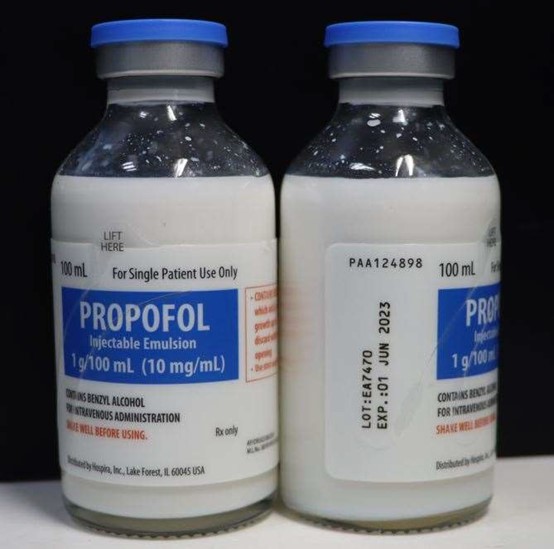
100mL Single Patient Use Glass Fliptop Vial; lot EA7470, to the user level due to visible particulates observed in two vials during annual examination of retention samples.
Risk Statement: Patients receiving the impacted product have a remote probability of experiencing potential adverse events, such as blockage of blood vessels, including decreased blood flow to the brain, heart attack, pulmonary embolus, and tissue necrosis. Hypersensitivity reactions and transmission of infectious disease can also occur.
To date, Pfizer has not received reports of any adverse events associated with this issue for this lot.
Propofol is an intravenous general anesthetic and sedation drug for use in the induction and maintenance of anesthesia or sedation. Propofol Injectable Emulsion is a terminally sterilized (TS) product. It is a sterile, nonpyrogenic emulsion containing 10 mg/mL of propofol suitable for intravenous administration supplied in a Single Patient Use Glass Fliptop Vial.
The NDC, Lot Number, Expiration Date, and Configuration details for Propofol Injectable Emulsion are indicated below. The product lot was distributed nationwide to wholesalers/hospitals in the United States from July 16, 2020 through July 24, 2020.
| Product | NDC | Lot Number | Expiration Date | Presentation | Configuration/ Count |
| Propofol Injectable Emulsion, 100 mL Single Patient Use Glass Fliptop Vial |
Vial: 0409-4699-54 Tray: 0409-4699-24 |
EA7470 | 01 JUNE 2023 | 1g/100 mL, Single Patient Use Glass Fliptop Vial |
Tray of 10 Units |
Pfizer places the utmost emphasis on patient safety and product quality at every step in the manufacturing and supply chain process. Pfizer has notified direct consignees by letter to arrange for return of any recalled product.
Wholesalers or hospitals with an existing inventory of the lot, which is being recalled, should discontinue use, stop distribution and quarantine immediately. If you have further distributed the recalled product, please notify your accounts and/or any additional locations which may have received the recalled product. Hospitals/Institutions should inform Healthcare Professionals in your organization of this recall. For additional assistance, call Sedgwick Inc. at 1-800-805-3093 between the hours of 8 a.m. to 5 p.m. ET, Monday through Friday.
Healthcare Professionals with questions regarding this recall can contact Pfizer using the below information.
| Contact Center | Contact Information | Area of Support |
| Pfizer Medical Information | 1-800-438-1985, option 3 (8am to 9pm ET Monday through Friday) www.pfizermedinfo.com | For medical questions regarding the product |
| Pfizer Drug Safety | 1-800-438-1985, option 1 (24 hours a day; 7 days a week) | To report adverse events and product complaints |
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA's MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
- Complete and submit the report Online: www.fda.gov/medwatch/report.htm
- Regular Mail or Fax: Download form www.fda.gov/MedWatch/getforms.htm or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178.
This recall is being executed with the knowledge of the U.S. Food and Drug Administration.
PROPOFOL Injectable Emulsion, USP (contains benzyl alcohol)
100 mL Single Patient Use, Glass Fliptop Vial