema, headache, and dyspnea; none of the reactions were life‑threatening. In Trial 4 (intravenous dosing in adult Black subjects), anti‑belimumab antibodies were detected in 2 of 321 (0.6%) subjects receiving BENLYSTA 10 mg/kg during the 52‑week, placebo‑controlled period. In Trial 5 (intravenous dosing in adults with active lupus nephritis), there was no formation of anti‑belimumab antibodies in 224 subjects receiving BENLYSTA 10 mg/kg plus standard therapy during the 104‑week, placebo‑controlled period. In Trial 6 (intravenous dosing in pediatric subjects with active SLE), there was no formation of anti‑belimumab antibodies in 53 subjects receiving BENLYSTA 10 mg/kg plus standard therapy during the 52‑week, placebo‑controlled period. In Trial 7 (subcutaneous dosing in adults with active SLE), there was no formation of anti‑belimumab antibodies in 556 subjects receiving BENLYSTA 200 mg during the 52‑week, placebo‑controlled period. In a 52‑week, open‑label, pediatric pharmacokinetic trial (subcutaneous dosing in pediatric subjects with active SLE), none of the 25 subjects developed anti‑belimumab antibodies.
The clinical relevance of the presence of anti‑belimumab antibodies is not known.
14. Clinical Studies
14.1 Overview of Clinical Trials
The safety and effectiveness of BENLYSTA administered:
- •
- Intravenously plus standard therapy were evaluated in 4 randomized, double‑blind, placebo‑controlled trials with 2,581 adult subjects with active SLE (Trial 1, NCT00071487, Trial 2, NCT00410384, Trial 3, NCT00424476, and Trial 4 NCT01632241) [see Clinical Studies (14.2)], and one randomized, double‑blind, placebo‑controlled trial with 93 pediatric subjects 5 years of age and older with active SLE (Trial 6, NCT01649765) [see Clinical Studies (14.4)], according to the American College of Rheumatology criteria. In these trials, subjects with severe active lupus nephritis and severe active CNS lupus were excluded. Subjects were on a stable standard therapy SLE treatment regimen comprising any of the following (alone or in combination): corticosteroids, antimalarials, NSAIDs, and immunosuppressives. Use of other biologics and intravenous cyclophosphamide was not permitted.
- •
- Intravenously plus standard therapy were evaluated in a randomized, double‑blind, placebo‑controlled trial in 448 adult subjects with active lupus nephritis (Trial 5; NCT01639339) [see Clinical Studies (14.3)].
- •
- Subcutaneously plus standard therapy in adults were evaluated in a randomized, double‑blind, placebo‑controlled trial in 836 adult subjects with active SLE (Trial 7; NCT01484496) [see Clinical Studies (14.5)].
14.2 Intravenous Administration in Adults with Active SLE
Trial 1: Active SLE – BENLYSTA 1 mg/kg, 4 mg/kg, 10 mg/kg - Intravenous
Trial 1 enrolled 449 adult subjects and evaluated doses of 1, 4, and 10 mg/kg BENLYSTA plus standard therapy compared with placebo plus standard therapy over 52 weeks in subjects with SLE. Subjects had to have a Safety of Estrogens in Lupus Erythematosus National Assessment‑Systemic Lupus Erythematosus Disease Activity Index (SELENA‑SLEDAI) score of ≥4 at baseline and a history of autoantibodies (anti‑nuclear antibody [ANA] and/or anti–double‑stranded DNA [anti‑dsDNA]), but 28% of the population was autoantibody negative at baseline. The co-primary endpoints were percent change in SELENA-SLEDAI score at Week 24 and time to first flare over 52 weeks. No significant differences between any of the groups receiving BENLYSTA and the group receiving placebo were observed. Exploratory analysis of this trial identified a subgroup of subjects (72%) who were autoantibody positive in whom BENLYSTA appeared to offer benefit. The results of this trial informed the design of Trials 2 and 3 and led to the selection of a target population limited to autoantibody‑positive SLE patients.
Trials 2, 3, and 4: Active SLE – BENLYSTA 1 mg/kg and 10 mg/kg - Intravenous
Trials 2 and 3 were randomized, double‑blind, placebo‑controlled trials in adult subjects with SLE that were similar in design except duration; Trial 2 (N = 819) was 76 weeks’ duration and Trial 3 (N = 865) was 52 weeks’ duration. Subjects had active SLE disease with a SELENA‑SLEDAI score ≥6 and positive autoantibody test results at screening. Subjects were excluded from the trial if they had ever received treatment with a B‑cell–targeted agent or if they were currently receiving other biologic agents. Intravenous cyclophosphamide was not permitted within the previous 6 months or during the trial. Trial 2 was conducted primarily in North America and Europe. Trial 3 was conducted in South America, Eastern Europe, Asia, and Australia.
Baseline concomitant medications included corticosteroids (Trial 2: 76%, Trial 3: 96%), immunosuppressives (Trial 2: 56%, Trial 3: 42%; including azathioprine, methotrexate, and mycophenolate), and antimalarials (Trial 2: 63%, Trial 3: 67%). Most subjects (>70%) were receiving 2 or more classes of SLE medications.
In Trial 2 and Trial 3, more than 50% of subjects had 3 or more active organ systems involved at baseline. The most common active organ systems at baseline based on SELENA‑SLEDAI were mucocutaneous (82% in both trials), immune (Trial 2: 74%, Trial 3: 85%), and musculoskeletal (Trial 2: 73%, Trial 3: 59%). Less than 16% of subjects had some degree of renal activity and less than 7% of subjects had activity in the vascular, cardio‑respiratory, or CNS systems.
At screening, subjects were stratified by disease severity based on their SELENA‑SLEDAI score (≤9 vs. ≥10), proteinuria level (<2 g/24 h vs. ≥2 g/24 h), and race (African or Indigenous‑American descent vs. other), and then randomly assigned to receive BENLYSTA 1 mg/kg, BENLYSTA 10 mg/kg, or placebo in addition to standard therapy. The subjects were administered trial medication intravenously over a 1‑hour period on Days 0, 14, 28, and then every 28 days for 48 weeks in Trial 3 and for 72 weeks in Trial 2.
The primary efficacy endpoint was a composite endpoint (SLE Responder Index‑4 or SRI‑4) that defined response as meeting each of the following criteria at Week 52 compared with baseline:
- •
- ≥4‑point reduction in the SELENA‑SLEDAI score, and
- •
- no new British Isles Lupus Assessment Group (BILAG) A organ domain score or 2 new BILAG B organ domain scores, and
- •
- no worsening (<0.30‑point increase) in Physician’s Global Assessment (PGA) score.
The SRI uses the SELENA‑SLEDAI score as an objective measure of reduction in global disease activity; the BILAG index to ensure no significant worsening in any specific organ system; and the PGA to ensure that improvements in disease activity are not accompanied by worsening of the patient’s condition overall.
In both Trials 2 and 3, the proportion of subjects with SLE achieving an SRI‑4 response, as defined for the primary endpoint, was significantly higher in the group receiving BENLYSTA 10 mg/kg plus standard therapy than in the group receiving placebo plus standard therapy. The effect on the SRI-4 was not consistently significantly different for subjects receiving BENLYSTA 1 mg/kg plus standard therapy relative to placebo plus standard therapy in both trials. The 1‑mg/kg dose is not recommended. The trends in comparisons between the treatment groups for the rates of response for the individual components of the endpoint were generally consistent with that of the SRI‑4 (Table 5). At Week 76 in Trial 2, the SRI‑4 response rate with BENLYSTA 10 mg/kg was not significantly different from that of placebo (39% and 32%, respectively).
| a The 1-mg/kg dose is not recommended. b Subjects dropping out of the trial early or experiencing certain increases in background medication were considered as failures in these analyses. In both trials, a higher proportion of placebo subjects were considered as failures for this reason compared with the groups receiving BENLYSTA. |
||||||
|
Response |
Trial 2 |
Trial 3 |
||||
|
Placebo + Standard Therapy (n = 275) |
BENLYSTA 1 mg/kg + Standard Therapya (n = 271) |
BENLYSTA 10 mg/kg + Standard Therapy (n = 273) |
Placebo + Standard Therapy (n = 287) |
BENLYSTA 1 mg/kg + Standard Therapya (n = 288) |
BENLYSTA 10 mg/kg + Standard Therapy (n = 290) |
|
|
SLE Responder Index-4 (SRI-4)b |
34% |
41% |
43% |
44% |
51% |
58% |
|
Odds Ratio (95% CI) vs. placebo |
1.3 (0.9, 1.9) |
1.5 (1.1, 2.2) |
1.6 (1.1, 2.2) |
1.8 (1.3, 2.6) |
||
|
Components of SLE Responder Index-4 (SRI-4) |
||||||
|
Percent of subjects with reduction in SELENA-SLEDAI ≥4 |
36% |
43% |
47% |
46% |
53% |
58% |
|
Percent of subjects with no worsening by BILAG index |
65% |
75% |
69% |
73% |
79% |
81% |
|
Percent of subjects with no worsening by PGA |
63% |
73% |
69% |
69% |
79% |
80% |
The reduction in disease activity seen in the SRI-4 was related primarily to improvement in the most commonly involved organ systems; namely, mucocutaneous, musculoskeletal, and immune.
Effect in Black/African-American Subjects: In Trials 2 and 3, exploratory sub‑group analyses of SRI-4 response rate in Black subjects (n = 148) were performed. The SRI‑4 response rate in Black subjects in groups receiving BENLYSTA plus standard therapy was less than that in the group receiving placebo plus standard therapy (22/50 or 44% for placebo, 15/48 or 31% for BENLYSTA 1 mg/kg, and 18/50 or 36% for BENLYSTA 10 mg/kg).
Trial 4 included 448 Black subjects and was conducted in North America, South America, Europe, and Africa. It had the same trial design as Trials 2 and 3 with exceptions of subjects having a baseline SELENA‑SLEDAI score of ≥8 and using the modified SLEDAI‑2K scoring for proteinuria. The population had a mean age of 39 years (range: 18 to 71) and 97% were female. The proportion of Black subjects achieving an SRI‑S2K response at Week 52 (primary endpoint), and the individual components of the endpoint, were higher in the group receiving BENLYSTA 10 mg/kg plus standard therapy relative to the group receiving placebo plus standard therapy. However, the treatment difference was not statistically significant (Table 6).
| a Analyses excluded any subject missing a baseline assessment for any of the components (1 for belimumab). | ||
| b Subjects dropping out of the trial early or experiencing certain increases in background medication were considered as failures in these analyses. A higher proportion of subjects receiving placebo were considered as failures for this reason compared with the group receiving BENLYSTA. | ||
|
Responsea |
Placebo + Standard Therapy (n = 149) |
BENLYSTA 10 mg/kg + Standard Therapy (n = 298) |
|
SLE Responder Index (SRI-S2K)b |
42% |
49% |
|
Odds Ratio (95% CI) vs. placebo |
1.4 (0.9, 2.1) P = 0.107 |
|
|
Components of SLE Responder Index (SRI-S2K) |
||
|
Percent of subjects with reduction in SELENA‑SLEDAI-S2K ≥4 |
42% |
50% |
|
Odds Ratio (95% CI) vs. placebo |
1.5 (1.0, 2.2) |
|
|
Percent of subjects with no worsening by BILAG index |
62% |
68% |
|
Odds Ratio (95% CI) vs. placebo |
1.2 (0.8, 1.9) |
|
|
Percent of subjects with no worsening by PGA |
64% |
70% |
|
Odds Ratio (95% CI) vs. placebo |
1.3 (0.8, 1.9) |
|
Effect on Concomitant Steroid Treatment: In Trial 2 and Trial 3, 46% and 69% of subjects, respectively, were receiving prednisone at doses >7.5 mg/day at baseline. The proportion of subjects able to reduce their average prednisone dose by at least 25% to ≤7.5 mg/day during Weeks 40 through 52 was not consistently significantly different for BENLYSTA plus standard therapy relative to placebo plus standard therapy in both trials. In Trial 2, 17% of subjects receiving BENLYSTA 10 mg/kg plus standard therapy and 19% of subjects receiving BENLYSTA 1 mg/kg plus standard therapy achieved this level of steroid reduction compared with 13% of subjects receiving placebo plus standard therapy. In Trial 3, 19%, 21%, and 12% of subjects receiving BENLYSTA 10 mg/kg, BENLYSTA 1 mg/kg, and placebo, respectively, plus standard therapy achieved this level of steroid reduction.
Effect on Severe SLE Flares: The probability of experiencing a severe SLE flare, as defined by a modification of the SELENA Trial flare criteria, which excluded severe flares triggered only by an increase of the SELENA‑SLEDAI score to >12, was calculated for both Trials 2 and 3. The proportion of subjects having at least 1 severe flare over 52 weeks was not consistently significantly different for BENLYSTA plus standard therapy relative to placebo plus standard therapy in both trials. In Trial 2, 18% of subjects receiving BENLYSTA 10 mg/kg plus standard therapy and 16% of subjects receiving BENLYSTA 1 mg/kg plus standard therapy had a severe flare compared with 24% of subjects receiving placebo plus standard therapy. In Trial 3, 14%, 18%, and 23% of subjects receiving BENLYSTA 10 mg/kg, BENLYSTA 1 mg/kg and placebo, respectively, plus standard therapy had a severe flare.
14.3 Intravenous Administration in Adults with Active Lupus Nephritis
Trial 5: Active Lupus Nephritis – BENLYSTA 10 mg/kg - Intravenous
The safety and effectiveness of BENLYSTA 10 mg/kg administered intravenously over 1 hour on Days 0, 14, 28, and then every 28 days plus standard therapy were evaluated in a 104‑week, randomized, double‑blind, placebo‑controlled trial in 448 adult subjects with active proliferative and/or membranous lupus nephritis (Trial 5). The subjects had a clinical diagnosis of SLE according to American College of Rheumatology classification criteria; biopsy‑proven lupus nephritis Class III, IV, and/or V; and had active renal disease at screening requiring standard therapy: corticosteroids with 1) mycophenolate for induction followed by mycophenolate for maintenance, or 2) cyclophosphamide for induction followed by azathioprine for maintenance. This trial was conducted in Asia, North America, South America, and Europe. The mean age of subjects was 33 years (range: 18 to 77); the majority (88%) were female.
The primary efficacy endpoint was Primary Efficacy Renal Response (PERR) at Week 104, defined as a response at Week 100 confirmed by a repeat measurement at Week 104 of the following parameters: urine protein:creatinine ratio (uPCR) ≤0.7 g/g and estimated glomerular filtration rate (eGFR) ≥60 mL/min/1.73 m2 or no decrease in eGFR of >20% from pre‑flare value.
The major secondary endpoints included:
- •
- Complete Renal Response (CRR) defined as a response at Week 100 confirmed by a repeat measurement at Week 104 of the following parameters: uPCR <0.5 g/g and eGFR ≥90 mL/min/1.73 m2 or no decrease in eGFR of >10% from pre-flare value.
- •
- PERR at Week 52.
- •
- Time to renal-related event or death (renal-related event defined as first event of end-stage renal disease, doubling of serum creatinine, renal worsening [defined by quantified increase in proteinuria and/or impaired renal function], or receipt of renal disease-related prohibited therapy due to inadequate lupus nephritis control or renal flare management).
The proportion of subjects achieving PERR at Week 104 was significantly higher in subjects receiving BENLYSTA plus standard therapy compared with placebo plus standard therapy (Table 7). The major secondary endpoints also showed significant improvement with BENLYSTA plus standard therapy compared with placebo plus standard therapy (Table 7 and Table 8).
| eGFR = Estimated glomerular filtration rate. a PERR at Week 104 was the primary efficacy analysis; CRR at Week 104 and PERR at Week 52 were included in pre-specified testing hierarchy. b In order to be considered a responder, steroid treatment had to be reduced to ≤10 mg/day from Week 24. Subjects who discontinued treatment early, received prohibited medication or increases in background standard therapy, or withdrew from the trial were considered non-responders. Prohibited medications and increases in background standard therapy were defined as: 1) use of corticosteroids above that allowed by protocol; 2) additional immunosuppressive agents (except topicals) beyond their induction/maintenance regimens; 3) angiotensin converting enzyme inhibitors (ACE) inhibitors, angiotensin II receptor blockers (ARBs), or antimalarials initiated after Week 24; 4) exceeding protocol-permitted doses for standard therapy (cyclophosphamide, azathioprine, mycophenolate); or 5) other biologics, intravenous immunoglobulin (IVIG), or plasmapheresis. c The percentage of subjects who did not take prohibited medications or have an increase in background standard therapy at Week 104 was 83% for BENLYSTA and 74% for placebo. |
|||
|
Efficacy Endpointa |
Placebo +
|
BENLYSTA +
|
Odds Ratio (OR) vs. Placebo
|
|
Primary Efficacy Renal Response (PERR) at Week 104b,c (responders) |
32% |
43% |
1.6 (1.0, 2.3) P = 0.031 |
|
Components of PERR |
|||
|
Urine protein:creatinine ratio ≤0.7 g/g |
34% |
44% |
1.5 (1.0, 2.3) |
|
eGFR ≥60 mL/min/1.73 m2 or no decrease in eGFR from pre-flare value of >20% |
50% |
57% |
1.3 (0.9, 1.9) |
|
Complete Renal Response (CRR) at Week 104b,c (responders) |
20% |
30% |
1.7 (1.1, 2.7) P = 0.017 |
|
Components of CRR |
|||
|
Urine protein:creatinine ratio <0.5 g/g |
29% |
39% |
1.6 (1.1, 2.4) |
|
eGFR ≥90 mL/min/1.73 m2 or no decrease in eGFR from pre-flare value of >10% |
40% |
47% |
1.3 (0.9, 2.0) |
|
PERR at Week 52b (responders) |
35% |
47% |
1.6 (1.1, 2.4) P = 0.025 |
In descriptive subgroup analyses, the PERR and CRR rates were examined by induction therapy (mycophenolate or cyclophosphamide), biopsy class (Class III or IV, Class III + V or Class IV + V, or Class V), and uPCR levels at baseline (<3 g/g or ≥3 g/g; post-hoc analysis) (Figure 1).
Figure 1. Odds Ratio of PERR and CRR at Week 104 across Subgroupsa, b in Adult Subjects with Active Lupus Nephritis (Intravenous Treatment) (Trial 5)
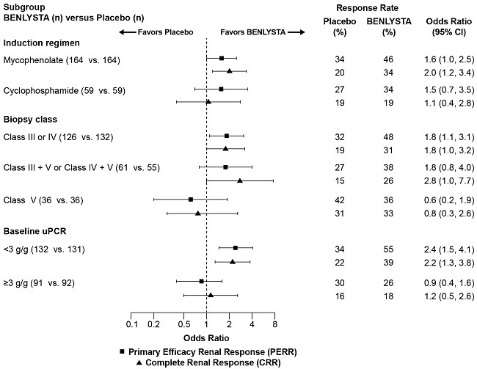
a Class III = Focal proliferative lupus nephritis; Class IV = Diffuse proliferative lupus nephritis; Class V = Membranous lupus nephritis; Class III + V = Mixed membranous-focal proliferative lupus nephritis; Class IV + V = Mixed membranous-diffuse proliferative lupus nephritis.
b Baseline urine protein:creatinine ratio (uPCR) was a post-hoc analysis.
The proportion of responders for PERR by visit through Week 104 is shown in Figure 2.
Figure 2. Primary Efficacy Renal Response (PERR) in Adult Subjects with Active Lupus Nephritis (+/- Standard Error) by Visita (Intravenous Treatment) (Trial 5)
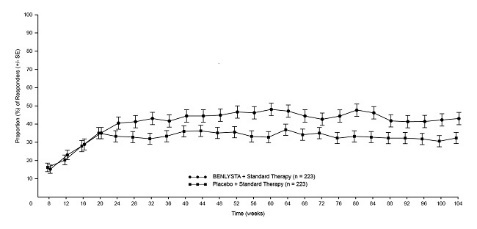
a Analysis is descriptive; bars represent standard error. The same subjects may not have responded at each timepoint.
In Trial 5, subjects who received BENLYSTA were significantly less likely to experience a renal-related event or death compared with placebo (Table 8).
| Placebo +
Standard Therapy n = 223 | BENLYSTA +
Standard Therapy n = 223 | Hazard Ratio (HR) vs. Placebo
(95% CI) |
|
|---|---|---|---|
| a Time to renal-related event or death included in pre-specified testing hierarchy. b When excluding deaths from analysis (1 for BENLYSTA; 2 for placebo), the percentage of subjects with a renal-related event was 15% for BENLYSTA compared with 27% for placebo (HR = 0.5; 95% CI: 0.3, 0.8). c Subjects who discontinued treatment, were withdrawn from the trial, lost to follow-up, or had a treatment failure not related to renal disease were censored on the date of the event. Subjects who completed the 104-week treatment period were censored at the Week 104 visit. Time to event was defined as (event date minus treatment start date plus 1 day). d Subjects could have had more than one event; the first event contributed to the overall endpoint. e Renal worsening was prospectively defined as the development of increased proteinuria and/or impaired renal function defined as: 1) Increased proteinuria (using spot urine): a reproducible increase in 24-hour urine protein levels to >1 g/g if the baseline value was <0.2 g/g or >2 g/g if the baseline value was between 0.2 g/g and 1 g/g or more than twice the value at baseline if the baseline value was >1 g/g; and 2) Impaired renal function: a reproducible decrease in eGFR of >20% accompanied by at least 1 of the following: proteinuria (>1 g/g), red blood cell casts, or white blood cell casts. f Renal-related treatment failure was prospectively defined as intake of prohibited medications for adjudicated inadequate lupus nephritis control or renal flare management. |
|||
|
Percentage (Number) of subjects with eventb |
28% (63) |
16% (35) | |
|
Time to eventc |
0.5 (0.3, 0.8) P = 0.001 |
||
|
Components of endpointd (percentage of subjects with event) |
|||
|
End-stage renal disease (ESRD) |
0.4% |
1% | |
|
Doubling of serum creatinine from baseline |
4% |
1% | |
|
Renal worseninge |
18% |
8% | |
|
Renal-related treatment failuref |
16% |
9% | |
|
Death |
1% |
0.4% | |
In descriptive subgroup analyses of time to renal-related event or death, results were consistent with the overall endpoint regardless of induction therapy (mycophenolate or cyclophosphamide), biopsy class (Class III or IV, Class III + V or Class IV + V, or Class V; post-hoc analysis), and baseline proteinuria (<3 g/g or ≥3 g/g; post-hoc analysis). The treatment difference was primarily driven by the renal worsening and renal-related treatment failure components of the endpoint.
14.4 Intravenous Administration in Pediatric Subjects with Active SLE
Trial 6: Active SLE - BENLYSTA 10 mg/kg in Pediatric Subjects - Intravenous
The safety and efficacy of BENLYSTA was evaluated in an international, randomized, double‑blind, placebo‑controlled, 52‑week, pharmacokinetics (PK), efficacy and safety trial (Trial 6) conducted in 93 pediatric subjects with a clinical diagnosis of SLE according to the American College of Rheumatology classification criteria. Subjects had active SLE disease, defined as a SELENA-SLEDAI score ≥6 and positive autoantibodies at screening as defined in the adult trials. Subjects were on a stable SLE treatment regimen (standard of care) and had similar inclusion and exclusion criteria as in the adult studies. The median age was 15 years (range: 6 to 17). The majority (95%) of subjects were female. More than 50% of subjects had 3 or more active organ systems involved at baseline. The most common active organ systems at baseline based on SELENA-SLEDAI were mucocutaneous (91%), immunologic (74%), and musculoskeletal (73%). Overall, 19% of pediatric subjects had some degree of renal activity and less than 7% had activity in the cardio‑respiratory, hematologic, CNS or vascular systems. Randomization into age‑related treatment cohorts was stratified by screening SELENA-SLEDAI scores (6 to 12 vs >13) and age (5 to 11 years vs 12 to 17 years).
The primary efficacy endpoint was the SLE Responder Index (SRI-4) at Week 52, as described in the adult intravenous trials. There was a numerically higher proportion of pediatric subjects achieving a response in SRI‑4 and its components in pediatric subjects receiving BENLYSTA plus standard therapy compared with placebo plus standard therapy (Table 9).
| a Based on a non-powered trial. | ||
| b Analyses excluded any subject missing a baseline assessment for any of the components (1 for placebo). | ||
|
Responseb |
Placebo + Standard Therapy (n = 39) |
BENLYSTA 10 mg/kg + Standard Therapy (n = 53) |
|
SLE Responder Index |
44% |
53% |
|
Odds Ratio (95% CI) vs. Placebo |
1.49 (0.64, 3.46) |
|
|
Components of SLE Responder Index |
||
|
Percent of subjects with reduction in SELENA‑SLEDAI ≥4 |
44% |
55% |
|
Percent of subjects with no worsening by BILAG index |
62% |
74% |
|
Percent of subjects with no worsening by PGA |
67% |
76% |
|
Other Endpoints |
||
|
SRI-6 using SELENA SLEDAI ≥6-point reduction |
34% |
41% |
|
Proportion of subjects with a sustained SRI response |
41% |
43% |
Effect on Concomitant Steroid Treatment: At baseline, 95% of pediatric subjects were receiving prednisone. Among those pediatric subjects, 20% of pediatric subjects receiving BENLYSTA plus standard therapy reduced their average prednisone dose by at least 25% per day during Weeks 44 through 52 compared with 21% of pediatric subjects on placebo plus standard therapy.
Effect on Severe SLE Flares: In Trial 6, the probability of experiencing a severe SLE flare, as measured by the modified SELENA-SLEDAI Flare Index, excluding severe flares triggered only by an increase of the SELENA‑SLEDAI score to >12, was calculated. The proportion of pediatric subjects reporting at least one severe flare during the trial was numerically lower in pediatric subjects receiving BENLYSTA plus standard therapy (17%) compared with those receiving placebo plus standard therapy (35%). Pediatric subjects receiving BENLYSTA 10 mg/kg plus standard therapy had a 64% lower risk of experiencing a severe flare during the 52 weeks of observation, relative to the placebo plus standard therapy group. Of the pediatric subjects experiencing a severe flare, the median time to the first severe flare was 150 days in pediatric subjects receiving BENLYSTA plus standard therapy compared with 113 days in pediatric subjects receiving placebo plus standard therapy.
14.5 Subcutaneous Administration in Adults with Active SLE
Trial 7: Active SLE – BENLYSTA 200 mg - Subcutaneous
The safety and effectiveness of BENLYSTA administered subcutaneously were evaluated in a randomized, double‑blind, placebo‑controlled trial involving 836 adult subjects with SLE according to the American College of Rheumatology criteria (Trial 7, NCT01484496). Subjects with severe active lupus nephritis and severe active CNS lupus were excluded. The trial (2:1 randomization) evaluated BENLYSTA 200 mg once weekly plus standard therapy (n = 556) compared with placebo once weekly plus standard therapy (n = 280) over 52 weeks in subjects with active SLE disease. Subjects had to have a SELENA‑SLEDAI score of ≥8 and positive autoantibody test (anti‑nuclear antibody [ANA] and/or anti‑double–stranded DNA [anti‑dsDNA]) results at screening.
No significant differences in baseline subject characteristics were observed between treatment groups. In some countries, treatment with a B‑cell–targeted agent was permitted if received a year or more prior to baseline; otherwise, treatment with a B‑cell–targeted agent was not permitted. Subjects were excluded from the trial if they were currently receiving other biologic agents. Anti‑tumor necrosis factor therapy, intravenous cyclophosphamide, interleukin‑1 receptor antagonist, IVIG, prednisone >100 mg/day, and plasmapheresis were not permitted within the previous 3 months or during the trial. The trial was conducted in North America, South America, Europe, and Asia. Baseline concomitant medications included corticosteroids (86%), antimalarials (69%), and immunosuppressives (46%, including azathioprine, methotrexate, and mycophenolate). Most subjects (approximately 80%) were receiving 2 or more classes of SLE medications.
More than 50% of subjects had 3 or more active organ systems involved at baseline. The most common active organ systems at baseline based on SELENA‑SLEDAI were mucocutaneous (88%), musculoskeletal (78%), and immunologic (76%). Overall, 12% of subjects had some degree of renal activity and less than 15% of subjects had activity in the vascular, cardio‑respiratory, or CNS systems. Subjects were stratified by disease severity based on their SELENA‑SLEDAI score (≤9 vs. ≥10), complement level (C3 and/or C4 low vs. other), and race (Black vs. other), and then randomly assigned to receive BENLYSTA 200 mg plus standard therapy or placebo once weekly plus standard therapy.
The primary efficacy endpoint was the SLE Responder Index‑4 (SRI‑4) at Week 52 as described in the intravenous trials. Secondary efficacy endpoints included time to first severe flare (as measured by the modified SELENA‑SLEDAI SLE Flare Index) and the proportion of subjects receiving prednisone >7.5 mg/day at baseline whose average prednisone dose had been reduced by ≥25% to ≤7.5 mg/day during Weeks 40 through 52.
The proportion of subjects achieving an SRI‑4 response was significantly higher in subjects receiving BENLYSTA plus standard therapy compared with placebo plus standard therapy. The trends comparing the treatment groups with respect to the probability of response for the individual components of the endpoint were consistent with that of the SRI‑4 (Table 10).
| a Analyses excluded any subject missing a baseline assessment for any of the components (1 for placebo; 2 for belimumab). | ||
| b Subjects dropping out of the trial early or experiencing certain increases in background medication were considered as failures in these analyses. A higher proportion of subjects receiving placebo plus standard therapy were considered as failures for this reason compared with the group receiving BENLYSTA plus standard therapy. | ||
|
Responsea |
Placebo + Standard Therapy (n = 279) |
BENLYSTA + Standard Therapy (n = 554) |
|
SLE Responder Index-4 (SRI-4)b |
48% |
61% P = 0.0006 |
|
Odds Ratio (95% CI) vs. placebo |
1.7 (1.3, 2.3) |
|
|
Components of SLE Responder Index-4 (SRI-4) |
||
|
Percent of subjects with reduction in SELENA-SLEDAI ≥4 |
49% |
62% |
|
Percent of subjects with no worsening by BILAG index |
74% |
81% |
|
Percent of subjects with no worsening by PGA |
73% |
81% |
The reduction in disease activity seen in the SRI-4 was related primarily to improvement in the most commonly involved organ systems, namely, mucocutaneous, musculoskeletal, immunologic, and vascular.
The proportion of SRI-4 responders by visit through Week 52 is shown in Figure 3.
Figure 3. Proportion (%) of SRI-4 Responders (+/- Standard Error) by Visita (Subcutaneous Treatment in Adult Subjects with Active SLE) (Trial 7)
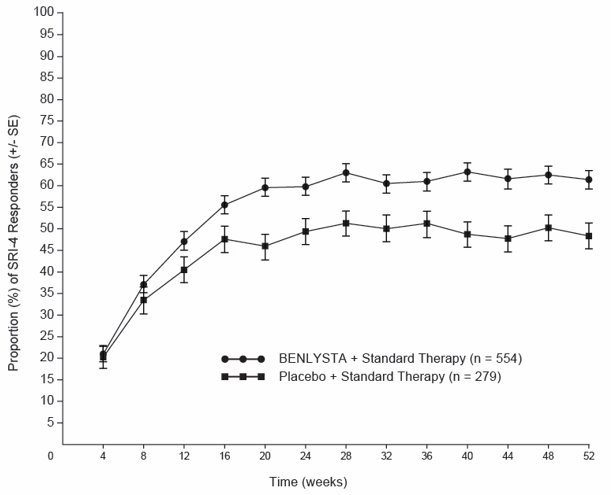
a The same subjects may not have responded at each timepoint.
Effect in Black/African‑American Subjects: Exploratory sub‑group analyses of SRI‑4 response rate in Black subjects (n = 91) were performed. The SRI‑4 response rate in Black subjects receiving BENLYSTA plus standard therapy was 45% (26/58) compared with 39% (13/33) in the group receiving placebo plus standard therapy [see Use in Specific Populations (8.8)].
Effect on Concomitant Steroid Treatment: At baseline, 60% of subjects were receiving prednisone at doses >7.5 mg/day. Among these subjects, 18% of subjects receiving BENLYSTA plus standard therapy reduced their average prednisone dose by at least 25% to ≤7.5 mg/day during Weeks 40 through 52 compared with 12% of subjects on placebo plus standard therapy; this difference was not statistically significant (OR = 1.65 [95% CI: 0.95, 2.84]).
Effect on Severe SLE Flares: The probability of experiencing a severe SLE flare, as measured by the modified SELENA‑SLEDAI SLE Flare Index, excluding severe flares triggered only by an increase of the SELENA‑SLEDAI score to >12, was calculated. The proportion of subjects reporting at least 1 severe flare during the trial was lower in subjects treated with BENLYSTA plus standard therapy (11%) compared with those receiving placebo plus standard therapy (18%). Subjects treated with BENLYSTA plus standard therapy had a 49% lower risk of experiencing at least 1 severe flare during the 52 weeks of observation, relative to the subjects receiving placebo plus standard therapy (HR = 0.51 [95% CI: 0.35, 0.74]). Of the subjects experiencing a severe flare, the median time to the first severe flare was delayed in subjects receiving BENLYSTA plus standard therapy compared with placebo plus standard therapy (171 days vs. 118 days).
16. How is Benlysta supplied
16.1 Intravenous Infusion
BENLYSTA (belimumab) for injection is a sterile, preservative-free, lyophilized powder for reconstitution and dilution prior to intravenous infusion provided in single‑dose glass vials with a rubber stopper (not made with natural rubber latex) and a flip‑off seal. Each 5‑mL vial contains 120 mg of belimumab. Each 20‑mL vial contains 400 mg of belimumab.
BENLYSTA vials are supplied as follows:
120 mg belimumab in a 5‑mL single-dose vial (NDC 49401-101-01)
400 mg belimumab in a 20‑mL single-dose vial (NDC 49401-102-01)
Refrigerate vials at 36°F to 46°F (2°C to 8°C). Store vials in the original carton until use to protect from light. Do not freeze. Avoid exposure to heat.
16.2 Subcutaneous Injection
BENLYSTA (belimumab) injection is a clear to opalescent, and colorless to pale yellow solution for subcutaneous use. Each single-dose prefilled autoinjector or single-dose prefilled syringe is designed to deliver 200 mg of belimumab in 1 mL of solution and is supplied as follows:
200 mg/mL single-dose prefilled autoinjector with 27-gauge, half-inch needle attached (NDC 49401-088-01) in a carton of 4 (NDC 49401-088-35).
200 mg/mL single-dose prefilled glass syringe with 27-gauge, half-inch needle attached (NDC 49401-088-42) in a carton of 4 (NDC 49401-088-47).
Prior to Dispensing
Refrigerate prefilled autoinjectors and prefilled syringes at 36°F to 46°F (2°C to 8°C). Keep the product in the original carton to protect from light until the time of use. Do not freeze. Do not shake. Avoid exposure to heat.
Following Dispensing
Refrigerate prefilled autoinjectors and prefilled syringes at 36°F to 46°F (2°C to 8°C). Keep the product in the original carton to protect from light until the time of use. Do not freeze. Do not shake. Avoid exposure to heat.
BENLYSTA may be stored outside of the refrigerator up to 86°F (30°C) for up to 12 hours if protected from sunlight. Do not use and do not place back in refrigerator if left out for more than 12 hours.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use). It is important that the patient’s overall health be assessed at each visit and any questions resulting from the patient’s reading of the Medication Guide and Instructions for Use be discussed.
For patients receiving BENLYSTA, give patients the Medication Guide for BENLYSTA.
Serious Infections
Inform patients that BENLYSTA may decrease their ability to fight infections, and that serious infections, including some fatal ones, occurred in patients receiving BENLYSTA in clinical trials. Instruct patients to tell their healthcare provider if they develop signs or symptoms of an infection [see Warnings and Precautions (5.1)].
Progressive Multifocal Leukoencephalopathy
Advise patients to contact their healthcare professional if they experience new or worsening neurological symptoms such as memory loss, confusion, dizziness or loss of balance, difficulty talking or walking, or vision problems [see Warnings and Precautions (5.1)].
Hypersensitivity Reactions/Anaphylaxis
Educate patients on the signs and symptoms of hypersensitivity reactions and infusion-related reactions, including wheezing, difficulty breathing, angioedema, rash, hypotension, bradycardia, and headache. Instruct patients to immediately tell their healthcare provider if they experience symptoms of an allergic reaction during or after the administration of BENLYSTA. Inform patients about possible delayed reactions that may include a combination of symptoms such as rash, nausea, fatigue, muscle aches, headache, and/or facial swelling that may occur after administration of BENLYSTA and advise them to contact their healthcare provider [see Warnings and Precautions (5.2)].
Depression and Suicidality
Instruct patients (and caregivers if applicable) to contact their healthcare provider if they experience new or worsening depression, suicidal thoughts, or other mood changes [see Warnings and Precautions (5.3)].
Immunizations
Inform patients that they should not receive live vaccines while taking BENLYSTA. Response to vaccinations could be impaired by BENLYSTA [see Warnings and Precautions (5.5)].
Pregnancy Registry
Inform patients that there is a pregnancy registry to evaluate fetal outcomes of pregnant women with lupus exposed to BENLYSTA [see Use in Specific Populations (8.1)].
Pregnancy
Inform female patients of reproductive potential that BENLYSTA may impact the immune system in infants of treated mothers and to inform their prescriber of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Trademarks are owned by or licensed to the GSK group of companies.
Manufactured by GlaxoSmithKline LLC
Philadelphia, PA 19104, U.S. License No. 1727, and
Distributed by GlaxoSmithKline
Durham, NC 27701
©2025 GSK group of companies or its licensor.
BNL:21PI
DETACH HERE AND GIVE MEDICATION GUIDE TO PATIENT.
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
|
MEDICATION GUIDE |
||
|
BENLYSTA (ben-LIST-ah) (belimumab) for injection, for intravenous use |
BENLYSTA (ben-LIST-ah) (belimumab) injection, for subcutaneous use |
|
|
What is the most important information I should know about BENLYSTA? Immunosuppressive agents, including BENLYSTA can cause serious side effects. Some of these side effects may cause death. The following are serious adverse reactions that have occurred in patients receiving BENLYSTA: |
||
|
||
|
|
|
|
||
|
|
|
|
||
|
|
|
|
What is BENLYSTA?
|
||
|
Do not use BENLYSTA if you:
|
||
|
Before you receive BENLYSTA, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Know the medicines you take. Keep a list of your medicines with you to show to your healthcare provider and pharmacist when you get a new medicine. |
||
|
How will I receive BENLYSTA? When given in a vein (intravenously)
When given under the skin (subcutaneously)
|
||
|
What are the possible side effects of BENLYSTA? BENLYSTA can cause serious side effects, including: See “What is the most important information I should know about BENLYSTA?”
|
||
|
|
|
The most common side effects of BENLYSTA include: |
||
|
|
|
|
These are not all the possible side effects of BENLYSTA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1‑800‑FDA‑1088. |
||
|
How should I store BENLYSTA?
Keep BENLYSTA and all medicines out of the reach of children. |
||
|
General information about the safe and effective use of BENLYSTA. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use BENLYSTA for a condition for which it was not prescribed. Do not give BENLYSTA to other people, even if they have the same symptoms that you have. It may harm them. This Medication Guide summarizes the most important information about BENLYSTA. You can ask your healthcare provider or pharmacist for information about BENLYSTA that is written for health professionals. |
||
|
What are the ingredients in BENLYSTA? Active ingredient: belimumab. Inactive ingredients (intravenous): citric acid, polysorbate 80, sodium citrate, sucrose. Inactive Ingredients (subcutaneous): L-arginine hydrochloride, L-histidine, L-histidine monohydrochloride, polysorbate 80, sodium chloride. |
||
|
For more information, go to www.BENLYSTA.com or call 1-877-423-6597. Trademarks are owned by or licensed to the GSK group of companies. Manufactured by GlaxoSmithKline LLC Philadelphia, PA 19104, U.S. License No. 1727, and Distributed by GlaxoSmithKline Durham, NC 27701 ©2025 GSK group of companies or its licensor. BNL:16MG |
||
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: June 2025
Instructions for Use
BENLYSTA (ben-LIST-ah)
(belimumab)
injection, for subcutaneous use
Prefilled Syringe
Read These Sections First
Read this Instructions for Use before you start to use BENLYSTA and each time you refill your prescription. There may be new information. You should also receive training from your healthcare provider on how to use the prefilled syringe the right way. If you do not follow these instructions the prefilled syringe may not work properly. BENLYSTA is for use under the skin only (subcutaneous).
Important Storage Information
- •
- Keep refrigerated until 30 minutes before use.
- •
- Keep in the original package until time of use to protect from light.
- •
- Do not freeze BENLYSTA. If the prefilled syringe has been frozen, do not use the prefilled syringe even if it is thawed.
- •
- Keep away from heat and sunlight.
- •
- Do not use and do not place back in the refrigerator if BENLYSTA is left out for more than 12 hours.
- •
- Keep out of the reach of children.
Important Warnings
- •
- The prefilled syringe should be used only 1 time and then thrown away. See below, “Step 6 - Throw away (dispose of) used syringe.”
- ○Do not share your BENLYSTA prefilled syringe with other people. You may give other people a serious infection or get a serious infection from them.
- ○Do not shake the prefilled syringe.
- ○Do not use if dropped onto a hard surface.
- ○Do not remove the Needle Cap until right before the injection.
- ○Do not use in children less than 18 years of age. It is not known if the prefilled syringe is safe and effective in children less than 18 years of age.
BENLYSTA Prefilled Syringe Parts
Before use
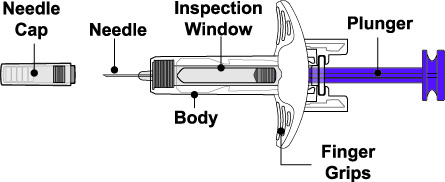
After Use – Needle is covered by Needle Guard
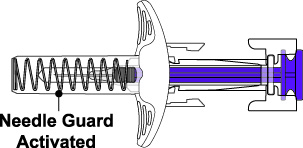
Supplies needed for the injection
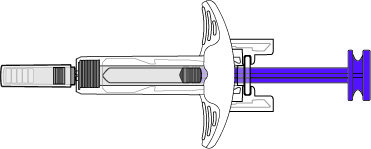
BENLYSTA Prefilled Syringe

Alcohol Swab (not included)
Gauze Pad or Cotton Ball (not included)
Sharps Container (not included)
Step 1 Gather and check supplies
Gather supplies
- •
- Remove 1 sealed tray containing a prefilled syringe from the refrigerator.
- •
- Place any remaining prefilled syringes back into the refrigerator.
- •
- Find a comfortable, well-lit, and clean surface and place the following supplies within reach:
- ○BENLYSTA Prefilled Syringe
- ○Alcohol swab (not included)
- ○Gauze pad or cotton ball (not included)
- ○Sharps container (not included)
- •
- Do not give the injection if you do not have all the supplies listed.
Take out the prefilled syringe
- •
- Peel back the film from the corner of the tray. See Figure A.
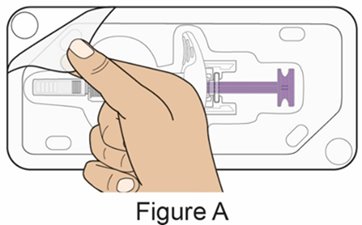
- •
- Holding the middle of the prefilled syringe (near the inspection window), carefully take the prefilled syringe out of the tray. See Figure B.
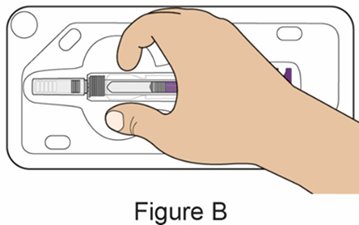
Check expiration (Exp) date
- •
- Check the expiration date on the prefilled syringe. See Figure C.
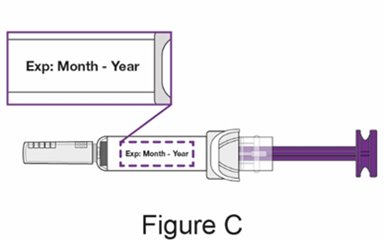
- •
- Do not use if the expiration date has passed.
Step 2 Prepare and inspect the BENLYSTA Prefilled Syringe
Allow to come to room temperature
- •
- Allow the prefilled syringe to sit at room temperature for 30 minutes. See Figure D.

- ○Do not warm the prefilled syringe in any other way. For example, do not warm in a microwave oven, hot water, or in direct sunlight.
- ○Do not remove the Needle Cap during this step.
Inspect BENLYSTA solution
- •
- Look in the Inspection Window to check that the BENLYSTA solution is colorless to slightly yellow in color. See Figure E.
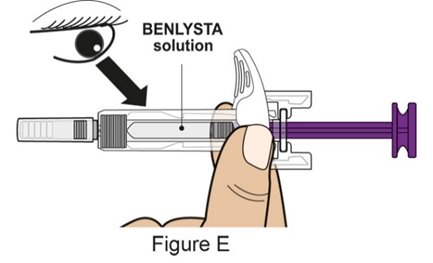
- •
- It is normal to see 1 or more air bubbles in the solution.
- •
- Do not use if the solution looks cloudy, discolored, or has particles.
Step 3 Choose and clean the injection site
Choose the injection site
- •
- Choose where to inject (abdomen or thigh). See Figure F.
- •
- If you need 2 injections to complete your dose, leave at least 2 inches between each injection if using the same site.
- •
- Avoid injecting into the same site each time or in areas where the skin is tender, bruised, red, or hard.
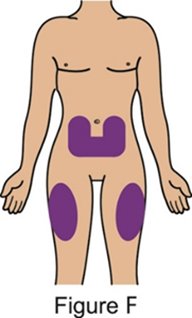
- •
- Do not inject within 2 inches of the belly button.
Clean the injection site
- •
- Wash your hands.
- •
- Clean the injection site by wiping it with an alcohol swab. Allow the skin to air dry. See Figure G.
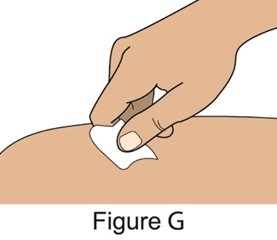
- •
-
Do not touch this area again before giving the injection.
- Step 4 Prepare for the injection
- Remove the Needle Cap
- •
- Do not remove the Needle Cap until right before you give the injection.
- •
- Hold the prefilled syringe by the body and with the Needle facing away from you. Remove the Needle Cap by pulling it straight off. See Figure H.
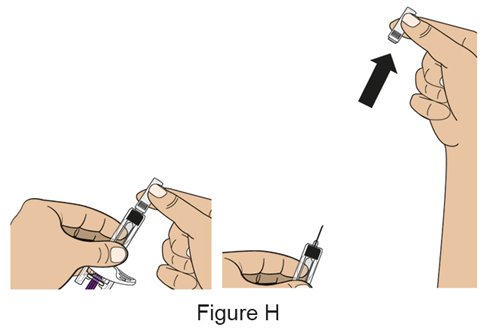
- •
- You may see a drop of liquid at the end of the Needle. This is normal.
- •
- Do not let the Needle touch any surface.
- •
- Do not push any air bubbles out of the prefilled syringe.
- •
- Do not put the Needle Cap back onto the prefilled syringe.
- •
- Keep your hands away from the Plunger to avoid pushing it before injecting.
Step 5 Inject BENLYSTA and inspect
Insert the Needle
- •
- Hold the prefilled syringe in one hand and use your free hand to gently pinch the skin around the injection site. See Figure I.
- •
- Insert the entire Needle into the pinched area of the skin at a slight 45-degree angle using a dart-like motion.
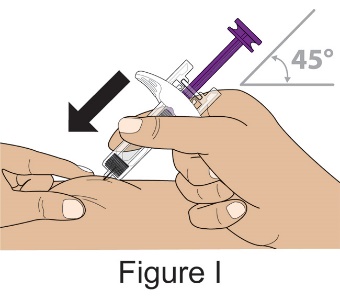
- •
- After the Needle is completely inserted, release the pinched skin.
Complete the injection
- •
- Push the Plunger all the way down until all of the solution is injected. See Figure J.
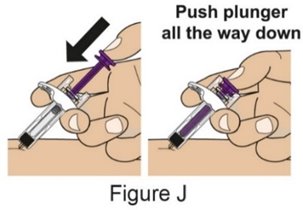
- •
- While keeping your hold on the syringe, slowly move your thumb back, allowing the Plunger to rise up. The Needle will automatically rise up into the Needle Guard. See Figure K.
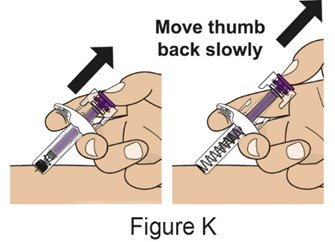
Inspect the injection site
- •
- There may be a small amount of blood at the injection site. If needed, press a cotton ball or gauze pad on the injection site.
- •
- Do not rub the injection site.
Step 6 Throw away (dispose of) used syringe
Dispose of the used syringe
Throw away (dispose of) the used syringe and Needle Cap in a Sharps Container. See Figure L.
- •
- Put your used syringe and sharps in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
- •
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that:
- ○is made of a heavy-duty plastic,
- ○can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- ○is upright and stable during use,
- ○is leak-resistant, and
- ○properly labeled to warn of hazardous waste inside the container.
- •
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to: http://www.fda.gov/safesharpsdisposal.
- •
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
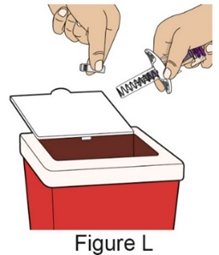
Additional information
For more information about BENLYSTA, go to www.BENLYSTA.com or call 1-877-423-6597.
Trademarks are owned by or licensed to the GSK group of companies.
Manufactured by GlaxoSmithKline LLC
Philadelphia, PA 19104, U.S. License No. 1727, and
Distributed by GlaxoSmithKline
Durham, NC 27701
©2024 GSK group of companies or its licensor.
BNL:7IFU-S
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised June 2024
Instructions for Use
BENLYSTA (ben-LIST-ah)
(belimumab)
injection, for subcutaneous use
Prefilled Autoinjector
Read These Sections First
Read this Instructions for Use before you start to use BENLYSTA and each time you refill your prescription. There may be new information. You should also receive training from your healthcare provider on how to use the autoinjector the right way. If you do not follow these instructions the autoinjector may not work properly. BENLYSTA is for use under the skin only (subcutaneous).
Important Storage Information
- •
- Keep refrigerated until 30 minutes before use.
- •
- Keep in the original package until time of use to protect from light.
- •
- Do not freeze BENLYSTA. If the autoinjector has been frozen, do not use the autoinjector even if it is thawed.
- •
- Keep away from heat and sunlight.
- •
- Do not use and do not place back in the refrigerator if BENLYSTA is left out for more than 12 hours.
- •
- Keep out of the reach of children.
Important Warnings
- •
- The autoinjector should be used only 1 time and then thrown away. See below, “Step 6 – Throw away (dispose of) used autoinjector.”
- ○Do not share your BENLYSTA Autoinjector with other people. You may give other people a serious infection or get a serious infection from them.
- ○Do not shake the autoinjector.
- ○Do not use if dropped onto a hard surface.
- ○Do not remove the Ring Cap until right before the injection.
- ○For children less than 10 years of age, BENLYSTA must be given by a healthcare provider or a trained caregiver.
BENLYSTA Autoinjector Parts
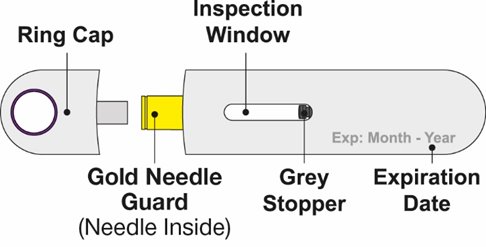
Supplies needed for the injection

BENLYSTA Autoinjector
Alcohol Swab (not included)
Gauze Pad or Cotton Ball (not included)
Sharps Container (not included)
Step 1 Gather and check supplies
Gather supplies
- •
- Remove 1 sealed tray containing an autoinjector from the refrigerator.
- •
- Place any remaining autoinjectors back into the refrigerator.
- •
- Find a comfortable, well-lit, and clean surface and place the following supplies within reach:
- ○BENLYSTA Autoinjector
- ○Alcohol swab (not included)
- ○Gauze pad or cotton ball (not included)
- ○Sharps container (not included)
- •
- Do not give the injection if you do not have all the supplies listed.
Take out the autoinjector
- •
- Peel back the film from the corner of the tray. See Figure A.
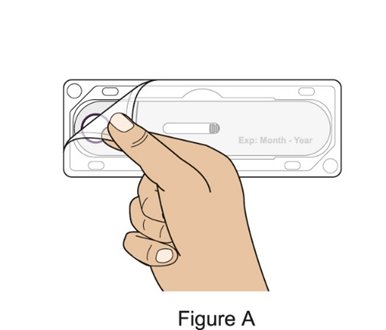
- •
- Holding the middle of the autoinjector (near the inspection window), carefully take the autoinjector out of the tray. See Figure B.
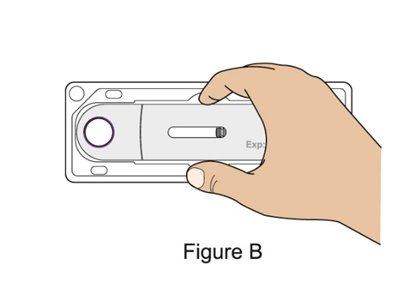
Check expiration (Exp) date
- •
- Check the expiration date on the autoinjector. See Figure C.
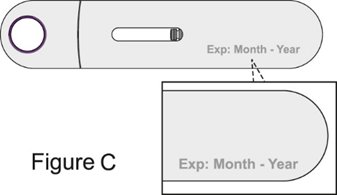
- •
- Do not use if the expiration date has passed.
Step 2 Prepare and inspect the BENLYSTA Autoinjector
Allow to come to room temperature
- •
- Allow the autoinjector to sit at room temperature for 30 minutes. See Figure D.

- ○Do not warm the autoinjector in any other way. For example, do not warm in a microwave oven, hot water, or in direct sunlight.
- ○Do not remove the Ring Cap during this step.
Inspect BENLYSTA solution
- •
- Look in the Inspection Window to check that the BENLYSTA solution is colorless to slightly yellow in color. See Figure E.
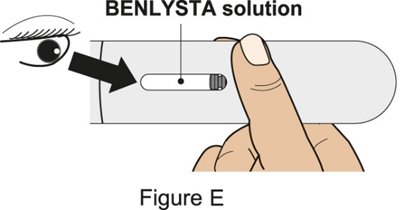
- •
- It is normal to see 1 or more air bubbles in the solution.
- •
- Do not use if the solution looks cloudy, discolored, or has particles.
Step 3 Choose and clean the injection site
Choose the injection site
- •
- Choose where to inject (abdomen or thigh). See Figure F.
- •
- If you need 2 injections to complete your dose, leave at least 2 inches between each injection if using the same site.
- •
- Avoid injecting into the same site each time or in areas where the skin is tender, bruised, red, or hard.
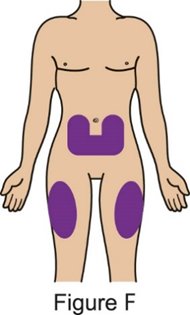
- •
- Do not inject within 2 inches of the belly button.
Clean the injection site
- •
- Wash your hands.
- •
- Clean the injection site by wiping it with an alcohol swab. Allow the skin to air dry. See Figure G.
- •
- Do not touch this area again before giving the injection.
Step 4 Prepare for the injection
Remove the Ring Cap
- •
- Do not remove the Ring Cap until right before you give the injection.
- •
- Remove the Ring Cap by pulling or twisting it off. The Ring Cap may be twisted off in either a clockwise or counterclockwise direction. See Figure H.
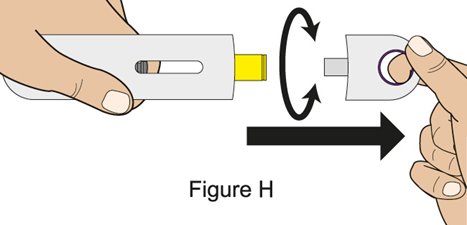
- •
- Do not put the Ring Cap back onto the autoinjector.
Position the autoinjector
- •
- Hold the autoinjector comfortably so that you can see the Inspection Window. It is important that you can look at the Inspection Window while giving the injection. You will look at the Inspection Window to:
- ○see that the purple indicator is moving while you are injecting.
- ○see that the purple indicator has stopped to make sure that the dose is complete. See Figure I.
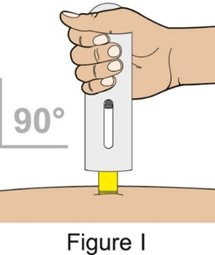
- •
- Position the autoinjector straight over the injection site at a 90-degree angle. Make sure the Gold Needle Guard is flat on the skin.
Step 5 Inject BENLYSTA and inspect
Start the injection
- •
- Firmly press the autoinjector all the way down onto the injection site and hold in place. See Figure J.
- •
- This will insert the needle and start the injection.
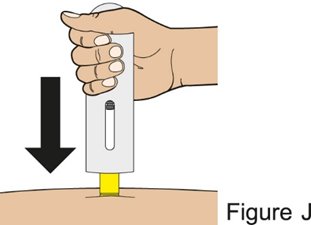
- •
- You may hear a “first click” at the start of the injection and see the Purple Indicator start to move through the Inspection Window. See Figure K.
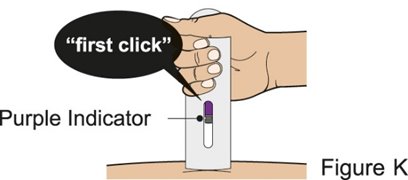
Complete the injection
- •
- Continue to hold the autoinjector down until you see that the Purple Indicator has stopped moving.
- •
- You may hear a "second click" a few seconds before the Purple Indicator stops moving. See Figure L.
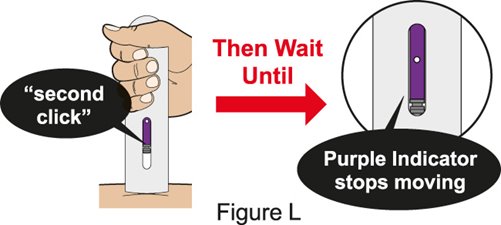
- •
- The injection may take up to 15 seconds to complete.
- •
- When the injection is complete, lift the autoinjector from the injection site.
Inspect the injection site
- •
- There may be a small amount of blood at the injection site. If needed, press a cotton ball or gauze pad on the injection site.
- ○Do not rub the injection site
Step 6 Throw away (dispose of) used autoinjector
Dispose of the used autoinjector
- •
- Throw away (dispose of) the used autoinjector and Ring Cap in a Sharps Container. See Figure M.
- •
- Put your used autoinjector in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) in your household trash.
- •
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that:
- ○is made of a heavy-duty plastic,
- ○can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- ○is upright and stable during use,
- ○is leak-resistant, and
- ○properly labeled to warn of hazardous waste inside the container.
- •
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to: http://www.fda.gov/safesharpsdisposal.
- •
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
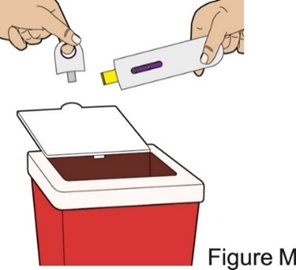
Additional information
For more information about BENLYSTA, go to www.BENLYSTA.com or call 1-877-423-6597.
Trademarks are owned by or licensed to the GSK group of companies.
Manufactured by GlaxoSmithKline LLC
Philadelphia, PA 19104, U.S. License No. 1727, and
Distributed by GlaxoSmithKline
Durham, NC 27701
©2024 GSK group of companies or its licensor.
BNL:6IFU-A
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised May 2024
PRINCIPAL DISPLAY PANEL
NDC 49401-101-01
Benlysta
(belimumab)
for Injection
120 mg/vial
GSK
For Intravenous Infusion after dilution only. Single-dose vial.
Discard unused portion
Reconstitution: Reconstitute with 1.5 mL of Sterile Water for Injection, USP. After reconstitution, the concentration of BENLYSTA is 80 mg/mL.
Dilution: Further dilute to 250 mL of one of the following solutions before use:
- •
- 0.9% Sodium Chloride Injection, USP
- •
- 0.45% Sodium Chloride Injection, USP
- •
- Lactated Ringer’s Injection, USP
Federal Law requires dispensing of BENLYSTA with the Medication Guide provided with this carton.
Rx only
©2022 GSK group of companies or its licensor.
Rev. 9/22
62000000082255

PRINCIPAL DISPLAY PANEL
NDC 49401-102-01
Benlysta
(belimumab)
for Injection
400 mg/vial
GSK
For Intravenous Infusion after dilution only. Single-dose vial.
Discard unused portion.
Reconstitution: Reconstitute with 4.8 mL of Sterile Water for Injection, USP. After reconstitution, the concentration of BENLYSTA is 80 mg/mL.
Dilution: Further dilute to 250 mL of one of the following solutions before use:
- •
- 0.9% Sodium Chloride Injection, USP
- •
- 0.45% Sodium Chloride Injection, USP
- •
- Lactated Ringer’s Injection, USP
Federal Law requires dispensing of BENLYSTA with the Medication Guide provided with this carton.
Rx only
©2022 GSK group of companies or its licensor.
Rev. 9/22
62000000082242

PRINCIPAL DISPLAY PANEL
NDC 49401-088-35
Benlysta
(belimumab)
Injection
200 mg/mL
Rx only
GSK
For Subcutaneous Use
Contents:
- •
- 4 Single-Dose 1-mL Prefilled Autoinjectors
- •
- Instructions for Use (Read carefully)
- •
- Medication Guide
- •
- Prescribing Information
Federal Law requires dispensing of BENLYSTA with the Medication Guide provided with this package.
©2024 GSK group of companies or is licensor.
Rev. 8/24
62000000095407

| BENLYSTA
belimumab injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| BENLYSTA
belimumab injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| BENLYSTA
belimumab solution |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Labeler - GlaxoSmithKline LLC (167380711) |
Biological Products Related to Benlysta
Find detailed information on biosimilars for this medication.
Frequently asked questions
- What is the difference between Benlysta and Saphnelo?
- What's the difference between Lupkynis and Benlysta?
- Does Benlysta cause weight gain?
- Where do you inject Benlysta?
- Can Benlysta cause an allergic reaction?
- How does Benlysta help lupus?
- How long does a Benlysta infusion take?
- Does Benlysta cause hair loss?
- Does Benlysta help with fatigue?
More about Benlysta (belimumab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (65)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- Support group
- FDA approval history
- Drug class: selective immunosuppressants
- Breastfeeding
- En español