Anavip: Package Insert / Prescribing Info
Package insert / product label
Generic name: crotalidae immune f(ab)2(equine)
Dosage form: injection, powder, lyophilized, for solution
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
ANAVIP(R)
crotalidae immune F(ab')2 (equine)
Lyophilized Powder for Solution for Injection
For Intravenous Use Only
Initial U.S. Approval: 2015
Recent Major Changes
Indication and Use (1) April 2021
Dosage and Administration (2) April 2021
Animal Toxicology and/or Pharmacology (13.2) April 2021
Indications and Usage for Anavip
ANAVIP [crotalidae immune F(ab')2 (equine)] is an equine-derived antivenin indicated for the management of adult and pediatric patients with North American Pit Viper envenomation. (1)
Anavip Dosage and Administration
Intravenous use only.
| Dose | No. of Vials | Infusion Rate |
|---|---|---|
| Initial Dose | 10 vials | Infuse intravenously over 60 minutes. |
| Additional dose(s) to achieve initial control | 10 vials (as needed) | Infuse intravenously over 60 minutes. |
| Observation and late dosing | 4 vials | Infuse intravenously over 60 minutes. |
- Initiate administration as soon as possible after North American Pit Viper bite in patients who develop signs of envenomation (e.g., local injury, coagulation abnormality, or systemic signs of envenomation). (2)
- Monitor patients in a health care setting at least 18 hours following initial control of signs and symptoms. Re-emerging symptoms including coagulapathies may be suppressed with an additional 4 vial doses of ANAVIP as needed. (2)
- Reconstitute each vial with 10 milliliters (mL) of sterile normal saline (0.9% NaCl). (2)
- Combine and further dilute to a total of 250 mL with sterile normal saline (0.9% NaCl). (2)
Dosage Forms and Strengths
Each vial contains a sterile, lyophilized preparation containing not more than 120 milligrams total protein and not less than the indicated number of mouse LD50 neutralizing units (3):
| Snake Species Used for Standardization | Minimum Mouse LD50 Units per Vial |
|---|---|
| Bothrops asper
(Terciopelo or fer-de-lance)1 | 780 |
| Crotalus simus (formerly Crotalus durissus)
(Central American Rattlesnake)1 | 790 |
| Crotalus adamanteus
(Eastern Diamondback Rattlesnake) | 244 |
| Crotalus atrox
(Western Diamondback Rattlesnake) | 147 |
| Crotalus scutulatus
(Mohave Rattlesnake)2 | 185 |
| Agkistrodon contortrix
(Copperhead) | 28 |
| Agkistrodon piscivorus
(Cottonmouth or Water Moccasin) | 61 |
1The venom of these species is used in the manufacture of ANAVIP.
2The Mohave phenotype A is used for standardization.
Contraindications
None. (4)
Warnings and Precautions
- ANAVIP may cause allergic reactions.
- Patients with known allergies to horse protein are particularly at risk for an anaphylactic reaction. If signs or symptoms of anaphylaxis or hypersensitivity reactions (including urticaria, rash, tightness of the chest, wheezing, hypotension) occur, discontinue immediately and institute appropriate treatment.
- Monitor patients with follow-up visits for signs and symptoms of delayed allergic reactions or serum sickness (rash, fever, myalgia, arthralgia, pruritus, urticarial rash) and treat appropriately if necessary. (5.1)
Adverse Reactions/Side Effects
The most common adverse reactions (>2%) in the clinical trials were: pruritus, nausea, rash,
arthralgia, peripheral edema, myalgia, headache, pain in extremity, vomiting and erythema. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Rare Disease Therapeutics, Inc., at 1-844-472-7389, or FDA at 1-800-FDA-1088
or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2021
Full Prescribing Information
2. Anavip Dosage and Administration
For Intravenous use only.
Administer ANAVIP as soon as possible after North American Pit Viper bite in patients who develop any signs of envenomation (e.g., local injury, coagulation abnormality, or systemic signs of envenomation).
The amount of antivenin required to treat a snake bitten patient is highly variable owing in part to the venom burden, the potency of the venom and the time to health care presentation.
Use supportive measures to treat certain manifestations of North American Pit Viper envenomation, such as pain, swelling, hypotension, and wound infection. Contact the local poison control centers for additional individual treatment advice.
Prior to initiating treatment, perform laboratory analyses, including complete blood count, platelet count, PT, PTT, serum fibrinogen level, and routine serum chemistries. Repeat testing at regular intervals to gauge response to therapy and anticipate additional dosing.
Initial Dose: 10 vials
- The initial dose of ANAVIP is 10 vials.
- Reconstitute the contents of each vial with 10 milliliters (mL) of sterile normal saline (0.9% NaCl). Average reconstitution time is 11.8 seconds (range 8-26 seconds) per vial when using continuous gentle swirling.
- Inspect the solution visually for particulate matter and discoloration prior to administration. The solution is expected to be clear to yellow/green and opalescent. Do not use if otherwise discolored or turbid.
- Combine the contents of the reconstituted vials promptly and further dilute to a total volume of 250 mL with sterile normal saline (0.9% NaCl). Fluid volumes may need to be adjusted for very small children or infants. Use reconstituted and diluted product within 6 hours. Discard partially or unused reconstituted and diluted product.
- Infuse intravenously over 60 minutes.
- For the first 10 minutes infuse at a 25-50 mL/hour rate, carefully monitoring for any allergic reactions, including any anaphylactic reactions. Discontinue the infusion if any allergic reaction occurs and institute appropriate emergency treatment. Reassess the risk to benefit before continuing the infusion.
- If no reactions occur, the infusion rate may be incrementally increased to the full 250 mL/hour rate until completion. If there is any allergic reaction at any time, stop the infusion, treat accordingly, and reassess the need to continue ANAVIP.
- Following the completion of infusion, monitor the patient for at least 60 minutes for any allergic reaction and to determine that local signs of envenomation are not progressing (leading edge of local injury not progressing), systemic symptoms are resolved and coagulation parameters have normalized or are trending toward normal.
Additional Dosing to Achieve Initial Control
- Administer additional 10 vial doses if needed to arrest the progressive symptoms and repeat every hour until local signs of envenomation are not progressing, systemic symptoms are resolved and coagulation parameters have normalized or are trending toward normal. There is no known maximum dose.
- Prepare additional loading doses as described above for initial dose.
- Once initial control has been achieved, observe the patient to determine any need for further dosing, as described below.
Observation and late Dosing
- Monitor patients in a health care setting at least 18 hours following initial control of signs and symptoms. Re-emerging symptoms including coagulopathies may be suppressed with additional 4 vial doses of ANAVIP as needed. Reconstitute each vial with 10 mL of sterile normal saline (0.9% NaCl). Combine and further dilute to a total of 250 mL. Infuse intravenously over 60 minutes.
3 DOSAGE FORMS AND STRENGTHS
ANAVIP is supplied as a sterile, lyophilized powder. Each vial contains not more than 120 milligrams (mg) total protein and not less than the indicated number of mouse LD50 neutralizing units:
| Snake Species Used for Standardization | Minimum Mouse LD50 Units per Vial |
|---|---|
| Bothrops asper
(Terciopelo or fer-de-lance)1 | 780 |
| Crotalus simus (formerly Crotalus durissus)
(Central American Rattlesnake)1 | 790 |
| Crotalus adamanteus
(Eastern Diamondback Rattlesnake) | 244 |
| Crotalus atrox
(Western Diamondback Rattlesnake) | 147 |
| Crotalus scutulatus
(Mohave Rattlesnake)2 | 185 |
| Agkistrodon contortrix
(Copperhead) | 28 |
| Agkistrodon piscivorus
(Cottonmouth or Water Moccasin) | 61 |
1The venom of these species is used in the manufacture of ANAVIP.
2The Mohave phenotype A is used for standardization.
5.1 Hypersensitivity
ANAVIP may cause allergic reactions.
- Patients with known allergies to horse protein are particularly at risk for an anaphylactic reaction. If signs or symptoms of anaphylaxis or hypersensitivity reactions (including urticaria, rash, tightness of the chest, wheezing, hypotension) occur, discontinue immediately and institute appropriate treatment.
- Monitor patients with follow-up visits for signs and symptoms of delayed allergic reactions or serum sickness (rash, fever, myalgia, arthralgia, pruritus, urticarial rash) and treat appropriately if necessary.4
5.2 Transmissible Infectious Agents
ANAVIP is made from equine (horse) plasma and may therefore carry a risk of transmitting infectious agents, e.g., viruses.
5.3 Reactions to Cresol
Trace amounts of cresol from the manufacturing process are contained in ANAVIP. Localized reactions and generalized myalgias have been reported with the use of cresol as an injectable excipient.
6 ADVERSE REACTIONS
The most common adverse reactions observed in more than 2 percent (2%) of patients in the clinical trials for ANAVIP were: pruritus, nausea, rash, arthralgia, peripheral edema, erythema, headache, myalgia, pain in extremity, and vomiting.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
A total of 86 patients were treated with ANAVIP, ranging from 2 to 80 years old. The patient population was comprised of 60 males and 26 females.
Patients were monitored for signs and symptoms of adverse reactions including acute hypersensitivity reactions and serum sickness.
Follow-up interviews were conducted at 5, 8, 15 and 22 days after treatment to assess symptoms of ongoing venom effect, serum sickness, and any other adverse reactions.
Table 2 shows the adverse reactions occurring in patients across all clinical trials for ANAVIP. Seventy-six percent (65/86) of patients receiving ANAVIP reported at least one adverse reaction.
| Adverse Reaction | ANAVIP [n=86] (%) |
| Patients Reporting at Least One Adverse Reactions | 65 (76%) |
| Skin and subcutaneous tissue disorders | 47 (55%) |
| Pruritus | 37 (43%) |
| Rash | 10 (12%) |
| Blister | 4 (5%) |
| Erythema | 3 (4%) |
| Gastrointestinal disorders | 28 (33%) |
| Nausea | 20 (23%) |
| Vomiting | 5 (6%) |
| Musculoskeletal and connective tissue disorders | 19 (22%) |
| Arthralgia | 9 (11%) |
| Myalgia | 6 (7%) |
| Pain in extremity | 5 (6%) |
| General disorders and administration site conditions | 21 (24%) |
| Edema peripheral | 7 (8%) |
| Chills | 3 (4%) |
| Pyrexia | 4 (5%) |
| Nervous system disorders | 12 (14%) |
| Headache | 5 (6%) |
| Psychiatric disorders | 4 (5%) |
| Anxiety | 2 (2%) |
| Insomnia | 2 (2%) |
| Metabolism and nutrition disorders | 4 (5%) |
| Dehydration | 2 (2%) |
| Respiratory, thoracic and mediastinal disorders | 5 (6%) |
| Dyspnea | 1 (1%) |
| Blood and lymphatic system disorders | 2 (2%) |
| Thrombocytopenia | 1 (1%) |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of ANAVIP. Because these reactions are reported as monotherapy or in combination with other drugs and voluntarily from a global population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Chest Pain/Discomfort
- Flushing
- Hypersensitivity
- Late Thrombocytopenia
- Necrosis
- Prolonged Prothrombin Time
- Tachycardia
- Treatment failure resulting in death
- Urticaria
8.1 Pregnancy
Risk Summary
Animal reproduction studies have not been conducted with ANAVIP. It is also not known
whether ANAVIP can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20% respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of ANAVIP in human milk, its effects on the breastfed infant, or its effect on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ANAVIP and any potential adverse effects on the breastfed infant from ANAVIP or from the underlying material condition.
8.4 Pediatric Use
Twenty-four percent (21/86) of patients studied in clinical trials were 16 years of age or younger (6 patients were 2 years of age to 5 years of age, 15 patients ranged from at least 5 years of age to 16 years of age). None of the pediatric patients in the phase 3 study experienced a recurrent coagulopathic effect. All adverse reactions in the pediatric patients were non-serious. The most frequent adverse reactions among pediatric patients were nausea and vomiting, itching, and fever. Thus, the safety and efficacy in the pediatric population was not different from adults.
11. Anavip Description
ANAVIP [crotalidae immune F(ab')2 (equine)] is a sterile, lyophilized, polyvalent preparation of equine immunoglobulin F(ab')2 fragments,
manufactured from the plasma of horses immunized with venom of
Bothrops asper and Crotalus simus (formerly Crotalus durissus). The product is obtained by pepsin digestion of horse plasma to remove the Fc portion of immunoglobulin, followed by fractionation and purification steps.
The F(ab')2 content is not less than 85%, F(ab') content is not more than 7%, and the product contains less than 5% intact immunoglobulin.
Each vial of ANAVIP contains 25.2-56.8 mg of sodium chloride, 47.30 - 91.35 mg of sucrose, and 95.03-183.15 mg of glycine as stabilizers. Trace amounts of pepsin (≤50 ng/vial), cresol (≤0.058 mg/vial), borates (≤1 mg/vial), and sulfates (≤1.7 mg/vial) may be present from the manufacturing process.
Each vial contains no more than 120 mg of protein and will be neutralize not less than 780 times the LD50 of
Bothrops asper (Terciopelo or fer-de-lance) venom, 790 times the LD50 of Crotalus simus (formerly Crotalus durissus) (Central American Rattlesnake) venom, 244 times the LD50 of Crotalus adamanteus (Eastern Diamondback Rattlesnake) venom, 147 times the LD50 of Crotalus atrox (Western Diamondback Rattlesnake) venom, 185 times the LD50 of Crotalus scutulatus (Mohave Rattlesnake) venom, 28 times the LD50 of Agkistrodon contortrix (Copperhead) venom and 61 times the LD50 of Agkistrodon piscivorus (Cottonmouth or Water Moccasin) venom in a mouse neutralization assay.
The manufacturing procedures that contribute to the reduction of risk of viral transmission include pepsin digestion, ammonium sulfate precipitation/heat treatment
and nanofiltration.
12. Anavip - Clinical Pharmacology
12.1 Mechanism of Action
ANAVIP contains venom-specific F(ab')2 fragments of immunoglobulin G (IgG) that bind and neutralize venom toxins, facilitating redistribution away from target tissues and elimination from the body.1,2
12.3 Pharmacokinetics
Thirteen healthy volunteers each received an intravenous (IV) dose of one vial (one vial = 81.86 mg) of an antivenin comparable to ANAVIP both in composition and manufacturing. On the first day of antivenin administration, blood samples were collected from all subjects at 16 specific time points: 0 (prior to drug infusion), 5, 10, 20, 30, 45, 60, 120, and 480 minutes after drug infusion. Additional samples were drawn just prior to discharge (Day 1), and on days 3, 5, 7, 9, 11, and 21. A two-compartment model best described the concentration-time data. The pharmacokinetic parameters of the antivenin are summarized in Table 3.
| Parameters | Units | Mean | SD |
| Area under plasma concentration vs time curve | AUC0-∞(µg•h/mL) | 4144 | 670 |
| Steady-state volume of distribution | Vss (L) | 3.3 | 0.9 |
| Mean residence time | MRT (h) | 157 | 40 |
| Elimination half-life | Beta-HL (h) | 133 | 53 |
| Total clearance | CL (mL/h) | 22 | 7 |
13. Nonclinical Toxicology
In a published non-Good Laboratory Practice study3, ANAVIP and another licensed North American Pit Viper antivenin were tested and cross-reactivity to the venoms of multiple different North American Pit Vipers was demonstrated in rabbits and mice. Animal studies to determine a no observed adverse effect level were unsuccessful due to technical limitations that prevented determination of a systematically toxic dose.
13.2 Animal Toxicology and/or Pharmacology
ANAVIP is standardized by its ability to neutralize the lethal action of each of the 7 venom immunogens following intravenous injection in mice.
The potency of the product will vary from batch to batch; however, a minimum number of mouse LD50 neutralizing units against each of the 7 venoms is included in every vial of final product, as shown in Table 1 [see Dosage Forms and Strengths (3)].
ANAVIP was effective in neutralizing the venoms of 7 clinically important North American Pit Vipers in a murine lethality model see Table 1 [see Dosage Forms and Strengths(3)]. The ED50 data demonstrate the number of milligrams of antivenom necessary to neutralize a milligram of venom.
The amount of Anavip necessary to neutralize one milligram of venom was determined in a murine model. The dynamic between the venom and antivenom in a murine model provides a controlled environment to test the cross reactivity of antivenom against a variety of different snake venoms. Unlike the potency assays performed, as shown in Table 1 [see Dosage Forms and Strengths (3)], the effective dose study provides a mg per mg comparison for venoms from 7 species commonly associated with North American Pit Viper envenomations. The results for each species are shown in Table 4.
| Challenge Venom | ED50 (mg antivenin/mg venom) |
| B. asper (Terciopelo or fer-de-lance) | 1.7 |
| C. simus (formerly C. durissus) (Central American Rattlesnake) | 3.4 |
| C. adamanteus (Eastern Diamondback Rattlesnake) | 2.4 |
| C. atrox (Western Diamondback Rattlesnake) | 2.9 |
| C. scutulatus A1 (Mohave Rattlesnake) | 14.6 |
| A. contortrix (Copperhead) | 8.0 |
| A. piscivorus (Cottonmouth or Water Moccasin) | 5.0 |
1C. scutulatus A is the Mohave phenotype containing neurotoxic venom
14. Clinical Studies
Study 1 was a randomized, prospective, open-label, controlled, comparative, multicenter study was conducted in 12 patients aged 18 to 70 years of age with signs of North American Pit Viper envenomation.6
The subjects received either a licensed North American Pit Viper antivenin as an active control, or ANAVIP. The subjects were dosed until initial control was achieved, followed by maintenance doses.
All patients in both treatment groups achieved initial control of local injury and coagulopathy following early antivenin treatment.
In the active control group, at the end of maintenance dosing, 5 of 6 subjects had platelet counts above 150,000/mm3, and all 6 had fibrinogen levels above 150 mg/dL.
During the follow-up phase two patients exhibited platelets below 150,000/mm3 and fibrinogen below 150 mg/dL, leading to inpatient management with administration of additional doses
(one subject received an additional 6 doses (12 vials) and one subject received an additional 4 doses (8 vials)).
In the ANAVIP arm, at the end of maintenance dosing, 5 of 6 subjects had platelet counts above 150,000/mm3. One subject’s platelets were 114,000/mm3 and were trending upward, and all 6 had fibrinogen levels above 150 mg/dL.
During the follow-up phase, 5 of 6 subjects had platelet counts above 150,000/mm3, with no downward trend; 1 subject’s platelet count was 127,000/mm3 on follow-up Day 1, reached 160,000/mm3 on Day 4, and continued trending upward.
All 6 subjects in the ANAVIP group had fibrinogen levels above 150 mg/dL during the follow-up phase. None in the ANAVIP group required rehospitalization or retreatment with ANAVIP.
Study 2 was a randomized, prospective, blinded, controlled, comparative, multicenter study, comparing two ANAVIP regimens with a licensed North American Pit Viper antivenin (comparator) conducted in patients with North American Pit Viper envenomation at 16 sites in the United States.
The study had an in-hospital Acute Treatment Phase that included screening and baseline assessments, initial and maintenance dosing, and an outpatient Follow-up Phase that included 4 follow-up visits on Days 5, 8, 15 and 22.
Patients were randomized in a 1:1:1 ratio to one of three groups: ANAVIP with ANAVIP maintenance therapy (Group 1), ANAVIP with placebo maintenance therapy (Group 2), or Comparator product with Comparator product maintenance therapy (Group 3).
Initial dosing consisted of sequential intravenous (IV) doses infused to achieve initial control. If initial control of envenomation was not achieved, treatment was repeated until initial control was attained. Maintenance dosing (4 vials of ANAVIP or placebo [normal saline (0.9% NaCl)], or 2 vials of comparator product) was initiated 6 hours after the start of the last dose required to achieve initial control, and continued every 6 hours for 3 doses.
The Follow-up Phase began immediately after the third maintenance dose. Patients returned to the clinical site on Days 5, 8, and 15 for scheduled follow-up visits. Patients with ongoing signs of envenomation received 4 vials of ANAVIP or 2 vials of Comparator product. Dosing was provided as needed until the patient was stabilized. One hundred twenty-one (121) patients received blinded study drug and were analyzed for safety and efficacy.
The efficacy endpoint was the proportion of patients experiencing a coagulopathic effect as measured on Study Day 5 or 8. Patients were assessed as experiencing a coagulopathic effect if they had any one of the following: absolute platelet levels < 150,000/mm3 as measured on either
Study Day 5 (±1 day) or 8 (±1 day); absolute fibrinogen levels <150 mg/dL as measured on either Study Day 5 (±1 day) or 8 (±1 day); or clinical coagulopathy between end of maintenance dosing and Study Day 5 requiring additional antivenin.
The comparison of coagulopathic effect proportions between treatment groups was tested using an exact logistic regression model with terms for treatment and region.
Comparisons of the proportion of coagulopathic effect for two levels of ANAVIP versus Comparator product were performed in the following order: ANAVIP with ANAVIP maintenance dose versus Comparator product; then ANAVIP with Placebo maintenance dose versus Comparator product. The number and percentage of patients who experienced a coagulopathic effect is summarized by treatment group in Table 5.
The efficacy analysis did not meet the pre-specified statistically defined superiority criterion. However, the percentages of subjects showing prespecified criteria for coagulopathic effect on either Day 5 and/or Day 8 were 10.3% and 5.3% in the Groups 1 and 2 when compared to 29.7% in Group 3 indicating efficacy of ANAVIP in management of coagulopathic effect in patients with North American Pit Viper envenomation.
| Experienced Coagulopathic Effect on Either Day 5 or Day 8 | Group 1 ANAVIP/ANAVIP (n=39) | Group 2 ANAVIP/Placebo (n=38) | Group 3 Comparator product (n=37) |
| Yes | 4 (10.3%) | 2 (5.3%) | 11 (29.7%) |
| No | 35 (89.7%) | 36 (94.7%) | 26 (70.3%) |
| Treatment Group (vs. Group 3) Odds Ratio (95% Cl1) | 0.275 (0.058, 1.048) | 0.135 (0.014, 0.686) |
1Cl= confidence interval
FDA conducted a post hoc analysis to assess the outcomes of the patients who presented with or without baseline coagulopathic effect in the three treatment groups.
Using the pre-specified criteria for coagulopathic effect, it was found that ANAVIP/ANAVIP (Group 1) had the highest percentage of baseline coagulopathic subjects among the three groups [41.5% compared with 17.5% and 32.5% for the ANAVIP/Placebo (Group 2) and Comparator product (Group 3), respectively].
Thirty-three percent (33%) of all baseline coagulopathic subjects also experienced coagulopathic effect on either Day 5 or 8, compared to only 6% for baseline non-coagulopathic subjects.
Only 18% of the subjects with baseline coagulopathic effect in Group 1 continued to remain coagulopathic at Days 5 or 8 compared to 58% in Group 3 (Table 6).
| Baseline Coagulopathy and Experienced coagulopathy on either Day 5 or 8 | Group 1 ANAVIP/ANAVIP | Group 2 ANAVIP/Placebo | Group 3 Comparator product | Total |
| Number of subjects | n=17 | n=7 | n=12 | n=36 |
| Yes | 3 (17.65%) | 2 (28.57%) | 7 (58.33%) | 12 (33.3%) |
| No | 14 (82.35%) | 5 (71.43%) | 5 (41.67%) | 24 (66.7%) |
| No Baseline Coagulopathy and Experienced coagulopathy on either Day 5 or 8 | Group 1 ANAVIP/ANAVIP | Group 2 ANAVIP/Placebo | Group 3 Comparator product | Total |
| Number of Subjects | n=22 | n=31 | n=25 | n=78 |
| Yes | 1 (4.55%) | 0 (0%) | 4 (16%) | 5 (6.4%) |
| No | 21 (95.45%) | 31 (100%) | 21 (84%) | 73 (93.6%) |
An exact logistic regression analysis adjusting for baseline coagulopathic effect and region was conducted and showed that treatment effect for both Groups 1 and 2 is statistically significant (Table 7). This analysis provides supportive evidence of the efficacy of ANAVIP.
| Group 1 ANAVIP/ANAVIP (n=39) | Group 2 ANAVIP/Placebo (n=38) | |
| Treatment Group (vs Comparator product) Odds ration (95% Cl1) | 0.184 (0.033, 0.794) | 0.121 (0.010, 0.764) |
1Cl= confidence interval
Analysis by snakebite type was performed but was limited due to the number of unknown snakebite types (n=43). However, 57 subjects who were envenomated by rattlesnakes showed more severe coagulopathic effects and resolution of these effects after treatment with ANAVIP as compared to 21 subjects who were envenomated by copperhead snakes.
Due to limited coagulopathic effects in the copperhead snake bite subgroup, efficacy outcomes for late coagulopathies could not be evaluated.
15. References
1. Seifert SA and Boyer LV: Recurrence phenomena after immunoglobulin therapy for snake envenomation: Part 1. Pharmacokinetics and pharmacodynamics of immunoglobulin antivenoms and related antibodies. Annals of Emergency Medicine 37(2):189-195; 2001.
2. Boyer LV, Seifert SA and Cain JS: Recurrence phenomena after immunoglobulin therapy for snake envenomation: Part 2. Guidelines for clinical management with crotaline Fab antivenom. Annals of Emergency Medicine 37(2):196-201; 2001.
3. Sanchez EE, Galan JA, Perez JC, et al. The efficacy of two antivenoms against the venom of North American snakes, Toxicon 41: 357-365; 2003.
4. Gold BS, Dart RC and Barish RA: Bites of venomous snakes. New England Journal of Medicine 347(5):347-56; 2002.
5. Boyer LV, Seifert SA, Clark RF, et al: Recurrent and persistent coagulopathy following Pit Viper envenomation. Archives of Internal Medicine 159:706-710; 1999.
6. Boyer LV, Chase PB, Degan JA, et al: Subacute coagulopathy in a randomized, comparative trial of Fab and F(ab’)2 antivenoms. Toxicon 74: 101-108; 2013.
16. How is Anavip supplied
ANAVIP is supplied as a sterile, lyophilized preparation in a single-dose vial. When reconstituted with 10 mL of 0.9% NaCl solution, each vial contains not more than 12 mg per mL of protein, and will neutralize not less than 780 times the LD50 of Bothrops asper (Terciopelo or fer-de-lance) venom and 790 times the LD50 of Crotalus simus (formerly Crotalus durissus) (Central American Rattlesnake) venom, 244 times the LD50 of Crotalus adamanteus (Eastern Diamondback Rattlesnake) venom, 147 times the LD50 of Crotalus atrox (Western Diamondback Rattlesnake) venom, 185 times the LD50 of Crotalus scutulatus (Mohave Rattlesnake) venom, 28 times the LD50 of Agkistrodon contortrix (Copperhead) venom and 61 times the LD50 of Agkistrodon piscivorus (Cottonmouth or Water Moccasin) venom in a mouse neutralization assay.
Each carton NDC 66621-0790-2 contains 1 vial of ANAVIP NDC 66621-0790-1.
- Store at room temperature (up to 25 ºC (77 ºF)). Brief temperature excursions are permitted up to 40 ºC (104ºF).
- DO NOT FREEZE.
- Use within 6 hours after reconstitution.
- Discard partially used vials.
17. Patient Counseling Information
- Advise patients to contact their physician immediately if they experience unusual bruising or bleeding (e.g., nosebleeds, excessive bleeding after brushing teeth, the appearance of blood in stools or urine, excessive menstrual bleeding, petechiae, excessive bruising or persistent oozing from superficial injuries) after hospital discharge.5
- Serious Allergic Reactions
o Advise patients to contact their physician immediately if they experience any signs and symptoms of delayed allergic reactions or serum sickness (e.g., rash, fever, myalgias, arthralgia, pruritus, urticaria) after hospital discharge [see Hypersensitivity (5.1)].4
Manufactured by:
Laboratorios Silanes S.A. de C.V.
Toluca, Estado de Mexico, Mexico
Silanes® and the Silanes logo are registered trademarks in Mexico of Laboratorios Silanes, S.A. de C.V.
![]()
Manufactured for:
Rare Disease Therapeutics, Inc.
2550 Meridian Blvd., Suite 150
Franklin, TN 37067
www.raretx.com
RDT® and the RDT logo are registered trademarks of Rare Disease Therapeutics, Inc.
![]()
U.S. License No. 1860
RDT Part No. ANV-PI-009
Silanes Part number: 360893-8
PACKAGE LABEL
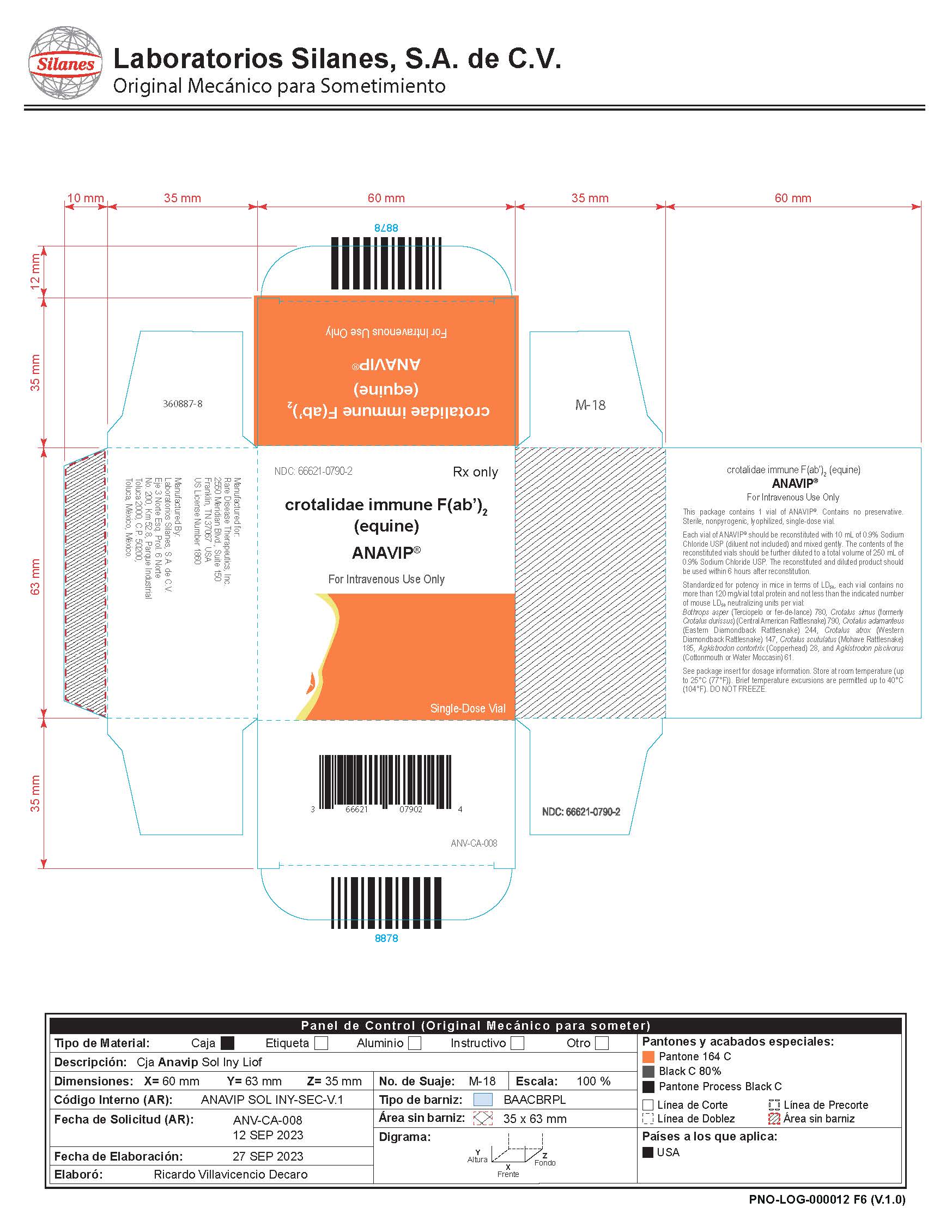


| ANAVIP
crotalidae immune f(ab)2(equine) injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Rare Disease Therapeutics, Inc (966133100) |
| Registrant - Rare Disease Therapeutics, Inc (966133100) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Laboratorios Silanes S.A. de C.V. | 588387584 | manufacture(66621-0790) | |