Onivyde: Package Insert / Prescribing Info
Package insert / product label
Generic name: irinotecan hydrochloride, liposomal
Dosage form: injection, powder, for solution
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
ONIVYDE® (irinotecan liposome injection), for intravenous use
Initial U.S. Approval: 1996
WARNING: SEVERE NEUTROPENIA and SEVERE DIARRHEA
See full prescribing information for complete boxed warning
Neutropenia
- Severe or life-threatening neutropenia , including fatal neutropenic sepsis and fatal neutropenic fever, has occurred in patients receiving ONIVYDE in combination with oxaliplatin, fluorouracil and leucovorin and in combination with fluorouracil and leucovorin. Withhold ONIVYDE for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment (2.2), (5.1).
Diarrhea
- Severe and life-threatening diarrhea has occurred in patients receiving ONIVYDE in combination with oxaliplatin, fluorouracil and leucovorin and in combination with fluorouracil and leucovorin. Do not administer ONIVYDE to patients with bowel obstruction. Withhold ONIVYDE for diarrhea of Grade 2-4 severity. Administer loperamide for late diarrhea of any severity. Administer atropine, if not contraindicated, for early diarrhea of any severity (2.2), (5.2).
Recent Major Changes
Indications and Usage for Onivyde
ONIVYDE is a topoisomerase inhibitor indicated:
- in combination with oxaliplatin, fluorouracil and leucovorin, for the first-line treatment of adult patients with metastatic pancreatic adenocarcinoma, (1)
- in combination with fluorouracil and leucovorin, for the treatment of adult patients with metastatic pancreatic adenocarcinoma after disease progression following gemcitabine-based therapy. (1)
Limitation of Use: ONIVYDE is not indicated as a single agent for the treatment of patients with metastatic pancreatic adenocarcinoma. (1)
Onivyde Dosage and Administration
- Do not substitute ONIVYDE for other drugs containing irinotecan HCl. (2.1)
- ONIVYDE in combination with oxaliplatin, fluorouracil and leucovorin:
- Recommended dose of ONIVYDE is 50 mg/m2 intravenous infusion over 90 minutes every two weeks. (2.2)
- Recommended starting dose of ONIVYDE in patients homozygous for UGT1A1*28 is 50 mg/m2 every two weeks. (2.2)
- There is no recommended dose of ONIVYDE for patients with serum bilirubin above the upper limit of normal. (2.2)
- ONIVYDE in combination with fluorouracil and leucovorin:
- Recommended dose of ONIVYDE is 70 mg/m2 intravenous infusion over 90 minutes every two weeks. (2.2)
- Recommended starting dose of ONIVYDE in patients homozygous for UGT1A1*28 is 50 mg/m2 every two weeks. (2.2)
- There is no recommended dose of ONIVYDE for patients with serum bilirubin above the upper limit of normal. (2.2)
- Premedicate with a corticosteroid and an anti-emetic 30 minutes prior to ONIVYDE. (2.2)
Dosage Forms and Strengths
Injection: 43 mg/10 mL (4.3 mg/mL) single dose vial (3)
Warnings and Precautions
- Interstitial lung disease (ILD): Fatal ILD has occurred in patients receiving irinotecan including ONIVYDE. Discontinue ONIVYDE if ILD is diagnosed. (5.3)
- Severe hypersensitivity reaction: Permanently discontinue ONIVYDE for severe hypersensitivity reactions. (5.4, 4)
- Embryo-fetal toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.5, 8.1, 8.3)
Adverse Reactions/Side Effects
The most common adverse reactions (reported in ≥ 20% of patients) were for:
- ONIVYDE in combination with oxaliplatin, fluorouracil and leucovorin: diarrhea, fatigue, nausea, vomiting, decreased appetite, abdominal pain, mucosal inflammation, constipation, and decreased weight. The most common laboratory abnormalities (≥ 10% Grade 3 or 4) were decreased neutrophils, decreased potassium, decreased lymphocytes, and decreased hemoglobin. (6.1)
- ONIVYDE in combination with fluorouracil and leucovorin: diarrhea, fatigue/asthenia, vomiting, nausea, decreased appetite, stomatitis, and pyrexia. The most common laboratory abnormalities (≥ 10% Grade 3 or 4) were lymphopenia and neutropenia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Ipsen Biopharmaceuticals, Inc.at 1-855-463-5127 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Strong CYP3A4 Inducers: Avoid the use of strong CYP3A4 inducers if possible. Substitute non-enzyme inducing therapies at least 2 weeks prior to initiation of ONIVYDE. (7.1)
- Strong CYP3A4 Inhibitors: Avoid the use of strong CYP3A4 or UGT1A1 inhibitors, if possible; discontinue strong CYP3A4 inhibitors at least 1 week prior to starting therapy. (7.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2024
Full Prescribing Information
WARNING: SEVERE NEUTROPENIA AND SEVERE DIARRHEA
Neutropenia
- Severe and life-threatening neutropenia, including fatal neutropenic sepsis and fatal neutropenic fever, has occurred in patients receiving ONIVYDE in combination with oxaliplatin, fluorouracil and leucovorin and in combination with fluorouracil and leucovorin. Withhold ONIVYDE for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
Diarrhea
- Severe and life-threatening diarrhea has occurred in patients receiving ONIVYDE in combination with oxaliplatin, fluorouracil and leucovorin and in combination with fluorouracil and leucovorin. Do not administer ONIVYDE to patients with bowel obstruction. Withhold ONIVYDE for diarrhea of Grade 2-4 severity. Administer loperamide for late diarrhea of any severity. Administer atropine, if not contraindicated, for early diarrhea of any severity [see Dosage and Administration (2.2) and Warnings and Precautions (5.2)].
1. Indications and Usage for Onivyde
- ONIVYDE is indicated, in combination with oxaliplatin, fluorouracil and leucovorin for the first-line treatment of adult patients with metastatic pancreatic adenocarcinoma.
- ONIVYDE is indicated, in combination with fluorouracil and leucovorin, for the treatment of adult patients with metastatic pancreatic adenocarcinoma after disease progression following gemcitabine-based therapy.
Limitations of Use: ONIVYDE is not indicated as a single agent for the treatment of patients with metastatic pancreatic adenocarcinoma. [see Clinical Studies (14)].
2. Onivyde Dosage and Administration
2.2 Recommended Dosage
In combination with oxaliplatin, fluorouracil and leucovorin for the first-line treatment of patients with metastatic pancreatic adenocarcinoma
Administer ONIVYDE prior to oxaliplatin, fluorouracil and leucovorin [see Clinical Studies (14)].
- The recommended dosage of ONIVYDE regardless of UGT1A1*28 allele genotype is 50 mg/m2 administered by intravenous infusion over 90 minutes every 2 weeks.
- There is no recommended dosage of ONIVYDE for patients with serum bilirubin above the upper limit of normal [see Adverse Reactions (6.1) and Clinical Studies (14)].
In combination with fluorouracil and leucovorin for the treatment of patients with metastatic pancreatic adenocarcinoma after disease progression following gemcitabine-based therapy
Administer ONIVYDE prior to fluorouracil and leucovorin [see Clinical Studies (14)].
- The recommended dosage of ONIVYDE is 70 mg/m2 administered by intravenous infusion over 90 minutes every 2 weeks.
- The recommended starting dose of ONIVYDE in patients known to be homozygous for the UGT1A1*28 allele is 50 mg/m2 administered by intravenous infusion over 90 minutes. Increase the dose of ONIVYDE to 70 mg/m2 as tolerated in subsequent cycles.
- There is no recommended dosage of ONIVYDE for patients with serum bilirubin above the upper limit of normal [see Adverse Reactions (6.1) and Clinical Studies (14)].
2.3 Dosage Modifications for Adverse Reactions
Recommended dosage modifications for ONIVYDE are in Table 1 and Table 2.
| Toxicity* | Occurrence | ONIVYDE adjustment in patients receiving 50mg/m2 |
|---|---|---|
|
||
| Grade 3 or 4 Adverse reactions† | Withhold ONIVYDE Upon recovery to ≤ Grade 1‡,§,¶, resume ONIVYDE at: |
|
| First | 40 mg/m2 | |
| Second | 32.5 mg/m2 | |
| Third | 25 mg/m2 | |
| Fourth | Discontinue ONIVYDE | |
| Grade 3 or 4 Hand foot syndrome | First | Discontinue ONIVYDE |
| Any grade neurocerebellar toxicity | First | Discontinue ONIVYDE |
| Grade ≥ 2 cardiac toxicity | First | Discontinue ONIVYDE |
| Interstitial lung disease | First | Discontinue ONIVYDE |
| Anaphylactic reaction | First | Discontinue ONIVYDE |
| Toxicity NCI CTCAE v4.0* | Occurrence | ONIVYDE adjustment in patients receiving 70 mg/m2 | Patients homozygous for UGT1A1*28 without previous increase to 70 mg/m2 |
|---|---|---|---|
|
|||
| Grade 3 or 4 adverse reactions | Withhold ONIVYDE. Upon recovery to ≤ Grade 1, resume ONIVYDE at: |
||
| First | 50 mg/m2 | 43 mg/m2 | |
| Second | 43 mg/m2 | 35 mg/m2 | |
| Third | Discontinue ONIVYDE | Discontinue ONIVYDE | |
| Interstitial Lung Disease | First | Discontinue ONIVYDE | Discontinue ONIVYDE |
| Anaphylactic Reaction | First | Discontinue ONIVYDE | Discontinue ONIVYDE |
For recommended dose modifications of fluorouracil (FU) or leucovorin (LV), refer to the Full Prescribing Information; refer to Clinical Studies (14).
2.4 Preparation and Administration
ONIVYDE is a hazardous drug. Follow applicable special handling and disposal procedures.1
Preparation
- Withdraw the calculated volume of ONIVYDE from the vial. Dilute ONIVYDE in 500 mL 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP and mix diluted solution by gentle inversion. Discard vials with any unused portion.
- Protect diluted solution from light.
- Administer diluted solution within 4 hours of preparation when stored at room temperature or within 24 hours of preparation when stored under refrigerated conditions [2ºC to 8ºC (36ºF to 46ºF)]. Allow diluted solution to come to room temperature prior to administration.
- Do NOT freeze.
3. Dosage Forms and Strengths
Injection: 43 mg/10 mL irinotecan free base as a white to slightly yellow, opaque, liposomal dispersion in a single-dose vial.
4. Contraindications
ONIVYDE is contraindicated in patients who have experienced a severe hypersensitivity reaction or anaphylaxis to ONIVYDE or irinotecan HCl. [see Warnings and Precautions (5.4), Adverse Reactions (6.2)].
5. Warnings and Precautions
5.1 Severe Neutropenia
ONIVYDE can cause severe or life-threatening neutropenia and fatal neutropenic sepsis.
In NAPOLI 3, Grade 3 and 4 neutropenia occurred in 26% of patients receiving ONIVYDE in combination with oxaliplatin, fluorouracil, and leucovorin (NALIRIFOX) and fatal neutropenic fever in 0.3% of patients [see Adverse Reactions (6.1)]. In NAPOLI-1, Grade 3 and 4 neutropenia occurred in 20% of patients receiving ONIVYDE in combination with fluorouracil and leucovorin (ONIVYDE/FU/LV). Neutropenic sepsis occurred in 3% and fatal neutropenic sepsis in 0.8% [see Adverse Reactions (6.1)].
In NAPOLI 3, the incidence of Grade 3 or 4 neutropenia was similar among Asian patients [6 of 20 (30%)] compared to White patients [76 of 289 (26%)] receiving ONIVYDE in combination with oxaliplatin, fluorouracil, and leucovorin. Neutropenic fever was reported in 5% of Asian patients (1 of 20) compared to 2.3% of White patients (7 of 306). In NAPOLI-1, the incidence of Grade 3 or 4 neutropenia was higher among Asian patients [18 of 33 (55%)] compared to White patients [13 of 73 (18%)] receiving ONIVYDE/FU/LV. Neutropenic fever/neutropenic sepsis was reported in 6% of Asian patients compared to 1% of White patients [see Clinical Pharmacology (12.3)].
Monitor complete blood cell counts on Days 1 and 8 of every cycle and more frequently if clinically indicated. Withhold ONIVYDE if the absolute neutrophil count (ANC) is below 1500/mm3 or if neutropenic fever occurs. Resume ONIVYDE when the ANC is 1500/mm3 or above. Reduce ONIVYDE dose for Grade 3-4 neutropenia or neutropenic fever following recovery in subsequent cycles [see Dosage and Administration (2.2)].
5.2 Severe Diarrhea
ONIVYDE can cause severe and life-threatening diarrhea. Do not administer ONIVYDE to patients with a bowel obstruction. Severe or life-threatening diarrhea can follow one of two patterns: late onset diarrhea (onset more than 24 hours following chemotherapy) and early onset diarrhea (onset within 24 hours of chemotherapy, sometimes occurring with other symptoms of cholinergic reaction) [see Adverse Reactions (6.1)]. An individual patient may experience both early and late-onset diarrhea.
In NAPOLI 3, Grade 3 and 4 diarrhea (early and late-onset) occurred in 20% receiving ONIVYDE in combination with oxaliplatin, fluorouracil, and leucovorin (NALIRIFOX). In NAPOLI-1, Grade 3 or 4 diarrhea occurred in 13% receiving ONIVYDE/FU/LV. The incidence of Grade 3 or 4 late onset diarrhea was 9% in patients receiving ONIVYDE/FU/LV. The incidence of Grade 3 or 4 early onset diarrhea was 3% in patients receiving ONIVYDE/FU/LV. Of patients receiving ONIVYDE/FU/LV in NAPOLI-1, 34% received loperamide for late-onset diarrhea and 26% received atropine for early-onset diarrhea.
To reduce the risk of severe diarrhea, patients should stop lactose-containing products, eat a low-fat diet and maintain hydration during treatment with ONIVYDE. Withhold ONIVYDE for Grade 2-4 diarrhea. Administer intravenous or subcutaneous atropine 0.25 to 1 mg (unless clinically contraindicated) for early onset diarrhea of any severity. Initiate loperamide for late onset diarrhea of any severity. Local institutional guidelines should be followed for the treatment of diarrhea that does not improve within 48 hours and may include the addition of diphenoxylate hydrochloride plus atropine sulfate or octreotide. Following recovery to Grade 1 diarrhea, resume ONIVYDE at a reduced dose [see Dosage and Administration (2.3)].
5.3 Interstitial Lung Disease
ONIVYDE can cause severe and fatal interstitial lung disease (ILD), including pneumonitis. Postmarketing cases of severe and fatal ILD have been reported with ONIVYDE. Risk factors include pre-existing lung disease, use of pneumotoxic medicinal products, colony stimulating factors or having previously received radiation therapy. Patients with risk factors should be closely monitored for respiratory symptoms before and during ONIVYDE therapy. Withhold ONIVYDE in patients with new or progressive dyspnea, cough, and fever, pending diagnostic evaluation. Discontinue ONIVYDE in patients with a confirmed diagnosis of ILD.
5.4 Severe Hypersensitivity Reaction
Irinotecan including ONIVYDE can cause severe hypersensitivity reactions, including anaphylactic reactions. Permanently discontinue ONIVYDE in patients who experience a severe hypersensitivity reaction [see Contraindications (4), Adverse reactions (6.2)].
5.5 Embryo-Fetal Toxicity
Based on animal data with irinotecan HCl and the mechanism of action of ONIVYDE, ONIVYDE can cause fetal harm when administered to a pregnant woman. Embryotoxicity and teratogenicity were observed following treatment with irinotecan HCl, at doses resulting in irinotecan exposures lower than those achieved with ONIVYDE 70 mg/m2 in humans, administered to pregnant rats and rabbits during organogenesis. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ONIVYDE and for seven months following the last dose [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
6. Adverse Reactions/Side Effects
The following adverse drug reactions are discussed in greater detail in other sections of the label:
- Severe Neutropenia [see Warnings and Precautions (5.1)]
- Severe Diarrhea [see Warnings and Precautions (5.2)]
- Interstitial Lung Disease [see Warnings and Precautions (5.3)]
- Severe Hypersensitivity Reactions [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of ONIVYDE cannot be directly compared to rates in clinical trials of other drugs and may not reflect the rates observed in practice.
Pancreatic Adenocarcinoma
In Combination with Oxaliplatin, Fluorouracil and Leucovorin for First-Line Treatment
The safety of ONIVYDE in patients with metastatic pancreatic adenocarcinoma who had not previously received chemotherapy was evaluated in NAPOLI 3 [see Clinical Studies (14)]. Patients received ONIVYDE 50 mg/m2 in combination with oxaliplatin 60 mg/m2, leucovorin 400 mg/m2 and fluorouracil 2400 mg/m2 over 46 hours every 2 weeks (NALIRIFOX; N=383) or nab-paclitaxel 125 mg/m2 over 35 minutes and gemcitabine 1000 mg/m2 over 30 minutes on Day 1, 8 and 15 of each 28-day cycle (Gem+NabP; N=387). The median duration of exposure to ONIVYDE in combination with oxaliplatin, fluorouracil, and leucovorin was 24 weeks (range: 0 to 101 weeks).
Serious adverse reactions occurred in 54% of patients who received ONIVYDE in combination with oxaliplatin, fluorouracil and leucovorin. Serious adverse reactions in ≥2% of patients included infections including COVID-19 (14%), diarrhea (9%), vomiting (6%), nausea (4.9%), fatigue (3.8%), embolism (3.5%), gastrointestinal tract stenosis or obstruction (3.5%), hemorrhage (3%), abdominal pain (2.7%), cerebrovascular accident (2.7%), dehydration (2.7%), liver function test abnormalities (2.2%), and pyrexia (2.2%). Fatal adverse reactions occurred in 6% of patients who received ONIVYDE in combination with oxaliplatin, fluorouracil and leucovorin including cerebrovascular accident (1.1%), hemorrhage (0.5%), pneumonia (0.5%), sepsis (0.5%) and sudden death (0.5%).
Permanent discontinuation of ONIVYDE due to an adverse reaction occurred in 17% of patients. Adverse reactions which resulted in permanent discontinuation of ONIVYDE in ≥1% of patients included neutropenia, thrombocytopenia, diarrhea, fatigue, infections and cerebrovascular accident.
Dosage reduction of ONIVYDE due to an adverse reaction occurred in 52% of patients. Adverse reactions which required dosage reduction in ≥1% of patients included anemia, decreased appetite, diarrhea, fatigue, febrile neutropenia, hypokalemia, liver function test abnormalities, nausea, mucosal inflammation, neutropenia, peripheral neuropathy, vomiting, thrombocytopenia and weight decreased.
Dosage interruptions of ONIVYDE due to an adverse reaction occurred in 1.9% of patients. Adverse reactions which required dosage interruption in ≥0.5% of patients included hypersensitivity and infusion related reaction.
The most common adverse reactions (≥20% with a difference between arms of ≥ 5% for all grades or ≥ 2% for Grades 3 or 4 compared to Gem+NabP) of ONIVYDE in combination with oxaliplatin, fluorouracil, and leucovorin were diarrhea, fatigue, nausea, vomiting, decreased appetite, abdominal pain, mucosal inflammation, constipation and decreased weight. The most common laboratory abnormalities (≥10% Grade 3 or 4) were decreased neutrophils, decreased potassium, decreased lymphocyte and decreased hemoglobin.
Table 3 and 4 summarize the adverse reactions and laboratory abnormalities, respectively, in the NAPOLI 3 study.
| Adverse Reaction | NALIRIFOX N=370 | Gem+NabP N=379 |
||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or4 (%) | All Grades (%) | Grade 3 or4 (%) |
|
| Gastrointestinal disorders | ||||
| Diarrhea‡ | 72 | 22 | 37 | 5 |
| Nausea | 59 | 12 | 43 | 2.6 |
| Vomiting‡ | 40 | 7 | 27 | 2.1 |
| Abdominal pain‡ | 35 | 4.3 | 25 | 4.7 |
| Constipation | 25 | 0.8 | 30 | 2.1 |
| General disorders and administration site condition | ||||
| Fatigue‡ | 62 | 15 | 63 | 10 |
| Mucosal inflammation‡ | 28 | 3.8 | 17 | 0.8 |
| Peripheral edema‡ | 16 | 0.3 | 34 | 2.4 |
| Pyrexia‡ | 11 | 0.8 | 24 | 1.6 |
| Investigations | ||||
| Weight decreased | 22 | 3 | 9 | 0.3 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 37 | 9 | 28 | 2.6 |
| Dehydration | 11 | 3.2 | 9 | 1.1 |
| Skin and subcutaneous tissue disorders | ||||
| Alopecia | 14 | 0 | 31 | 0.5 |
| Rash‡ | 11 | 0.3 | 22 | 1.6 |
| Nail disorder | 0.3 | 0 | 7 | 0.3 |
| Vascular disorders | ||||
| Hemorrhage‡ | 11 | 2.4 | 18 | 3.4 |
| Embolism‡ | 11 | 7 | 11 | 5 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Dyspnea‡ | 8 | 0.5 | 13 | 2.1 |
| Musculoskeletal and connective tissue disorders | ||||
| Musculoskeletal pain‡ | 18 | 1.6 | 27 | 1.1 |
| Infections and infestations | ||||
| Pneumonia | 2.4 | 1.6 | 6 | 4 |
| Sepsis‡ | 1.6 | 1.1 | 6 | 3.4 |
| Laboratory abnormality | NALIRIFOX | Gem-NabP | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3- or 4 (%) | All Grades (%) | Grade 3 or4 (%) |
|
|
||||
| Hematology | ||||
| Hemoglobin decreased | 91 | 10 | 96 | 15 |
| Lymphocytes decreased | 64 | 11 | 76 | 19 |
| Leukocytes decreased | 62 | 8 | 77 | 28 |
| Neutrophils decreased | 56 | 26 | 65 | 37 |
| Platelets decreased | 55 | 1.7 | 75 | 7 |
| Hepatic | ||||
| Alkaline phosphatase increased | 45 | 2.9 | 35 | 2.7 |
| Alanine aminotransferase increased | 40 | 2.6 | 56 | 4.6 |
| Aspartate aminotransferase increased | 38 | 2 | 49 | 2.4 |
| Metabolic | ||||
| Potassium decreased | 62 | 22 | 29 | 8 |
| Sodium increased | 11 | 0 | 5 | 0.3 |
| Potassium increased | 8 | 0.6 | 21 | 3 |
In Combination with Fluorouracil and Leucovorin after Progresssion on Gemcitabine or Gemcitabine-based Therapy
The safety data described below are derived from patients with metastatic pancreatic adenocarcinoma previously treated with gemcitabine-based therapy who received any part of protocol-specified therapy in NAPOLI-1, an international, randomized, active-controlled, open-label trial. Protocol-specified therapy consisted of ONIVYDE 70 mg/m2 with leucovorin 400 mg/m2 and fluorouracil 2400 mg/m2 over 46 hours every 2 weeks (ONIVYDE/FU/LV; N=117), ONIVYDE 100 mg/m2 every 3 weeks (N=147), or leucovorin 200 mg/m2 and fluorouracil 2000 mg/m2 over 24 hours weekly for 4 weeks followed by 2 week rest (FU/LV; N=134) [see Clinical Studies (14)]. Serum bilirubin within the institutional normal range, albumin ≥ 3 g/dL, and Karnofsky Performance Status (KPS) ≥ 70 were required for study entry. The median duration of exposure was 9 weeks in the ONIVYDE/FU/LV arm, 9 weeks in the ONIVYDE monotherapy arm, and 6 weeks in the FU/LV arm.
The most common adverse reactions (≥ 20%) of ONIVYDE were diarrhea, fatigue/asthenia, vomiting, nausea, decreased appetite, stomatitis, and pyrexia. The most common, severe laboratory abnormalities (≥ 10% Grade 3 or 4) were lymphopenia and neutropenia. The most common serious adverse reactions (≥ 2%) of ONIVYDE were diarrhea, vomiting, neutropenic fever or neutropenic sepsis, nausea, pyrexia, sepsis, dehydration, septic shock, pneumonia, acute renal failure, and thrombocytopenia.
Adverse reactions led to permanent discontinuation of ONIVYDE in 11% of patients receiving ONIVYDE/FU/LV; the most frequent adverse reactions resulting in discontinuation of ONIVYDE were diarrhea, vomiting, and sepsis. Dose reductions of ONIVYDE for adverse reactions occurred in 33% of patients receiving ONIVYDE/FU/LV; the most frequent adverse reactions requiring dose reductions were neutropenia, diarrhea, nausea, and anemia. ONIVYDE was withheld or delayed for adverse reactions in 62% of patients receiving ONIVYDE/FU/LV; the most frequent adverse reactions requiring interruption or delays were neutropenia, diarrhea, fatigue, vomiting, and thrombocytopenia.
Table 5 provides the frequency and severity of adverse reactions in NAPOLI-1 that occurred with higher incidence (≥5% difference for Grades 1-4 or ≥2% difference for Grades 3-4) in patients who received ONIVYDE/FU/LV compared to patients who received FU/LV.
| Adverse Reaction | ONIVYDE/FU/LV N=117 | FU/LV N=134 |
||
|---|---|---|---|---|
| Grades 1-4 (%) | Grades 3-4 (%) | Grades 1-4 (%) | Grades 3-4 (%) |
|
| Gastrointestinal disorders | ||||
| Diarrhea | 59 | 13 | 26 | 4 |
| Early diarrhea† | 30 | 3 | 15 | 0 |
| Late diarrhea‡ | 43 | 9 | 17 | 4 |
| Vomiting | 52 | 11 | 26 | 3 |
| Nausea | 51 | 8 | 34 | 4 |
| Stomatitis§ | 32 | 4 | 12 | 1 |
| Infections and infestations | 38 | 17 | 15 | 10 |
| Sepsis | 4 | 3 | 2 | 1 |
| Neutropenic fever/neutropenic sepsis¶ | 3 | 3 | 1 | 0 |
| Gastroenteritis | 3 | 3 | 0 | 0 |
| Intravenous catheter-related infection | 3 | 3 | 0 | 0 |
| General disorders and administration site conditions | ||||
| Fatigue/asthenia | 56 | 21 | 43 | 10 |
| Pyrexia | 23 | 2 | 11 | 1 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 44 | 4 | 32 | 2 |
| Weight loss | 17 | 2 | 7 | 0 |
| Dehydration | 8 | 4 | 7 | 2 |
| Skin and subcutaneous tissue disorders | ||||
| Alopecia | 14 | 1 | 5 | 0 |
Other Adverse Reactions
Clinically relevant adverse reactions occurring in <5% of patients include:
Cholinergic Reactions: ONIVYDE can cause cholinergic reactions manifesting as rhinitis, increased salivation, flushing, bradycardia, miosis, lacrimation, diaphoresis, and intestinal hyperperistalsis with abdominal cramping and early onset diarrhea. In NAPOLI-1, Grade 1 or 2 cholinergic symptoms other than early diarrhea occurred in 12 (4.5%) ONIVYDE-treated patients. Six of these 12 patients received atropine and in 1 of the 6 patients, atropine was administered for cholinergic symptoms other than diarrhea.
Infusion Reactions: Infusion reactions, consisting of rash, urticaria, periorbital edema, or pruritus, occurring on the day of ONIVYDE administration were reported in 3% of patients receiving ONIVYDE or ONIVYDE/FU/LV.
Laboratory abnormalities that occurred with higher incidence in the ONIVYDE/FU/LV arm compared to the FU/LV arm (≥5% difference) are summarized in the following table.
| Laboratory abnormality | ONIVYDE/FU/LV N=117 | FU/LV N=134 |
||
|---|---|---|---|---|
| Grades 1-4 (%) | Grades 3-4 (%) | Grades 1-4 (%) | Grades 3-4 (%) |
|
| Hematology | ||||
| Anemia | 97 | 6 | 86 | 5 |
| Lymphopenia | 81 | 27 | 75 | 17 |
| Neutropenia | 52 | 20 | 6 | 2 |
| Thrombocytopenia | 41 | 2 | 33 | 0 |
| Hepatic | ||||
| Increased alanine aminotransferase (ALT) | 51 | 6 | 37 | 1 |
| Hypoalbuminemia | 43 | 2 | 30 | 0 |
| Metabolic | ||||
| Hypomagnesemia | 35 | 0 | 21 | 0 |
| Hypokalemia | 32 | 2 | 19 | 2 |
| Hypocalcemia | 32 | 1 | 20 | 0 |
| Hypophosphatemia | 29 | 4 | 18 | 1 |
| Hyponatremia | 27 | 5 | 12 | 3 |
| Renal | ||||
| Increased creatinine | 18 | 0 | 13 | 0 |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of ONIVYDE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune system disorders: Hypersensitivity (including Anaphylactic reaction and Angioedema)
7. Drug Interactions
7.1 Strong CYP3A4 Inducers
Following administration of non-liposomal irinotecan (i.e., irinotecan HCl), exposure to irinotecan or its active metabolite, SN-38, is substantially reduced in adult and pediatric patients concomitantly receiving the CYP3A4 enzyme-inducing anticonvulsants phenytoin and strong CYP3A4 inducers. Avoid the use of strong CYP3A4 inducers (e.g., rifampin, phenytoin, carbamazepine, rifabutin, rifapentine, phenobarbital, St. John's wort) if possible. Substitute non-enzyme inducing therapies at least 2 weeks prior to initiation of ONIVYDE therapy [see Clinical Pharmacology (12.3)].
7.2 Strong CYP3A4 or UGT1A1 Inhibitors
Following administration of non-liposomal irinotecan (i.e., irinotecan HCl), patients receiving concomitant ketoconazole, a CYP3A4 and UGT1A1 inhibitor, have increased exposure to irinotecan and its active metabolite SN-38. Co-administration of ONIVYDE with other inhibitors of CYP3A4 (e.g., clarithromycin, indinavir, itraconazole, lopinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telaprevir, voriconazole) or UGT1A1 (e.g., atazanavir, gemfibrozil, indinavir) may increase systemic exposure to irinotecan or SN-38. Avoid the use of strong CYP3A4 or UGT1A1 inhibitors if possible. Discontinue strong CYP3A4 inhibitors at least 1 week prior to starting ONIVYDE therapy [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on animal data with irinotecan HCl and the mechanism of action of ONIVYDE, ONIVYDE can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data in pregnant women. Embryotoxicity and teratogenicity were observed following treatment with irinotecan HCl, at doses resulting in irinotecan exposures lower than those achieved with ONIVYDE 70 mg/m2 in humans, administered to pregnant rats and rabbits during organogenesis [see Data]. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
No animal studies have been conducted to evaluate the effect of irinotecan liposome on reproduction and fetal development; however, studies have been conducted with irinotecan HCl. Irinotecan crosses the placenta of rats following intravenous administration. Intravenous administration of irinotecan at a dose of 6 mg/kg/day to rats and rabbits during the period of organogenesis resulted in increased post-implantation loss and decreased numbers of live fetuses. In separate studies in rats, this dose resulted in an irinotecan exposure of approximately 0.002 times the exposure of irinotecan based on area under the curve (AUC) in patients administered ONIVYDE at the 70 mg/m2 dose. Administration of irinotecan HCl resulted in structural abnormalities and growth delays in rats at doses greater than 1.2 mg/kg/day (approximately 0.0002 times the clinical exposure to irinotecan in ONIVYDE based on AUC). Teratogenic effects included a variety of external, visceral, and skeletal abnormalities. Irinotecan HCl administered to rat dams for the period following organogenesis through weaning at doses of 6 mg/kg/day caused decreased learning ability and decreased female body weights in the offspring.
8.2 Lactation
Risk Summary
There is no information regarding the presence of irinotecan liposome, irinotecan, or SN-38 (an active metabolite of irinotecan) in human milk, or the effects on the breastfed infant or on milk production. Irinotecan is present in rat milk [see Data].
Because of the potential for serious adverse reactions in breastfed infants from ONIVYDE, advise a nursing woman not to breastfeed during treatment with ONIVYDE and for one month after the last dose.
8.3 Females and Males of Reproductive Potential
Contraception
Females
ONIVYDE can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with ONIVYDE and for seven months after the last dose.
Males
Because of the potential for genotoxicity, advise males with female partners of reproductive potential to use condoms during treatment with ONIVYDE and for four months after the last dose [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of ONIVYDE have not been established in pediatric patients.
8.5 Geriatric Use
Of the 634 patients who received ONIVYDE as a single agent, in combination with FU and leucovorin or in combination with oxaliplatin, FU and leucovorin in NAPOLI-1 and NAPOLI 3, 49% were ≥ 65 years old and 10% were ≥ 75 years old. No overall differences in safety and effectiveness were observed between these patients and younger patients.
10. Overdosage
There are no treatment interventions known to be effective for management of overdosage of ONIVYDE.
11. Onivyde Description
ONIVYDE is formulated with irinotecan hydrochloride trihydrate, a topoisomerase inhibitor, into a liposomal dispersion for intravenous use. The chemical name of irinotecan hydrochloride trihydrate is (S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo1H-pyrano[3',4':6,7]-indolizino[1,2-b]quinolin-9-yl-[1,4'bipiperidine]-1'-carboxylate, monohydrochloride, trihydrate. The empirical formula is C33H38N4O6∙HCl∙3H2O and the molecular weight is 677.19 g/mole. The molecular structure is:
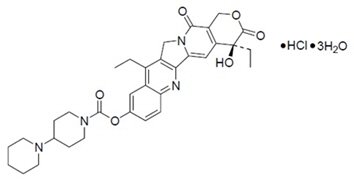
ONIVYDE is a sterile, white to slightly yellow opaque isotonic liposomal dispersion. Each 10 mL single-dose vial contains 43 mg irinotecan free base at a concentration of 4.3 mg/mL. The liposome is a unilamellar lipid bilayer vesicle, approximately 110 nm in diameter, which encapsulates an aqueous space containing irinotecan in a gelated or precipitated state as the sucrose octasulfate salt. The vesicle is composed of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) 6.81 mg/mL, cholesterol 2.22 mg/mL, and methoxy-terminated polyethylene glycol (MW 2000)-distearoylphosphatidyl ethanolamine (MPEG-2000-DSPE) 0.12 mg/mL. Each mL also contains 2-[4-(2-hydroxyethyl) piperazin-1-yl]ethanesulfonic acid (HEPES) as a buffer 4.05 mg/mL and sodium chloride as an isotonicity reagent 8.42 mg/mL.
12. Onivyde - Clinical Pharmacology
12.1 Mechanism of Action
Irinotecan liposome injection is a topoisomerase 1 inhibitor encapsulated in a lipid bilayer vesicle or liposome. Topoisomerase 1 relieves torsional strain in DNA by inducing single-strand breaks. Irinotecan and its active metabolite SN-38 bind reversibly to the topoisomerase 1-DNA complex and prevent re-ligation of the single-strand breaks, leading to exposure time-dependent double-strand DNA damage and cell death. In mice bearing human tumor xenografts, irinotecan liposome administered at irinotecan HCl-equivalent doses 5-fold lower than irinotecan HCl achieved similar intratumoral exposure of SN-38.
12.3 Pharmacokinetics
The plasma pharmacokinetics of total irinotecan and total SN-38 were evaluated in patients with cancer who received ONIVYDE, as a single agent or as part of combination chemotherapy, at doses between 35 mg/m2 and 155 mg/m2 and concentration proportional to dose was observed.
The pharmacokinetic parameters of total irinotecan and total SN-38 following the administration of ONIVYDE 70 mg/m2 as a single agent or part of combination chemotherapy are presented in Table 7.
| Dose (mg/m2) | Descriptive Statistics | Total Irinotecan | Total SN-38 | |||||
|---|---|---|---|---|---|---|---|---|
| Cmax
[µg/mL] | AUCSS
[day∙µg/mL] | t1/2
[day] | V [L] | Cmax
[ng/mL] | AUCSS
[day∙ng/mL] | V [L] |
||
| AUCSS: Area under the plasma concentration curve at steady-state | ||||||||
| t1/2: Terminal elimination half-life | ||||||||
| V: Volume of distribution | ||||||||
| 50 | Geometric Mean | 25.1 | 37.8 | 1.93 | 3.63 | 2.09 | 12.1 | 3.46 |
| CV (%) | 18.5 | 73.6 | 14 | 33.5 | 42.1 | 46.6 | 35.5 | |
| 70 | Geometric Mean | 30.8 | 50.4 | 1.87 | 4.23 | 2.64 | 14.7 | 4.06 |
| CV (%) | 19.7 | 75.3 | 26.4 | 28.1 | 64.5 | 58 | 29.4 | |
Distribution
Direct measurement of irinotecan liposome showed that 95% of irinotecan remains liposome -encapsulated, and the ratios between total and encapsulated forms did not change with time from 0 to 170 hours post-dose. The mean volume of distribution is summarized in Table 7.
Plasma protein binding is <0.44% of the total irinotecan in ONIVYDE.
Elimination
Metabolism
The metabolism of irinotecan liposome has not been evaluated. Irinotecan is subject to extensive metabolic conversion by various enzyme systems, including carboxylesterases to form the active metabolite SN-38, and UGT1A1 mediating glucuronidation of SN-38 to form the inactive glucuronide metabolite SN-38G. Irinotecan can also undergo CYP3A4-mediated oxidative metabolism to several inactive oxidation products, one of which can be hydrolyzed by carboxylesterase to release SN-38. In the population PK analysis of irinotecan liposome, UGT1A1*28 7/7 homozygous status (10.6%) had no effect on SN-38 clearance compared with patients not homozygous for UGT1A1*28 7/7.
Excretion
The disposition of ONIVYDE has not been elucidated in humans. Following administration of irinotecan HCl, the urinary excretion of irinotecan is 11 to 20%; SN-38, <1%; and SN-38 glucuronide, 3%. The cumulative biliary and urinary excretion of irinotecan and its metabolites (SN-38 and SN-38 glucuronide), over a period of 48 hours following administration of irinotecan HCl in two patients, ranged from approximately 25% (100 mg/m2) to 50% (300 mg/m2).
Specific Populations
Age, Sex, Ethnicity, Renal and Hepatic Impairment:
The population pharmacokinetic analyses suggest that age (20 to 87 years) and BSA (1.15 to 2.88 m2) had no clinically meaningful effect on the exposure of irinotecan and SN-38.
Irinotecan and SN-38 AUC in female patients were 28% and 32% higher, respectively, than those in male patients. Irinotecan AUC in patients of Asian ethnicity were 32% lower than that in non-Asian patients. The exposures of irinotecan and SN-38 in patients with mild or moderate renal impairment were comparable to patients with normal renal function after adjusting for BSA. The exposures of irinotecan and SN-38 in patients with mild hepatic impairment (based on NCI score) were comparable to patients with normal hepatic function. There was insufficient data in patients with severe renal impairment (CLcr < 30 mL/min) or in patients with moderate and severe hepatic impairment to assess their effects on the exposures of irinotecan and SN-38. Increased AST/ALT had no effect on irinotecan clearance; however, increased bilirubin level was associated with lower SN‑38 clearance. SN-38 AUC was increased by 32% in patients with bilirubin level of 1.14 mg/dL (95th of the overall population) compared with that of median bilirubin level of 0.44 mg/dL. No data are available in patients with bilirubin >2.8 mg/dL.
Drug Interactions
In a population pharmacokinetic analysis, the pharmacokinetics of total irinotecan and total SN-38 were not altered by the co-administration of fluorouracil/leucovorin. In Study MM‑398‑07‑02‑03 and NAPOLI-3, irinotecan AUC was decreased by 33% and SN-38 Cmax increased by 23% following co-administration with oxaliplatin.
Following administration of irinotecan HCl, dexamethasone (moderate CYP3A4 inducer) does not alter the pharmacokinetics of irinotecan.
In vitro studies indicate that irinotecan, SN-38 and another metabolite, aminopentane carboxylic acid (APC), do not inhibit cytochrome P-450 isozymes.
12.5 Pharmacogenomics
Individuals who are homozygous for the UGT1A1*28 allele are at increased risk for neutropenia from irinotecan HCl. In NAPOLI-1, patients homozygous for the UGT1A1*28 allele (N=7) initiated ONIVYDE at a reduced dose of 50 mg/m2 in combination with FU/LV. The frequency of Grade 3 or 4 neutropenia in these patients [2 of 7 (28.6%)] was similar to the frequency in patients not homozygous for the UGT1A1*28 allele who received a starting dose of ONIVYDE of 70 mg/m2 [30 of 110 (27.3%)]. In NAPOLI-3, patients homozygous for the UGT1A1*28 allele (N = 39) initiated ONIVYDE at the same starting dose of 50 mg/m2 as patients not homozygous for the UGT1A1*28 allele (N = 328). The frequency of Grade 3 or 4 neutropenia was 23% in patients homozygous for the UGT1A1*28 allele and 13% in patients not homozygous for the UGT1A1*28 allele. The frequency of dose reduction of ONIVYDE due to treatment-emergent adverse effects was 59% versus 51% in patients homozygous versus non-homozygous for the UGT1A1*28 allele.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been performed to assess the potential of irinotecan liposome for carcinogenicity, genotoxicity or impairment of fertility. Intravenous administration of irinotecan hydrochloride to rats once weekly for 13 weeks followed by a 91-week recovery period resulted in a significant linear trend between irinotecan HCl dosage and the incidence of combined uterine horn endometrial stromal polyps and endometrial stromal sarcomas. Irinotecan HCl was clastogenic both in vitro (chromosome aberrations in Chinese hamster ovary cells) and in vivo (micronucleus test in mice). Neither irinotecan nor its active metabolite, SN-38, was mutagenic in the in vitro Ames assay.
Dedicated fertility studies have not been performed with irinotecan liposome injection. Atrophy of male and female reproductive organs was observed in dogs receiving irinotecan liposome injection every 3 weeks at doses equal to or greater than 15 mg/kg, (approximately 3 times the clinical exposure of irinotecan following administration to ONIVYDE dosed at 70 mg/m2) for a total of 6 doses. No significant adverse effects on fertility and general reproductive performance were observed after intravenous administration of irinotecan HCl in doses of up to 6 mg/kg/day to rats; however, atrophy of male reproductive organs was observed after multiple daily irinotecan HCl doses both in rodents at 20 mg/kg (approximately 0.007 times the clinical irinotecan exposure following ONIVYDE administration at 70 mg/m2) and in dogs at 0.4 mg/kg (0.0007 times the clinical exposure to irinotecan following administration of ONIVYDE).
14. Clinical Studies
Pancreatic Adenocarcinoma
In Combination with Oxaliplatin, Fluorouracil and Leucovorin for First-Line Treatment of Metastatic Pancreatic Adenocarcinoma
The efficacy of ONIVYDE in combination with oxaliplatin, fluorouracil and leucovorin (NALIRIFOX) was evaluated in NAPOLI 3 (NCT04083235), a randomized, multicenter, open-label, active-controlled trial in 770 patients with metastatic pancreatic adenocarcinoma who had not previously received chemotherapy in the metastatic setting. Randomization was stratified by region, liver metastases and ECOG performance status. Patients were randomized (1:1) to receive one of the following treatment arm:
- NALIRIFOX: ONIVYDE 50 mg/m2 as an intravenous infusion over 90 minutes, followed by oxaliplatin 60 mg/m2 as an intravenous infusion over 120 minutes, followed by leucovorin 400 mg/m2 intravenously over 30 minutes, followed by fluorouracil 2400 mg/m2 intravenously over 46 hours, every 2 weeks.
- Gem+NabP: Nab-paclitaxel 125 mg/m2 as an intravenous infusion over 35 minutes, followed by gemcitabine 1000 mg/m2 intravenously over 30 minutes on days 1, 8 and 15 of each 28-day cycle.
Patients homozygous for the UGT1A1*28 allele initiated ONIVYDE at the same dose (50 mg/m2 ONIVYDE). Treatment continued until RECIST v1.1 defined disease progression or unacceptable toxicity. Tumor status assessments were conducted at baseline and every 8 weeks thereafter as assessed by the investigator according to RECIST v1.1.
The main efficacy outcome measure was overall survival (OS). Additional efficacy measures were investigator-assessed progression-free survival (PFS) and objective response rate (ORR).
Baseline demographic and patient characteristics were: median age of 65 years (range: 20-85); 50% age 65 or older; 56% male; 83% White, 4.9% Asian, 2.5% Black or African American, 0.4% multiple race, 0.3% American Indian or Alaska Native; 0.1% Native Hawaiian or other Pacific Islander, 1.7% other, 7% not reported; and 82% non-Hispanic, 10% Hispanic, 8% not reported. ECOG performance status was 0 or 1 in 44% and 56% of patients, respectively; 80% had liver metastases.
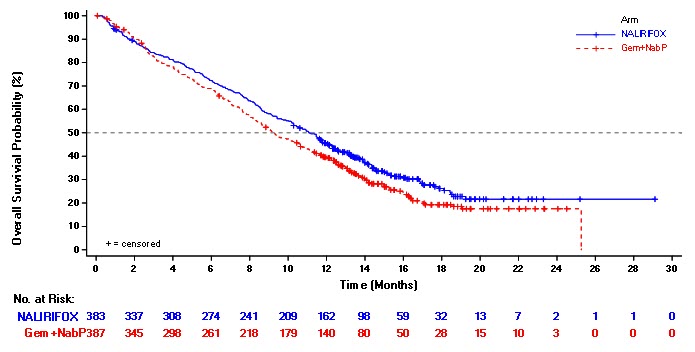
NAPOLI 3 demonstrated a statistically significant improvement in OS and PFS for the NALIRIFOX arm over Gem+NabP arm as summarized in Table 8 and Figure 1.
| NALIRIFOX*
(N=383) | Gem+NabP (N=387) |
|
|---|---|---|
| **Based on the stratified Cox proportional hazard model; stratified by ECOG PS (0 vs. 1), region (North America vs. East Asia vs. Rest of the world), and liver metastases (yes vs. no) per interaction web response system | ||
| Overall Survival | ||
| Number of Deaths, n (%) | 259 (68) | 285 (74) |
| Median Overall Survival (months) | 11.1 | 9.2 |
| (95% CI) | (10.0, 12.1) | (8.3, 10.6) |
| Hazard Ratio (95% CI) * | 0.84 (0.71, 0.99) | |
| p-value † | 0.0403 | |
| Progression-Free Survival | ||
| Death or Progression, n (%) | 249 (65) | 259 (67) |
| Median Progression-Free Survival (months) | 7.4 | 5.6 |
| (95% CI) | (6.0, 7.7) | (5.3, 5.8) |
| Hazard Ratio (95% CI) * | 0.70 (0.59, 0.85) | |
| P-value † | 0.0001 | |
| Objective Response Rate ‡ | ||
| ORR (95% CI) | 41.8 (36.8, 46.9) | 36.2 (31.4, 41.2) |
| CR, n (%) | 1 (0.3) | 1 (0.3) |
| PR, n (%) | 159 (41.5) | 139 (35.9) |
Figure 1 Kaplan-Meier Curve for Overall Survival in all randomized Patients in NAPOLI 3
Previously treated metastatic pancreatic adenocarcinoma in combination with fluorouracil and leucovorin
The efficacy of ONIVYDE was evaluated in NAPOLI-1 (NCT01494506), a three-arm, randomized, open-label trial in patients with metastatic pancreatic adenocarcinoma with documented disease progression, after gemcitabine or gemcitabine-based therapy. Key eligibility criteria included Karnofsky Performance Status (KPS) ≥70, serum bilirubin within institution limits of normal, and albumin ≥3.0 g/dL. Patients were randomized to receive ONIVYDE plus fluorouracil/leucovorin (ONIVYDE/FU/LV), ONIVYDE, or fluorouracil/leucovorin (FU/LV). Randomization was stratified by ethnicity (White vs. East Asian vs. other), KPS (70-80 vs. 90-100), and baseline albumin level (≥ 4 g/dL vs. 3.0-3.9 g/dL). Patients randomized to ONIVYDE/FU/LV received ONIVYDE 70 mg/m2 as an intravenous infusion over 90 minutes, followed by leucovorin 400 mg/m2 intravenously over 30 minutes, followed by fluorouracil 2400 mg/m2 intravenously over 46 hours, every 2 weeks. The ONIVYDE dose of 70 mg/m2 is based on irinotecan free base (equivalent to 80 mg/m2 of irinotecan as the hydrochloride trihydrate). Patients randomized to ONIVYDE as a single agent received ONIVYDE 100 mg/m2 as an intravenous infusion over 90 minutes every 3 weeks. Patients randomized to FU/LV received leucovorin 200 mg/m2 intravenously over 30 minutes, followed by fluorouracil 2000 mg/m2 intravenously over 24 hours, administered on Days 1, 8, 15 and 22 of a 6-week cycle. Patients homozygous for the UGT1A1*28 allele initiated ONIVYDE at a reduced dose (50 mg/m2 ONIVYDE, if given with FU/LV or 70 mg/m2 ONIVYDE as a single agent). When ONIVYDE was withheld or discontinued for adverse reactions, FU was also withheld or discontinued. When the dose of ONIVYDE was reduced for adverse reactions, the dose of FU was reduced by 25%. Treatment continued until disease progression or unacceptable toxicity.
The major efficacy outcome measure was overall survival (OS) with two pair-wise comparisons: ONIVYDE versus FU/LV and ONIVYDE/FU/LV versus FU/LV. Additional efficacy outcome measures were progression-free survival (PFS) and objective response rate (ORR). Tumor status assessments were conducted at baseline and every 6 weeks thereafter. The trial was initiated as a two-arm study and amended after initiation to include a third arm (ONIVYDE/FU/LV). The comparisons between the ONIVYDE/FU/LV and the FU/LV arms are limited to patients enrolled in the FU/LV arm after this protocol amendment.
Four hundred seventeen patients were randomized to: ONIVYDE/FU/LV (N=117), ONIVYDE (N=151), or FU/LV (N=149). Baseline demographics and tumor characteristics for the 236 patients randomized to ONIVYDE/FU/LV or FU/LV (N=119) after the addition of the third arm to the study were a median age of 63 years (range 34-81 years) and with 41% ≥ 65 years of age; 58% were men; 63% were White, 30% were Asian, 3% were Black or African American, and 5% were other. Mean baseline albumin level was 3.97 g/dL, and baseline KPS was 90-100 in 53% of patients. Disease characteristics included liver metastasis (67%) and lung metastasis (31%). A total of 13% of patients received gemcitabine in the neoadjuvant/adjuvant setting only, 55% of patients had 1 prior line of therapy for metastatic disease, and 33% of patients had 2 or more prior lines of therapy for metastatic disease. All patients received prior gemcitabine (alone or in combination with another agent), 54% received prior gemcitabine in combination with another agent, and 13% received prior gemcitabine in combination with nab-paclitaxel.
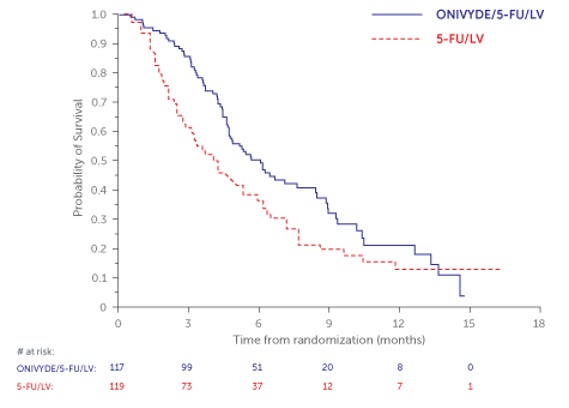
NAPOLI-1 demonstrated a statistically significant improvement in overall survival for the ONIVYDE/FU/LV arm over the FU/LV arm as summarized in Table 9 and Figure 2.
There was no improvement in overall survival for the ONIVYDE arm over the FU/LV arm (hazard ratio=1.00, p-value=0.97 (two-sided log-rank test)).
| ONIVYDE/FU/LV (N=117) | FU/LV (N=119) |
|
|---|---|---|
|
||
| Overall Survival | ||
| Number of Deaths, n (%) | 77 (66) | 86 (72) |
| Median Overall Survival (months) | 6.1 | 4.2 |
| (95% CI) | (4.8, 8.5) | (3.3, 5.3) |
| Hazard Ratio (95% CI) | 0.68 (0.50, 0.93) | |
| p-value (log-rank test) | 0.014 | |
| Progression-Free Survival | ||
| Death or Progression, n (%) | 83 (71) | 94 (79) |
| Median Progression-Free Survival (months) | 3.1 | 1.5 |
| (95% CI) | (2.7, 4.2) | (1.4, 1.8) |
| Hazard Ratio (95% CI) | 0.55 (0.41, 0.75) | |
| Objective Response Rate | ||
| Confirmed Complete or Partial Response n (%) | 9 (7.7%) | 1 (0.8%) |
Figure 2 Kaplan-Meier Curve for Overall Survival in all randomized Patients in NAPOLI-1
16. How is Onivyde supplied
17. Patient Counseling Information
Advise patients of the following:
Severe Neutropenia
Advise patients of the risk of neutropenia leading to severe and life-threatening infections and the need for monitoring of blood counts. Instruct patients to contact their healthcare provider immediately if experiencing signs of infection, such as fever, chills, dizziness, or shortness of breath [see Warnings and Precautions (5.1)].
Severe Diarrhea
Inform patients of the risk of severe and life-threatening diarrhea. Advise patients to stop lactose-containing products, maintain hydration, and eat frequent small meals with a low-fat diet. Advise patients to contact their healthcare provider if they experience persistent vomiting or diarrhea; black or bloody stools; or symptoms of dehydration such as lightheadedness, dizziness, or faintness [see Warnings and Precautions (5.2)].
Interstitial Lung Disease
Inform patients of the potential risk of ILD. Advise patients to contact their healthcare provider as soon as possible for new onset cough or dyspnea [see Interstitial Lung Disease (5.3)].
Hypersensitivity to irinotecan HCl or ONIVYDE
Advise patients of the potential risk of severe hypersensitivity and that ONIVYDE is contraindicated in patients with a history of severe allergic reactions with irinotecan HCl or ONIVYDE. Instruct patients to seek immediate medical attention for signs of severe hypersensitivity reaction such as chest tightness; shortness of breath; wheezing; dizziness or faintness; or swelling of the face, eyelids, or lips [see Contraindications (4) and Warnings and Precautions (5.4)].
Females and males of reproductive potential
Embryo-fetal toxicity: Inform females of reproductive potential of the potential risk to a fetus, to use effective contraception during treatment and for seven months after the last dose, and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.5), Use in Specific Populations (8.1, 8.3)].
Contraception: Advise male patients with female partners of reproductive potential to use condoms during treatment with ONIVYDE and for four months after the last dose [see Females and Males of Reproductive Potential (8.3)].
Lactation
Advise women not to breastfeed during treatment with ONIVYDE and for one month after the last dose [see Use in Special Populations (8.2)].
PRINCIPAL DISPLAY PANEL - 43 mg/10 mL Vial Label
NDC 15054-0043-1
Rx Only
Onivyde®
(irinotecan liposome injection)
43 mg/10 mL
(4.3 mg/mL)
For Intravenous Infusion After Dilution
Single Dose Vial
Discard Unused Portion
Liposomal Formulation
Do Not Substitute For
Irinotecan Hydrochloride

PRINCIPAL DISPLAY PANEL - 43 mg/10 mL Vial Carton
NDC 15054-0043-1
Rx Only
onivyde®
(irinotecan liposome injection)
43 mg/10 mL
(4.3 mg/mL)
For Intravenous Infusion After Dilution
Warning: Hazardous drug
Single Dose Vial
Discard Unused Portion
Liposomal Formulation
Do Not Substitute For Irinotecan Hydrochloride
| ONIVYDE
irinotecan hydrochloride injection, powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Ipsen Biopharmaceuticals, Inc. (118461578) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| ScinoPharm Taiwan Ltd. | 657484726 | API MANUFACTURE(15054-0043) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Hubei Haosun Pharmaceuticals Co. Ltd | 527128525 | API MANUFACTURE(15054-0043) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Ipsen Pharma Biotech | 502570286 | MANUFACTURE(15054-0043) , ANALYSIS(15054-0043) , LABEL(15054-0043) , PACK(15054-0043) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Boston Analytical | 181194176 | ANALYSIS(15054-0043) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Quality Chemical Laboratories (QCL) | 071344167 | ANALYSIS(15054-0043) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Avista | 079509111 | ANALYSIS(15054-0043) | |