Doxil: Package Insert / Prescribing Info
Package insert / product label
Generic name: doxorubicin hydrochloride
Dosage form: injection, suspension, liposomal
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
DOXIL® (doxorubicin hydrochloride liposome injection), for intravenous use
Initial U.S. Approval: 1995
WARNING: CARDIOMYOPATHY and INFUSION-RELATED REACTIONS
See full prescribing information for complete boxed warning.
- DOXIL can cause myocardial damage, including acute left ventricular failure. The risk of cardiomyopathy was 11% when the cumulative anthracycline dose was between 450 mg/m2 to 550 mg/m2. Assess left ventricular cardiac function prior to initiation of DOXIL, during treatment, and after treatment (5.1).
- Serious, life-threatening, and fatal infusion-related reactions can occur. Acute infusion-related reactions occurred in 11% of patients with solid tumors. Withhold DOXIL for infusion-related reactions and resume at a reduced rate. Discontinue DOXIL infusion for serious or life-threatening infusion-related reactions (5.2).
Indications and Usage for Doxil
DOXIL is an anthracycline topoisomerase inhibitor indicated for:
- Ovarian cancer: After failure of platinum-based chemotherapy (1.1)
- AIDS-related Kaposi's Sarcoma: After failure of prior systemic chemotherapy or intolerance to such therapy (1.2).
- Multiple Myeloma: In combination with bortezomib in patients who have not previously received bortezomib and have received at least one prior therapy (1.3).
Doxil Dosage and Administration
Administer DOXIL at an initial rate of 1 mg/min to minimize the risk of infusion reactions. If no infusion-related reactions occur, increase rate of infusion to complete administration over 1 hour. Do not administer as bolus injection or undiluted solution (2).
Dosage Forms and Strengths
Doxorubicin hydrochloride liposome injection: 20 mg/10 mL (2 mg/mL) and 50 mg/25 mL (2 mg/mL) in single-dose vials (3)
Contraindications
Warnings and Precautions
Adverse Reactions/Side Effects
Most common adverse reactions (>20%) are asthenia, fatigue, fever, anorexia, nausea, vomiting, stomatitis, diarrhea, constipation, hand-foot syndrome, rash, neutropenia, thrombocytopenia, and anemia (6).
To report SUSPECTED ADVERSE REACTIONS contact Janssen Products, LP at 1-800-JANSSEN (1-800-526-7736) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2019
Full Prescribing Information
WARNING: CARDIOMYOPATHY and INFUSION-RELATED REACTIONS
- DOXIL can cause myocardial damage, including acute left ventricular failure. The risk of cardiomyopathy was 11% when the cumulative anthracycline dose was between 450 mg/m2 to 550 mg/m2. Assess left ventricular cardiac function prior to initiation of DOXIL and during and after treatment [see Warnings and Precautions (5.1)].
- Serious, life-threatening, and fatal infusion-related reactions can occur with DOXIL. Acute infusion-related reactions occurred in 11% of patients with solid tumors. Withhold DOXIL for infusion-related reactions and resume at a reduced rate. Discontinue DOXIL for serious or life-threatening infusion-related reactions [see Warnings and Precautions (5.2)].
1. Indications and Usage for Doxil
1.1 Ovarian Cancer
DOXIL is indicated for the treatment of patients with ovarian cancer whose disease has progressed or recurred after platinum-based chemotherapy.
2. Doxil Dosage and Administration
2.1 Important Use Information
Do not substitute DOXIL for other doxorubicin hydrochloride products.
Do not administer as an undiluted suspension or as an intravenous bolus [see Warnings and Precautions (5.2)].
2.2 Ovarian Cancer
The recommended dose of DOXIL is 50 mg/m2 intravenously over 60 minutes every 28 days until disease progression or unacceptable toxicity.
2.3 AIDS-Related Kaposi's Sarcoma
The recommended dose of DOXIL is 20 mg/m2 intravenously over 60 minutes every 21 days until disease progression or unacceptable toxicity.
2.4 Multiple Myeloma
The recommended dose of DOXIL is 30 mg/m2 intravenously over 60 minutes on day 4 of each 21-day cycle for eight cycles or until disease progression or unacceptable toxicity. Administer DOXIL after bortezomib on day 4 of each cycle [see Clinical Studies (14.3)].
2.5 Dose Modifications for Adverse Reactions
Do not increase DOXIL after a dose reduction for toxicity.
| Toxicity | Dose Adjustment |
|---|---|
| Hand-Foot Syndrome (HFS) | |
| Grade 1: Mild erythema, swelling, or desquamation not interfering with daily activities |
|
| Grade 2: Erythema, desquamation, or swelling interfering with, but not precluding normal physical activities; small blisters or ulcerations less than 2 cm in diameter |
|
| Grade 3: Blistering, ulceration, or swelling interfering with walking or normal daily activities; cannot wear regular clothing |
|
| Grade 4: Diffuse or local process causing infectious complications, or a bed ridden state or hospitalization |
|
| Stomatitis | |
| Grade 1: Painless ulcers, erythema, or mild soreness |
|
| Grade 2: Painful erythema, edema, or ulcers, but can eat |
|
| Grade 3: Painful erythema, edema, or ulcers, and cannot eat |
|
| Grade 4: Requires parenteral or enteral support |
|
| Neutropenia or Thrombocytopenia | |
| Grade 1 | No dose reduction |
| Grade 2 | Delay until ANC ≥ 1,500 and platelets ≥ 75,000; resume treatment at previous dose |
| Grade 3 | Delay until ANC ≥ 1,500 and platelets ≥ 75,000; resume treatment at previous dose |
| Grade 4 | Delay until ANC ≥ 1,500 and platelets ≥ 75,000; resume at 25% dose reduction or continue previous dose with prophylactic granulocyte growth factor |
| Toxicity | DOXIL |
|---|---|
| Fever ≥38°C and ANC <1,000/mm3 |
|
On any day of drug administration after Day 1 of each cycle:
|
|
| Grade 3 or 4 non-hematologic drug related toxicity | Do not dose until recovered to Grade <2, then reduce dose by 25%. |
For neuropathic pain or peripheral neuropathy, no dosage adjustments are required for DOXIL. Refer to bortezomib manufacturer's prescribing information.
2.6 Preparation and Administration
Preparation
Dilute DOXIL doses up to 90 mg in 250 mL of 5% Dextrose Injection, USP prior to administration. Dilute doses exceeding 90 mg in 500 mL of 5% Dextrose Injection, USP prior to administration. Refrigerate diluted DOXIL at 2°C to 8°C (36°F to 46°F) and administer within 24 hours.
Administration
Inspect parenteral drug products visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if a precipitate or foreign matter is present.
Do not use with in-line filters.
Administer the first dose of DOXIL at an initial rate of 1 mg/min. If no infusion-related adverse reactions are observed, increase the infusion rate to complete the administration of the drug over one hour [see Warnings and Precautions (5.2)]. Do not rapidly flush the infusion line.
Do not mix DOXIL with other drugs.
Management of Suspected Extravasation
Discontinue DOXIL for burning or stinging sensation or other evidence indicating perivenous infiltration or extravasation. Manage confirmed or suspected extravasation as follows:
- Do not remove the needle until attempts are made to aspirate extravasated fluid
- Do not flush the line
- Avoid applying pressure to the site
- Apply ice to the site intermittently for 15 min 4 times a day for 3 days
- If the extravasation is in an extremity, elevate the extremity
3. Dosage Forms and Strengths
Doxorubicin hydrochloride liposome injection: 20 mg/10 mL (2 mg/mL) and 50 mg/25 mL (2 mg/mL) in single-dose vials. The drug product appears as a translucent, red liposomal dispersion.
4. Contraindications
DOXIL is contraindicated in patients who have a history of severe hypersensitivity reactions, including anaphylaxis, to doxorubicin hydrochloride [see Warnings and Precautions (5.2)].
5. Warnings and Precautions
5.1 Cardiomyopathy
Doxorubicin hydrochloride can cause myocardial damage, including acute left ventricular failure. The risk of cardiomyopathy with doxorubicin hydrochloride is generally proportional to the cumulative exposure. Include prior use of other anthracyclines or anthracenediones in calculations of cumulative dose. The risk of cardiomyopathy may be increased at lower cumulative doses in patients with prior mediastinal irradiation.
In a clinical study in 250 patients with advanced cancer who were treated with DOXIL, the risk of cardiomyopathy was 11% when the cumulative anthracycline dose was between 450 mg/m2 to 550 mg/m2. Cardiomyopathy was defined as >20% decrease in resting left ventricular ejection fraction (LVEF) from baseline where LVEF remained in the normal range or a >10% decrease in LVEF from baseline where LVEF was less than the institutional lower limit of normal. Two percent of patients developed signs and symptoms of congestive heart failure without documented evidence of cardiomyopathy.
Assess left ventricular cardiac function (e.g. MUGA or echocardiogram) prior to initiation of DOXIL, during treatment to detect acute changes, and after treatment to detect delayed cardiomyopathy. Administer DOXIL to patients with a history of cardiovascular disease only when the potential benefit of treatment outweighs the risk.
5.2 Infusion-Related Reactions
Serious, life-threatening, and fatal infusion-related reactions characterized by one or more of the following symptoms can occur with DOXIL: flushing, shortness of breath, facial swelling, headache, chills, chest pain, back pain, tightness in the chest and throat, fever, tachycardia, pruritus, rash, cyanosis, syncope, bronchospasm, asthma, apnea, and hypotension. Of 239 patients with ovarian cancer treated with DOXIL in Trial 4, 7% of patients experienced acute infusion-related reactions resulting in dose interruption. All occurred during cycle 1 and none during subsequent cycles. Across multiple studies of DOXIL monotherapy including this and other studies enrolling 760 patients with various solid tumors, 11% of patients had infusion-related reactions. The majority of infusion-related events occurred during the first infusion.
Ensure that medications to treat infusion-related reactions and cardiopulmonary resuscitative equipment are available for immediate use prior to initiation of DOXIL. Initiate DOXIL infusions at a rate of 1 mg/min and increase rate as tolerated [see Dosage and Administration (2.6)]. Withhold DOXIL for Grade 1, 2, or 3 infusion-related reactions and resume at a reduced infusion rate. Discontinue DOXIL for serious or life-threatening infusion-related reactions.
5.3 Hand-Foot Syndrome (HFS)
In Trial 4, the incidence of HFS was 51% of patients in the DOXIL arm and 0.9% of patients in the topotecan arm, including 24% Grade 3 or 4 cases of HFS in DOXIL-treated patients and no Grade 3 or 4 cases in topotecan-treated patients. HFS or other skin toxicity required discontinuation of DOXIL in 4.2% of patients.
HFS was generally observed after 2 or 3 cycles of treatment but may occur earlier. Delay DOXIL for the first episode of Grade 2 or greater HFS [see Dosage and Administration (2.5)]. Discontinue DOXIL if HFS is severe and debilitating.
5.4 Secondary Oral Neoplasms
Secondary oral cancers, primarily squamous cell carcinoma, have been reported from post-marketing experience in patients with long-term (more than one year) exposure to DOXIL. These malignancies were diagnosed both during treatment with DOXIL and up to 6 years after the last dose. Examine patients at regular intervals for the presence of oral ulceration or with any oral discomfort that may be indicative of secondary oral cancer.
The altered pharmacokinetics and preferential tissue distribution of liposomal doxorubicin that contributes to enhanced skin toxicity and mucositis compared to free doxorubicin may play a role in the development of oral secondary malignancies with long-term use.
5.5 Embryo-Fetal Toxicity
Based on findings in animals and its mechanism of action, DOXIL can cause fetal harm when administered to a pregnant woman; avoid the use of DOXIL during the 1st trimester. Available human data do not establish the presence or absence of major birth defects and miscarriage related to the use of doxorubicin hydrochloride during the 2nd and 3rd trimesters. At doses approximately 0.12 times the recommended clinical dose, DOXIL was embryotoxic and abortifacient in rabbits. Advise pregnant women of the potential risk to a fetus. Advise females and males of reproductive potential to use effective contraception during and for 6 months after treatment with DOXIL [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in more detail in other sections of the labeling.
- Cardiomyopathy [see Warnings and Precautions (5.1)]
- Infusion-Related Reactions [see Warnings and Precautions (5.2)]
- Hand-Foot Syndrome [see Warnings and Precautions (5.3)]
- Secondary Oral Neoplasms [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates on other clinical trials and may not reflect the rates observed in clinical practice.
The safety data reflect exposure to DOXIL in 1310 patients including: 239 patients with ovarian cancer, 753 patients with AIDS-related Kaposi's sarcoma, and 318 patients with multiple myeloma. The most common adverse reactions (>20%) observed with DOXIL are asthenia, fatigue, fever, nausea, stomatitis, vomiting, diarrhea, constipation, anorexia, hand-foot syndrome, rash and neutropenia, thrombocytopenia and anemia.
The following tables present adverse reactions from clinical trials of single-agent DOXIL in ovarian cancer and AIDS-Related Kaposi's sarcoma.
Patients With Ovarian Cancer
The safety data described below are from Trial 4, which included 239 patients with ovarian cancer treated with DOXIL 50 mg/m2 once every 4 weeks for a minimum of four courses in a randomized, multicenter, open-label study. In this trial, patients received DOXIL for a median number of 3.2 months (range 1 day to 25.8 months). The median age of the patients is 60 years (range 27 to 87), with 91% Caucasian, 6% Black, and 3% Hispanic or Other.
Table 3 presents the hematologic adverse reactions from Trial 4.
| DOXIL Patients (n=239) | Topotecan Patients (n=235) |
|
|---|---|---|
| Neutropenia | ||
| 500 – <1000/mm3 | 8% | 14% |
| <500/mm3 | 4.2% | 62% |
| Anemia | ||
| 6.5 – <8 g/dL | 5% | 25% |
| < 6.5 g/dL | 0.4% | 4.3% |
| Thrombocytopenia | ||
| 10,000 – <50,000/mm3 | 1.3% | 17% |
| <10,000/mm3 | 0.0% | 17% |
Table 4 presents the non-hematologic adverse reactions from Trial 4.
| Non-Hematologic Adverse Reaction 10% or Greater | DOXIL (%) treated (n=239) | Topotecan (%) treated (n=235) |
||
|---|---|---|---|---|
| All grades | Grades 3–4 | All grades | Grades 3–4 | |
| Body as a Whole | ||||
| Asthenia | 40 | 7 | 52 | 8 |
| Fever | 21 | 0.8 | 31 | 6 |
| Mucous Membrane Disorder | 14 | 3.8 | 3.4 | 0 |
| Back Pain | 12 | 1.7 | 10 | 0.9 |
| Infection | 12 | 2.1 | 6 | 0.9 |
| Headache | 11 | 0.8 | 15 | 0 |
| Digestive | ||||
| Nausea | 46 | 5 | 63 | 8 |
| Stomatitis | 41 | 8 | 15 | 0.4 |
| Vomiting | 33 | 8 | 44 | 10 |
| Diarrhea | 21 | 2.5 | 35 | 4.2 |
| Anorexia | 20 | 2.5 | 22 | 1.3 |
| Dyspepsia | 12 | 0.8 | 14 | 0 |
| Nervous | ||||
| Dizziness | 4.2 | 0 | 10 | 0 |
| Respiratory | ||||
| Pharyngitis | 16 | 0 | 18 | 0.4 |
| Dyspnea | 15 | 4.1 | 23 | 4.3 |
| Cough increased | 10 | 0 | 12 | 0 |
| Skin and Appendages | ||||
| Hand-foot syndrome | 51 | 24 | 0.9 | 0 |
| Rash | 29 | 4.2 | 12 | 0.4 |
| Alopecia | 19 | N/A | 52 | N/A |
The following additional adverse reactions were observed in patients with ovarian cancer with doses administered every four weeks (Trial 4).
Incidence 1% to 10%
Cardiovascular: vasodilation, tachycardia, deep vein thrombosis, hypotension, cardiac arrest.
Digestive: oral moniliasis, mouth ulceration, esophagitis, dysphagia, rectal bleeding, ileus.
Hematologic and Lymphatic: ecchymosis.
Metabolic and Nutritional: dehydration, weight loss, hyperbilirubinemia, hypokalemia, hypercalcemia, hyponatremia.
Nervous: somnolence, dizziness, depression.
Respiratory: rhinitis, pneumonia, sinusitis, epistaxis.
Skin and Appendages: pruritus, skin discoloration, vesiculobullous rash, maculopapular rash, exfoliative dermatitis, herpes zoster, dry skin, herpes simplex, fungal dermatitis, furunculosis, acne.
Special Senses: conjunctivitis, taste perversion, dry eyes.
Urinary: urinary tract infection, hematuria, vaginal moniliasis.
Patients With AIDS-Related Kaposi's Sarcoma
The safety data described is based on the experience reported in 753 patients with AIDS-related Kaposi's sarcoma (KS) enrolled in four open-label, uncontrolled trials of DOXIL administered at doses ranging from 10 to 40 mg/m2 every 2 to 3 weeks. Demographics of the population were: median age 38.7 years (range 24–70); 99% male; 88% Caucasian, 6% Hispanic, 4% Black, and 2% Asian/other/unknown. The majority of patients were treated with 20 mg/m2 of DOXIL every 2 to 3 weeks with a median exposure of 4.2 months (range 1 day to 26.6 months). The median cumulative dose was 120 mg/m2 (range 3.3 to 798.6 mg/m2); 3% received cumulative doses of greater than 450 mg/m2.
Disease characteristics were: 61% poor risk for KS tumor burden, 91% poor risk for immune system, and 47% poor risk for systemic illness; 36% were poor risk for all three categories; median CD4 count 21 cells/mm3 (51% less than 50 cells/mm3); mean absolute neutrophil count at study entry approximately 3,000 cells/mm3.
Of the 693 patients with concomitant medication information, 59% were on one or more antiretroviral medications [35% zidovudine (AZT), 21% didanosine (ddI), 16% zalcitabine (ddC), and 10% stavudine (D4T)]; 85% received PCP prophylaxis (54% sulfamethoxazole/trimethoprim); 85% received antifungal medications (76% fluconazole); 72% received antivirals (56% acyclovir, 29% ganciclovir, and 16% foscarnet) and 48% patients received colony-stimulating factors (sargramostim/filgrastim) during their course of treatment.
Adverse reactions led to discontinuation of treatment in 5% of patients with AIDS-related Kaposi's sarcoma and included myelosuppression, cardiac adverse reactions, infusion-related reactions, toxoplasmosis, HFS, pneumonia, cough/dyspnea, fatigue, optic neuritis, progression of a non-KS tumor, allergy to penicillin, and unspecified reasons. Tables 5 and 6 summarize adverse reactions reported in patients treated with DOXIL for AIDS-related Kaposi's sarcoma in a pooled analysis of the four trials.
| Patients With Refractory or Intolerant AIDS-Related Kaposi's Sarcoma (n=74*) | Total Patients With AIDS-Related Kaposi's Sarcoma (n=720†) |
|
|---|---|---|
|
||
| Neutropenia | ||
| < 1000/mm3 | 46% | 49% |
| < 500/mm3 | 11% | 13% |
| Anemia | ||
| < 10 g/dL | 58% | 55% |
| < 8 g/dL | 16% | 18% |
| Thrombocytopenia | ||
| < 150,000/mm3 | 61% | 61% |
| < 25,000/mm3 | 1.4% | 4.2% |
| Adverse Reactions | Patients With Refractory or Intolerant AIDS-Related Kaposi's Sarcoma (n=77*) | Total Patients With AIDS-Related Kaposi's Sarcoma (n=705†) |
|---|---|---|
|
||
| Nausea | 18% | 17% |
| Asthenia | 7% | 10% |
| Fever | 8% | 9% |
| Alopecia | 9% | 9% |
| Alkaline Phosphatase Increase | 1.3% | 8% |
| Vomiting | 8% | 8% |
| Diarrhea | 5% | 8% |
| Stomatitis | 5% | 7% |
| Oral Moniliasis | 1.3% | 6% |
The following additional adverse reactions were observed in 705 patients with AIDS-related Kaposi's sarcoma.
Incidence 1% to 5%
Body as a Whole: headache, back pain, infection, allergic reaction, chills.
Cardiovascular: chest pain, hypotension, tachycardia.
Cutaneous: herpes simplex, rash, itching.
Digestive: mouth ulceration, anorexia, dysphagia.
Metabolic and Nutritional: SGPT increase, weight loss, hyperbilirubinemia.
Other: dyspnea, pneumonia, dizziness, somnolence.
Incidence Less Than 1%
Body As A Whole: sepsis, moniliasis, cryptococcosis.
Cardiovascular: thrombophlebitis, cardiomyopathy, palpitation, bundle branch block, congestive heart failure, heart arrest, thrombosis, ventricular arrhythmia.
Digestive: hepatitis.
Metabolic and Nutritional Disorders: dehydration.
Respiratory: cough increase, pharyngitis.
Skin and Appendages: maculopapular rash, herpes zoster.
Special Senses: taste perversion, conjunctivitis.
Patients With Multiple Myeloma
The safety data described are from 318 patients treated with DOXIL (30 mg/m2) administered on day 4 following bortezomib (1.3 mg/m2 i.v. bolus on days 1, 4, 8 and 11) every 3 weeks, in a randomized, open-label, multicenter study (Trial 6). In this trial, patients in the DOXIL + bortezomib combination group were treated for a median number of 4.5 months (range 21 days to 13.5 months). The population was 28 to 85 years of age (median age 61), 58% male, 90% Caucasian, 6% Black, and 4% Asian and Other. Table 7 lists adverse reactions reported in 10% or more of patients treated with DOXIL in combination with bortezomib for multiple myeloma.
| Adverse Reaction | DOXIL + bortezomib (n=318) | Bortezomib (n=318) |
||
|---|---|---|---|---|
| Any (%) | Grade 3–4 | Any (%) | Grade 3–4 | |
|
||||
| Blood and lymphatic system disorders | ||||
| Neutropenia | 36 | 32 | 22 | 16 |
| Thrombocytopenia | 33 | 24 | 28 | 17 |
| Anemia | 25 | 9 | 21 | 9 |
| General disorders and administration site conditions | ||||
| Fatigue | 36 | 7 | 28 | 3 |
| Pyrexia | 31 | 1 | 22 | 1 |
| Asthenia | 22 | 6 | 18 | 4 |
| Gastrointestinal disorders | ||||
| Nausea | 48 | 3 | 40 | 1 |
| Diarrhea | 46 | 7 | 39 | 5 |
| Vomiting | 32 | 4 | 22 | 1 |
| Constipation | 31 | 1 | 31 | 1 |
| Mucositis/Stomatitis | 20 | 2 | 5 | <1 |
| Abdominal pain | 11 | 1 | 8 | 1 |
| Infections and infestations | ||||
| Herpes zoster | 11 | 2 | 9 | 2 |
| Herpes simplex | 10 | 0 | 6 | 1 |
| Investigations | ||||
| Weight decreased | 12 | 0 | 4 | 0 |
| Metabolism and Nutritional disorders | ||||
| Anorexia | 19 | 2 | 14 | <1 |
| Nervous system disorders | ||||
| Peripheral Neuropathy* | 42 | 7 | 45 | 11 |
| Neuralgia | 17 | 3 | 20 | 4 |
| Paresthesia/dysesthesia | 13 | <1 | 10 | 0 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Cough | 18 | 0 | 12 | 0 |
| Skin and subcutaneous tissue disorders | ||||
| Rash† | 22 | 1 | 18 | 1 |
| Hand-foot syndrome | 19 | 6 | <1 | 0 |
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during postapproval use of DOXIL. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Musculoskeletal and Connective Tissue Disorders: muscle spasms
Respiratory, Thoracic and Mediastinal Disorders: pulmonary embolism (in some cases fatal)
Hematologic Disorders: Secondary acute myelogenous leukemia
Skin and Subcutaneous Tissue Disorders: erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, lichenoid keratosis
Secondary Oral Neoplasms: [see Warnings and Precautions (5.4)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings in animals and its mechanism of action, DOXIL can cause fetal harm when administered to a pregnant woman; avoid the use of DOXIL during the 1st trimester. In animal reproduction studies, DOXIL was embryotoxic in rats and abortifacient in rabbits following intravenous administration during organogenesis at doses approximately 0.12 times the recommended clinical dose (see Data). Available human data do not establish the presence or absence of major birth defects and miscarriage related to the use of doxorubicin hydrochloride during the 2nd and 3rd trimesters. Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated populations are unknown. However, the background risk in the U.S. general population of major birth defects is 2–4% and of miscarriage is 15–20% of clinically recognized pregnancies.
Data
Animal Data
DOXIL was embryotoxic at doses of 1 mg/kg/day in rats and was embryotoxic and abortifacient at 0.5 mg/kg/day in rabbits (both doses are about 0.12 times the recommended dose of 50 mg/m2 human dose on a mg/m2 basis). Embryotoxicity was characterized by increased embryo-fetal deaths and reduced live litter sizes.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating DOXIL.
Contraception
Females
DOXIL can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during and for 6 months after treatment with DOXIL.
Males
DOXIL may damage spermatozoa and testicular tissue, resulting in possible genetic fetal abnormalities. Males with female sexual partners of reproductive potential should use effective contraception during and for 6 months after treatment with DOXIL [see Non-clinical Toxicology (13.1)].
Infertility
Females
In females of reproductive potential, DOXIL may cause infertility and result in amenorrhea. Premature menopause can occur with doxorubicin hydrochloride. Recovery of menses and ovulation is related to age at treatment.
Males
DOXIL may result in oligospermia, azoospermia, and permanent loss of fertility. Sperm counts have been reported to return to normal levels in some men. This may occur several years after the end of therapy [see Non-clinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of DOXIL in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of DOXIL conducted in patients with either epithelial ovarian cancer (Trial 4) or with AIDS-related Kaposi's sarcoma (Trial 5) did not contain sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger subjects.
In Trial 6, of 318 patients treated with DOXIL in combination with bortezomib for multiple myeloma, 37% were 65 years of age or older and 8% were 75 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients.
10. Overdosage
Acute overdosage with doxorubicin hydrochloride causes increased risk of severe mucositis, leukopenia, and thrombocytopenia.
11. Doxil Description
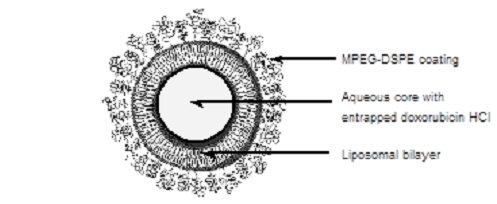
The active ingredient in DOXIL is doxorubicin hydrochloride, an anthracycline topoisomerase inhibitor, that is encapsulated in STEALTH® liposomes for intravenous use.
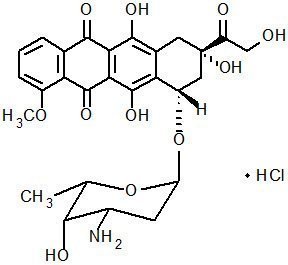
The chemical name of doxorubicin hydrochloride is (8S,10S)-10-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-8-glycolyl-7,8,9,10-tetrahydro-6,8,11-trihydroxy-1-methoxy-5,12-naphthacenedione hydrochloride. The molecular formula is C27H29NO11∙HCl and the molecular weight is 579.99.
The structural formula is:
DOXIL is a sterile, translucent, red liposomal dispersion. Each single-dose vial contains 20 mg or 50 mg doxorubicin hydrochloride at a concentration of 2 mg/mL (equivalent to 1.87 mg/mL of doxorubicin) and a pH of 6.5. The STEALTH liposome carriers are composed of cholesterol, 3.19 mg/mL; fully hydrogenated soy phosphatidylcholine (HSPC), 9.58 mg/mL; and N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine sodium salt (MPEG-DSPE), 3.19 mg/mL. Each mL also contains ammonium sulfate, approximately 0.6 mg; histidine as a buffer; hydrochloric acid and/or sodium hydroxide for pH control; and sucrose to maintain isotonicity. Greater than 90% of the drug is encapsulated in the STEALTH liposomes.
MPEG-DSPE has the following structural formula:
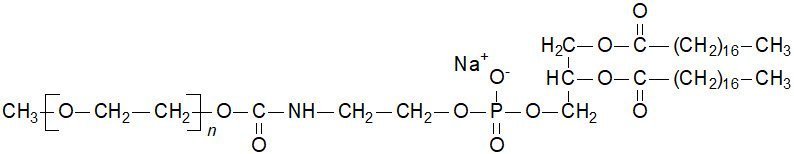
n = ca. 45
HSPC has the following structural formula:
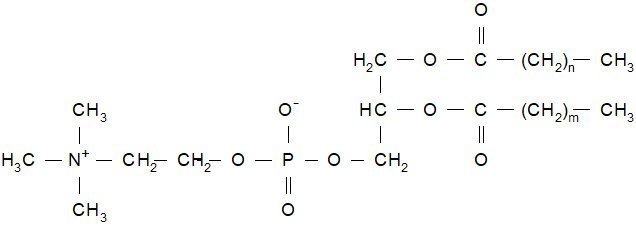
m, n=14 or 16
Representation of a STEALTH liposome:
12. Doxil - Clinical Pharmacology
12.1 Mechanism of Action
The active ingredient of DOXIL is doxorubicin hydrochloride. The mechanism of action of doxorubicin hydrochloride is thought to be related to its ability to bind DNA and inhibit nucleic acid synthesis. Cell structure studies have demonstrated rapid cell penetration and perinuclear chromatin binding, rapid inhibition of mitotic activity and nucleic acid synthesis, and induction of mutagenesis and chromosomal aberrations.
12.3 Pharmacokinetics
The pharmacokinetic parameters for total doxorubicin following a single dose of DOXIL infused over 30 minutes are presented in Table 8.
| Dose | ||
|---|---|---|
| Parameter (units) | 10 mg/m2 | 20 mg/m2 |
| N=23 Mean ± Standard Error |
||
| Peak Plasma Concentration (µg/mL) | 4.12 ± 0.215 | 8.34 ± 0.49 |
| Plasma Clearance (L/h/m2) | 0.056 ± 0.01 | 0.041 ± 0.004 |
| Steady State Volume of Distribution (L/m2) | 2.83 ± 0.145 | 2.72 ± 0.120 |
| AUC (µg/mL∙h) | 277 ± 32.9 | 590 ± 58.7 |
| First Phase (λ1) Half-Life (h) | 4.7 ± 1.1 | 5.2 ± 1.4 |
| Second Phase (λ1) Half-Life (h) | 52.3 ± 5.6 | 55.0 ± 4.8 |
DOXIL displayed linear pharmacokinetics over the range of 10 to 20 mg/m2. Relative to DOXIL doses at or below 20 mg/m2, the pharmacokinetics of total doxorubicin following a 50 mg/m2 DOXIL dose are nonlinear. At this dose, the elimination half-life of DOXIL is longer and the clearance lower compared to a 20 mg/m2 dose.
Distribution
Direct measurement of liposomal doxorubicin shows that at least 90% of the drug (the assay used cannot quantify less than 5–10% free doxorubicin) remains liposome-encapsulated during circulation.
In contrast to doxorubicin, which displays a large volume of distribution (range 700 to 1100 L/m2), the small steady state volume of distribution of liposomal doxorubicin suggests that DOXIL is largely confined to vascular fluid. Doxorubicin becomes available after the liposomes are extravasated. Plasma protein binding of DOXIL has not been determined; the plasma protein binding of doxorubicin is approximately 70%.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Mutagenicity or carcinogenicity studies have not been conducted with DOXIL, however doxorubicin was shown to be mutagenic in the in vitro Ames assay, and clastogenic in multiple in vitro assays (CHO cell, V79 hamster cell, human lymphoblast, and SCE assays) and the in vivo mouse micronucleus assay. The possible adverse effects on fertility in animals have not been adequately evaluated. DOXIL resulted in mild to moderate ovarian and testicular atrophy in mice after administration of a single dose of 36 mg/kg (about 2 times the 50 mg/m2 human dose on a mg/m2 basis). Decreased testicular weights and hypospermia were observed in rats after repeat doses ≥ 0.25 mg/kg/day (about 0.03 times the 50 mg/m2 human dose on a mg/m2 basis), and diffuse degeneration of the seminiferous tubules and a marked decrease in spermatogenesis were observed in dogs after repeat doses of 1 mg/kg/day (about 0.4 times the 50 mg/m2 human dose on a mg/m2 basis).
14. Clinical Studies
14.1 Ovarian Cancer
DOXIL was studied in three open-label, single-arm, clinical studies of 176 patients with metastatic ovarian cancer (Trials 1, 2, and 3). One hundred forty-five of these patients were refractory to both paclitaxel- and platinum-based chemotherapy regimens, defined as disease progression while on treatment or relapse within 6 months of completing treatment. Patients received DOXIL at 50 mg/m2 every 3 or 4 weeks for 3–6+ cycles in the absence of dose-limiting toxicity or disease progression.
The median age at diagnosis ranged from 52 to 64 years in the 3 studies, and the range was 22 to 85. Most patients had International Federation of Obstetricians and Gynecologists (FIGO) stage III or IV disease (ranging from 83% to 93%). Approximately one third of the patients had three or more prior lines of therapy (ranging from 22% to 33%).
The primary outcome measure was confirmed response rate based on Southwestern Oncology Group (SWOG) criteria for patients refractory to both paclitaxel- and a platinum-containing regimen. Secondary efficacy parameters were time to response, duration of response, and time to progression.
The response rates for the individual single arm trials are given in Table 9 below.
| Trial 1 (U.S.) N=27 | Trial 2 (U.S.) N=82 | Trial 3 (non-U.S.) N=36 |
|
|---|---|---|---|
| Response Rate | 22.2% | 17.1% | 0% |
| 95% Confidence Interval | 8.6% – 42.3% | 9.7% – 27.0% | 0.0% – 9.7% |
In a pooled analysis of Trials 1–3, the response rate for all patients refractory to paclitaxel and platinum agents was 13.8% (95% CI 8.1% to 19.3%). The median time to progression was 15.9 weeks, the median time to response was 17.6 weeks, and the duration of response was 39.4 weeks.
In Trial 4, a randomized, multicenter, open-label, trial in 474 patients with epithelial ovarian cancer after platinum-based chemotherapy, patients were randomized to receive either DOXIL 50 mg/m2 every 4 weeks (n=239) or topotecan 1.5 mg/m2 daily for 5 consecutive days every 3 weeks (n=235). Patients were stratified according to platinum sensitivity (response to initial platinum-based therapy and a progression-free interval of greater than 6 months off treatment) and the presence of bulky disease (tumor mass greater than 5 cm in size). The primary outcome measure was time to progression (TTP). Other endpoints included overall survival and objective response rate.
Of the 474 patients, the median age at diagnosis was 60 years (range 25 to 87), 90% were FIGO stage III and IV; 46% were platinum sensitive; and 45% had bulky disease.
There was no statistically significant difference in TTP between the two arms. Results are provided in Table 10.
| Protocol Defined ITT Population | ||
|---|---|---|
| DOXIL (n=239) | Topotecan (n=235) |
|
|
||
| TTP (Protocol Specified Primary Endpoint) | ||
| Median (Months)† | 4.1 | 4.2 |
| p-value‡ | 0.62 | |
| Hazard Ratio§ | 0.96 | |
| 95% CI for Hazard Ratio | (0.76, 1.20) | |
| Overall Survival | ||
| Median (Months) † | 14.4 | 13.7 |
| p-value¶ | 0.05 | |
| Hazard Ratio§ | 0.82 | |
| 95% CI for Hazard Ratio | (0.68, 1.00) | |
| Response Rate | ||
| Overall Response n (%) | 47 (19.7) | 40 (17.0) |
| Complete Response n (%) | 9 (3.8) | 11 (4.7) |
| Partial Response n (%) | 38 (15.9) | 29 (12.3) |
| Median Duration of Response (Months) † | 6.9 | 5.9 |
14.2 AIDS-Related Kaposi's Sarcoma
DOXIL was studied in an open-label, single-arm, multicenter study at a dose of 20 mg/m2 every 3 weeks, until disease progression or unacceptable toxicity (Trial 5).
Data is described for a cohort of 77 patients retrospectively identified as having disease progression on prior systemic combination chemotherapy (at least two cycles of a regimen containing at least two of three treatments: bleomycin, vincristine or vinblastine, or doxorubicin) or as being intolerant to such therapy. Forty-nine of the 77 (64%) patients had received prior doxorubicin hydrochloride.
The median time on study was 5.1 months (range 1 day to 15 months). The median cumulative dose of DOXIL was 154 mg/m2 (range 20 to 620 mg/m2). Among the 77 patients, mean age was 38 years (range 24 to 54); 87% were Caucasian, 5% Hispanic, 4% Black, and 4% Asian/Other/Unknown; median CD4 count was 10 cells/mm3; ACTG staging criteria were 78% poor risk for tumor burden, 96% poor risk for immune system, and 58% poor risk for systemic illness at baseline; and mean Karnofsky status score was 74%. All patients had cutaneous or subcutaneous lesions, 40% also had oral lesions, 26% pulmonary lesions, and 14% had lesions of the stomach/intestine.
Two analyses of tumor response were used: one based on investigator assessment of changes in lesions based on modified ACTG criteria (partial response defined as no new lesions, sites of disease, or worsening edema; flattening of ≥50% of previously raised lesions or area of indicator lesions decreasing by ≥50%; and response lasting at least 21 days with no prior progression), and one based on changes in up to five prospectively indentified representative indicator lesions (partial response defined as flattening of ≥50% of previously raised indicator lesions, or >50% decrease in the area of indicator lesions and lasting at least 21 days with no prior progression).
Of the 77 patients, 34 were evaluable for investigator assessment and 42 were evaluable for indicator lesion assessment; analyses of tumor responses are shown in Table 11.
| Investigator Assessment | All Evaluable Patients (n=34) | Evaluable Patients Who Received Prior Doxorubicin (n=20) |
| Response† | ||
| Partial (PR) | 27% | 30% |
| Stable | 29% | 40% |
| Progression | 44% | 30% |
| Duration of PR (Days) | ||
| Median | 73 | 89 |
| Range | 42+ – 210+ | 42+ – 210+ |
| Time to PR (Days) | ||
| Median | 43 | 53 |
| Range | 15 – 133 | 15 – 109 |
| Indicator Lesion Assessment | All Evaluable Patients (n=42) | Evaluable Patients Who Received Prior Doxorubicin (n=23) |
| Response† | ||
| Partial (PR) | 48% | 52% |
| Stable | 26% | 30% |
| Progression | 26% | 17% |
| Duration of PR (Days) | ||
| Median | 71 | 79 |
| Range | 22+ – 210+ | 35 – 210+ |
| Time to PR (Days) | ||
| Median | 22 | 48 |
| Range | 15 – 109 | 15 – 109 |
Retrospective efficacy analyses were performed in two trials that had subsets of patients who received single-agent DOXIL and who were on stable antiretroviral therapy for at least 60 days prior to enrollment and until a response was demonstrated. In one trial, 7 of 17 (40%) patients had a durable response (median duration not reached but was longer than 11.6 months). In the second trial, 4 of 11 patients (40%) on a stable antiretroviral therapy demonstrated durable responses.
14.3 Multiple Myeloma
The efficacy of DOXIL in combination with bortezomib was evaluated in Trial 6, a randomized, open-label, international, multicenter study in 646 patients who had not previously received bortezomib and whose disease progressed during or after at least one prior therapy. Patients were randomized (1:1) to receive either DOXIL (30 mg/m2) administered IV on day 4 following bortezomib (1.3 mg/m2 IV on days 1, 4, 8 and 11) or bortezomib alone every 3 weeks for up to 8 cycles or until disease progression or unacceptable toxicity. Patients who maintained a response were allowed to receive further treatment. The median number of cycles in each treatment arm was 5 (range 1–18).
The baseline demographics and clinical characteristics of the patients with multiple myeloma were similar between treatment arms (Table 12).
| Patient Characteristics | DOXIL + bortezomib n=324 | bortezomib n=322 |
|---|---|---|
| Median age in years (range) | 61 (28, 85) | 62 (34, 88) |
| % Male/female | 58 / 42 | 54 / 46 |
| % Caucasian/Black/other | 90 / 6 / 4 | 94 / 4 / 2 |
| Disease Characteristics | ||
| % with IgG/IgA/Light chain | 57 / 27 / 12 | 62 / 24 / 11 |
| % β2-microglobulin group | ||
| ≤2.5 mg/L | 14 | 14 |
| >2.5 mg/L and ≤5.5 mg/L | 56 | 55 |
| >5.5 mg/L | 30 | 31 |
| Serum M-protein (g/dL): Median (Range) | 2.5 (0–10.0) | 2.7 (0–10.0) |
| Urine M-protein (mg/24 hours): Median (Range) | 107 (0–24883) | 66 (0–39657) |
| Median Months Since Diagnosis | 35.2 | 37.5 |
| % Prior Therapy | ||
| One | 34 | 34 |
| More than one | 66 | 66 |
| Prior Systemic Therapies for Multiple Myeloma | ||
| Corticosteroid (%) | 99 | >99 |
| Anthracyclines | 68 | 67 |
| Alkylating agent (%) | 92 | 90 |
| Thalidomide/lenalidomide (%) | 40 | 43 |
| Stem cell transplantation (%) | 57 | 54 |
The primary outcome measure was time to progression (TTP). TTP was defined as the time from randomization to the first occurrence of progressive disease or death due to progressive disease. The combination arm demonstrated significant improvement in TTP. As the prespecified primary objective was achieved at the interim analysis, patients in the bortezomib monotherapy group were then allowed to receive the DOXIL + bortezomib combination. Efficacy results are as shown in Table 13 and Figure 1.
| Endpoint | DOXIL + bortezomib n=324 | Bortezomib n=322 |
|---|---|---|
| Time to Progression* | ||
| Progression or death due to progression (n) | 99 | 150 |
| Censored (n) | 225 | 172 |
| Median in days (months) | 282 (9.3) | 197 (6.5) |
| 95% CI | 250; 338 | 170; 217 |
| Hazard ratio† | 0.55 | |
| (95% CI) | (0.43, 0.71) | |
| p-value‡ | <0.001 | |
| Response (n)§ | 303 | 310 |
| % Complete Response (CR) | 5 | 3 |
| % Partial Response (PR) | 43 | 40 |
| % CR + PR | 48 | 43 |
| p-value¶ | 0.25 | |
| Median Duration of Response (months) | 10.2 | 7.0 |
| (95% CI) | (10.2; 12.9) | (5.9; 8.3) |
Figure 1- Time to Progression Kaplan-Meier Curve
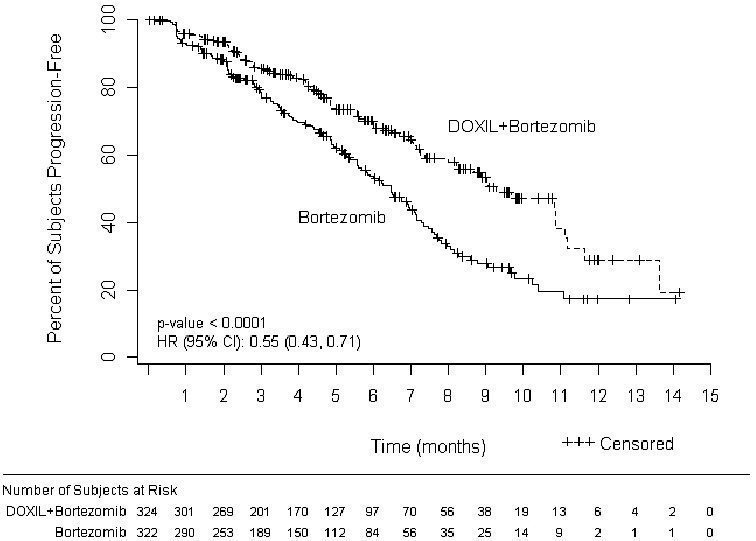
At the final analysis of survival, 78% of subjects in the DOXIL and bortezomib combination therapy group and 80% of subjects in the bortezomib monotherapy group had died after a median follow up of 8.6 years. The median survival was 33 months in the DOXIL and bortezomib combination therapy group and 31 months in the bortezomib monotherapy group. There was no difference observed in overall survival at the final analysis [HR for DOXIL + bortezomib vs. bortezomib = 0.96 (95% CI 0.80, 1.14)].
Seventy-eight percent of subjects in the DOXIL and bortezomib combination therapy group and 80% of subjects in the bortezomib monotherapy group had received subsequent therapy.
16. How is Doxil supplied
DOXIL is a sterile, translucent, red liposomal dispersion in 10-mL or 30-mL glass, single-dose vials.
The following individually cartoned vials are available:
| mg in vial | fill volume | vial size | NDC #s |
|---|---|---|---|
| 20 mg vial | 10-mL | 10-mL | 59676-960-01 |
| 50 mg vial | 25-mL | 30-mL | 59676-960-02 |
17. Patient Counseling Information
Cardiomyopathy
Advise patients to contact their healthcare provider if they develop symptoms of heart failure [see Warnings and Precautions (5.1)].
Infusion-Related Reactions
Advise patients about the symptoms of infusion-related reactions and to seek immediate medical attention if they develop any of these symptoms [see Warnings and Precautions (5.2)].
Myelosuppression
Advise patients to contact their healthcare provider for a new onset fever or symptoms of infection.
Hand-Foot Syndrome
Advise patients to notify their healthcare provider if they experience tingling or burning, redness, flaking, bothersome swelling, small blisters, or small sores on the palms of their hands or soles of their feet (symptoms of Hand-Foot Syndrome) [see Warnings and Precautions (5.3)].
Stomatitis
Advise patients to notify their healthcare provider if they develop painful redness, swelling, or sores in the mouth (symptoms of stomatitis).
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider with a known or suspected pregnancy [see Warnings and Precautions (5.5) and Use in Specific Populations (8.1)].
Advise females and males of reproductive potential to use effective contraception during and for 6 months following treatment with DOXIL [see Use in Specific Populations (8.3)].
Lactation
Advise females not to breastfeed during treatment with DOXIL [see Use in Specific Populations (8.2)].
Infertility
Advise females and males of reproductive potential that DOXIL may cause temporary or permanent infertility [see Use in Specific Populations (8.3)].
Manufactured by:
TTY Biopharm Company Limited
Taoyuan City, 32069, Taiwan
or
GlaxoSmithKline Manufacturing S.p.A.
Parma, Italy
Manufactured for:
Janssen Products, LP
Horsham, PA 19044
© Janssen Products, LP 2010
An Alza Corporation STEALTH® Technology Product.
STEALTH® and DOXIL® are registered trademarks of Alza Corporation.
PRINCIPAL DISPLAY PANEL - 20 mg Vial Carton
NDC 59676-960-01
DOXIL®
(DOXOrubicin HCl
liposome injection)
20 mg in 10 mL
(2 mg/mL)
Single-Use Vial. Discard unused portion.
LIPOSOMAL FORMULATION -
DO NOT SUBSTITUTE
FOR DOXORUBICIN HCL
FOR INTRAVENOUS
INFUSION ONLY
Refrigerate, 2°-8°C
(36°-46°F). Do Not Freeze.
Cytotoxic
Rx only
janssen
An Alza Corporation STEALTH®
Technology Product
PRINCIPAL DISPLAY PANEL - 50 mg Vial Carton
NDC 59676-960-02
DOXIL®
(DOXOrubicin HCl
liposome injection)
50 mg in 25 mL
(2 mg/mL)
Single-Use Vial. Discard unused portion.
LIPOSOMAL FORMULATION -
DO NOT SUBSTITUTE
FOR DOXORUBICIN HCL
FOR INTRAVENOUS
INFUSION ONLY
Refrigerate, 2°-8°C
(36°-46°F). Do Not Freeze.
Cytotoxic
Rx only
janssen
An Alza Corporation STEALTH®
Technology Product
| DOXIL
doxorubicin hydrochloride injection, suspension, liposomal |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Janssen Products, LP (804684207) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Meiji Seika Pharma Co., Ltd. | 695400580 | API MANUFACTURE(59676-960) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| TTY Biopharm Company Limited (Chung-Li Factory) | 658865659 | MANUFACTURE(59676-960) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| GlaxoSmithKline | 338471078 | MANUFACTURE(59676-960) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Pharmaceutica NV | 370005019 | PACK(59676-960) | |