Terbinafine Prescribing Information
Package insert / product label
Generic name: terbinafine hydrochloride
Dosage form: tablet
Drug class: Miscellaneous antifungals
Medically reviewed by Drugs.com. Last updated on Apr 23, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
TERBINAFINE tablets, for oral use
Initial U.S. Approval: 1992
Indications and Usage for Terbinafine
Terbinafine tablets are an allylamine antifungal indicated for the treatment of onychomycosis of the toenail or fingernail due to dermatophytes (tinea unguium). (1)
Terbinafine Dosage and Administration
Dosage Forms and Strengths
Tablet, 250 mg (3)
Contraindications
Warnings and Precautions
- Liver failure, sometimes leading to liver transplant or death, has occurred with the use of oral terbinafine. Obtain pretreatment serum transaminases. Prior to initiating treatment and periodically during therapy, assess liver function tests. Discontinue terbinafine tablets if liver injury develops. (5.1)
- Taste disturbance, including taste loss, has been reported with the use of terbinafine tablets. Taste disturbance can be severe, may be prolonged, or may be permanent. Discontinue terbinafine tablets if taste disturbance occurs. (5.2)
- Smell disturbance, including loss of smell, has been reported with the use of terbinafine tablets. Smell disturbance may be prolonged, or may be permanent. Discontinue terbinafine tablets if smell disturbance occurs. (5.3)
- Depressive symptoms have been reported with terbinafine use. Prescribers should be alert to the development of depressive symptoms. (5.4)
- Severe neutropenia has been reported. If the neutrophil count is less than or equal to 1000 cells/mm3, terbinafine tablets should be discontinued. (5.5)
- Stevens-Johnson syndrome, toxic epidermal necrolysis, erythema multiforme, exfoliative dermatitis, bullous dermatitis, and drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome have been reported with oral terbinafine use. If signs or symptoms of drug reaction occur, treatment with terbinafine tablets should be discontinued. (5.6)
Adverse Reactions/Side Effects
Common (greater than 2% of patients treated with terbinafine tablets) reported adverse events include headache, diarrhea, rash, dyspepsia, liver enzyme abnormalities, pruritus, taste disturbance, nausea, abdominal pain, and flatulence. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Aurobindo Pharma USA, Inc. at 1-866-850-2876 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Terbinafine is an inhibitor of CYP450 2D6 isozyme and has an effect on metabolism of desipramine. Drug interactions have also been noted with cimetidine, fluconazole, cyclosporine, rifampin, and caffeine. (7.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2022
Related/similar drugs
nystatin topical, clotrimazole topical, ketoconazole topical, itraconazole, miconazole topical, Lamisil, Lotrisone
Full Prescribing Information
1. Indications and Usage for Terbinafine
Terbinafine tablets are indicated for the treatment of onychomycosis of the toenail or fingernail due to dermatophytes (tinea unguium).
Prior to initiating treatment, appropriate nail specimens for laboratory testing [potassium hydroxide (KOH) preparation, fungal culture, or nail biopsy] should be obtained to confirm the diagnosis of onychomycosis.
2. Terbinafine Dosage and Administration
2.1 Assessment Prior to Initiation
Before administering terbinafine tablets, evaluate patients for evidence of chronic or active liver disease [see Contraindications (4) and Warnings and Precautions (5.1)].
2.2 Dosage
Fingernail onychomycosis: One 250 mg tablet once daily for 6 weeks.
Toenail onychomycosis: One 250 mg tablet once daily for 12 weeks.
The optimal clinical effect is seen some months after mycological cure and cessation of treatment. This is related to the period required for outgrowth of healthy nail.
3. Dosage Forms and Strengths
Tablet, 250 mg white to off-white, round uncoated, biconvex beveled edge tablets having ‘D’ debossed on one side and ‘74’ on the other side.
4. Contraindications
Terbinafine tablets are contraindicated in patients with:
- Chronic or active liver disease [see Warnings and Precautions (5.1)]
- History of allergic reaction to oral terbinafine because of the risk of anaphylaxis [see Adverse Reactions (6.2)]
5. Warnings and Precautions
5.1 Hepatotoxicity
Terbinafine tablets are contraindicated for patients with chronic or active liver disease. Before prescribing terbinafine tablets, perform liver function tests because hepatotoxicity may occur in patients with and without preexisting liver disease. Cases of liver failure, some leading to liver transplant or death, have occurred with the use of terbinafine tablets in individuals with and without preexisting liver disease.
In the majority of liver cases reported in association with use of terbinafine tablets, the patients had serious underlying systemic conditions. The severity of hepatic events and/or their outcome may be worse in patients with active or chronic liver disease. Periodic monitoring of liver function tests is recommended. Discontinue terbinafine tablets if biochemical or clinical evidence of liver injury develops.
Warn patients prescribed terbinafine tablets and/or their caregivers to report immediately to their healthcare providers any symptoms or signs of persistent nausea, anorexia, fatigue, vomiting, right upper abdominal pain or jaundice, dark urine, or pale stools. Advise patients with these symptoms to discontinue taking oral terbinafine, and immediately evaluate the patient’s liver function.
5.2 Taste Disturbance Including Loss of Taste
Taste disturbance, including taste loss, has been reported with the use of terbinafine tablets. It can be severe enough to result in decreased food intake, weight loss, anxiety, and depressive symptoms. Taste disturbance may resolve within several weeks after discontinuation of treatment, but may be prolonged (greater than 1 year), or may be permanent. If symptoms of a taste disturbance occur, terbinafine tablets should be discontinued.
5.3 Smell Disturbance Including Loss of Smell
Smell disturbance, including loss of smell, has been reported with the use of terbinafine tablets. Smell disturbance may resolve after discontinuation of treatment, but may be prolonged (greater than 1 year), or may be permanent. If symptoms of a smell disturbance occur, terbinafine tablets should be discontinued.
5.4 Depressive Symptoms
Depressive symptoms have occurred during postmarketing use of terbinafine tablets. Prescribers should be alert to the development of depressive symptoms, and patients should be instructed to report depressive symptoms to their physician.
5.5 Hematologic Effects
Transient decreases in absolute lymphocyte counts (ALCs) have been observed in controlled clinical trials. In placebo-controlled trials, 8/465 subjects receiving terbinafine tablets (1.7%) and 3/137 subjects receiving placebo (2.2%) had decreases in ALC to below 1000/mm3 on 2 or more occasions. In patients with known or suspected immunodeficiency, physicians should consider monitoring complete blood counts if treatment continues for more than 6 weeks. Cases of severe neutropenia have been reported. These were reversible upon discontinuation of terbinafine tablets, with or without supportive therapy. If clinical signs and symptoms suggestive of secondary infection occur, a complete blood count should be obtained. If the neutrophil count is less than or equal to 1000 cells/mm3, terbinafine tablets should be discontinued and supportive management started.
5.6 Serious Skin/Hypersensitivity Reactions
There have been postmarketing reports of serious skin/hypersensitivity reactions [e.g., Stevens-Johnson syndrome, toxic epidermal necrolysis, erythema multiforme, exfoliative dermatitis, bullous dermatitis, and drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome]. Manifestations of DRESS syndrome may include cutaneous reaction (such as rash or exfoliative dermatitis), eosinophilia, and one or more organ complications such as hepatitis, pneumonitis, nephritis, myocarditis, and pericarditis. If progressive skin rash or signs/symptoms of the above drug reactions occur, treatment with terbinafine tablets should be discontinued.
5.7 Lupus Erythematosus
During postmarketing experience, precipitation and exacerbation of cutaneous and systemic lupus erythematosus have been reported in patients taking terbinafine tablets. Terbinafine tablets should be discontinued in patients with clinical signs and symptoms suggestive of lupus erythematosus.
5.8 Thrombotic Microangiopathy
Cases of thrombotic microangiopathy (TMA), including thrombotic thrombocytopenic purpura and hemolytic uremic syndrome, some fatal, have been reported with terbinafine. Discontinue terbinafine if clinical symptoms and laboratory findings consistent with TMA occur. The findings of unexplained thrombocytopenia and anemia should prompt further evaluation and consideration of diagnosis of TMA.
6. Adverse Reactions/Side Effects
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most frequently reported adverse events observed in the 3 U.S./Canadian placebo-controlled trials are listed in the Table 1. The adverse events reported encompass gastrointestinal symptoms (including diarrhea, dyspepsia, and abdominal pain), liver test abnormalities, rashes, urticaria, pruritus, and taste disturbances. Changes in the ocular lens and retina have been reported following the use of terbinafine tablets in controlled trials. The clinical significance of these changes is unknown. In general, the adverse events were mild, transient, and did not lead to discontinuation from study participation.
Table 1. Most frequently reported adverse events observed in the 3 U.S./Canadian placebo-controlled trials
| Adverse Event | Discontinuation | |||
|---|---|---|---|---|
| Terbinafine Tablets (%) n=465 | Placebo (%) n=137 | Terbinafine Tablets (%) n=465 | Placebo (%) n=137 |
|
| * Liver enzyme abnormalities greater than or equal to 2 times the upper limit of normal range. |
||||
| Headache | 12.9 | 9.5 | 0.2 | 0 |
| Gastrointestinal Symptoms: Diarrhea Dyspepsia Abdominal Pain Nausea Flatulence | 5.6 4.3 2.4 2.6 2.2 | 2.9 2.9 1.5 2.9 2.2 | 0.6 0.4 0.4 0.2 0 | 0 0 0 0 0 |
| Dermatological Symptoms: Rash Pruritus Urticaria | 5.6 2.8 1.1 | 2.2 1.5 0 | 0.9 0.2 0 | 0.7 0 0 |
| Liver Enzyme Abnormalities* | 3.3 | 1.4 | 0.2 | 0 |
| Taste Disturbance | 2.8 | 0.7 | 0.2 | 0 |
| Visual Disturbance | 1.1 | 1.5 | 0.9 | 0 |
6.2 Postmarketing Experience
The following adverse events have been identified during post-approval use of terbinafine tablets. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: Pancytopenia, agranulocytosis, severe neutropenia, thrombocytopenia, anemia, thrombotic microangiopathy (TMA), including thrombotic thrombocytopenic purpura and hemolytic uremic syndrome [see Warnings and Precautions (5.5, 5.8)]
Immune system disorders: Serious hypersensitivity reactions e.g., angioedema and allergic reactions (including anaphylaxis), precipitation and exacerbation of cutaneous and systemic lupus erythematosus [see Warnings and Precautions (5.7)], serum sickness-like reaction
Psychiatric disorders: Anxiety and depressive symptoms independent of taste disturbance have been reported with use of terbinafine tablets. In some cases, depressive symptoms have been reported to subside with discontinuance of therapy and to recur with reinstitution of therapy [see Warnings and Precautions (5.4)].
Nervous system disorders: Cases of taste disturbance, including taste loss, have been reported with the use of terbinafine tablets. It can be severe enough to result in decreased food intake, weight loss, anxiety, and depressive symptoms. Cases of smell disturbance, including smell loss, have been reported with the use of terbinafine tablets [see Warnings and Precautions (5.2, 5.3)]. Cases of paresthesia and hypoesthesia have been reported with the use of terbinafine tablets.
Eye disorders: Visual field defects, reduced visual acuity
Ear and labyrinth disorders: Hearing impairment, vertigo, tinnitus
Vascular disorders: Vasculitis
Gastrointestinal disorders: Pancreatitis, vomiting
Hepatobiliary disorders: Cases of liver failure some leading to liver transplant or death [see Warnings and Precautions (5.1)], idiosyncratic and symptomatic hepatic injury. Cases of hepatitis, cholestasis, and increased hepatic enzymes [see Warnings and Precautions (5.1)] have been seen with the use of terbinafine tablets.
Skin and subcutaneous tissue disorders: Serious skin reactions [e.g., Stevens-Johnson syndrome, toxic epidermal necrolysis, erythema multiforme, exfoliative dermatitis, bullous dermatitis, and drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome] [see Warnings and Precautions (5.6)], acute generalized exanthematous pustulosis, psoriasiform eruptions or exacerbation of psoriasis, photosensitivity reactions, hair loss
Musculoskeletal and connective tissue disorders: Rhabdomyolysis, arthralgia, myalgia
General disorders and administration site conditions: Malaise, fatigue, influenza-like illness, pyrexia
Investigations: Altered prothrombin time (prolongation and reduction) in patients concomitantly treated with warfarin and increased blood creatine phosphokinase have been reported
7. Drug Interactions
7.1 Drug-Drug Interactions
In vivo studies have shown that terbinafine is an inhibitor of the CYP450 2D6 isozyme. Drugs predominantly metabolized by the CYP450 2D6 isozyme include the following drug classes: tricyclic antidepressants, selective serotonin reuptake inhibitors, beta-blockers, antiarrhythmics class 1C (e.g., flecainide and propafenone) and monoamine oxidase inhibitors Type B. Coadministration of terbinafine tablets should be done with careful monitoring and may require a reduction in dose of the 2D6-metabolized drug. In a study to assess the effects of terbinafine on desipramine in healthy volunteers characterized as normal metabolizers, the administration of terbinafine resulted in a 2-fold increase in Cmax and a 5-fold increase in area under the curve (AUC). In this study, these effects were shown to persist at the last observation at 4 weeks after discontinuation of terbinafine tablets. In studies in healthy subjects characterized as extensive metabolizers of dextromethorphan (antitussive drug and CYP2D6 probe substrate), terbinafine increases the dextromethorphan/dextrorphan metabolite ratio in urine by 16- to 97-fold on average. Thus, terbinafine may convert extensive CYP2D6 metabolizers to poor metabolizer status.
In vitro studies with human liver microsomes showed that terbinafine does not inhibit the metabolism of tolbutamide, ethinylestradiol, ethoxycoumarin, cyclosporine, cisapride and fluvastatin. In vivo drug-drug interaction studies conducted in healthy volunteer subjects showed that terbinafine does not affect the clearance of antipyrine or digoxin. Terbinafine decreases the clearance of caffeine by 19%. Terbinafine increases the clearance of cyclosporine by 15%.
The influence of terbinafine on the pharmacokinetics of fluconazole, cotrimoxazole (trimethoprim and sulfamethoxazole), zidovudine or theophylline was not considered to be clinically significant.
Coadministration of a single dose of fluconazole (100 mg) with a single dose of terbinafine resulted in a 52% and 69% increase in terbinafine Cmax and AUC, respectively. Fluconazole is an inhibitor of CYP2C9 and CYP3A enzymes. Based on this finding, it is likely that other inhibitors of both CYP2C9 and CYP3A4 (e.g., ketoconazole, amiodarone) may also lead to a substantial increase in the systemic exposure (Cmax and AUC) of terbinafine when concomitantly administered.
There have been spontaneous reports of increase or decrease in prothrombin times in patients concomitantly taking oral terbinafine and warfarin, however, a causal relationship between terbinafine tablets and these changes has not been established.
Terbinafine clearance is increased 100% by rifampin, a CYP450 enzyme inducer, and decreased 33% by cimetidine, a CYP450 enzyme inhibitor. Terbinafine clearance is unaffected by cyclosporine. There is no information available from adequate drug-drug interaction studies with the following classes of drugs: oral contraceptives, hormone replacement therapies, hypoglycemics, phenytoins, thiazide diuretics, and calcium channel blockers.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Available data from postmarketing cases on the use of terbinafine tablets in pregnant women are insufficient to evaluate a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes.
In animal reproduction studies, terbinafine did not cause malformations or any harm to the fetus when administered to pregnant rabbits and rats during the period of organogenesis at oral doses up to 12 and 23 times the maximum recommended human dose (MRHD) of 250 mg/day, respectively (see data).
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown; however, in the U.S. general population, the estimated background risk of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
In embryo-fetal development studies in rats and rabbits, pregnant animals received orally (by gavage) doses of terbinafine up to 300 mg/kg/day, during the period of organogenesis. There were no maternal or embryo-fetal effects in either species up to the maximum dose tested. The 300 mg/kg/day dose level in rats and rabbits corresponds to 23 and 12 times the MRHD [based on body surface area (BSA) comparisons], respectively.
In a rat peri- and postnatal development study, terbinafine doses of up to 300 mg/kg/day (12 times the MRHD based on BSA comparisons) given by oral gavage during late pregnancy and lactation (Day 15 of gestation to day 20 post-partum) had no adverse effects on parturition and lactation.
8.2 Lactation
Risk Summary
After oral administration, terbinafine is present in human milk. However, there are no data on the effects on the breastfed child or on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for terbinafine tablets and any potential adverse effects on the breastfed child from terbinafine tablets or from the underlying maternal condition.
8.4 Pediatric Use
The safety and efficacy of terbinafine tablets have not been established in pediatric patients with onychomycosis.
8.5 Geriatric Use
Clinical studies of terbinafine tablets did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
In patients with renal impairment (creatinine clearance less than or equal to 50 mL/min), the use of terbinafine tablets has not been adequately studied.
8.7 Hepatic Impairment
Terbinafine tablets are contraindicated for patients with chronic or active liver disease [see Contraindications (4) and Warnings and Precautions (5.1)]. Cases of liver failure, some leading to liver transplant or death, have occurred with the use of terbinafine tablets in individuals with and without preexisting liver disease. The severity of hepatic events and/or their outcome may be worse in patients with active or chronic liver disease.
10. Overdosage
Clinical experience regarding overdose with oral terbinafine is limited. Doses up to 5 grams (20 times the therapeutic daily dose) have been taken without inducing serious adverse reactions. The symptoms of overdose included nausea, vomiting, abdominal pain, dizziness, rash, frequent urination, and headache.
11. Terbinafine Description
Terbinafine tablets, USP contain the synthetic allylamine antifungal compound terbinafine hydrochloride USP.
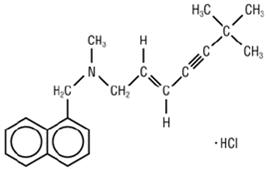
Chemically, terbinafine hydrochloride is (E)-N-(6,6-dimethyl-2-hepten-4-ynyl)-N-methyl-1-naphthalenemethanamine hydrochloride. The molecular formula C21H26ClN with a molecular weight of 327.90, and the following structural formula:
Terbinafine hydrochloride USP is a white to off-white fine crystalline powder. It is freely soluble in methanol and methylene chloride, soluble in ethanol, and slightly soluble in water.
Each tablet contains:
Active Ingredient: Terbinafine hydrochloride USP (equivalent to 250 mg of terbinafine)
Inactive Ingredients: Microcrystalline cellulose, sodium starch glycolate, colloidal silicon dioxide, hypromellose, and magnesium stearate.
12. Terbinafine - Clinical Pharmacology
12.1 Mechanism of Action
Terbinafine is an allylamine antifungal [see Clinical Pharmacology (12.4)].
12.3 Pharmacokinetics
Following oral administration, terbinafine is well absorbed (greater than 70%) and the bioavailability of terbinafine tablets as a result of first-pass metabolism is approximately 40%. Peak plasma concentrations of 1 mcg/mL appear within 2 hours after a single 250 mg dose; the AUC is approximately 4.56 mcg•h/mL. An increase in the AUC of terbinafine of less than 20% is observed when terbinafine tablets are administered with food.
In plasma, terbinafine is greater than 99% bound to plasma proteins and there are no specific binding sites. At steady-state, in comparison to a single dose, the peak concentration of terbinafine is 25% higher and plasma AUC increases by a factor of 2.5; the increase in plasma AUC is consistent with an effective half-life of ~36 hours. Terbinafine is distributed to the sebum and skin. A terminal half-life of 200 to 400 hours may represent the slow elimination of terbinafine from tissues such as skin and adipose. Prior to excretion, terbinafine is extensively metabolized by at least 7 CYP isoenzymes with major contributions from CYP2C9, CYP1A2, CYP3A4, CYP2C8, and CYP2C19. No metabolites have been identified that have antifungal activity similar to terbinafine. Approximately 70% of the administered dose is eliminated in the urine.
In patients with renal impairment (creatinine clearance less than or equal to 50 mL/min) or hepatic cirrhosis, the clearance of terbinafine is decreased by approximately 50% compared to normal volunteers. No effect of gender on the blood levels of terbinafine was detected in clinical trials. No clinically relevant age-dependent changes in steady-state plasma concentrations of terbinafine have been reported.
12.4 Microbiology
Terbinafine, an allylamine antifungal, inhibits biosynthesis of ergosterol, an essential component of fungal cell membrane, via inhibition of squalene epoxidase enzyme. This results in fungal cell death primarily due to the increased membrane permeability mediated by the accumulation of high concentrations of squalene but not due to ergosterol deficiency. Depending on the concentration of the drug and the fungal species test in vitro, terbinafine hydrochloride may be fungicidal. However, the clinical significance of in vitro data is unknown.
Terbinafine has been shown to be active against most strains of the following microorganisms both in vitro and in clinical infections:
Trichophyton mentagrophytes
Trichophyton rubrum
The following in vitro data are available, but their clinical significance is unknown. In vitro, terbinafine exhibits satisfactory MIC’s against most strains of the following microorganisms; however, the safety and efficacy of terbinafine in treating clinical infections due to these microorganisms have not been established in adequate and well-controlled clinical trials:
Candida albicans
Epidermophyton floccosum
Scopulariopsis brevicaulis
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 28-month oral carcinogenicity study in rats, an increase in the incidence of liver tumors was observed in males at the highest dose tested, 69 mg/kg/day (2 times the MRHD based on AUC comparisons of the parent terbinafine); however, even though dose-limiting toxicity was not achieved at the highest tested dose, higher doses were not tested.
The results of a variety of in vitro (mutations in E. coli and S. typhimurium, DNA repair in rat hepatocytes, mutagenicity in Chinese hamster fibroblasts, chromosome aberration, and sister chromatid exchanges in Chinese hamster lung cells), and in vivo (chromosome aberration in Chinese hamsters, micronucleus test in mice) genotoxicity tests gave no evidence of a mutagenic or clastogenic potential.
Oral reproduction studies in rats at doses up to 300 mg/kg/day (12 times the MRHD based on BSA comparisons) did not reveal any specific effects on fertility or other reproductive parameters. Intravaginal application of terbinafine hydrochloride at 150 mg/day in pregnant rabbits did not increase the incidence of abortions or premature deliveries nor affect fetal parameters.
13.2 Animal Toxicology and/or Pharmacology
A wide range of in vivo studies in mice, rats, dogs, and monkeys, and in vitro studies using rat, monkey, and human hepatocytes suggest that peroxisome proliferation in the liver is a rat-specific finding. However, other effects, including increased liver weights and APTT, occurred in dogs and monkeys at doses giving Css trough levels of the parent terbinafine 2 to 3 times those seen in humans at the MRHD. In a 52-week oral toxicology study conducted in juvenile maturing dogs, increased heart and liver weights were noted in males and signs of CNS disturbance including 3 cases of single episodes of seizures were noted in females at the highest dose tested, 100 mg/kg/day [19 times (males) and 10 times (females) the MRHD based on AUC comparisons of the parent terbinafine]. No treatment related findings were noted at 30 mg/kg/day [1.6 times (males) and 1.9 times (females) the MRHD based on AUC comparisons of the parent terbinafine] in this study.
14. Clinical Studies
The efficacy of terbinafine tablets in the treatment of onychomycosis is illustrated by the response of subjects with toenail and/or fingernail infections who participated in 3 U.S./Canadian placebo-controlled clinical trials.
Results of the first toenail trial, as assessed at week 48 (12 weeks of treatment with 36 weeks follow-up after completion of therapy), demonstrated mycological cure, defined as simultaneous occurrence of negative KOH plus negative culture, in 70% of subjects. Fifty-nine percent (59%) of subjects experienced effective treatment (mycological cure plus 0% nail involvement or greater than 5mm of new unaffected nail growth); 38% of subjects demonstrated mycological cure plus clinical cure (0% nail involvement).
In a second toenail trial of dermatophytic onychomycosis, in which nondermatophytes were also cultured, similar efficacy against the dermatophytes was demonstrated. The pathogenic role of the nondermatophytes cultured in the presence of dermatophytic onychomycosis has not been established. The clinical significance of this association is unknown.
Results of the fingernail trial, as assessed at week 24 (6 weeks of treatment with 18 weeks follow-up after completion of therapy), demonstrated mycological cure in 79% of subjects, effective treatment in 75% of the subjects, and mycological cure plus clinical cure in 59% of the subjects.
The mean time to overall success was approximately 10 months for the first toenail trial and 4 months for the fingernail trial. In the first toenail trial, for subjects evaluated at least 6 months after achieving clinical cure and at least 1 year after completing therapy with terbinafine tablets, the clinical relapse rate was approximately 15%.
16. How is Terbinafine supplied
Terbinafine Tablets USP, 250 mg are supplied as white to off-white, round uncoated, biconvex beveled edge tablets having ‘D’ debossed on one side and ‘74’ on the other side.
Bottles of 30 NDC 65862-079-30
Bottles of 100 NDC 65862-079-01
Bottles of 1,000 NDC 65862-079-99
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Protect from light.
17. Patient Counseling Information
Advise the patient to read the FDA-Approved Medication Guide.
Patients taking terbinafine tablets should receive the following information and instructions:
- Advise patients to immediately report to their physician or get emergency help if they experience any of the following symptoms: hives, mouth sores, blistering and peeling of skin, swelling of face, lips, tongue, or throat, difficulty swallowing or breathing. Terbinafine tablets treatment should be discontinued.
- Advise patients to immediately report to their physician any symptoms of persistent nausea, anorexia, fatigue, vomiting, right upper abdominal pain, jaundice, dark urine, or pale stools. Terbinafine tablets treatment should be discontinued.
- Advise patients to report to their physician any signs of taste disturbance, smell disturbance and/or depressive symptoms, fever, skin eruption, lymph node enlargement, erythema, scaling, loss of pigment, and unusual photosensitivity that can result in a rash. Terbinafine tablets treatment should be discontinued.
- Advise patients to minimize exposure to natural and artificial sunlight (tanning beds or UVA/B treatment) while using terbinafine tablets.
- Advise patients that if they forget to take terbinafine tablets, to take their tablets as soon as they remember, unless it is less than 4 hours before the next dose is due.
- Advise patients to call their physician if they take too many terbinafine tablets.
- Advise patients to call their physician if they become pregnant during treatment with terbinafine tablets.
Dispense with Medication Guide available at: www.aurobindousa.com/medication-guides
Distributed by:
Aurobindo Pharma USA, Inc.
279 Princeton-Hightstown Road
East Windsor, NJ 08520
Manufactured by:
Aurobindo Pharma Limited
Hyderabad-500 032, India
Revised: 01/2022
Medication Guide
Terbinafine Tablets, USP
[Terbinafine (ter BIN na feen)]
What is the most important information I should know about terbinafine tablets?
Terbinafine tablets may cause serious side effects, including:
-
Liver problems that can lead to the need for a liver transplant or death. This can happen in people who have liver problems and in people who have never had liver problems. Tell your doctor right away if you get any of these symptoms of liver problems:
- nausea
- poor appetite
- tiredness
- vomiting
- upper right stomach-area (abdomen) pain
- yellowing of your skin or eyes (jaundice)
- dark (tea-colored) urine
- pale or light colored stools
Your doctor should do a blood test to check you for liver problems before you start treatment with terbinafine tablets. Your doctor may also check you for liver problems during treatment, and tell you to stop taking terbinafine tablets if you develop liver problems.
What are terbinafine tablets?
Terbinafine tablets are a prescription medicine used to treat fungal infections of the fingernails and toenails (onychomycosis).
Your doctor should do tests to check you for fungal infection of your nails before you start terbinafine tablets.
It is not known if terbinafine tablets are safe and effective in children for the treatment of onychomycosis.
Who should not take terbinafine tablets?
Do not take terbinafine tablets if you:
- have had a severe allergic reaction to terbinafine hydrochloride when taken by mouth.
- have had liver disease for a long time (chronic) or have active liver disease.
What should I tell my doctor before taking terbinafine tablets?
Before taking terbinafine tablets, tell your doctor about all of your medical conditions, including if you:
- have or had liver problems
- have a weakened immune system (immunocompromised)
- have lupus (an autoimmune disease)
- are pregnant or plan to become pregnant. It is not known if terbinafine tablets may harm your unborn baby.
Tell your doctor right away if you become pregnant during treatment with terbinafine tablets.
- are breastfeeding or plan to breastfeed. Terbinafine hydrochloride passes into your breast milk and may harm your baby. Talk to your doctor about the best way to feed your baby if you take terbinafine tablets.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Terbinafine tablets may affect the way other medicines work and other medicines may affect how terbinafine tablets work.
How should I take terbinafine tablets?
- Take terbinafine tablets exactly as your doctor tells you to take them.
- Terbinafine hydrochloride comes as a tablet that you take by mouth.
-
Terbinafine tablets are usually taken:
- 1 time each day for 6 weeks to treat fungal infections of your fingernail, or
- 1 time each day for 12 weeks to treat fungal infections of your toenail
- Terbinafine tablets can be taken with or without food.
- If you miss a dose of terbinafine tablets, take it as soon as you remember. If it is less than 4 hours before your next dose, skip the missed dose. Just take the next dose at your regular time.
If you take too many terbinafine tablets, call your doctor. You may have the following symptoms:
- nausea
- vomiting
- stomach-area (abdomen) pain
- dizziness
- rash
- frequent urination
- headache
What should I avoid while taking terbinafine tablets?
Avoid sunlight. Terbinafine tablets can make your skin sensitive to the sun and the light from sunlamps and tanning beds. You can get a severe sunburn. Use sunscreen and wear a hat and clothes that cover your skin if you have to be in sunlight. Talk to your doctor if you get sunburn.
What are the possible side effects of terbinafine tablets?
Terbinafine tablets may cause serious side effects, including:
- See “What is the most important information I should know about terbinafine tablets?”
-
Change in your sense of taste or loss of taste is common with terbinafine tablets, but can also be severe. This may improve within several weeks after you stop taking terbinafine tablets, but may last for a long time or may become permanent. Tell your doctor if you develop any of the following:
- change in your sense of taste or loss of taste
- poor appetite
- change in your mood or depressive symptoms.
- weight loss
- anxiousness
See the list of depressive symptoms below.
- Change in your sense of smell or loss of smell may happen with terbinafine tablets. This may improve after you stop taking terbinafine tablets, but may last for a long time or may become permanent. Tell your doctor if you have a change in your sense of smell or loss of smell.
-
Depressive symptoms. Tell your doctor right away if you develop any of these signs or symptoms:
- feel sad or worthless
- change in sleep pattern
- loss of energy or interest in daily activities
- restlessness
- mood changes
- Low white blood cell count. Terbinafine tablets may decrease your white blood cell count, especially neutrophils. You may have a higher risk of getting an infection when your white blood cell count is low.
-
Serious skin or allergic reactions, which may include problems with some of your body organs. Tell your doctor right away or get emergency medical help if you get any of these symptoms:
- skin rash
- hives
- sores in your mouth, or your skin blisters and peels
- swelling of your face, eyes, lips, tongue or throat
- trouble swallowing or breathing
- fever
- swollen lymph glands
Also tell your doctor about any new symptoms, such as cough, chest pain, fast heartbeat, or blood in your urine.
-
New or worsening lupus. Stop taking terbinafine tablets and tell your doctor if you get any of the following:
- a skin rash that gets worse (progresses), is scaly, red, shows scarring, or loss of skin color
- unusual sensitivity to the sun that can cause a rash
- Blood clotting problems. When taking terbinafine tablets, you may develop a blood clotting problem, that can sometimes cause death. Tell your doctor, if you get any unexplained bleeding or bruising.
The most common side effects of terbinafine tablets include:
- headache
- diarrhea
- rash
- upset stomach
- abnormal liver function tests
- itching
- nausea
- stomach-area (abdomen) pain
- gas
These are not all of the possible side effects of terbinafine tablets.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store terbinafine tablets?
- Store terbinafine tablets at 20° to 25°C (68° to 77°F).
- Keep terbinafine tablets in a tightly closed container and keep out of the light.
Keep terbinafine tablets and all medicines out of the reach of children.
General information about the safe and effective use of terbinafine tablets.
Medicines are sometimes prescribed for purposes other than those listed in Medication Guide. Do not use terbinafine tablets for a condition for which it was not prescribed. Do not give terbinafine tablets to other people, even if they have the same symptoms that you have. They may harm them. You can ask your pharmacist or doctor for information about terbinafine tablets that is written for health professionals.
What are the ingredients in terbinafine tablets?
Active ingredient: Terbinafine hydrochloride
Inactive ingredients: Microcrystalline cellulose, sodium starch glycolate, colloidal silicon dioxide, hypromellose, and magnesium stearate.
This Medication Guide has been approved by the U.S. Food and Drug Administration
Dispense with Medication Guide available at: www.aurobindousa.com/medication-guides
Distributed by:
Aurobindo Pharma USA, Inc.
279 Princeton-Hightstown Road
East Windsor, NJ 08520
Manufactured by:
Aurobindo Pharma Limited
Hyderabad-500 032, India
Revised: 01/2022

| TERBINAFINE
terbinafine hydrochloride tablet |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Aurobindo Pharma Limited (650082092) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Aurobindo Pharma Limited | 918917642 | ANALYSIS(65862-079) , MANUFACTURE(65862-079) | |
Frequently asked questions
- What are the most common skin conditions? (with photos)
- What is the best over-the-counter nail fungus treatment?
- What home remedies work well for toenail fungus?
- How do I get rid of nail fungus?
More about terbinafine
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (482)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Support group
- Drug class: miscellaneous antifungals
- Breastfeeding
- En español
