Epinephrine FDA Alerts
The FDA Alerts below may be specifically about epinephrine or relate to a group or class of drugs which include epinephrine.
MedWatch Safety Alerts are distributed by the FDA and published by Drugs.com. Following is a list of possible medication recalls, market withdrawals, alerts and warnings.
Recent FDA Alerts for epinephrine
FDA Warns Health Care Professionals Not to Use Epinephrine Nasal Solutions from BPI Labs and Endo USA
January 16, 2025 -- FDA is warning health care professionals not to use unapproved epinephrine nasal solutions manufactured by BPI Labs LLC, in Largo, Fla., and Endo USA, in Malvern, Pa. Health care professionals have confused these products with FDA-approved injectable epinephrine products for intravenous use. BPI Labs and Endo USA nasal solutions products should never be injected intravenously.
The nasal solution and injectable products have similar packaging and containers and are manufactured by the same companies. The similarities of the bottle and packaging labels between the nasal product and the sterile injectable make it difficult to distinguish them from each other which can lead to health care professionals accidentally injecting the nasal solution instead of the injection product.
Unlike an injectable drug, nasal solutions are not required to be sterile. Injecting a non-sterile drug can lead to infection, which can be life threatening for certain patients.
Health care professionals use both products in hospitals and health care settings.
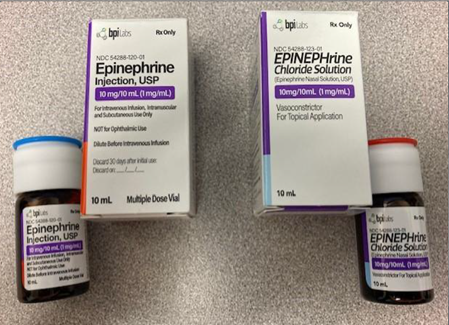
Figure 1: Left images: BPI Labs’ FDA-approved Epinephrine Injection with blue lid on the bottle
Right images: BPI Labs’ unapproved EPINEPHrine Chloride Nasal Solution with red lid on the bottle
Photograph submitted to FDA via an adverse event report
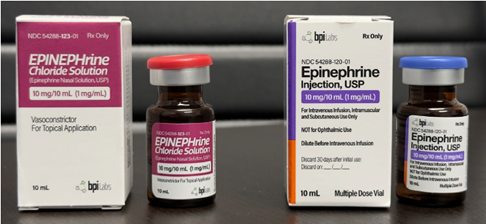
Figure 2: Left images: BPI Labs recently updated its label and packaging. The new unapproved EPINEPHrine Chloride Nasal Solution with a red bottle lid and maroon label in a maroon box.
Right images: BPI Labs’ FDA-approved Epinephrine Injection with blue lid on the bottle and purple highlight on the bottle and package.
Photograph provided to FDA by BPI Labs
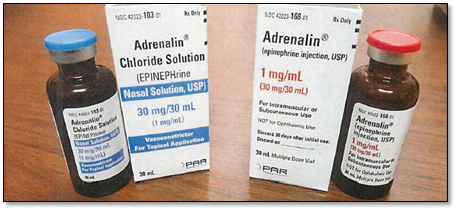
Figure 3: Left images: Endo USA’s unapproved Adrenalin Chloride (epinephrine) nasal solution, labeled as Par Pharmaceuticals, with a blue lid on the bottle
Right images: Endo USA’s FDA-approved Adrenalin (epinephrine) injection, labeled as Par Pharmaceuticals, with red lid on the bottle.
Photograph submitted to FDA via an adverse event report
Endo USA voluntarily recalled on Dec. 20, 2024, its unapproved Adrenalin Chloride Solution (Epinephrine Nasal Solution, USP) due to the potential for health care professionals accidentally injecting the nasal solution instead of the injection product.
FDA recommended BPI Labs recall its unapproved EPINEPHrine Nasal Solution on Dec. 12, 2024. The agency followed up with the company several times to reiterate this recommendation. The latest adverse event report FDA received involved the product with BPI Labs’ most recently revised label (figure 2, left images). The company has not acted to remove its unapproved drug from the market.
FDA has received more than 25 reports since 2016 stating confusion between unapproved epinephrine nasal solution and approved epinephrine injection involving similarities in product labels and containers. Recently in 2024, the agency received a report involving a patient who received the nasal solution as an injection.
FDA encourages health care professionals and patients to report adverse events or quality problems experienced with the use of any medication to FDA’s MedWatch Adverse Event Reporting program:
- Complete and submit the report online; or
- Download and complete the form, then submit it via fax at 1-800-FDA-0178
Source: FDA
Spectrum Laboratory Products, Inc. Issues Voluntary Worldwide Recall of Epinephrine (L-Adrenaline) USP Bulk Active Pharmaceutical Ingredient (API) Due to Discoloration of Product
January 9, 2023 -- Spectrum Laboratory Products, Inc. is voluntarily recalling three lots of Epinephrine (L-Adrenaline) USP, a bulk active pharmaceutical ingredient (API) used to manufacture or compound prescription products, to the user level. Customer complaints have found the product to be discolored.
Risk Statement: Epinephrine is a critical medication used during life-threatening conditions which can affect any age and any person. The use of a finished dose product manufactured or compounded with this recalled product could result in less-effective product, and incomplete treatment of life-threatening conditions including, low blood pressure, heart failure, anaphylaxis, irregular heartbeat, and heart attack. Treatment with a less-effective product, essentially underdosing epinephrine, could result in death. Spectrum Laboratory Products, Inc. has not received any reports of adverse events related to this recall.
Epinephrine (L-Adrenaline) USP bulk API Powder, is used in manufacturing and compounding of finished dose epinephrine prescription products which can be used to treat a variety of medical conditions including anaphylaxis and other severe immediate hypersensitivity reactions, asthma, bronchospasm, airway edema, nasal congestion, dilation during intraocular surgery, vasoconstrictor with local anesthetics, hypotension or shock, heart failure, bradycardia or atrioventricular block, and sudden cardiac arrest. The Epinephrine (L-Adrenaline) USP bulk API Powder is packaged in amber glass bottles enclosed in a vacuum sealed pouch. NDC’s, Package sizes, lot numbers and expiration dates can be found in the table below. The affected Epinephrine, USP product can be identified by Spectrum catalog number EP130. Product was distributed directly from Spectrum facilities nationwide in the USA and to Canada.
| Product | NDC | Package Size | Lot # | Exp Date |
|---|---|---|---|---|
| Epinephrine, USP (Product code EP130) | ||||
| 49452-2740-2 | 1 KG | 1KG0865 | 31-Mar-2023 | |
| 49452-2740-1 | 100 GM | 2KL0353 2KF0151 |
30-Sep-2023 31-Mar-2023 |
|
| 49452-2740-4 | 1 GM | |||
| 49452-2740-3 | 25 GM | |||
| 49452-2740-5 | 5 GM |
Spectrum Laboratory Products, Inc. is notifying its distributors and customers by certified mail, email and phone, and arranging for return of all recalled products. Consumers, distributors, or retail pharmacies that have Epinephrine, USP catalog number EP130, which is being recalled, should stop use immediately and return to place of purchase.
Consumers with questions regarding this recall can contact Spectrum Laboratory Products, Inc. by 800-772-8786 or compliance@spectrumchemical.com on Monday to Friday from 8 am to 5 pm, pacific time. Consumers should contact their physician or healthcare provider if they have experienced any problems that may be related to taking or using this drug product.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA's MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
- Complete and submit the report Online
- Regular Mail or Fax: Download form or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the U.S. Food and Drug Administration.
Source: FDA
FDA Alerts Patients and Health Care Professionals of Amneal and Impax Laboratories Epinephrine Auto-Injector Device Malfunctions
June 1, 2020 -- FDA is alerting patients, caregivers and health care professionals to immediately inspect certain lots of Amneal and Impax epinephrine auto-injector 0.3 mg to ensure the yellow “stop collar” in the device is present.
In letters to health care professionals and consumers, Impax Laboratories LLC, a subsidiary of Amneal Pharmaceuticals LLC, the manufacturer of the epinephrine auto-injector, detailed how certain lots of these devices might not contain the yellow “stop collar” component. If the auto-injector is missing the yellow “stop collar” component, the device has the potential safety risk of delivering a double dose of epinephrine to a patient. It is vital for lifesaving products to work as designed in an emergency situation.
Patients, pharmacists and health care professionals who have received Amneal or Impax’s epinephrine auto-injector after December 20, 2018, should immediately visually inspect the auto-injector to confirm the presence of the yellow “stop collar” by:
- Removing the auto-injector from the carrying case.
- Placing the auto-injector on a flat surface.
- Locating the edge of the label that states, “Peel here for further instructions.” Lift the label edge until you see the clear part of the auto-injector.
- Looking for the yellow “stop collar” inside the clear part of the auto-injector. If the yellow “stop collar” is not visible inside the clear part of the auto-injector, gently rotate the blue sheath remover, without pulling or removing the blue sheath remover, to observe if the yellow “stop collar” comes into view inside the clear part of the auto-injector.
- If yellow “stop collar” is present, then the product is safe to use, and no further action is necessary. Re-wrap the label to its original position and place the auto-injector into the carrying case.
Patients and health care professionals should contact the Amneal Drug Safety Department at 1(877) 835-5472 to assist in determining if the yellow “stop collar” is missing and to make arrangements to return defective devices and obtain a replacement at no additional cost. Patients should contact their pharmacy regarding a replacement epinephrine auto-injector before returning the defective device to Amneal.
FDA recommends that patients inspect their auto-injector devices as soon as possible and immediately contact the Amneal Drug Safety Department if they have questions about inspecting their auto-injector device, or if they’re unsure if the yellow “stop collar” is missing.
Pharmacists should inspect the products before dispensing them to patients to ensure the yellow “stop collar” is present. If the yellow “stop collar” is missing, pharmacists should not dispense the product and should contact Amneal.
The yellow “stop collar” is one of several components that work together to assure proper dosing of the auto-injector. While some patients require a second dose of epinephrine, an epinephrine overdose has the potential to cause severe patient harm or death. As stated on the product label, consumers should always seek emergency medical help right away after using their epinephrine auto-injector.
FDA reminds patients and health care professionals that epinephrine auto-injectors are available through additional manufacturers.
FDA is notifying patients and caregivers that epinephrine auto-injectors are not being recalled. We urge patients and caregivers to use the epinephrine auto-injector they have on hand and be aware of the potential issues outlined in the statement above.
FDA is continuously monitoring adverse events reported with epinephrine auto-injector products. FDA asks health care professionals and consumers to report any adverse events or quality problems to the FDA’s MedWatch program:
- Complete and submit the report online at www.fda.gov/medwatch/report.htm.
- Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
Source: FDA
FDA Alerts Patients and Health Care Professionals that Some EpiPen Auto-Injectors May Not Readily Slide Out of Carrier Tube
[11/2/2018] FDA is alerting patients, caregivers and health care professionals that the labels attached to some EpiPen 0.3mg and EpiPen Jr 0.15mg auto-injectors, and the authorized generic versions, may block access to the auto-injector and prevent the ability to easily access the product.
In a letter to health care professionals from Pfizer, the manufacturer of the Mylan EpiPen, the label sticker on the auto-injector unit may have been improperly applied, causing resistance when removing it from the carrier tube. The carrier tube is the immediate package in which the auto-injector is contained. In some cases, the patient or caregiver may not be able to quickly remove the epinephrine auto-injector from the carrier tube.
The auto-injector device and the epinephrine it delivers are not affected by this issue and can be used as prescribed. It is vital for lifesaving products to work as designed in an emergency situation, and patients and caregivers should inspect their epinephrine auto-injector prior to needing it to ensure they can quickly access the product.
The letter also describes how to inspect potentially affected products and explains that patients should contact Mylan Customer Relations at 800-796-9526 if an auto-injector does not slide out easily from the carrier tube OR the label is not fully adhered to the auto-injector. Pharmacists should inspect the products before dispensing them to patients to ensure quick access to the auto-injector and should not dispense any product which does not easily slide out of its carrier tube.
FDA is not aware of any adverse event reports associated with improperly applied EpiPen or EpiPen Jr auto-injectors, or their authorized generics label. As stated on the product label, consumers should always seek emergency medical help right away after using their epinephrine auto-injector.
More epinephrine resources
- Epinephrine injection Consumer Information
- Epinephrinesnap-EMS Consumer Information
- Epinephrinesnap-V Consumer Information
- Epinephrine inhalation Consumer Information
- Epinephrine nasal Consumer Information
- Epinephrine (Inhalation) Advanced Consumer Information
- EPINEPHrine Bitartrate, EPINEPHrine Hydrochloride, Epinephryl Borate (Mydriatic) (EENT) AHFS DI Monograph
- EPINEPHrine Hydrochloride (Vasoconstrictor) (EENT) AHFS DI Monograph
- Epinephrine AHFS DI Monograph
- Epinephrine Auto-Injector and Prefilled Syringe Consumer Information
- Epinephrine Inhalation Aerosol Consumer Information
- Epinephrine Injection Solution Consumer Information
- Epinephrine Concentrate Injection Prescribing Information
- Epinephrine Injection Prescribing Information
- Epy Kit Prescribing Information