Viltepso: Package Insert / Prescribing Info
Package insert / product label
Generic name: viltolarsen
Dosage form: injection, solution
Drug class: Miscellaneous uncategorized agents
J Code (medical billing code): J1427 (10 mg, injection)
Medically reviewed by Drugs.com. Last updated on Jul 27, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
VILTEPSO (viltolarsen) injection, for intravenous use
Initial U.S. Approval: 2020
Recent Major Changes
Indications and Usage for Viltepso
VILTEPSO is an antisense oligonucleotide indicated for the treatment of Duchenne muscular dystrophy (DMD) in patients who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping. This indication is approved under accelerated approval based on an increase in dystrophin production in skeletal muscle observed in patients treated with VILTEPSO. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. (1)
Viltepso Dosage and Administration
- Serum cystatin C, urine dipstick, and urine protein-to-creatinine ratio should be measured before starting VILTEPSO. (2.1)
- Recommended dosage is 80 milligrams per kilogram of body weight once weekly. (2.2)
- Administer as an intravenous infusion over 60 minutes. (2.2, 2.4)
- If the volume of VILTEPSO required is less than 100 mL, dilution in 0.9% Sodium Chloride Injection, USP, is required. (2.3)
Dosage Forms and Strengths
Injection: 250 mg/5 mL (50 mg/mL) in a single-dose vial (3)
Contraindications
None (4)
Warnings and Precautions
Kidney Toxicity: Based on animal data, may cause kidney toxicity. Kidney function should be monitored; creatinine may not be a reliable measure of renal function in DMD patients. (5.1, 13.2)
Adverse Reactions/Side Effects
The most common adverse reactions (incidence ≥15% in patients treated with VILTEPSO) were upper respiratory tract infection, injection site reaction, cough, and pyrexia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact NS Pharma at 1-866 NSPHARM (1-866-677-4276) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2025
Full Prescribing Information
1. Indications and Usage for Viltepso
VILTEPSO is indicated for the treatment of Duchenne muscular dystrophy (DMD) in patients who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping. This indication is approved under accelerated approval based on an increase in dystrophin production in skeletal muscle observed in patients treated with VILTEPSO [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
2. Viltepso Dosage and Administration
2.1 Monitoring to Assess Safety
Serum cystatin C, urine dipstick, and urine protein-to-creatinine ratio should be measured before starting VILTEPSO. Consider measurement of glomerular filtration rate prior to initiation of VILTEPSO. Monitoring for kidney toxicity during treatment is recommended. Obtain the urine samples prior to infusion of VILTEPSO or at least 48 hours after the most recent infusion [see Warnings and Precautions (5.1)].
2.2 Dosing Information
The recommended dosage of VILTEPSO is 80 mg/kg administered once weekly as a 60-minute intravenous infusion.
If a dose of VILTEPSO is missed, it should be administered as soon as possible after the scheduled dose time.
2.3 Preparation Instructions
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Prepare the VILTEPSO dose using aseptic technique.
- Calculate the total dose of VILTEPSO to be administered based on the patient's weight and the recommended dosage of 80 mg/kg. Determine the volume of VILTEPSO needed and the correct number of vials to supply the full calculated dose.
- Allow vials to warm to room temperature. Mix the contents of each vial by gently inverting 2 to 3 times. Do not shake.
- Visually inspect each vial of VILTEPSO. VILTEPSO is a clear and colorless solution. Do not use if the solution in the vials is discolored or particulate matter is present.
- Withdraw the calculated volume of VILTEPSO from the appropriate number of vials.
- If the volume of VILTEPSO required is less than 100 mL, dilution in 0.9% Sodium Chloride Injection, USP is required. Withdraw from the 100-mL infusion bag a volume of 0.9% Sodium Chloride Injection, USP, equivalent to the calculated volume of VILTEPSO and inject the VILTEPSO into the infusion bag, such that the total volume in the bag is 100 mL.
- If the volume of VILTEPSO required is 100 mL or more, dilution is not required, and the required amount of VILTEPSO should be placed into an empty infusion bag.
- Visually inspect the infusion bag containing the solution for particulates. Gently invert the infusion bag to ensure equal distribution of product. Do not shake.
- VILTEPSO contains no preservatives. Infusion should begin as soon as possible, but no more than 5 hours after preparation of VILTEPSO, and be completed within 6 hours of preparation (allowing for 1 hour of infusion time), if diluted solution is stored at 20°C to 26°C (68°F to 79°F). If immediate use is not possible, the solution may be stored for up to 24 hours at 2°C to 8°C (36°F to 46°F). Do not freeze.
- VILTEPSO is supplied in single-dose vials. Discard unused VILTEPSO.
2.4 Administration Instructions
VILTEPSO is administered via intravenous infusion using a peripheral or central venous catheter. Flush the intravenous access line with 0.9% Sodium Chloride Injection, USP, after infusion. Filtration of VILTEPSO is not required.
Infuse VILTEPSO over 60 minutes. Do not mix other medications with VILTEPSO or infuse other medications concomitantly via the same intravenous access line. VILTEPSO should be mixed with 0.9% Sodium Chloride Injection, USP, only.
3. Dosage Forms and Strengths
VILTEPSO is a clear and colorless solution available as follows:
- Injection: 250 mg/5 mL (50 mg/mL) solution in a single-dose vial
5. Warnings and Precautions
5.1 Kidney Toxicity
Kidney toxicity was observed in animals who received viltolarsen [see Use in Specific Populations (8.4)]. Although kidney toxicity was not observed in the clinical studies with VILTEPSO, the clinical experience with VILTEPSO is limited, and kidney toxicity, including potentially fatal glomerulonephritis, has been observed after administration of some antisense oligonucleotides. Kidney function should be monitored in patients taking VILTEPSO. Because of the effect of reduced skeletal muscle mass on creatinine measurements, serum creatinine may not be a reliable measure of kidney function in DMD patients. Serum cystatin C, urine dipstick, and urine protein-to-creatinine ratio should be measured before starting VILTEPSO. Consider also measuring glomerular filtration rate using an exogenous filtration marker before starting VILTEPSO. During treatment, monitor urine dipstick every month, and serum cystatin C and urine protein-to-creatinine ratio every three months. Only urine expected to be free of excreted VILTEPSO should be used for monitoring of urine protein. Urine obtained on the day of VILTEPSO infusion prior to the infusion, or urine obtained at least 48 hours after the most recent infusion, may be used. Alternatively, use a laboratory test that does not use the reagent pyrogallol red, as this reagent has the potential to cross react with any VILTEPSO that is excreted in the urine and thus lead to a false positive result for urine protein.
If a persistent increase in serum cystatin C or proteinuria is detected, refer to a pediatric nephrologist for further evaluation.
6. Adverse Reactions/Side Effects
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In clinical trials with VILTEPSO, 32 patients have been exposed to VILTEPSO once weekly, ranging between 40 mg/kg (0.5 times the recommended dosage) and 80 mg/kg (the recommended dosage), including 16 patients treated for greater than 12 months and 8 patients treated for greater than 24 months as part of an ongoing open-label extension study. All patients were male and had genetically confirmed DMD.
Study 1 was a multicenter, 2-period, dose-finding study conducted in the United States and Canada in males 4 years to less than 10 years of age on a stable corticosteroid regimen for at least 3 months. During the initial period (first 4 weeks) of Study 1, patients were randomized (double-blind) to VILTEPSO or placebo. All patients then received 20 weeks of VILTEPSO 40 mg/kg once weekly (0.5 times the recommended dose) (N=8), or 80 mg/kg once weekly (N=8) [see Clinical Studies (14)].
Study 2 was a multicenter, parallel-group, open-label, dose-finding study conducted in Japan. Eligible patients included ambulatory and non-ambulatory males 5 years to less than 18 years of age who were assigned to receive intravenous VILTEPSO 40 mg/kg once weekly (0.5 times the recommended dose) (N=8) or 80 mg/kg once weekly (N=8) for 24 weeks.
Adverse reactions reported in ≥10% of patients treated with VILTEPSO 80 mg/kg/wk in pooled Studies 1 and 2 are displayed in Table 1. The most common adverse reactions (incidence ≥15% in patients treated with VILTEPSO) were upper respiratory tract infection, injection site reaction, cough, and pyrexia. Patients in the pooled analysis were treated with VILTEPSO for 20 to 24 weeks.
|
* Upper respiratory tract infection includes the following terms: upper respiratory tract infection, nasopharyngitis, and rhinorrhea. |
|
|
** Injection site reaction includes the following terms: injection site bruising, injection site erythema, injection site reaction, and injection site swelling. |
|
| Adverse Reaction | VILTEPSO
80 mg/kg Once Weekly (n=16) % |
| Upper respiratory tract infection* | 63 |
| Injection site reaction** | 25 |
| Cough | 19 |
| Pyrexia | 19 |
| Contusion | 13 |
| Arthralgia | 13 |
| Diarrhea | 13 |
| Vomiting | 13 |
| Abdominal pain | 13 |
| Ejection fraction decreased | 13 |
| Urticaria | 13 |
6.2 Immunogenicity
As with all oligonucleotides, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies may be misleading.
For Study 1, samples collected from all 16 patients at Day 1 (pre-dose), Week 5, Week 13, and Week 24 were assessed for anti-viltolarsen antibodies. All samples were determined to be antibody negative. For the same study, serum samples collected from all 16 patients at Day 1 (pre-dose), Week 13, and Week 24 were analyzed for anti-dystrophin antibodies. Anti-dystrophin antibodies were detected in 1 out of 16 patients (6.25%) at Weeks 13 and 24; however, at Weeks 37, 49, 73, and 97, no anti-dystrophin antibodies were detected in the same patient. Further, this patient achieved a change from baseline in dystrophin levels that was comparable to the mean change in his dosage group (80 mg/kg/week) and there were no adverse events reported with this antibody production. For Study 2, all samples collected from the 16 patients were determined to be both anti-viltolarsen antibody and anti-dystrophin antibody negative. Overall, there was a lack of observed immunogenicity, which indicates that viltolarsen is not highly immunogenic.
Related/similar drugs
8. Use In Specific Populations
8.2 Lactation
Risk Summary
There are no human or animal data to assess the effect of VILTEPSO on milk production, the presence of viltolarsen in milk, or the effects of VILTEPSO on the breastfed infant.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VILTEPSO and any potential adverse effects on the breastfed infant from VILTEPSO or from the underlying maternal condition.
8.4 Pediatric Use
VILTEPSO is indicated for the treatment of DMD in patients who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping, including pediatric patients [see Clinical Studies (14)].
Juvenile Animal Toxicity Data
Viltolarsen (0, 15, 60, 240, or 1200 mg/kg) was administered to juvenile male mice by subcutaneous injection on postnatal day (PND) 7 and by intravenous injection weekly from PND 14 to PND 70. The highest dose resulted in deaths because of renal toxicity. In surviving animals at 240 and 1200 mg/kg, there was a dose-dependent increase in the incidence and severity of renal tubular effects (including degeneration), which were not accompanied by clinical pathology correlates. Reduced body weight gain and delayed sexual maturation were observed at the highest dose tested. At the no-effect dose for renal toxicity (60 mg/kg), plasma exposures were similar to that in humans at the recommended human dose of 80 mg/kg/week.
8.5 Geriatric Use
DMD is largely a disease of children and young adults; therefore, there is no geriatric experience with VILTEPSO.
8.6 Patients with Renal Impairment
VILTEPSO has not been studied in patients with renal impairment. Viltolarsen is mostly excreted unchanged in the urine, and renal impairment may increase its exposure. However, because of the effect of reduced skeletal muscle mass on creatinine measurements in DMD patients, no specific dosage adjustment can be recommended for DMD patients with renal impairment based on estimated glomerular filtration rate. Patients with known renal function impairment should be closely monitored during treatment with VILTEPSO.
11. Viltepso Description
VILTEPSO (viltolarsen) injection is a sterile, preservative-free, aqueous solution for intravenous administration. VILTEPSO is a clear and colorless solution. VILTEPSO is supplied in single-dose vials containing 250 mg/5 mL viltolarsen (50 mg/mL) in 0.9% sodium chloride. Each milliliter of VILTEPSO contains 50 mg viltolarsen and 9 mg sodium chloride in water for injection. The final product is adjusted to a pH ranging between 7.0 and 7.5 using hydrochloric acid and/or sodium hydroxide.
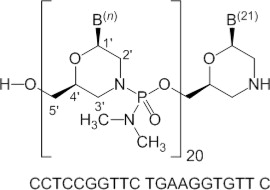
Viltolarsen is an antisense oligonucleotide of the phosphorodiamidate morpholino oligomer (PMO) subclass. PMOs are synthetic molecules in which the five-membered ribofuranosyl rings found in natural DNA and RNA are replaced by a six-membered morpholino ring. Each morpholino ring is linked through an uncharged phosphorodiamidate moiety rather than the negatively charged phosphate linkage that is present in natural DNA and RNA. Each phosphorodiamidate morpholino subunit contains one of the heterocyclic bases found in DNA (adenine, cytosine, guanine, or thymine). Viltolarsen contains 21 linked subunits. The molecular formula of viltolarsen is C244H381N113O88P20 and the molecular weight is 6924.82 daltons. The structure and base sequence of viltolarsen are shown in Figure 1.
12. Viltepso - Clinical Pharmacology
12.1 Mechanism of Action
VILTEPSO is designed to bind to exon 53 of dystrophin pre-mRNA resulting in exclusion of this exon during mRNA processing in patients with genetic mutations that are amenable to exon 53 skipping. Exon 53 skipping is intended to allow for production of an internally truncated dystrophin protein in patients with genetic mutations that are amenable to exon 53 skipping.
12.2 Pharmacodynamics
After treatment with VILTEPSO 80 mg/kg once weekly, all patients evaluated (N=8) were found to produce mRNA for a truncated dystrophin protein, as measured by reverse transcription polymerase chain reaction (RT-PCR), and demonstrated exon 53 skipping, as measured by DNA sequence analysis.
In Study 1, all patients who received VILTEPSO 80 mg/kg once weekly for 20 to 24 weeks showed an increase from baseline in dystrophin protein expression, as quantified by a validated Western blot method (mean 5.3%; median 3.8%; range 0.7% to 13.9% of normal levels when normalized to myosin heavy chain; p-value 0.01). Mass spectrometry, immunofluorescence staining, and RT-PCR results were supportive of the Western blot data [see Clinical Studies (14)]. Expected localization of truncated dystrophin to the sarcolemma in muscle fibers of patients treated with viltolarsen was confirmed by immunofluorescence staining.
12.3 Pharmacokinetics
The pharmacokinetics of viltolarsen was evaluated in DMD patients following administration of intravenous (IV) doses ranging from 1.25 mg/kg/week (0.016 times the recommended dosage) to 80 mg/kg/week (the recommended dosage). Viltolarsen exposure increased proportionally with dose, with minimal accumulation with once-weekly dosing. Inter-subject variability (as %CV) for Cmax and AUC ranged from 16% to 27% respectively.
VILTEPSO is administered as an IV infusion over 60 minutes. Bioavailability is assumed to be 100%, and median Tmax was around 1 hour (end of infusion).
Distribution
The mean viltolarsen steady-state volume of distribution was 300 mL/kg (%CV=14 at a dose of 80 mg/kg. Viltolarsen plasma protein binding ranged from 39% to 40% and is not concentration dependent.
Elimination
Specific Populations
Age, Sex & Race
The pharmacokinetics of viltolarsen have been evaluated only in male pediatric DMD patients.There is no experience with VILTEPSO in patients 65 years of age or older. No marked differences in any PK parameters were observed between White and Asian patients.
Patients with Renal or Hepatic Impairment
VILTEPSO has not been studied in patients with renal or hepatic impairment. Viltolarsen was found to be metabolically stable, and hepatic metabolism does not contribute to the elimination of viltolarsen. In addition, viltolarsen was mainly excreted unchanged in the urine. Viltolarsen is eliminated renally, and renal impairment is expected to result in increasing exposure of viltolarsen. However, because of the effect of reduced skeletal muscle mass on creatinine measurements in DMD patients, no specific dosage adjustment can be recommended for DMD patients with renal impairment based on glomerular filtration rate estimated by serum creatinine [see Use in Specific Populations (8.6)].
In Vitro Drug Interaction Studies
Viltolarsen did not inhibit CYP3A4/5, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, UGT1A1, or UGT2B7. Viltolarsen did not induce CYP1A2, CYP2B6, or CYP3A4.
Viltolarsen is not metabolized by CYP enzymes and is not a substrate of transporters BCRP, BSEP, MDR1, OAT1, OAT3, OCT1, OCT2, MATE1, or MATE2-K. Viltolarsen did not inhibit the transporters tested (OATP1B1, OATP1B3, OAT3, BCRP, MDR1, BSEP, OAT1, OCT1, OCT2, MATE1, and MATE2-K).
Based on in vitro data, viltolarsen has a low potential for drug-drug interactions with major CYP enzymes and drug transporters in humans.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Mutagenesis
Viltolarsen was negative for genotoxicity in in vitro (bacterial reverse mutation, chromosomal aberration in Chinese hamster lung cells) and in vivo (mouse bone marrow micronucleus) assays.
Impairment of Fertility
Intravenous administration of viltolarsen (0, 60, 240, or 1000 mg/kg) to male mice weekly prior to and during mating to untreated females did not have adverse effects on fertility. Plasma exposure (AUC) at the highest dose was approximately 18 times that in humans at the recommended human dose of 80 mg/kg/week.
14. Clinical Studies
The effect of VILTEPSO on dystrophin production was evaluated in one study in DMD patients with a confirmed mutation of the DMD gene that is amenable to exon 53 skipping (Study 1; NCT02740972).
Study 1 was a multicenter, 2-period, dose-finding study conducted in the United States and Canada.
During the initial period (first 4 weeks) of Study 1, patients were randomized (double blind) to VILTEPSO or placebo. All patients then received 20 weeks of open-label VILTEPSO 40 mg/kg once weekly (0.5 times the recommended dosage) (N=8) or 80 mg/kg once weekly (N=8). Study 1 enrolled ambulatory male patients 4 years to less than 10 years of age (median age 7 years) on a stable corticosteroid regimen for at least 3 months.
Efficacy was assessed based on change from baseline in dystrophin protein level (measured as % of the dystrophin level in healthy subjects, i.e., % of normal) at Week 25. Muscle biopsies (left or right biceps brachii) were collected from patients at baseline and following 24 weeks of VILTEPSO treatment, and analyzed for dystrophin protein level by Western blot normalized to myosin heavy chain (primary endpoint) and mass spectrometry (secondary endpoint).
In patients who received VILTEPSO 80 mg/kg once weekly, mean dystrophin levels increased from 0.6% (SD 0.8) of normal at baseline to 5.9% (SD 4.5) of normal by Week 25, with a mean change in dystrophin of 5.3% (SD 4.5) of normal levels (p=0.01) as assessed by validated Western blot (normalized to myosin heavy chain); the median change from baseline was 3.8%. All patients demonstrated an increase in dystrophin levels over their baseline values. As assessed by mass spectrometry (normalized to filamin C), mean dystrophin levels increased from 0.6% (SD 0.2) of normal at baseline to 4.2% (SD 3.7) of normal by Week 25, with a mean change in dystrophin of 3.7% (SD 3.8) of normal levels (nominal p=0.03, not adjusted for multiple comparisons); the median change from baseline was 1.9%.
Individual patient dystrophin levels in patients evaluated in Study 1 are shown in Figure 2 and Table 2.
Figure 2: Dystrophin Expression in Individual Patients (Study 1)
Patients Treated With VILTEPSO 80 mg/kg/week (n=8)
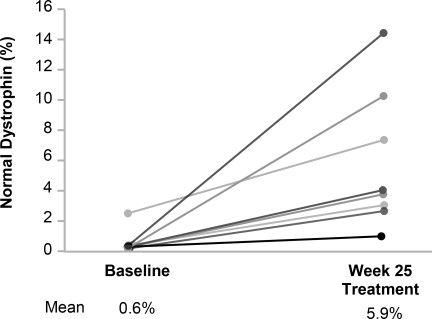
Note: Solid lines represent individual patient data. Dystrophin was measured using Western blot and normalized to myosin heavy chain.
|
a Data were normalized by myosin heavy chain |
|||
| Patient
Number | Western Blot % Normal Dystrophina | ||
| Baseline | Week 25 | Change from Baseline | |
| 1 | 0.46 | 1.14 | 0.69 |
| 2 | 0.40 | 3.97 | 3.57 |
| 3 | 0.46 | 2.97 | 2.51 |
| 4 | 0.09 | 10.40 | 10.31 |
| 5 | 0.51 | 14.42 | 13.91 |
| 6 | 2.61 | 7.40 | 4.79 |
| 7 | 0.43 | 3.06 | 2.63 |
| 8 | 0.09 | 4.07 | 3.98 |
16. How is Viltepso supplied
17. Patient Counseling Information
Kidney Toxicity
Inform patients nephrotoxicity has occurred with drugs similar to VILTEPSO. Advise patients of the importance of monitoring for kidney toxicity by their healthcare providers during treatment with VILTEPSO [see Warnings and Precautions (5.1)].
Manufactured for:
NS Pharma, Inc.
Paramus, NJ 07652

| VILTEPSO
viltolarsen injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - NS Pharma, Inc. (135666654) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Ajinomoto Co., Inc. | 706261286 | API MANUFACTURE(73292-011) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| AndersonBrecon Inc. | 053217022 | LABEL(73292-011) , PACK(73292-011) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Fuji Yakuhin Co., Ltd. | 713746022 | MANUFACTURE(73292-011) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| YMC Co., Ltd. | 718477255 | API MANUFACTURE(73292-011) | |
Frequently asked questions
- What are the latest drug treatments for DMD?
- How long does it take for Viltepso to work?
- What is Viltepso's mechanism of action?
More about Viltepso (viltolarsen)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: miscellaneous uncategorized agents
- En español