Perikabiven: Package Insert / Prescribing Info
Package insert / product label
Generic name: amino acids, electrolytes, dextrose and lipid
Dosage form: injection, emulsion
Drug class: Intravenous nutritional products
Medically reviewed by Drugs.com. Last updated on Aug 20, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
PERIKABIVEN® (amino acids, electrolytes, dextrose and lipid injectable emulsion), for intravenous use
Initial U.S. Approval: 2014
Recent Major Changes
Indications and Usage for Perikabiven
PERIKABIVEN is indicated as a source of calories, protein, electrolytes and essential fatty acids for adult patients requiring parenteral nutrition when oral or enteral nutrition is not possible, insufficient, or contraindicated.
PERIKABIVEN may be used to prevent essential fatty acid deficiency or treat negative nitrogen balance in adult patients. (1)
Limitations of Use:
Not recommended for use in pediatric patients <2 years including preterm infants because the fixed content of the formulation does not meet nutritional requirements in this age group. (1, 5.1, 8.4)
Perikabiven Dosage and Administration
Dosage Forms and Strengths
Contraindications
- Concomitant treatment with ceftriaxone in neonates (28 days of age or younger). (4)
- Known hypersensitivity to egg, soybean, peanut or any of the active ingredients or excipients. (4)
- Severe disorders of lipid metabolism characterized by hypertriglyceridemia (serum triglycerides >1,000 mg/dL). (4, 5.10)
- Inborn errors of amino acid metabolism. (4)
- Cardiopulmonary instability. (4)
- Hemophagocytic syndrome. (4)
Warnings and Precautions
- Clinical Decompensation with Rapid Infusion of Intravenous Lipid Emulsion in Neonates and Infants: Acute respiratory distress, metabolic acidosis, and death after rapid infusion of intravenous lipid emulsions have been reported. (5.1)
- Parenteral Nutrition-Associated Liver Disease: Increased risk in patients who receive parenteral nutrition for greater than 2 weeks. Monitor liver tests; if abnormalities occur, consider discontinuation or dosage reduction. (5.2)
- Pulmonary Embolism and Respiratory Distress due to Pulmonary Vascular Precipitates: If signs of pulmonary distress occur, stop the infusion and initiate a medical evaluation. (5.3)
- Hypersensitivity Reactions: Monitor for signs or symptoms and discontinue infusion if reactions occur. (5.4)
- Precipitation with Ceftriaxone: Do not administer ceftriaxone simultaneously with PERIKABIVEN via a Y-Site (4, 5.5, 8.4)
- Infection, fat overload, hyperglycemia and refeeding complications: Monitor for signs and symptoms; monitor laboratory parameters. (5.6, 5.7, 5.8, 5.9, 5.14)
Adverse Reactions/Side Effects
The most common adverse reactions (≥3%) are hyperglycemia, hypokalemia, pyrexia, and increased blood triglycerides. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Fresenius Kabi USA, LLC at 1-800-551-7176 or FDA at 1-800-FDA-1088 or
www.fda.gov/medwatch.
Drug Interactions
Coumarin and coumarin derivatives, including warfarin: Anticoagulant activity may be counteracted; monitor laboratory parameters. (7.1)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2023
Full Prescribing Information
1. Indications and Usage for Perikabiven
PERIKABIVEN is indicated as a source of calories, protein, electrolytes and essential fatty acids for adult patients requiring parenteral nutrition when oral or enteral nutrition is not possible, insufficient, or contraindicated. PERIKABIVEN may be used to prevent essential fatty acid deficiency or treat negative nitrogen balance in adult patients.
Limitations of Use:
PERIKABIVEN is not recommended for use in pediatric patients under the age of 2 years, including preterm infants because the fixed content of the formulation does not meet the nutritional requirements of this age group [see Warnings and Precautions (5.1) and Use in Specific Populations (8.4)].
2. Perikabiven Dosage and Administration
2.1 Administration
- PERIKABIVEN is for intravenous infusion into a peripheral or central vein [see Warnings and Precautions (5.11)].
- Use a 1.2 micron in-line filter.
- Use of a vented intravenous administration set with the vent in the open position could result in air embolism.
- Use a dedicated line without any connections. Multiple connections could result in air embolism due to residual air being drawn from the primary container before administration of the fluid from the secondary container is completed.
- Do not exceed the recommended maximum infusion rate of 3.7 mL/kg/hour [see Dosage and Administration (2.4) and Warnings and Precautions (5.1)].
- Ceftriaxone must not be administered simultaneously with calcium-containing intravenous solutions such as PERIKABIVEN via a Y-site due to precipitation. However, in patients other than neonates, ceftriaxone and PERIKABIVEN may be administered sequentially if the infusion lines are thoroughly flushed between infusions with a compatible fluid [see Contraindications (4), Warnings and Precautions (5.5)].
- Do not use administration sets and lines that contain di-2-ethylhexyl phthalate (DEHP). Administration sets that contain polyvinyl chloride (PVC) components have DEHP as a plasticizer.
2.2 Important Preparation Instructions
- Inspect the bag prior to activation. Discard the bag in the following situations:
- Evidence of damage to the bag
- More than one chamber is white
- Solution is yellow
- Any seal is already broken
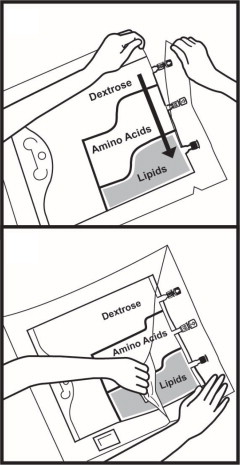
- Activate the bag [see Dosage and Administration (2.3)].
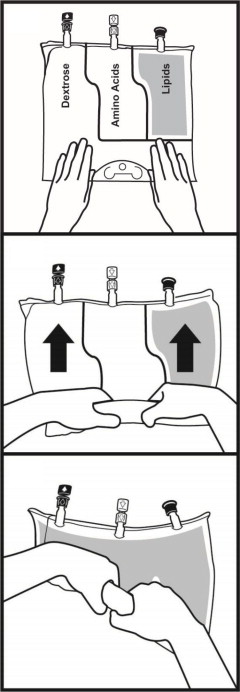
- Once the bag is activated, ensure the vertical seals between chambers are broken at least from the bend in the seals and down to the ports. The upper sections of the vertical seals above the bend and the horizontal seal may remain closed.
- It is recommended to mix the contents thoroughly by inverting the bag upside down to ensure a homogenous admixture.
- Ensure the vertical seals between chambers are broken and the contents of all three chambers are mixed together prior to infusion [see Dosage and Administration (2.3)].
- Use PERIKABIVEN immediately after mixing and the introduction of additives. If not used immediately, the storage time and conditions prior to use should not be longer than 24 hours at 2° to 8°C (36° to 46°F). After removal from storage at 2° to 8°C (36° to 46°F), the admixture should be infused within 24 hours. Any mixture remaining must be discarded.
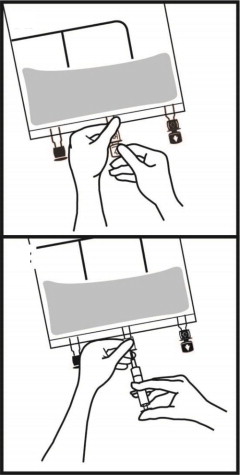
- For total parenteral nutrition add multivitamins and trace elements via the additive port. Any other additions to the bag should be evaluated by a pharmacist for compatibility. Questions about compatibility may be directed to Fresenius Kabi USA, LLC.
- When introducing additives, it is recommended to use 18 to 23 gauge needles with a maximum length of 1.5 inches (40 mm) and to mix thoroughly after each addition, use aseptic technique and add after the vertical seals have been broken (i.e., bag has been activated) and the three components are mixed [see Dosage and Administration (2.3)].
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Inspect PERIKABIVEN to ensure:
- Precipitates have not formed during the mixing or addition of additives.
- The emulsion has not separated. Separation of the emulsion can be visibly identified by a yellowish streaking or the accumulation of yellowish droplets in the mixed emulsion.
Discard the admixture if any of the above are observed.
2.3 Instructions for Use
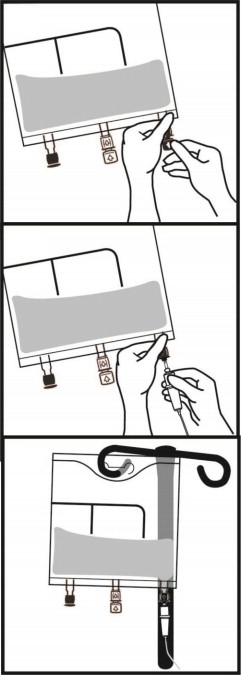
2.4 Dosing Considerations
The dosage of PERIKABIVEN should be individualized based on the patient's clinical condition (ability to adequately metabolize amino acids, dextrose and lipids), body weight and nutritional/fluid requirements, as well as additional energy given orally/enterally to the patient.
PERIKABIVEN is a combination of amino acids, electrolytes, dextrose, and lipids in a fixed volume and concentration. The dosage selection is based upon fluid requirements which can be used in conjunction with the nutritional requirements to determine final dosage [see Table 1].
PERIKABIVEN meets the total nutritional requirements for protein, dextrose and lipids in stable patients, and can be individualized to meet specific needs with the addition of nutrients. The maximum infusion rate is based upon the dextrose component.
Prior to administration of PERIKABIVEN, correct severe fluid, electrolyte and acid-base disorders. Before starting the infusion, obtain serum triglyceride levels to establish the baseline value.
Recommended Adult Dosage
The recommended dosage of PERIKABIVEN in adults is 27 to 40 mL/kg/day. The amount of macronutrients provided by PERIKABIVEN are shown in Table 1.
The maximum daily dosage of PERIKABIVEN in adults should not exceed 40 mL/kg/day.
In patients with serum triglyceride concentrations above 400 mg/dL, stop the PERIKABIVEN infusion and monitor serum triglyceride levels. Once the triglycerides are <400 mg/dL, restart PERIKABIVEN at a lower infusion rate and advance rate in smaller increments towards target dosage, checking the triglyceride levels prior to each adjustment [see Contraindications (4) and Warnings and Precautions (5.13)].
|
* Protein is provided as amino acids. When infused intravenously amino acids are metabolized and utilized as the building blocks of protein. |
|
|
** As Dextrose monohydrate |
|
| Nutrition Provided by PERIKABIVEN recommended dosage | |
| Fluid mL/kg/day | 27 to 40 |
| Protein* g/kg/day Nitrogen g/kg/day | 0.64 to 0.94 0.1 to 0.15 |
| Dextrose ** g/kg/day | 2.03 to 3 |
| Lipids g/kg/day | 0.95 to 1.4 |
| Total Energy Requirement kcal/kg/day | 18 to 27 |
Treatment with PERIKABIVEN may be continued for as long as is required by the patient's condition.
Dosing in Renal Impairment
In patients with renal impairment, the dosage of PERIKABIVEN should be the recommended adult dosage (see above). Prior to administration, correct severe fluid or electrolyte imbalances. Closely monitor serum electrolyte levels and adjust the volume of PERIKABIVEN administered as required [see Warnings and Precautions (5.12)].
Renal patients not needing dialysis require 0.6 to 0.8 g of protein/kg/day. Patients on dialysis or continuous renal replacement therapy should receive 1.2 to 1.8 g of protein/kg/day up to a maximum of 2.5 g of protein/kg/day based on nutritional status and estimated protein losses. The PERIKABIVEN dosage can be adjusted based on the treatment for the renal impairment, supplementing protein as indicated. Additional protein may be added to PERIKABIVEN bag or infused separately. If required, additional amino acids may be added to the PERIKABIVEN bag or infused separately.
Infusion Duration and Rate
The recommended duration of infusion for PERIKABIVEN is between 12 and 24 hours, depending on the clinical situation.
The maximum infusion rate of PERIKABIVEN is 3.7 mL/kg/hour. This corresponds to 0.09 g/kg/hour of amino acids, 0.28 g/kg/hour of dextrose (the rate limiting factor) and 0.13 g/kg/hour of lipids.
Dosing Instructions
- Determine the fluid requirements (27 to 40 mL/kg/day) and the patient's nutritional requirements to be delivered, then select the corresponding PERIKABIVEN bag.
- Determine the preferred duration of infusion (12 to 24 hours).
- Ensure that the rate of infusion (PERIKABIVEN dosage in mL/kg/day divided by the preferred duration of infusion (hours)) does not exceed the maximum infusion rate for the patient (i.e., 3.7 mL/kg/hour). The infusion rate may need to be reduced and duration of infusion increased in order not to exceed the maximum infusion rate.
- Once the infusion rate in mL/kg/hour has been selected, calculate the infusion rate (mL/hour) using the patient's weight.
- Compare the patient's nutrient requirements with the amount supplied by PERIKABIVEN. Discuss with a pharmacist any additions that may be required.
3. Dosage Forms and Strengths
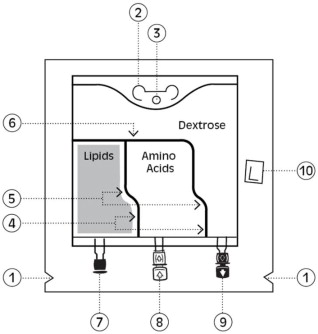
PERIKABIVEN is a sterile, hypertonic emulsion in a three chamber container. The individual chambers contain one of the following respectively: amino acids and electrolytes, dextrose, or lipid injectable emulsion. Table 2 describes the individual components of PERIKABIVEN.
|
1. Balanced by ions from amino acids |
||||
|
2. Contributed by sodium glycerophosphate and sodium acetate |
||||
|
3. Contributed by sodium glycerophosphate and phospholipids |
||||
|
4. Derived from sodium acetate and glacial acetic acid (for pH adjustment) |
||||
|
5. Contributed by calcium chloride, lysine hydrochloride, and potassium chloride |
||||
|
6. Derived from magnesium sulfate |
||||
|
7. Total caloric value including lipid, phospholipid and glycerin |
||||
|
8. pH of amino acid with electrolyte solution was adjusted with glacial acetic acid, USP and pH of lipid emulsion was adjusted with sodium hydroxide, USP |
||||
|
9. Calculated on the basis of 3.4 kcal/g of dextrose, monohydrate |
||||
| How Supplied | 1,440 mL | 1,920 mL | ||
| Composition of PERIKABIVEN | ||||
| Soybean Oil, USP (g/100 mL) | 3.5 | |||
| Dextrose Monohydrate, USP (g/100 mL) | 7.5 | |||
| Amino Acids, USP (g/100 mL) | 2.36 | |||
| Total Nitrogen (mg/100 mL) | 375 | |||
|
Essential amino acids (mg/100 mL) | Lysine, USP (added as the hydrochloride salt) | 187 | ||
| Phenylalanine, USP | 164 | |||
| Leucine, USP | 164 | |||
| Valine, USP | 152 | |||
| Histidine, USP | 141 | |||
| Threonine, USP | 116 | |||
| Methionine, USP | 116 | |||
| Isoleucine, USP | 116 | |||
| Tryptophan, USP | 40 | |||
|
Nonessential amino acids (mg/100 mL) | Alanine, USP | 333 | ||
| Arginine, USP | 235 | |||
| Glycine, USP | 164 | |||
| Proline, USP | 141 | |||
| Glutamic Acid | 116 | |||
| Serine, USP | 94 | |||
| Aspartic Acid, USP | 71 | |||
| Tyrosine, USP | 4.8 | |||
| Electrolytes (mg/100 mL) | Sodium Acetate Trihydrate, USP | 170 | ||
| Potassium Chloride, USP | 124 | |||
| Sodium Glycerophosphate Anhydrous | 105 | |||
| Magnesium Sulfate Heptahydrate, USP | 68 | |||
| Calcium Chloride Dihydrate, USP | 20 | |||
|
Electrolyte Profile1 (mEq/L) | Sodium2 | 22 (22 mmol/L) | ||
| Potassium | 17 (17 mmol/L) | |||
| Magnesium | 5.6 (2.8 mmol/L) | |||
| Calcium | 2.8 (1.4 mmol/L) | |||
| Phosphorous3 | N.A. (7.5 mmol/L) | |||
| Acetate4 | 27 (27 mmol/L) | |||
| Chloride5 | 32 (32 mmol/L) | |||
| Sulfate6 | 5.6 (2.8 mmol/L) | |||
| Calorie Content (kcal/L) | From Dextrose9 | 255 | ||
| From Lipid | 3507 | |||
| From Amino Acids | 95 | |||
| Total | 700 | |||
| pH8 | 5.6 | |||
| Osmolarity (mOsm/L) | 750 | |||
4. Contraindications
The use of PERIKABIVEN is contraindicated in:
- Neonates (28 days of age or younger) receiving concomitant treatment with ceftriaxone, even if separate infusion lines are used, due to the risk of fatal ceftriaxone calcium salt precipitation in the neonate's bloodstream [see Limitations of Use (1), Warnings and Precautions (5.5), Use in Specific Populations (8.4)].
- Patients with known hypersensitivity to egg, soybean, peanut or any of the active or inactive ingredients in PERIKABIVEN [see Warnings and Precautions (5.4)];
- Patients with severe disorders of lipid metabolism characterized by hypertriglyceridemia (serum triglyceride concentration >1,000 mg/dL) [see Warnings and Precautions (5.10)].
- Patients with inborn errors of amino acid metabolism
- Patients with cardiopulmonary instability (including pulmonary edema, cardiac insufficiency, myocardial infarction, acidosis and hemodynamic instability requiring significant vasopressor support)
- Patients with hemophagocytic syndrome
5. Warnings and Precautions
5.1 Clinical Decompensation with Rapid Infusion of Intravenous Lipid Emulsion in Neonates and Infants
In the postmarketing setting, serious adverse reactions including acute respiratory distress, metabolic acidosis, and death have been reported in neonates and infants after rapid infusion of intravenous lipid emulsions. Hypertriglyceridemia was commonly reported.
Preterm and small for gestational age infants have poor clearance of intravenous lipid emulsion and increased free fatty acid plasma levels following lipid emulsion infusion.
5.2 Parenteral Nutrition-Associated Liver Disease and Other Hepatobiliary Disorders
Risk of Parenteral Nutrition-Associated Liver Disease
Parenteral nutrition-associated liver disease (PNALD), also referred to as intestinal failure-associated liver disease (IFALD), can present as cholestasis or hepatic steatosis, and may progress to steatohepatitis with fibrosis and cirrhosis (possibly leading to chronic hepatic failure). The etiology of PNALD is multifactorial; however, intravenously administered phytosterols (plant sterols) contained in plant-derived lipid emulsions, such as Intralipid (included in PERIKABIVEN), have been associated with development of PNALD.
In a randomized study of neonates and infants expected to be treated with PN for at least 28 days, parenteral nutrition-associated cholestasis (PNAC), a precursor to PNALD, developed more frequently in Intralipid-treated patients than in patients treated with a 4-oil mixed lipid emulsion [see Adverse Reactions (6.1), Use in Specific Populations (8.4)].
Monitor liver tests in patients treated with PERIKABIVEN and consider discontinuation or dosage reduction if abnormalities occur.
Other Hepatobiliary Disorders
Hepatobiliary disorders including cholecystitis and cholelithiasis have developed in some PN-treated patients without preexisting liver disease.
Monitor liver tests when administering PERIKABIVEN. Patients developing signs of hepatobiliary disorders should be assessed early to determine whether these conditions are related to PERIKABIVEN use.
5.3 Pulmonary Embolism and Respiratory Distress due to Pulmonary Vascular Precipitates
Pulmonary vascular precipitates causing pulmonary emboli (including some fatalities) and respiratory distress have been reported in patients receiving parenteral nutrition.
Excessive addition of calcium and phosphate increases the risk of the formation of calcium phosphate precipitates; however, precipitates have been reported even in the absence of phosphate salt in the solution. Precipitation following passage through an in-line filter and suspected in vivo precipitate formation has also been reported.
Visually inspect the prepared solution, the infusion set, and catheter for precipitates, prior to administration as well as periodically during the administration. If signs of respiratory distress or pulmonary embolism occur, stop the PERIKABIVEN infusion and initiate a medical evaluation.
5.4 Hypersensitivity Reactions
PERIKABIVEN contains soybean oil, which may cause hypersensitivity reactions. Cross reactions have been observed between soybean and peanut. PERIKABIVEN is contraindicated in patients with known hypersensitivity to egg, soybean, peanut or any of the active or inactive ingredients in PERIKABIVEN. If a hypersensitivity reaction occurs, stop infusion of PERIKABIVEN immediately and initiate appropriate treatment and supportive measures.
5.5 Precipitation with Ceftriaxone
Precipitation of ceftriaxone-calcium can occur when ceftriaxone is mixed with calcium-containing parenteral nutrition solutions, such as PERIKABIVEN in the same intravenous administration line. Do not administer ceftriaxone simultaneously with PERIKABIVEN via a Y-site.
However, in patients other than neonates, ceftriaxone and PERIKABIVEN may be administered sequentially if the infusion lines are thoroughly flushed between infusions with a compatible fluid [see Dosage and Administration (2.1)].
Deaths have occurred in neonates (28 days of age or younger) who received concomitant intravenous calcium-containing solutions with ceftriaxone resulting from calcium-ceftriaxone precipitates in the lungs and kidneys, even when separate infusion lines were used [see Contraindications (4), Pediatric Use (8.4)].
5.6 Infections
Parenteral nutrition, such as PERIKABIVEN, can support microbial growth and is an independent risk factor for the development of catheter-related bloodstream infections. To decrease the risk of infectious complications, ensure aseptic techniques are used for catheter placement, catheter maintenance, and preparation and administration of PERIKABIVEN.
Monitor for signs and symptoms of infection including fever and chills, as well as laboratory test results that might indicate infection (including leukocytosis and hyperglycemia). Perform frequent checks of the intravenous catheter insertion site for edema, redness, and discharge.
5.7 Fat Overload Syndrome
Fat overload syndrome is a rare condition that has been reported with intravenous lipid formulations and is characterized by a sudden deterioration in the patient's condition (e.g., fever, anemia, leukopenia, thrombocytopenia, coagulation disorders, hyperlipidemia, hepatomegaly, deteriorating liver function, and central nervous system manifestations such as coma). A reduced or limited ability to metabolize lipids, accompanied by prolonged plasma clearance (resulting in higher lipid levels), may result in this syndrome. Although fat overload syndrome has been most frequently observed when the recommended lipid dose or infusion rate was exceeded, cases have also been described when the lipid formulation was administered according to instructions.
If signs or symptoms of fat overload syndrome occur, stop PERIKABIVEN. The syndrome is usually reversible when the infusion including the lipid emulsion is stopped.
5.8 Refeeding Syndrome
Administering PN to severely malnourished patients with parenteral nutrition may result in refeeding syndrome, characterized by the intracellular shift of potassium, phosphorus, and magnesium as patients become anabolic. Thiamine deficiency and fluid retention may also develop. To prevent these complications, closely monitor severely undernourished patients and slowly increase their nutrient intake.
5.9 Diabetes and Hyperglycemia
Administration of dextrose at a rate exceeding the patient's utilization rate may lead to hyperglycemia, hyperosmolar coma, and death. Monitor blood glucose levels and treat hyperglycemia to maintain optimal glucose levels while infusing PERIKABIVEN. Insulin may be administered or adjusted to maintain optimal blood glucose levels during PERIKABIVEN administration.
5.10 Hypertriglyceridemia
The use of PERIKABIVEN is contraindicated in patients with hypertriglyceridemia with serum triglyceride concentrations >1,000 mg/dL.
Patients with conditions such as inherited lipid disorders, obesity, diabetes mellitus, or metabolic syndromes have a higher risk of developing hypertriglyceridemia with the use of PERIKABIVEN. In addition, patients with hypertriglyceridemia may have worsening of their hypertriglyceridemia with administration of PERIKABIVEN. Excessive dextrose administration may further increase such risk.
Evaluate patients' capacity to eliminate and metabolize the infused lipid emulsion by measuring serum triglycerides before the start of infusion (baseline value), with each increase in dosage, and regularly throughout treatment. If triglyceride levels are above 400 mg/dL in adults, stop the PERIKABIVEN infusion and monitor serum triglyceride levels to avoid clinical consequences of hypertriglyceridemia such as pancreatitis.
To minimize the risk of new or worsening of hypertriglyceridemia, assess high-risk patients for their overall energy intake including other sources of lipid and dextrose, as well as concomitant drugs that may affect lipid and dextrose metabolism.
5.11 Vein Damage and Thrombosis
The infusion of hypertonic nutrient injections into a peripheral vein may result in vein irritation, vein damage, and/or thrombosis. PERIKABIVEN is indicated for peripheral administration, or may be infused into a central vein; however, peripheral catheters should not be used for solutions with osmolarity of ≥ 900 mOsm/L. The catheter should be removed as soon as possible if thrombophlebitis develops.
5.12 Electrolyte Imbalance and Fluid Overload in Patients with Decreased Renal Function
Patients with decreased renal function, including those with pre-renal azotemia, renal obstruction, or intrinsic renal disease, may be at increased risk of electrolyte and fluid volume imbalance when receiving PN, including PERIKABIVEN. In patients with decreased renal function with electrolyte imbalance or fluid overload, the PERIKABIVEN should be used with caution in patients with renal impairment. PERIKABIVEN dosage (e.g., fluid, protein, and electrolyte content) may require adjustment.
Monitor renal function parameters. Patients developing signs of decreased renal function should be assessed early by a clinician knowledgeable in renal disease in order to determine the appropriate PERIKABIVEN dosage and other treatment options.
5.13 Aluminum Toxicity
PERIKABIVEN contains no more than 25 mcg/L of aluminum.
The aluminum contained in PERIKABIVEN may reach toxic levels with prolonged parenteral administration in patients with impaired kidney function. Preterm infants are at greater risk because their kidneys are immature, and they require large amounts of calcium and phosphate solutions that contain aluminum. Patients with impaired kidney function, including preterm infants, who receive parenteral levels of aluminum at greater than 4 to 5 mcg/kg/day, accumulate aluminum at levels associated with central nervous system and bone toxicity. Tissue loading may occur at even lower rates of administration of total parenteral nutrition products.
5.14 Monitoring/Laboratory Tests
Monitor fluid status closely in patients with pulmonary edema or heart failure.
Throughout treatment, monitor serum triglycerides [see Warnings and Precautions (5.13)], fluid and electrolyte status, serum osmolarity, blood glucose, liver and kidney function, blood count (including platelets), and coagulation parameters.
PERIKABIVEN contains Vitamin K that may counteract anticoagulant activity [see Drug Interactions (7)].
The lipids contained in PERIKABIVEN may interfere with some laboratory tests (e.g., hemoglobin, triglycerides, lactate dehydrogenase, bilirubin, and oxygen saturation) if blood is sampled before the lipids in PERIKABIVEN have cleared from the bloodstream. Conduct these tests at least 6 hours after stopping the infusion.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are discussed in greater detail in other sections of the prescribing information.
- Clinical Decompensation with Rapid Infusion of Intravenous Lipid Emulsion in Neonates and Infants [see Warnings and Precautions (5.1)].
- Parenteral Nutrition-Associated Liver Disease and Other Hepatobiliary Disorders [see Warnings and Precautions (5.2)].
- Pulmonary Embolism and Respiratory Distress due to Pulmonary Vascular Precipitates [see Warnings and Precautions (5.3)].
- Hypersensitivity Reactions [see Warnings and Precautions (5.4)].
- Precipitation with Ceftriaxone [see Warnings and Precautions (5.5)].
- Infections [see Warnings and Precautions (5.6)].
- Fat Overload Syndrome [see Warnings and Precautions (5.7)].
- Refeeding Syndrome [see Warnings and Precautions (5.8)].
- Diabetes and Hyperglycemia [see Warnings and Precautions (5.9)].
- Hypertriglyceridemia [see Warnings and Precautions (5.10)].
- Vein Damage and Thrombosis [see Warnings and Precautions (5.11)].
- Electrolyte Imbalance and Fluid Overload in Patients with Decreased Renal Function [see Warnings and Precautions (5.12)].
- Aluminum Toxicity [see Warnings and Precautions (5.13)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The clinical data described for PERIKABIVEN reflects exposure in 93 patients exposed for 5 to 7 days in 4 active-controlled trials. The pooled population exposed to PERIKABIVEN was 18 to 87 years old, 48% female, 73% Caucasian. The enrolled patients had varied underlying conditions such as gastrointestinal disorders (55%), vascular disorders (30%), metabolism and nutrition disorders (28%), respiratory, thoracic, and mediastinal disorders (22%), and psychiatric disorders (20%). Most patients received peripheral intravenous infusion doses of ≥80% of their target mean daily exposure.
Adverse reactions occurring in at least 2% of patients who received PERIKABIVEN are shown in Table 3.
|
* Terms as reported in clinical studies |
|
| Adverse reaction | PERIKABIVEN N=93 (%) |
| Hyperglycemia* | 5 (5) |
| Hypokalemia | 4 (4) |
| Pyrexia | 4 (4) |
| Blood triglycerides increased | 3 (3) |
| Phlebitis | 2 (2) |
| Nausea | 2 (2) |
| Pruritus | 2 (2) |
| Gamma-glutamyltransferase increased | 2 (2) |
| Blood alkaline phosphatase increased | 2 (2) |
| Alanine aminotransferase increased | 2 (2) |
| Blood glucose increased* | 2 (2) |
| C-reactive protein increased | 2 (2) |
| Blood urea increased | 2 (2) |
| Hypoalbuminemia | 2 (2) |
Less common adverse reactions in ≤1% of patients who received PERIKABIVEN were hyperkalemia, hypomagnesaemia, hypernatremia, tachycardia, hypertension, thrombophlebitis, vomiting, jaundice, rash and increased blood bilirubin.
In a randomized active-controlled, double-blind, parallel-group, multi-center study that included 152 neonates and 9 patients ranging in age from 29 to 153 days who were expected to require PN for at least 28 days, parenteral nutrition-associated cholestasis (PNAC), a precursor to PNALD, developed more frequently in Intralipid-treated patients than in patients treated with a 4-oil mixed lipid emulsion. Intralipid is the lipid emulsion component of PERIKABIVEN.
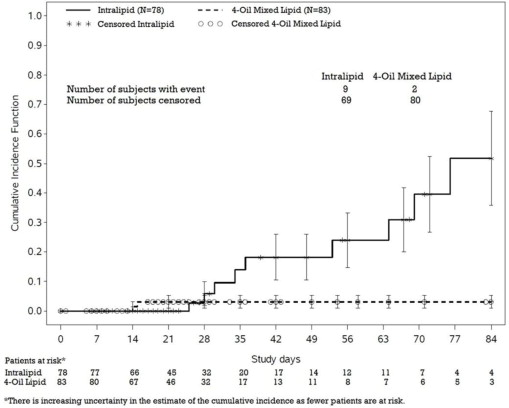
PNAC (defined as direct bilirubin >2mg/dl with a second confirmed elevation >2mg/dl at least 7 days later) occurred in 11.5% (9/78) of Intralipid-treated patients and 2.4% (2/83) of patients treated with a 4-oil mixed lipid emulsion. Most PNAC events occurred in patients who were treated for longer than 28 days.
The estimated cumulative incidence of PNAC is shown in the Kaplan-Meier cumulative incidence curve in Figure 1.
Figure 1: Cumulative Incidence Curve of Time to Parenteral Nutrition-Associated Cholestasis (PNAC) with Standard Error Bars
Monitor liver tests in patients treated with PERIKABIVEN and consider discontinuation or dosage reduction if abnormalities occur.
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during post-approval use of PERIKABIVEN in countries where it is registered. Because these reactions are reported voluntarily post-approval from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to product exposure.
- Gastrointestinal disorders: abdominal distension, abdominal pain
- General disorders and administration site conditions: chest tightness
- Hepatobiliary disorders: cholestasis
- Immune system disorders: allergic reaction, anaphylaxis
- Infections and infestations: infection
- Vascular disorders: flushed face
Related/similar drugs
7. Drug Interactions
7.1 Ceftriaxone
Precipitation of ceftriaxone-calcium can occur when ceftriaxone is mixed with calcium-containing parenteral nutrition solutions, such as PERIKABIVEN, in the same intravenous administration line. Do not administer ceftriaxone simultaneously with PERIKABIVEN, via a Y-site. However, ceftriaxone and PERIKABIVEN, may be administered sequentially if the infusion lines are thoroughly flushed between infusions with a compatible fluid [see Dosage and Administration (2.1)].
Deaths have occurred in neonates (28 days of age or younger) who received concomitant intravenous calcium-containing solutions with ceftriaxone resulting from calcium-ceftriaxone precipitates in the lungs and kidneys, even when separate infusion lines were used [see Contraindications (4), Use in Specific Populations (8.4)].
7.2 Coumarin and Coumarin Derivatives
The soybean oil present in PERIKABIVEN has vitamin K1. Vitamin K1 can reverse the anticoagulant activity of coumarin or coumarin derivatives, which work by blocking recycling of vitamin K1. Monitoring for anticoagulant activity is recommended in patients who are on both PERIKABIVEN and coumarin or coumarin derivatives.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
The limited available data on the use of PERIKABIVEN in pregnant women are not sufficient to inform a drug-associated risk. However, there are clinical considerations if PERIKABIVEN is used in pregnant women [see Clinical Considerations]. Animal reproduction studies have not been conducted with PERIKABIVEN.
The estimated background risk of major birth defects and miscarriage for the indicated population are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryofetal Risk
Severe malnutrition in a pregnant woman is associated with preterm delivery, low birth weight, intrauterine growth restriction, congenital malformations and perinatal mortality. Parenteral nutrition should be considered if a pregnant woman's nutritional requirements cannot be fulfilled by oral or enteral intake.
8.2 Lactation
Risk Summary
There are no data available to assess the presence of PERIKABIVEN and/or its active metabolite(s) in human milk, the effects on the breastfed child or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PERIKABIVEN, and any potential adverse effects of PERIKABIVEN on the breastfed child or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of PERIKABIVEN has not been established in pediatric patients of any age. In the postmarketing setting, clinical decompensation with rapid infusion of intravenous lipid emulsion in neonates and infants, sometimes fatal has been reported [see Warnings and Precautions (5.1)]. Patients, particularly preterm infants, are at risk for aluminum toxicity [see Warnings and Precautions (5.13)].
Deaths have occurred in neonates (28 days of age or younger) who received concomitant intravenous calcium-containing solutions with ceftriaxone resulting from calcium-ceftriaxone precipitates in the lungs and kidneys, even when separate infusion lines were used. [see Contraindications (4), Warnings and Precautions (5.5)].
PERIKABIVEN is not recommended for use in pediatric patients under the age of two years, including preterm infants, as the fixed content of the formulation does not meet the nutritional requirements of this age group due to the following reasons:
- Calcium and dextrose needs are not met and lipids, protein and magnesium exceed requirements.
- The product does not contain the amino acids cysteine and taurine, considered conditionally essential for neonates and infants.
Patients, including pediatric patients, may be at risk for PNALD [see Warnings and Precautions (5.2)].
Newborns – especially those born premature and with low birth weight – are at increased risk of developing hypo – or hyperglycemia and therefore need close monitoring during treatment with intravenous dextrose solutions to ensure adequate glycemic control in order to avoid potential long term adverse effects. Hypoglycemia in the newborn can cause prolonged seizures, coma and brain damage. Hyperglycemia has been associated with intraventricular hemorrhage, late onset bacterial and fungal infection, retinopathy of prematurity, necrotizing enterocolitis, bronchopulmonary dysplasia, prolonged length of hospital stay, and death.
8.5 Geriatric Use
Clinical studies of PERIKABIVEN did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from other younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or drug therapy.
10. Overdosage
In the event of overdose, serious adverse reactions may result [see Warnings and Precautions (5.1, 5.7)]. Stop the infusion of PERIKABIVEN to allow lipids to clear from serum. The effects are usually reversible after the lipid infusion is stopped. If medically appropriate, further intervention may be indicated. The lipid administered and fatty acids produced are not dialyzable.
11. Perikabiven Description
PERIKABIVEN is a sterile, hypertonic emulsion, for peripheral or central venous administration, in a Three Chamber Bag. The product contains no added sulfites.
Chamber 1 contains Dextrose monohydrate solution for fluid replenishment and caloric supply.
Chamber 2 contains the Amino Acid solution with Electrolytes, which comprises essential and nonessential amino acids provided with electrolytes.
Chamber 3 contains Intralipid® 20% (a 20% Lipid Injectable Emulsion), prepared for intravenous administration as a source of calories and essential fatty acids.
See below for formulations of each chamber and Table 2 for strength, pH, osmolarity, ionic concentration and caloric content of PERIKABIVEN when all the chambers are mixed together.
Chamber 1: Contains sterile, hypertonic solution of Dextrose, USP in water for injection with a pH range of 3.5 to 5.5. Dextrose, USP is chemically designated D-glucose, monohydrate (C6H12O6 • H2O) and has the following structure:
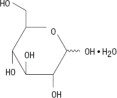
Dextrose is derived from corn.
Chamber 2: Contains a sterile solution of amino acids and electrolytes in water for injection. In addition, glacial acetic acid has been added to adjust the pH so that the final solution pH is 5.4 to 5.8. The formulas for the individual electrolytes and amino acids are as follows:
| Electrolytes | |
| Sodium Acetate Trihydrate, USP | CH3COONax3H2O |
| Potassium Chloride, USP | KCl |
| Sodium Glycerophosphate | C3H5(OH)2PO4Na2xH2O |
| Magnesium Sulfate Heptahydrate, USP | MgSO4x7H2O |
| Calcium Chloride Dihydrate, USP | CaCl2x2H2O |
| Essential Amino Acids | |
| Lysine (added as the hydrochloride salt) | H2N(CH2)4CH(NH2)COOH.HCl |
| Phenylalanine | |
| Leucine | (CH3)2CHCH2CH(NH2)COOH |
| Valine | (CH3)2CHCH(NH2)COOH |
| Histidine | |
| Threonine | CH3CH(OH)CH(NH2)COOH |
| Methionine | CH3S(CH2)2CH(NH2)COOH |
| Isoleucine | CH3CH2CH(CH3)CH(NH2)COOH |
| Tryptophan | |
| Nonessential Amino Acids | |
| Alanine | CH3CH(NH2)COOH |
| Arginine | H2NC(NH)NH(CH2)3CH(NH2)COOH |
| Glycine | H2NCH2COOH |
| Proline | |
| Glutamic Acid | HOOC(CH2)2CH(NH2)COOH |
| Serine | HOCH2CH(NH2)COOH |
| Aspartic Acid | HOOCCH2CH(NH2)COOH |
| Tyrosine |
 CH
CH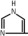 CH
CH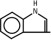 CH
CH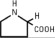
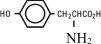
Chamber 3: Contains a 20% Lipid Injectable Emulsion (Intralipid® 20%) which is made up of 20% Soybean Oil, 1.2% Egg Yolk Phospholipids, 2.25% Glycerin, and water for injection. In addition, sodium hydroxide has been added to adjust the pH. The final product pH range is 6 to 9.
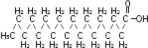
The soybean oil is a refined natural product consisting of a mixture of neutral triglycerides of predominantly unsaturated fatty acids with the following structure:
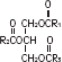
where 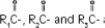 are saturated and unsaturated fatty acid residues. The major component fatty acids are linoleic (48 to 58 %), oleic (17 to 30%), palmitic (9 to 13%), linolenic (5 to 11%) and stearic acid (2.5 to 5%). These fatty acids have the following chemical and structural formulas:
are saturated and unsaturated fatty acid residues. The major component fatty acids are linoleic (48 to 58 %), oleic (17 to 30%), palmitic (9 to 13%), linolenic (5 to 11%) and stearic acid (2.5 to 5%). These fatty acids have the following chemical and structural formulas:
| Linoleic acid C18H32O2 | |
| Oleic acid C18H34O2 | |
| Palmitic acid C16H32O2 | |
| Linolenic acid C18H30O2 | |
| Stearic acid C18H36O2 |
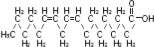
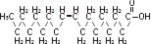
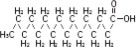
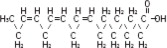
Purified egg phosphatides are a mixture of naturally occurring phospholipids which are isolated from the egg yolk. These phospholipids have the following general structure:
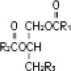
 contain saturated and unsaturated fatty acids that abound in neutral fats. R3 is primarily either the choline or ethanolamine ester of phosphoric acid.
contain saturated and unsaturated fatty acids that abound in neutral fats. R3 is primarily either the choline or ethanolamine ester of phosphoric acid.
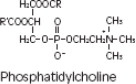
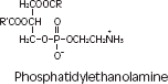
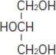
Glycerin is chemically designated C3H8O3 and is a clear colorless, hygroscopic syrupy liquid. It has the following structural formula:
The container-solution unit is a closed system and is not dependent upon entry of external air during administration. The container is overwrapped to provide protection from the physical environment and to provide an additional oxygen and moisture barrier when necessary. An oxygen absorber is placed between the inner bag and the overpouch.
The container is not made with natural rubber latex or polyvinyl chloride (PVC). PERIKABIVEN contains no more than 25 mcg/L of aluminum.
12. Perikabiven - Clinical Pharmacology
12.1 Mechanism of Action
PERIKABIVEN is used as a supplement or as the sole source of nutrition in patients, providing macronutrients (amino acids, dextrose and lipids) and micronutrients (electrolytes) parenterally.
The amino acids provide the structural units that make up proteins and are used to synthesize proteins and other biomolecules or are oxidized to urea and carbon dioxide as a source of energy.
The administered dextrose is oxidized to carbon dioxide and water, yielding energy.
Intravenously administered lipids provide a biologically utilizable source of calories and essential fatty acids. Fatty acids serve as an important substrate for energy production. The most common mechanism of action for energy derived from fatty acid metabolism is beta- oxidation. Fatty acids are important for membrane structure and function, precursors for bioactive molecules (such as prostaglandins), and as regulators of gene expression.
12.3 Pharmacokinetics
The infused lipid particles provided by PERIKABIVEN are expected to be cleared from the blood stream in a manner thought to be comparable to the clearing of chylomicrons. In healthy volunteers, the maximum clearance rate of the triglycerides after fasting overnight has been found to be 3.8 ± 1.5g/kg per 24 hours.
Both elimination and oxidation rates are dependent on the patient's clinical condition; elimination is faster and utilization is increased in postoperative patients, in sepsis, burns and trauma, while patients with renal impairment and hypertriglyceridemia may show lower utilization of exogenous lipid emulsions. Due to differences in elimination, patients with these conditions should be closely monitored during PERIKABIVEN administration [see Warnings and Precautions (5.10, 5.12)].
The disposition of infused amino acids, dextrose and electrolytes are essentially the same as those supplied by ordinary food.
A clinical study in healthy volunteers employing high intravenous doses (80 mmol) of either sodium glycerophosphate used in PERIKABIVEN or reference, inorganic sodium phosphate demonstrated that both compounds resulted in comparable serum inorganic phosphate concentrations after a single intravenous dose. Changes from baseline in the serum levels of sodium, potassium and total calcium were comparable across the two phosphate sources in this study.
16. How is Perikabiven supplied
PERIKABIVEN is a sterile emulsion available in the following 2 sizes:
| NDC | Volume |
| 63323-714-19 | 1,920 mL |
| 63323-714-14 | 1,440 mL |
Exposure of pharmaceutical products to heat should be minimized. Avoid excessive heat. Protect from freezing. If accidentally frozen, discard the bag. It is recommended that the product be stored at 5°C to 25°C (41°F to 77°F).
Do not remove container from overpouch until intended for use.
After breaking the vertical seals, chemical and physical in-use stability of the mixed three chamber bag has been demonstrated for 24 hours at 25°C (77°F).
The product should be used immediately after the introduction of additives. If not used immediately, the storage time and conditions prior to use should not be longer than 24 hours at 2° to 8°C (36° to 46°F). After removal from storage at 2° to 8°C (36° to 46°F), the admixture should be infused within 24 hours. Any mixture remaining must be discarded.
17. Patient Counseling Information
When initiating PERIKABIVEN administration, discuss the following information with the patient or caregiver:
Parenteral Nutrition-Associated Liver Disease and Other Hepatobiliary Disorders
Inform patients and caregivers that use of parenteral nutrition may result in parenteral nutrition- associated liver disease and/or other hepatobiliary disorders [see Warnings and Precautions (5.2)].
Pulmonary Embolism and Respiratory Distress due to Pulmonary Vascular Precipitates
Inform patients and caregivers that pulmonary vascular precipitates causing pulmonary emboli (including some fatalities) and presenting as respiratory distress have been reported in patients receiving parenteral nutrition. If PERIKABIVEN is infused at home, instruct patients or caregivers to visually inspect the prepared solution, the infusion set, and catheter for precipitates, prior to administration as well as periodically during the administration [see Warnings and Precautions (5.3)]
Hypersensitivity Reactions
Inform patients and caregivers that PERIKABIVEN may cause hypersensitivity reactions. If PERIKABIVEN is infused at home, instruct patients or caregivers to stop the infusion of PERIKABIVEN immediately and seek medical attention if a hypersensitivity reaction occurs [see Warnings and Precautions (5.4)].
Infections
Inform patients and caregivers that patients who receive PERIKABIVEN are at risk of infection. If PERIKABIVEN is infused at home, instruct patients or caregivers to ensure aseptic techniques are used for the preparation and administration of PERIKABIVEN and to monitor for signs and symptoms of infection [see Warnings and Precautions (5.6)].
Fat Overload Syndrome
Inform patients and caregivers that fat overload syndrome has been reported with the use of intravenous lipid emulsions. If PERIKABIVEN is infused at home, instruct patients or caregivers to stop PERIKABIVEN if signs or symptoms of fat overload syndrome occur [see Warnings and Precautions (5.7)].
Refeeding Syndrome
If the patient is severely malnourished, inform patients and caregivers that administering parenteral nutrition including PERIKABIVEN may result in refeeding syndrome [see Warnings and Precautions (5.8)].
Diabetes and Hyperglycemia
Inform patients and their caregivers that administration of dextrose at a rate exceeding the patient's utilization rate may lead to hyperglycemia, hyperosmolar coma, and death [see Warnings and Precautions (5.9)].
Hypertriglyceridemia
Inform patients and their caregivers about the risks of hypertriglyceridemia with PERIKABIVEN use [see Warnings and Precautions (5.10)].
Vein Damage and Thrombosis
Inform patients and caregivers that the infusion of hypertonic nutrient injections into a peripheral vein may result in vein irritation, vein damage, and/or thrombosis [see Warnings and Precautions (5.11)].
Electrolyte Imbalance and Fluid Overload in Patients with Decreased Renal Function
For patients with decreased renal function, inform them or their caregivers that the patient may be at increased risk of electrolyte and fluid volume imbalance when PERIKABIVEN is being administered [see Warnings and Precautions (5.12)].
Aluminum Toxicity
Inform patients and their caregivers that prolonged PN administration in patients with renal impairment, including preterm neonates, may result in aluminum reaching toxic levels associated with central nervous system and bone toxicity [see Warnings and Precautions (5.13)].
Preparation and Administration Instructions
If it is acceptable for a patient or caregiver to administer PERIKABIVEN at home, then the patient or caregiver must be trained on the following: how to inspect and prepare, add compatible additives (when appropriate), administer, and store PERIKABIVEN [see Dosage and Administration (2.1, 2.2)]. Inform patients or caregivers not to deviate from the administration instructions given by the healthcare provider.
Manufactured by:

Uppsala, Sweden
Fresenius Kabi, Perikabiven and Intralipid are registered trademarks of Fresenius Kabi.
www.freseniuskabinutrition.com/products/kabiven-perikabiven/
451207F
PACKAGE LABEL - PRINCIPAL DISPLAY PANEL - PERIKABIVEN ®1440 mL Bag Label
PERIKABIVEN ® 1440 mL
Amino Acids, Electrolytes, Dextrose, and Lipid Injectable Emulsion, for intravenous use
(2.4%, 0.5%*, 7.5% and 3.5%), No sulfites added PERIPHERAL INFUSION

PACKAGE LABEL - PRINCIPAL DISPLAY PANEL - PERIKABIVEN ®1440 mL Bag Shipper Label
NDC 63323-714-14
PERIKABIVEN ® 4 x 1440 mL
Amino Acids, Electrolytes, Dextrose, and Lipid Injectable Emulsion, for intravenous use
(2.4%, 0.5%*, 7.5% and 3.5%),
No sulfites added PERIPHERAL INFUSION
Rx only



| PERIKABIVEN
dextrose, soybean oil, electrolytes, lysine, phenylalanine, leucine, valine, threonine, methionine, isoleucine, tryptophan, alanine, arginine, glycine, proline, histidine, glutamic acid, serine, aspartic acid and tyrosine injection, emulsion |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Labeler - Fresenius Kabi USA, LLC (608775388) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Fresenius Kabi AB Uppsala | 559785113 | analysis(63323-714) , API manufacture(63323-714) , manufacture(63323-714) | |
More about Perikabiven (parenteral nutrition solution w/electrolytes)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- Drug class: intravenous nutritional products