Epirubicin: Package Insert / Prescribing Info
Package insert / product label
Generic name: epirubicin hydrochloride
Dosage form: injection, solution
Drug class: Antibiotics / antineoplastics
J Code (medical billing code): J9178 (2 mg, intravenous)
Medically reviewed by Drugs.com. Last updated on Aug 2, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
Initial U.S. Approval: 1999
WARNING: SEVERE OR LIFE-THREATENING HEMATOLOGICAL AND OTHER ADVERSE REACTIONS
See full prescribing information for complete boxed warning
- •Severe local tissue necrosis associated with extravasation during administration (5.9)
- •Myocardial toxicity, manifested in its most severe form by potentially fatal congestive heart failure (CHF) (5.3)
- •Secondary acute myelogenous leukemia (AML) (5.4)
- •Reduce dosage in patients with impaired hepatic function (5.5)
- •Severe myelosuppression (5.2)
- •Administer only under the supervision of a physician who is experienced in the use of cancer chemotherapeutic agents (5)
Indications and Usage for Epirubicin
Epirubicin Hydrochloride Injection, USP is an anthracycline topoisomerase II inhibitor indicated as a component of adjuvant therapy in patients with evidence of axillary node tumor involvement following resection of primary breast cancer (1) .
Epirubicin Dosage and Administration
- Administer intravenously in repeated 3 to 4 week cycles, either total dose on Day 1 of each cycle or divided equally and given on Days 1 and 8 of each cycle (2) .
- The recommended starting dose of epirubicin hydrochloride injection is 100 to 120 mg/m2. Dosage reductions are possible when given in certain combinations(2.1).
- Dosage adjustments after the first treatment cycle should be made based on hematologic and nonhematologic toxicities (2.2).
- Reduce dose in patients with hepatic impairment (2.2 , 8.6 , 12.3 ).
- Consider lower doses in patients with severe renal impairment (2.2, 8.7 , 12.3 ).
Dosage Forms and Strengths
Single use vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use solution (50 mg/25 mL and 200 mg/100 mL) (3)
Contraindications
Patients should not be treated with epirubicin hydrochloride injection if they have any of the following conditions: baseline neutrophil count < 1500 cells/mm3; cardiomyopathy and/or heart failure, recent myocardial infarction, severe arrhythmias; previous treatment with anthracyclines up to the maximum cumulative dose; hypersensitivity to epirubicin, other anthracyclines, or anthracenediones; or severe hepatic dysfunction (4).
Warnings and Precautions
- •A dose-dependent, reversible leukopenia and/or neutropenia is the predominant manifestation of hematologic toxicity associated with epirubicin hydrochloride and represents the most common acute dose-limiting toxicity (5.2).
- •Cardiotoxicity is a known risk of anthracycline treatment and may be manifested by early (or acute) or late (delayed) events (5.3 ).
- •The occurrence of secondary acute myelogenous leukemia, with or without a preleukemic phase, has been reported in patients treated with anthracyclines(5.4).
- •Serum total bilirubin and AST levels should be evaluated before and during treatment with epirubicin hydrochloride. Patients with elevated bilirubin or AST may experience slower clearance of drug with an increase in overall toxicity. Lower doses are recommended in these patients. Patients with severe hepatic impairment have not been evaluated (5.5).
- •Serum creatinine should be assessed before and during therapy. Dosage adjustment is necessary in patients with serum creatinine >5 mg/dL. Patients undergoing dialysis have not been studied (5.6).
- •Epirubicin hydrochloride may induce hyperuricemia as a consequence of the extensive purine catabolism that accompanies drug-induced rapid lysis of highly chemosensitive neoplastic cells (tumor-lysis syndrome)(5.7).
- •Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including epirubicin hydrochloride may result in serious or fatal infections (5.8 ).
- •Venous sclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Extravasation of epirubicin hydrochloride during the infusion may cause local pain, severe tissue lesions (vesication, severe cellulitis), and necrosis. Facial flushing, as well as local erythematous streaking along the vein, may be indicative of excessively rapid administration. It may precede local phlebitis or thrombophlebitis. Patients administered the 120 mg/m2 regimen of epirubicin hydrochloride as a component of combination chemotherapy should also receive prophylactic antibiotic therapy with trimethoprim-sulfamethoxazole (e.g., Septra®, Bactrim®) or a fluoroquinolone (5.9).
- •Epirubicin hydrochloride is emetigenic. Antiemetics may reduce nausea and vomiting; prophylactic use of antiemetics should be considered before administration of epirubicin hydrochloride, particularly when given in conjunction with other emetigenic drugs (5.10).
- •Administration of epirubicin hydrochloride after previous radiation therapy may induce an inflammatory recall reaction at the site of the irradiation (5.11).
- •Thrombophlebitis and thromboembolic phenomena, including pulmonary embolism (in some cases fatal) have been coincidentally reported with the use of epirubicin hydrochloride (5.12 ).
- •Epirubicin hydrochloride can cause fetal harm when administered to a pregnant woman. Advise women of potential risk to the fetus (5.12 ).
Adverse Reactions/Side Effects
- In early breast cancer, acute adverse events occurring in ≥10% of patients are leukopenia, neutropenia, anemia, thrombocytopenia, amenhorrhea, lethargy, nausea/vomiting, mucositis, diarrhea, infection, conjunctivitis/keratitis, alopecia, local toxicity and rash/itch (6).
To report SUSPECTED ADVERSE REACTIONS, contact West-Ward Pharmaceuticals Corp. at 1-877-845-0689, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Use In Specific Populations
-
Nursing Mothers: Discontinue nursing prior to taking epirubicin hydrochloride (8.3).
-
Pediatric Use: Safety and effectiveness of epirubicin hydrochloride in pediatric patients have not been established. Pediatric patients may be at greater risk for anthracycline-induced acute manifestations of cardiotoxicity and for chronic CHF(8.4).
-
Geriatric Use: Care should be taken in monitoring toxicity when epirubicin hydrochloride is administered to female patients > 70 years of age (8.5).
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2024
Full Prescribing Information
WARNING: RISK OF TISSUE NECROSIS, CARDIAC TOXICITY, SECONDARY ACUTE MYELOGENOUS LEUKEMIA, AND MYELOSUPPRESSION
- Severe local tissue necrosis will occur if there is extravasation during administration. Epirubicin hydrochloride must not be given by the intramuscular or subcutaneous route [see Warnings and Precautions (5.9) ].
- Cardiac toxicity, including fatal congestive heart failure (CHF), may occur either during therapy with epirubicin hydrochloride or months to years after termination of therapy. The probability of developing clinically evident CHF is estimated as approximately 0.9% at a cumulative dose of 550 mg/m2, 1.6% at 700 mg/m2, and 3.3% at 900 mg/m2. In the adjuvant treatment of breast cancer, the maximum cumulative dose used in clinical trials was 720 mg/m2. The risk of developing CHF increases rapidly with increasing total cumulative doses of epirubicin hydrochloride in excess of 900 mg/m2; this cumulative dose should only be exceeded with extreme caution. Active or dormant cardiovascular disease, prior or concomitant radiotherapy to the mediastinal/pericardial area, previous therapy with other anthracyclines or anthracenediones, or concomitant use of other cardiotoxic drugs may increase the risk of cardiac toxicity. Cardiac toxicity with epirubicin hydrochloride may occur at lower cumulative doses whether or not cardiac risk factors are present [see Warnings and Precautions (5.3)] .
- Secondary acute myelogenous leukemia (AML) has been reported in patients with breast cancer treated with anthracyclines, including epirubicin. The occurrence of refractory secondary leukemia is more common when such drugs are given in combination with DNA-damaging antineoplastic agents, when patients have been heavily pretreated with cytotoxic drugs, or when doses of anthracyclines have been escalated. The cumulative risk of developing treatment-related AML or myelodysplastic syndrome (MDS), in 7110 patients with breast cancer who received adjuvant treatment with epirubicin hydrochloride-containing regimens, was estimated as 0.27% at 3 years, 0.46% at 5 years, and 0.55% at 8 years [see Warnings and Precautions (5.4)].
- Severe myelosuppression may occur [see Warnings and Precautions (5.2)].
1. Indications and Usage for Epirubicin
Epirubicin Hydrochloride Injection, USP is indicated as a component of adjuvant therapy in patients with evidence of axillary node tumor involvement following resection of primary breast cancer [see Clinical Studies (14.1 )].
2. Epirubicin Dosage and Administration
When possible, to reduce the risk of developing cardiotoxicity in patients receiving epirubicin hydrochloride injection after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, epirubicin hydrochloride injection-based therapy should be delayed until the other agents have cleared from the circulation [see Warnings and Precautions (5.3)].
Administer epirubicin hydrochloride injection by intravenous infusion. Give epirubicin hydrochloride injection in repeated 3 to 4 week cycles. The total dose of epirubicin hydrochloride injection may be given on Day 1 of each cycle or divided equally and given on Days 1 and 8 of each cycle. The recommended dosages of epirubicin hydrochloride injection are as follows:
2.1 Recommended Dose
The recommended dose of epirubicin hydrochloride injection is 100 to 120 mg/m2. The following regimens are recommended:
| CEF-120: | Cyclophosphamide Epirubicin HCl 5-Fluorouracil Repeated every 28 days for 6 cycles | 75 mg/m2 PO D 1 to 14 60 mg/m2 IV D 1, 8 500 mg/m2 IV D 1, 8 |
| FEC-100: | 5-Fluorouracil Epirubicin HCl Cyclophosphamide | 500 mg/m2 100 mg/m2 500 mg/m2 |
All drugs administered intravenously on Day 1 and repeated every 21 days for 6 cycles.
Patients administered the 120 mg/m2 regimen of epirubicin hydrochloride injection should receive prophylactic antibiotic therapy.
2.2 Dose Modifications
Epirubicin hydrochloride injection dosage adjustments for hematologic and non-hematologic toxicities within a cycle of treatment, is based on nadir platelet counts <50,000/mm3, absolute neutrophil counts (ANC) <250/mm3, neutropenic fever, or Grades 3/4 nonhematologic toxicity. Reduce epirubicin hydrochloride injection Day 1 dose in subsequent cycles to 75% of the Day 1 dose given in the current cycle. Delay Day 1 chemotherapy in subsequent courses of treatment until platelet counts are ≥100,000/mm3, ANC ≥1500/mm3, and nonhematologic toxicities have recovered to ≤ Grade 1.
Bone Marrow Dysfunction
Consider administering a lower starting dose (75 to 90 mg/m2) for heavily pretreated patients, patients with preexisting bone marrow depression, or in the presence of neoplastic bone marrow infiltration [see Warnings andPrecautions (5)]. For patients receiving a divided dose of epirubicin hydrochloride injection (Day 1 and Day 8), the Day 8 dose should be 75% of Day 1 if platelet counts are 75,000 to 100,000/mm3 and ANC is 1000 to 1499/mm3. If Day 8 platelet counts are <75,000/mm3, ANC <1000/mm3, or Grades 3/4 nonhematologic toxicity has occurred, omit the Day 8 dose.
Hepatic Impairment
Recommendations regarding use of epirubicin hydrochloride injection in patients with hepatic impairment are not available because patients with hepatic abnormalities were not included in the adjuvant trials [see Warnings and Precautions (5.5 ) and Clinical Pharmacology (12.3)]. In patients with elevated serum AST or serum total bilirubin concentrations, the following dose reductions are recommended:
• Bilirubin 1.2 to 3 mg/dL or AST 2 to 4 times upper limit of normal 1/2 of recommended starting dose
• Bilirubin > 3 mg/dL or AST > 4 times upper limit of normal 1/4 of recommended starting dose
Renal Impairment
While no specific dose recommendation can be made based on the limited available data in patients with renal impairment, consider lower doses in patients with severe renal impairment (serum creatinine > 5 mg/dL) [see Warnings and Precautions (5.6) and Clinical Pharmacology (12.3) ].
2.3 Preparation and Administration Precautions
Storage of the solution for injection at refrigerated conditions can result in the formation of a gelled product. This gelled product will return to a slightly viscous to mobile solution after 2 to a maximum of 4 hours equilibration at controlled room temperature (15 to 25ºC).
Inspect parenteral drug products visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Procedures for proper handling and disposal of anticancer drugs should be used when handling and preparing epirubicin hydrochloride. Several guidelines on this subject have been published.1-4[see References (15)].
Protective Measures
Take the following protective measures when handling epirubicin hydrochloride injection:
• Train personnel in appropriate techniques for reconstitution and handling.
• Exclude pregnant staff from working with this drug.
• Wear protective clothing: goggles, gowns, and disposable gloves and masks when handling epirubicin hydrochloride injection.
• Define a designated area for syringe preparation (preferably under a laminar flow system), with the work surface protected by disposable, plastic-backed, absorbent paper.
• Place all items used for reconstitution, administration, or cleaning (including gloves) in high-risk, waste-disposal bags for high temperature incineration.
• Treat spillage or leakage with dilute sodium hypochlorite (1% available chlorine) solution, preferably by soaking, and then water. Place all contaminated and cleaning materials in high-risk, waste-disposal bags for incineration. Treat accidental contact with the skin or eyes immediately by copious lavage with water, or soap and water, or sodium bicarbonate solution. However, do not abrade the skin by using a scrub brush. Seek medical attention. Always wash hands after removing gloves.
Incompatibilities
Avoid prolonged contact with any solution of an alkaline pH as it will result in hydrolysis of the drug. Do not mix epirubicin hydrochloride injection with heparin or fluorouracil due to chemical incompatibility that may lead to precipitation.
Epirubicin hydrochloride injection can be used in combination with other antitumor agents, but do not mix with other drugs in the same syringe.
Preparation of Infusion Solution
Administer epirubicin hydrochloride injection into the tubing of a freely flowing intravenous infusion (0.9% sodium chloride or 5% glucose solution). Patients receiving initial therapy at the recommended starting doses of 100 to 120 mg/m2 should generally have epirubicin hydrochloride injection infused over 15 to 20 minutes. For patients who require lower epirubicin hydrochloride injection starting doses due to organ dysfunction or who require modification of epirubicin hydrochloride injection doses during therapy, the epirubicin hydrochloride injection infusion time may be proportionally decreased, but should not be less than 3 minutes. This technique is intended to minimize the risk of thrombosis or perivenous extravasation, which could lead to severe cellulitis, vesication, or tissue necrosis. A direct push injection is not recommended due to the risk of extravasation, which may occur even in the presence of adequate blood return upon needle aspiration. Venous sclerosis may result from injection into small vessels or repeated injections into the same vein [see Warnings and Precautions (5.9)]. Use epirubicin hydrochloride injection within 24 hours of first penetration of the rubber stopper. Discard any unused solution.
3. Dosage Forms and Strengths
Epirubicin Hydrochloride Injection, USP is provided in glass single-use vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use solution in the following sizes: 50 mg/25 mL and 200 mg/100 mL.
4. Contraindications
Patients should not be treated with epirubicin hydrochloride injection if they have any of the following conditions:
• cardiomyopathy and/or heart failure, recent myocardial infarction or severe arrhythmias [see Warnings and Precautions (5.3 )].
• Previous treatment with maximum cumulative dose of anthracyclines [see Warnings and Precautions (5)].
• Hypersensitivity to epirubicin hydrochloride, other anthracyclines, or anthracenediones [see Adverse Reactions (6.2)].
5. Warnings and Precautions
Administer epirubicin hydrochloride injection only under the supervision of qualified physicians experienced in the use of cytotoxic therapy. Before beginning treatment with epirubicin hydrochloride, patients should recover from acute toxicities (such as stomatitis, neutropenia, thrombocytopenia, and generalized infections) of prior cytotoxic treatment. Also, precede initial treatment with epirubicin hydrochloride by a careful baseline assessment of blood counts; serum levels of total bilirubin, AST, and creatinine; and cardiac function as measured by left ventricular ejection function (LVEF). Carefully monitor patients during treatment for possible clinical complications due to myelosuppression. Supportive care may be necessary for the treatment of severe neutropenia and severe infectious complications. Monitoring for potential cardiotoxicity is also important, especially with greater cumulative exposure to epirubicin hydrochloride.
5.1 Injection-Related Reactions
Epirubicin hydrochloride injection is administered by intravenous infusion. Venous sclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Extravasation of epirubicin hydrochloride during the infusion may cause local pain, severe tissue lesions (vesication, severe cellulitis), and necrosis. Administer epirubicin hydrochloride slowly into the tubing of a freely running intravenous infusion. Patients receiving initial therapy at the recommended starting doses of 100 to 120 mg/m2 should generally have epirubicin hydrochloride infused over 15 to 20 minutes. For patients who require lower epirubicin hydrochloride starting doses due to organ dysfunction or who require modification of epirubicin hydrochloride doses during therapy, the epirubicin hydrochloride infusion time may be proportionally decreased, but should not be less than 3 minutes [see Dosage and Administration (2.3)]. If possible, avoid veins over joints or in extremities with compromised venous or lymphatic drainage. Immediately terminate infusion and restart in another vein if a burning or stinging sensation indicates perivenous infiltration. Perivenous infiltration may occur without causing pain. Facial flushing, as well as local erythematous streaking along the vein, may be indicative of excessively rapid administration. It may precede local phlebitis or thrombophlebitis. Give prophylactic antibiotic therapy to patients administered the 120 mg/m2 regimen of epirubicin hydrochloride as a component of combination chemotherapy [see Clinical Studies (14.1 ) and Dosage and Administration 2.1 )].
5.2 Hematologic
Epirubicin hydrochloride can suppress bone marrow function as manifested by leukopenia, thrombocytopenia and anemia [see Adverse Reactions (6.1)], and myelosuppression is usually the dose-limiting toxicity. Patients should be monitored for myelosuppression during therapy [see Dosage and Administration (2.2 , 2.3 )].
5.3 Cardiac
Cardiotoxicity is a known risk of anthracycline treatment. Anthracycline-induced cardiac toxicity may be manifested by early (or acute) or late (delayed) events. Early cardiac toxicity of epirubicin hydrochloride consists mainly of sinus tachycardia and/or electrocardiogram (ECG) abnormalities such as non-specific ST-T wave changes, but tachyarrhythmias, including premature ventricular contractions and ventricular tachycardia, bradycardia, as well as atrioventricular and bundle-branch block have also been reported. These effects do not usually predict subsequent development of delayed cardiotoxicity, are rarely of clinical importance, and are generally not considered an indication for the suspension of epirubicin hydrochloride treatment. Delayed cardiac toxicity results from a characteristic cardiomyopathy that is manifested by reduced LVEF and/or signs and symptoms of congestive heart failure (CHF) such as tachycardia, dyspnea, pulmonary edema, dependent edema, hepatomegaly, ascites, pleural effusion, gallop rhythm. Life-threatening CHF is the most severe form of anthracycline-induced cardiomyopathy. This toxicity appears to be dependent on the cumulative dose of epirubicin hydrochloride and represents the cumulative dose-limiting toxicity of the drug. If it occurs, delayed cardiotoxicity usually develops late in the course of therapy with epirubicin hydrochloride or within 2 to 3 months after completion of treatment, but later events (several months to years after treatment termination) have been reported.
Given the risk of cardiomyopathy, exceed a cumulative dose of 900 mg/m2 epirubicin hydrochloride only with extreme caution. Risk factors [active or dormant cardiovascular disease, prior or concomitant radiotherapy to the mediastinal/pericardial area, previous therapy with other anthracyclines or anthracenediones, concomitant use of other drugs with the ability to suppress cardiac contractility or cardiotoxic drugs, especially those with long half-lives (e.g., trastuzumab)] may increase the risk of epirubicin hydrochloride cardiotoxicity [seeDrug Interaction (7.4 ) and Dosage and Administration (2 )]. Although not formally tested, it is probable that the toxicity of epirubicin hydrochloride and other anthracyclines or anthracenediones is additive. Cardiac toxicity with epirubicin hydrochloride may occur at lower cumulative doses whether or not cardiac risk factors are present.
Although endomyocardial biopsy is recognized as the most sensitive diagnostic tool to detect anthracycline-induced cardiomyopathy, this invasive examination is not practically performed on a routine basis. ECG changes such as dysrhythmias, a reduction of the QRS voltage, or a prolongation beyond normal limits of the systolic time interval may be indicative of anthracycline-induced cardiomyopathy, but ECG is not a sensitive or specific method for following anthracycline-related cardiotoxicity. The risk of serious cardiac impairment may be decreased through regular monitoring of LVEF during the course of treatment with prompt discontinuation of epirubicin hydrochloride at the first sign of impaired function. The preferred method for repeated assessment of cardiac function is evaluation of LVEF measured by multi-gated radionuclide angiography (MUGA) or echocardiography (ECHO). A baseline cardiac evaluation with an ECG and a MUGA scan or an ECHO is recommended, especially in patients with risk factors for increased cardiac toxicity. Perform repeated MUGA or ECHO determinations of LVEF, particularly with higher, cumulative anthracycline doses. The technique used for assessment should be consistent through follow-up. In patients with risk factors, particularly prior anthracycline or anthracenedione use, the monitoring of cardiac function must be particularly strict and the risk-benefit of continuing treatment with epirubicin hydrochloride in patients with impaired cardiac function must be carefully evaluated.
Do not administer epirubicin hydrochloride in combination with other cardiotoxic agents unless the patient’s cardiac function is closely monitored. Patients receiving epirubicin hydrochloride after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, may also be at an increased risk of developing cardiotoxicity [see Dosage and Administration (2)].
5.4 Secondary Leukemia
The occurrence of secondary acute myelogenous leukemia, with or without a preleukemic phase, has been reported in patients treated with anthracyclines. Secondary leukemia is more common when such drugs are given in combination with DNA-damaging antineoplastic agents, when patients have been heavily pretreated with cytotoxic drugs, or when doses of the anthracyclines have been escalated. These leukemias can have a short 1 to 3 year latency period.
Epirubicin hydrochloride is mutagenic, clastogenic, and carcinogenic in animals [see Nonclinical Toxicology (13.1)].
5.5 Hepatic
The major route of elimination of epirubicin is the hepatobiliary system [see Clinical Pharmacology (12.3)]. Evaluate serum total bilirubin and AST levels before and during treatment with epirubicin hydrochloride. Patients with elevated bilirubin or AST may experience slower clearance of drug with an increase in overall toxicity. Lower doses are recommended in these patients [seeDosage and Administration (2.2 )]. Patients with severe hepatic impairment have not been evaluated; therefore, do not use epirubicin hydrochloride in this patient population.
5.6 Renal
Assess serum creatinine before and during therapy. Dosage adjustment is necessary in patients with serum creatinine >5 mg/dL [see Dosage and Administration (2.2 )]. Patients undergoing dialysis have not been studied.
5.7 Tumor-Lysis Syndrome
As with other cytotoxic agents, epirubicin hydrochloride may induce hyperuricemia as a consequence of the extensive purine catabolism that accompanies drug-induced rapid lysis of highly chemosensitive neoplastic cells (tumor-lysis syndrome). Other metabolic abnormalities may also occur. While not generally a problem in patients with breast cancer, consider the potential for tumor-lysis syndrome in potentially susceptible patients and consider monitoring serum uric acid, potassium, calcium, phosphate, and creatinine immediately after initial chemotherapy administration. Hydration, urine alkalinization, and prophylaxis with allopurinol to prevent hyperuricemia may minimize potential complications of tumor-lysis syndrome.
5.8 Immunosuppressant Effects/Increased Susceptibility to Infections
Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including epirubicin, may result in serious or fatal infections. Avoid vaccination with a live vaccine in patients receiving epirubicin hydrochloride. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
5.9 Gastrointestinal
Epirubicin hydrochloride is emetigenic. Antiemetics may reduce nausea and vomiting; prophylactic use of antiemetics should be considered before administration of epirubicin hydrochloride, particularly when given in conjunction with other emetigenic drugs [see Adverse Reactions (6.2)].
5.10 Thrombophlebitis and Thromboembolic Phenomena
As with other cytotoxic agents, thrombophlebitis and thromboembolic phenomena, including pulmonary embolism (in some cases fatal) have been coincidentally reported with the use of epirubicin hydrochloride.
5.11 Coadministration with Cimetidine
Cimetidine increased the AUC of epirubicin by 50%. Stop cimetidine treatment during treatment with epirubicin hydrochloride [seeClinical Pharmacology (12.3) ].
5.12 Pregnancy
Epirubicin hydrochloride can cause fetal harm when administered to a pregnant woman. Epirubicin was embryolethal and teratogenic in rats and rabbits. There are no adequate and well-controlled studies of epirubicin hydrochloride in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus. Woman of child-bearing potential should be advised to avoid becoming pregnant during treatment and should use effective contraceptive methods [see Use in Specific Populations (8.1)].
5.13 Male Fertility and Reproductive Outcomes
Males with female sexual partners of childbearing potential should use contraception during and after cessation of epirubicin hydrochloride therapy. Epirubicin hydrochloride may damage testicular tissue and spermatozoa. Possible sperm DNA damage raises concerns about loss of fertility and genetic abnormalities in fetuses. The duration of this effect is uncertain [see Nonclinical Toxicology (13.1 )].
6. Adverse Reactions/Side Effects
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Integrated safety data are available from two studies (Studies MA-5 and GFEA-05) [seeClinical Studies (14.1 )] evaluating epirubicin hydrochloride-containing combination regimens in patients with early breast cancer. Of the 1260 patients treated in these studies, 620 patients received the higher-dose epirubicin hydrochloride regimen (FEC-100/CEF-120), 280 patients received the lower-dose epirubicin hydrochloride regimen (FEC-50), and 360 patients received CMF. Serotonin-specific antiemetic therapy and colony-stimulating factors were not used in these trials. Clinically relevant acute adverse events are summarized in Table 1.
Table 1. Clinically Relevant Acute Adverse Events in Patients with Early Breast Cancer
| Event | % of Patients |
|||||
| FEC-100/CEF-120 (N=620) | FEC-50 (N=280) | CMF (N=360) |
||||
| Grades 1 to 4 | Grades 3/4 | Grades 1 to 4 | Grades 3/4 | Grades 1 to 4 | Grades 3/4 |
|
| Hematologic Leukopenia Neutropenia Anemia Thrombocytopenia | 80.3 80.3 72.2 48.8 | 58.6 67.2 5.8 5.4 | 49.6 53.9 12.9 4.6 | 1.5 10.5 0 0 | 98.1 95.8 70.9 51.4 | 60.3 78.1 0.9 3.6 |
| Endocrine Amenorrhea Hot flashes | 71.8 38.9 | 0 4 0 | 69.3 5.4 | 0 0 | 67.7 69.1 | 0 6.4 |
| Body as a Whole Lethargy Fever | 45.8 5.2 | 1.9 0 | 1.1 1.4 | 0 0 | 72.7 4.5 | 0.3 0 |
| Gastrointestinal Nausea/vomiting Mucositis Diarrhea Anorexia | 92.4 58.5 24.8 2.9 | 25 8.9 0.8 0 | 83.2 9.3 7.1 1.8 | 22.1 0 0 0 | 85 52.9 50.7 5.8 | 6.4 1.9 2.8 0.3 |
| Infection Infection Febrile neutropenia | 21.5 NA | 1.6 6.1 | 15 0 | 0 0 | 25.9 NA | 0.6 1.1 |
| Ocular Conjunctivitis/keratitis | 14.8 | 0 | 1.1 | 0 | 38.4 | 0 |
| Skin Alopecia Local toxicity Rash/itch Skin changes | 95.5 19.5 8.9 4.7 | 56 60 30 30 | 69.6 2.5 1.4 0.7 | 19.3 0.4 0 0 | 84.4 8.1 14.2 7.2 | 6.7 0 0 0 |
FEC & CEF = cyclophosphamide + epirubicin hydrochloride + fluorouracil; CMF = cyclophosphamide + methotrexate + fluorouracil; NA = not available Grade 1 or 2 changes in transaminase levels were observed but were more frequently seen with CMF than with CEF.
Delayed Events
Table 2 describes the incidence of delayed adverse events in patients participating in the MA-5 and GFEA-05 trials.
Table 2. Long-Term Adverse Events in Patients with Early Breast Cancer
| Event | % of Patients |
||
| FEC-100/CEF-120 (N=620) | FEC-50 (N=280) | CMF (N=360) |
|
| Cardiac events Asymptomatic drops in LVEF CHF | 2.1* 1.5 | 1.4 0.4 | 0.8* 0.3 |
| Leukemia AML | 0.8 | 0 | 0.3 |
*In study MA-5, cardiac function was not monitored after 5 years.
Two cases of acute lymphoid leukemia (ALL) were also observed in patients receiving epirubicin hydrochloride. However, an association between anthracyclines such as epirubicin hydrochloride and ALL has not been clearly established.
6.2 Overview of Acute and Delayed Toxicities
Hematologic
Dose-dependent, reversible leukopenia and/or neutropenia is the predominant manifestation of hematologic toxicity associated with epirubicin hydrochloride and represents the most common acute dose-limiting toxicity of this drug. In most cases, the white blood cell (WBC) nadir is reached 10 to 14 days from drug administration. Leukopenia/neutropenia is usually transient, with WBC and neutrophil counts generally returning to normal values by Day 21 after drug administration. As with other cytotoxic agents, epirubicin hydrochloride at the recommended dose in combination with cyclophosphamide and fluorouracil can produce severe leukopenia and neutropenia. Severe thrombocytopenia and anemia may also occur. Clinical consequences of severe myelosuppression include fever, infection, septicemia, septic shock, hemorrhage, tissue hypoxia, symptomatic anemia, or death. If myelosuppressive complications occur, use appropriate supportive measures (e.g., intravenous antibiotics, colony-stimulating factors, transfusions). Myelosuppression requires careful monitoring. Assess total and differential WBC, red blood cell (RBC), and platelet counts before and during each cycle of therapy with epirubicin hydrochloride [see Warnings and Precautions (5.2 )].
Gastrointestinal
A dose-dependent mucositis (mainly oral stomatitis, less often esophagitis) may occur in patients treated with epirubicin hydrochloride. Clinical manifestations of mucositis may include a pain or burning sensation, erythema, erosions, ulcerations, bleeding, or infections. Mucositis generally appears early after drug administration and, if severe, may progress over a few days to mucosal ulcerations; most patients recover from this adverse event by the third week of therapy. Hyperpigmentation of the oral mucosa may also occur. Nausea, vomiting, and occasionally diarrhea and abdominal pain can also occur. Severe vomiting and diarrhea may produce dehydration. Antiemetics may reduce nausea and vomiting; consider prophylactic use of antiemetics before therapy [see Warnings and Precautions (5.9) ].
Cutaneous and Hypersensitivity Reactions
Alopecia occurs frequently, but is usually reversible, with hair regrowth occurring within 2 to 3 months from the termination of therapy. Flushes, skin and nail hyperpigmentation, photosensitivity, and hypersensitivity to irradiated skin (radiation-recall reaction) have been observed. Urticaria and anaphylaxis have been reported in patients treated with epirubicin hydrochloride; signs and symptoms of these reactions may vary from skin rash and pruritus to fever, chills, and shock.
Cardiovascular
In a retrospective survey, including 9144 patients, mostly with solid tumors in advanced stages, the probability of developing CHF increased with increasing cumulative doses of epirubicin hydrochloride (Figure 1). The estimated risk of epirubicin hydrochloride-treated patients developing clinically evident CHF was 0.9% at a cumulative dose of 550 mg/m2, 1.6% at 700 mg/m2, and 3.3% at 900 mg/m2. The risk of developing CHF in the absence of other cardiac risk factors increased steeply after an epirubicin hydrochloride cumulative dose of 900 mg/m2[see Warnings and Precautions (5.3)].
Figure 1. Risk of CHF in 9144 Patients Treated with Epirubicin Hydrochloride
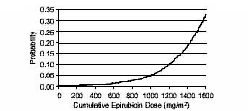
In another retrospective survey of 469 epirubicin hydrochloride-treated patients with metastatic or early breast cancer, the reported risk of CHF was comparable to that observed in the larger study of over 9000 patients [see Warnings and Precautions (5.3) ].
Other serious drug-related cardiovascular adverse events that occurred during clinical trials with epirubicin hydrochloride, administered in different indications, include ventricular tachycardia, AV block, bundle branch block, bradycardia and thromboembolism.
Secondary Leukemia
An analysis of 7110 patients who received adjuvant treatment with epirubicin hydrochloride in controlled clinical trials as a component of poly-chemotherapy regimens for early breast cancer, showed a cumulative risk of secondary acute myelogenous leukemia or myelodysplastic syndrome (AML/MDS) of about 0.27% (approximate 95% CI, 0.14 to 0.4) at 3 years, 0.46% (approximate 95% CI, 0.28 to 0.65) at 5 years, and 0.55% (approximate 95% CI, 0.33 to 0.78) at 8 years. The risk of developing AML/MDS increased with increasing epirubicin hydrochloride cumulative doses as shown in Figure 2.
Figure 2. Risk of AML/MDS in 7110 Patients Treated with Epirubicin Hydrochloride
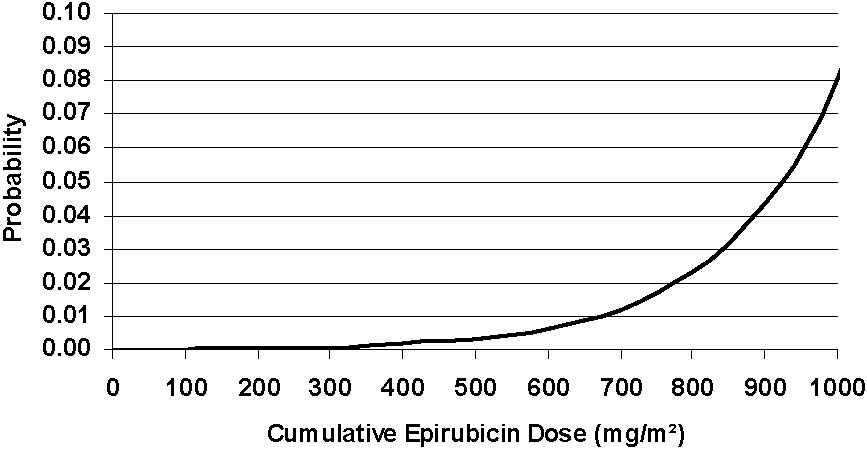
The cumulative probability of developing AML/MDS was found to be particularly increased in patients who received more than the maximum recommended cumulative dose of epirubicin HCl (720 mg/m2) or cyclophosphamide (6,300 mg/m2), as shown in Table 3.
Table 3. Cumulative Probability of AML/MDS in Relation to Cumulative Doses of Epirubicin Hydrochloride and Cyclophosphamide
| Years from Treatment Start | Cumulative Probability of Developing AML/MDS % (95% CI) |
|||
| Cyclophosphamide Cumulative Dose ≤6,300 mg/m2 | Cyclophosphamide Cumulative Dose >6,300 mg/m2 |
|||
| Epirubicin HCl Cumulative Dose ≤720 mg/m2 N=4760 | Epirubicin HCl Cumulative Dose >720 mg/m2 N=111 | Epirubicin HCl Cumulative Dose ≤720 mg/m2 N=890 | Epirubicin HCl Cumulative Dose >720 mg/m2 N=261 |
|
| 3 | 0.12 (0.01 to 0.22) | 0 (0 to 0) | 0.12 (0 to 0.37) | 4.37 (1.69 to 7.05) |
| 5 | 0.25 (0.08 to 0.42) | 2.38 (0 to 6.99) | 0.31 (0 to 0.75) | 4.97 (2.06 to 7.87) |
| 8 | 0.37 (0.13 to 0.61) | 2.38 (0 to 6.99) | 0.31 (0 to 0.75) | 4.97 (2.06 to 7.87) |
Injection-Site Reactions [see Warnings and Precautions (5.1)].
6.3 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of epirubicin hydrochloride. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Infections and infestations: sepsis, pneumonia
Immune system disorders: anaphylaxis
Metabolism and nutrition disorders: dehydration, hyperuricemia
Vascular disorders: shock, haemorrhage, embolism arterial, thrombophlebitis, phlebitis
Respiratory, thoracic and mediastinal disorders: pulmonary embolism
Gastrointestinal disorders: erosions, ulcerations, pain or burning sensation, bleeding, hyperpigmentation of the oral mucosa
Skin and subcutaneous tissue disorders: erythema, flushes, skin and nail hyperpigmentation, photosensitivity, hypersensitivity to irradiated skin (radiation-recall reaction), urticaria
Renal and urinary disorders: red coloration of urine for 1 to 2 days after administration
General disorders and administration site conditions: fever, chills
Injury, poisoning and procedural complications: chemical cystitis (following intravesical administration)
Related/similar drugs
7. Drug Interactions
7.1 Cardioactive Compounds
Do not administer epirubicin in combination with other cardiotoxic agents unless the patient’s cardiac function is closely monitored. Patients receiving epirubicin after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, may also be at an increased risk of developing cardiotoxicity. Avoid epirubicin-based therapy for up to 24 weeks after stopping trastuzumab when possible. If epirubicin is used before this time, monitor cardiac function carefully [see Dosage and Administration (2) and Warnings and Precautions (5.3)].
Concomitant use of epirubicin hydrochloride with other cardioactive compounds that could cause heart failure (e.g., calcium channel blockers), requires close monitoring of cardiac function throughout treatment.
7.2 Cimetidine
Cimetidine increases the exposure to epirubicin [see Clinical Pharmacology (12.3)]. Stop cimetidine during treatment with epirubicin hydrochloride.
7.3 Other Cytotoxic Drugs
Epirubicin hydrochloride used in combination with other cytotoxic drugs may show on-treatment additive toxicity, especially hematologic and gastrointestinal effects.
Paclitaxel:
The administration of epirubicin immediately prior to or after paclitaxel increased the systemic exposure of epirubicin, epirubicinol and 7-deoxydoxorubicin aglycone [see Clinical Pharmacology (12.3)].
Docetaxel:
The administration of epirubicin immediately prior to or after docetaxel did not have an effect on the systemic exposure of epirubicin, but increased the systemic exposure of epirubicinol and 7-deoxydoxorubicin aglycone [seeClinical Pharmacology (12.3)].
7.4 Radiation Therapy
There are few data regarding the coadministration of radiation therapy and epirubicin hydrochloride. In adjuvant trials of epirubicin hydrochloride-containing CEF-120 or FEC-100 chemotherapies, breast irradiation was delayed until after chemotherapy was completed. This practice resulted in no apparent increase in local breast cancer recurrence relative to published accounts in the literature. A small number of patients received epirubicin hydrochloride-based chemotherapy concomitantly with radiation therapy but had chemotherapy interrupted in order to avoid potential overlapping toxicities. It is likely that use of epirubicin hydrochloride with radiotherapy may sensitize tissues to the cytotoxic actions of irradiation. Administration of epirubicin hydrochloride after previous radiation therapy may induce an inflammatory recall reaction at the site of the irradiation.
8. Use In Specific Populations
8.1 Pregnancy
Teratogenic Effects: Pregnancy Category D. See ‘Warnings and Precautions’ section.
Epirubicin hydrochloride can cause fetal harm when administered to a pregnant woman. Administration of 0.8 mg/kg/day intravenously of epirubicin to rats (about 0.04 times the maximum recommended single human dose on a body surface area basis) during Days 5 to 15 of gestation was embryotoxic (increased resorptions and post-implantation loss) and caused fetal growth retardation (decreased body weight), but was not teratogenic up to this dose. Administration of 2 mg/kg/day intravenously of epirubicin to rats (about 0.1 times the maximum recommended single human dose on a body surface area basis) on Days 9 and 10 of gestation was embryotoxic (increased late resorptions, post-implantation losses, and dead fetuses; and decreased live fetuses), retarded fetal growth (decreased body weight), and caused decreased placental weight. This dose was also teratogenic, causing numerous external (anal atresia, misshapen tail, abnormal genital tubercle), visceral (primarily gastrointestinal, urinary, and cardiovascular systems), and skeletal (deformed long bones and girdles, rib abnormalities, irregular spinal ossification) malformations. Administration of intravenous epirubicin to rabbits at doses up to 0.2 mg/kg/day (about 0.02 times the maximum recommended single human dose on a body surface area basis) during Days 6 to 18 of gestation was not embryotoxic or teratogenic, but a maternally toxic dose of 0.32 mg/kg/day increased abortions and delayed ossification. Administration of a maternally toxic intravenous dose of 1 mg/kg/day epirubicin to rabbits (about 0.1 times the maximum recommended single human dose on a body surface area basis) on Days 10 to 12 of gestation induced abortion, but no other signs of embryofetal toxicity or teratogenicity were observed. When doses up to 0.5 mg/kg/day epirubicin were administered to rat dams from Day 17 of gestation to Day 21 after delivery (about 0.025 times the maximum recommended single human dose on a body surface area basis), no permanent changes were observed in the development, functional activity, behavior, or reproductive performance of the offspring.
There are no adequate and well-controlled studies of epirubicin hydrochloride in pregnant women. Two pregnancies have been reported in women taking epirubicin. A 34 year old woman, 28 weeks pregnant at her diagnosis of breast cancer, was treated with cyclophosphamide and epirubicin every 3 weeks for 3 cycles. She received the last dose at 34 weeks of pregnancy and delivered a healthy baby at 35 weeks. A second 34 year old woman with breast cancer metastatic to the liver was randomized to FEC-50 but was removed from study because of pregnancy. She experienced a spontaneous abortion. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant [see Warnings and Precautions (5.12) ].
8.3 Nursing Mothers
Epirubicin was excreted into the milk of rats treated with 0.5 mg/kg/day of epirubicin during peri- and postnatal periods. It is not known whether this drug is excreted in human milk. Because many drugs, including other anthracyclines, are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from epirubicin hydrochloride, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
Safety and effectiveness of epirubicin hydrochloride have not been established in pediatric patients. Pediatric patients may be at greater risk for anthracycline-induced acute manifestations of cardiotoxicity and for chronic CHF. The pharmacokinetics of epirubicin in pediatric patients have not been evaluated.
8.5 Geriatric Use
Although a lower starting dose of epirubicin hydrochloride was not used in trials in elderly female patients, particular care should be taken in monitoring toxicity when epirubicin hydrochloride is administered to female patients ≥ 70 years of age [see Clinical Pharmacology (12.3 )].
8.6 Hepatic Impairment
Epirubicin is eliminated by both hepatic metabolism and biliary excretion and clearance is reduced in patients with hepatic dysfunction. Do not treat patients with severe hepatic impairment with epirubicin hydrochloride. Reduce the starting dose for patients with less severe hepatic impairment [see Dosage and Administration (2.2 ) and Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No significant alterations in the pharmacokinetics of epirubicin or its major metabolite, epirubicinol, have been observed in patients with serum creatinine < 5 mg/dL. Consider lower doses in patients with severe renal impairment (serum creatinine > 5 mg/dL), as a reduction in plasma clearance was reported in these patients [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)]. Patients on dialysis have not been studied.
10. Overdosage
There is no known antidote for overdoses of epirubicin hydrochloride. A 36 year old man with non-Hodgkin’s lymphoma received a daily 95 mg/m2 dose of epirubicin hydrochloride injection for 5 consecutive days. Five days later, he developed bone marrow aplasia, grade 4 mucositis, and gastrointestinal bleeding. No signs of acute cardiac toxicity were observed. He was treated with antibiotics, colony-stimulating factors, and antifungal agents, and recovered completely. A 63 year old woman with breast cancer and liver metastasis received a single 320 mg/m2 dose of epirubicin hydrochloride. She was hospitalized with hyperthermia and developed multiple organ failure (respiratory and renal), with lactic acidosis, increased lactate dehydrogenase, and anuria. Death occurred within 24 hours after administration of epirubicin hydrochloride. Additional instances of administration of doses higher than recommended have been reported at doses ranging from 150 to 250 mg/m2. The observed adverse events in these patients were qualitatively similar to known toxicities of epirubicin. Most of the patients recovered with appropriate supportive care.
If an overdose occurs, provide supportive treatment (including antibiotic therapy, blood and platelet transfusions, colony-stimulating factors, and intensive care as needed) until the recovery of toxicities. Delayed CHF has been observed months after anthracycline administration. Observe patients carefully over time for signs of CHF and provide with appropriate supportive therapy.
11. Epirubicin Description
Epirubicin Hydrochloride Injection, USP is an anthracycline cytotoxic agent, intended for intravenous administration. Epirubicin hydrochloride is supplied as a sterile, clear, red solution and is available in glass vials containing 50 and 200 mg of epirubicin hydrochloride as a preservative-free, ready-to-use solution. Each milliliter of solution contains 2 mg of epirubicin hydrochloride. Inactive ingredients include sodium chloride, and water for injection. The pH of the solution has been adjusted to 3.0 with hydrochloric acid.
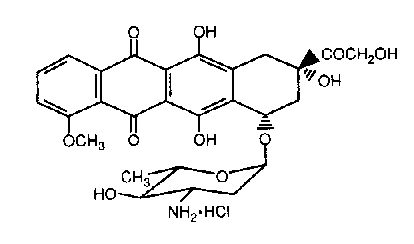
Epirubicin hydrochloride is the 4-epimer of doxorubicin and is a semi-synthetic derivative of daunorubicin. The chemical name is (1S,3S)-3-Glycoloyl-1,2,3,4,6,11-hexahydro-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1-naphthacenyl 3-amino-2,3,6-trideoxy- -L-arabino-hexopyranoside hydrochloride. The active ingredient is a red-orange hygroscopic powder, with the molecular formula C27H29NO11•HCl and a molecular weight of 579.98.
The structural formula is as follows:
12. Epirubicin - Clinical Pharmacology
12.1 Mechanism of Action
Epirubicin is an anthracycline cytotoxic agent. Although it is known that anthracyclines can interfere with a number of biochemical and biological functions within eukaryotic cells, the precise mechanisms of epirubicin’s cytotoxic and/or antiproliferative properties have not been completely elucidated.
Epirubicin forms a complex with DNA by intercalation of its planar rings between nucleotide base pairs, with consequent inhibition of nucleic acid (DNA and RNA) and protein synthesis.
Such intercalation triggers DNA cleavage by topoisomerase II, resulting in cytocidal activity. Epirubicin also inhibits DNA helicase activity, preventing the enzymatic separation of double-stranded DNA and interfering with replication and transcription. Epirubicin is also involved in oxidation/reduction reactions by generating cytotoxic free radicals. The antiproliferative and cytotoxic activity of epirubicin is thought to result from these or other possible mechanisms.
Epirubicin is cytotoxic in vitro to a variety of established murine and human cell lines and primary cultures of human tumors. It is also active in vivo against a variety of murine tumors and human xenografts in athymic mice, including breast tumors.
12.3 Pharmacokinetics
Epirubicin pharmacokinetics are linear over the dose range of 60 to 150 mg/m2 and plasma clearance is not affected by the duration of infusion or administration schedule. Pharmacokinetic parameters for epirubicin following 6 to 10 minute, single-dose intravenous infusions of epirubicin hydrochloride at doses of 60 to 150 mg/m2 in patients with solid tumors are shown in Table 4. The plasma concentration declined in a triphasic manner with mean half-lives for the alpha, beta, and gamma phases of about 3 minutes, 2.5 hours, and 33 hours, respectively.
Table 4. Summary of Mean (±SD) Pharmacokinetic Parameters in Patientsa with Solid Tumors Receiving Intravenous Epirubicin Hydrochloride 60 to 150 mg/m2
| Dose b (mg/m2 ) | Cmaxc (mcg/mL) | AUC d (mcg•h/mL) | t1/2e (hours) | CLf (L/hour) | Vssg (L/kg) |
| 60 | 5.7 ± 1.6 | 1.6 ± 0.2 | 35.3 ± 9 | 65 ± 8 | 21 ± 2 |
| 75 | 5.3 ± 1.5 | 1.7 ± 0.3 | 32.1 ± 5 | 83 ± 14 | 27 ± 11 |
| 120 | 9 ± 3.5 | 3.4 ± 0.7 | 33.7 ± 4 | 65 ± 13 | 23 ± 7 |
| 150 | 9.3 ± 2.9 | 4.2 ± 0.8 | 31.1 ± 6 | 69 ± 13 | 21 ± 7 |
a Advanced solid tumor cancers, primarily of the lung
b N=6 patients per dose level
c Plasma concentration at the end of 6 to 10 minute infusion
d Area under the plasma concentration curve
e Half-life of terminal phase
f Plasma clearance
g Steady state volume of distribution
Distribution
Following intravenous administration, epirubicin is rapidly and widely distributed into the tissues. Binding of epirubicin to plasma proteins, predominantly albumin, is about 77% and is not affected by drug concentration. Epirubicin also appears to concentrate in red blood cells; whole blood concentrations are approximately twice those of plasma.
Metabolism
Epirubicin is extensively and rapidly metabolized by the liver and is also metabolized by other organs and cells, including red blood cells. Four main metabolic routes have been identified:
(1) reduction of the C-13 keto-group with the formation of the 13(S)-dihydro derivative, epirubicinol; (2) conjugation of both the unchanged drug and epirubicinol with glucuronic acid; (3) loss of the amino sugar moiety through a hydrolytic process with the formation of the doxorubicin and doxorubicinol aglycones; and (4) loss of the amino sugar moiety through a redox process with the formation of the 7-deoxy-doxorubicin aglycone and 7-deoxy-doxorubicinol aglycone. Epirubicinol has in vitro cytotoxic activity one-tenth that of epirubicin. As plasma levels of epirubicinol are lower than those of the unchanged drug, they are unlikely to reach in vivo concentrations sufficient for cytotoxicity. No significant activity or toxicity has been reported for the other metabolites.
Excretion
Epirubicin and its major metabolites are eliminated through biliary excretion and, to a lesser extent, by urinary excretion. Mass-balance data from 1 patient found about 60% of the total radioactive dose in feces (34%) and urine (27%). These data are consistent with those from 3 patients with extrahepatic obstruction and percutaneous drainage, in whom approximately 35% and 20% of the administered dose were recovered as epirubicin or its major metabolites in bile and urine, respectively, in the 4 days after treatment.
Effect of Age
A population analysis of plasma data from 36 cancer patients (13 males and 23 females, 20 to 73 years) showed that age affects plasma clearance of epirubicin in female patients. The predicted plasma clearance for a female patient of 70 years of age was about 35% lower than that for a female patient of 25 years of age. An insufficient number of males > 50 years of age were included in the study to draw conclusions about age-related alterations in clearance in males. Although a lower epirubicin hydrochloride starting dose does not appear necessary in elderly female patients, and was not used in clinical trials, particular care should be taken in monitoring toxicity when epirubicin hydrochloride is administered to female patients > 70 years of age [see Patient Counseling Information (17)].
Effect of Gender
In patients ≤ 50 years of age, mean clearance values in adult male and female patients were similar. The clearance of epirubicin is decreased in elderly women.
Effect of Race
The influence of race on the pharmacokinetics of epirubicin has not been evaluated.
Effect of Hepatic Impairment
Epirubicin is eliminated by both hepatic metabolism and biliary excretion and clearance is reduced in patients with hepatic dysfunction. In a study of the effect of hepatic dysfunction, patients with solid tumors were classified into 3 groups. Patients in Group 1 (n=22) had serum AST (SGOT) levels above the upper limit of normal (median: 93 IU/L) and normal serum bilirubin levels (median: 0.5 mg/dL) and were given epirubicin hydrochloride doses of 12.5 to 90 mg/m2. Patients in Group 2 had alterations in both serum AST (median: 175 IU/L) and bilirubin levels (median: 2.7 mg/dL) and were treated with an epirubicin hydrochloride dose of 25 mg/m2 (n=8). Their pharmacokinetics were compared to those of patients with normal serum AST and bilirubin values, who received epirubicin hydrochloride doses of 12.5 to 120 mg/m2. The median plasma clearance of epirubicin was decreased compared to patients with normal hepatic function by about 30% in patients in Group 1 and by 50% in patients in Group 2. Patients with more severe hepatic impairment have not been evaluated [see Dosage and Administration (2.2) , and Warnings and Precautions (5.5)].
Effect of Renal Impairment
No significant alterations in the pharmacokinetics of epirubicin or its major metabolite, epirubicinol, have been observed in patients with serum creatinine < 5 mg/dL. A 50% reduction in plasma clearance was reported in four patients with serum creatinine ≥ 5 mg/dL [see Warnings and Precautions (5.6) and Dosing and Administration(2.2)]. Patients on dialysis have not been studied.
Effect of Paclitaxel
The administration of paclitaxel (175 to 225 mg/m2 as a 3 hour infusion) immediately before or after epirubicin (90 mg/m2 as bolus) caused variable increases in the systemic exposure (mean AUC) of epirubicin ranging from 5% to 109%. At same doses of epirubicin and paclitaxel, the mean AUC of the inactive metabolites of epirubicin (epirubicinol and 7-deoxy-aglycone) increased by 120% and 70%, respectively, when paclitaxel was immediately administered after epirubicin. Epirubicin had no effect on the exposure of paclitaxel whether it was administered before or after paclitaxel.
Effect of Docetaxel
The administration of docetaxel (70 mg/m2 as 1 hour infusion) immediately before or after epirubicin (90 mg/m2 as bolus) had no effect on the systemic exposure (mean AUC) of epirubicin. However, the mean AUC of epirubicinol and 7-deoxy-aglycone increased by 22.5% and 95%, respectively, when docetaxel was immediately administered after epirubicin compared to epirubicin alone. Epirubicin had no effect on the exposure of docetaxel whether it was administered before or after docetaxel.
Effect of Cimetidine
Coadministration of cimetidine (400 mg twice daily for 7 days starting 5 days before chemotherapy) increased the mean AUC of epirubicin (100 mg/m2) by 50% and decreased its plasma clearance by 30%.
Drugs Metabolized by Cytochrome P-450 Enzymes.
No systematic in vitro or in vivo evaluation has been performed to examine the potential for inhibition or induction by epirubicin of oxidative cytochrome P-450 isoenzymes.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Conventional long-term animal studies to evaluate the carcinogenic potential of epirubicin have not been conducted, but intravenous administration of a single 3.6 mg/kg epirubicin dose to female rats (about 0.2 times the maximum recommended human dose on a body surface area basis) approximately doubled the incidence of mammary tumors (primarily fibroadenomas) observed at 1 year. Administration of 0.5 mg/kg epirubicin intravenously to rats (about 0.025 times the maximum recommended human dose on a body surface area basis) every 3 weeks for ten doses increased the incidence of subcutaneous fibromas in males over an 18 month observation period. In addition, subcutaneous administration of 0.75 or 1 mg/kg/day (about 0.015 times the maximum recommended human dose on a body surface area basis) to newborn rats for 4 days on both the first and tenth day after birth for a total of eight doses increased the incidence of animals with tumors compared to controls during a 24-month observation period.
Epirubicin was mutagenic in vitro to bacteria (Ames test) either in the presence or absence of metabolic activation and to mammalian cells (HGPRT assay in V79 Chinese hamster lung fibroblasts) in the absence but not in the presence of metabolic activation. Epirubicin was clastogenic in vitro (chromosome aberrations in human lymphocytes) both in the presence and absence of metabolic activation and was also clastogenic in vivo (chromosome aberration in mouse bone marrow).
In fertility studies in rats, males were given epirubicin daily for 9 weeks and mated with females that were given epirubicin daily for 2 weeks prior to mating and through Day 7 of gestation. When 0.3 mg/kg/day (about 0.015 times the maximum recommended human single dose on a body surface area basis) was administered to both sexes, no pregnancies resulted. No effects on mating behavior or fertility were observed at 0.1 mg/kg/day, but male rats had atrophy of the testes and epididymis, and reduced spermatogenesis. The 0.1 mg/kg/day dose also caused embryolethality. An increased incidence of fetal growth retardation was observed in these studies at 0.03 mg/kg/day (about 0.0015 times the maximum recommended human single dose on a body surface area basis). Multiple daily doses of epirubicin to rabbits and dogs also caused atrophy of male reproductive organs. Single 20.5 and 12 mg/kg doses of intravenous epirubicin caused testicular atrophy in mice and rats, respectively (both approximately 0.5 times the maximum recommended human dose on a body surface area basis). A single dose of 16.7 mg/kg epirubicin caused uterine atrophy in rats.
14. Clinical Studies
14.1 Adjuvant Treatment of Breast Cancer
Two randomized, open-label, multicenter studies evaluated the use of epirubicin hydrochloride injection 100 to 120 mg/m2 in combination with cyclophosphamide and fluorouracil for the adjuvant treatment of patients with axillary-node positive breast cancer and no evidence of distant metastatic disease (Stage II or III). Study MA-5 evaluated 120 mg/m2 of epirubicin hydrochloride per course in combination with cyclophosphamide and fluorouracil (CEF-120 regimen). This study randomized premenopausal and perimenopausal women with one or more positive lymph nodes to an epirubicin hydrochloride-containing CEF-120 regimen or to a CMF regimen. Study GFEA-05 evaluated the use of 100 mg/m2 of epirubicin hydrochloride per course in combination with fluorouracil and cyclophosphamide (FEC-100). This study randomized pre- and postmenopausal women to the FEC-100 regimen or to a lower-dose FEC-50 regimen. In the GFEA-05 study, eligible patients were either required to have ≥ 4 nodes involved with tumor or, if only 1 to 3 nodes were positive, to have negative estrogen- and progesterone-receptors and a histologic tumor grade of 2 or 3. A total of 1281 women participated in these studies. Patients with T4 tumors were not eligible for either study. Table 5 shows the treatment regimens that the patients received. Relapse-free survival was defined as time to occurrence of a local, regional, or distant recurrence, or disease-related death. Patients with contralateral breast cancer, second primary malignancy, or death from causes other than breast cancer were censored at the time of the last visit prior to these events.
Table 5. Treatment Regimens Used in Phase 3 Studies of Patients with Early Breast Cancer
|
Treatment Groups |
Agent |
Regimen |
|
|
MA-5a |
CEF-120 |
Cyclophosphamide |
75 mg/m2 PO, d 1 to 14, q 28 days |
|
N=716 |
(total, 6 cycles)b N=356 |
Epirubicin HCl |
60 mg/m2 IV, d 1 & 8, q 28 days |
|
CMF (total, 6 cycles) N=360 |
Fluorouracil |
500 mg/m2 IV, d 1 & 8, q 28 days |
|
|
Cyclophosphamide |
100 mg/m2 PO, d 1 to 14, q 28 days |
||
|
Methotrexate |
40 mg/m2 IV, d 1 & 8, q 28 days |
||
|
Fluorouracil |
600 mg/m2 IV, d 1 & 8, q 28 days |
||
|
GFEA-05c N=565 |
FEC-100 (total, 6 cycles) N=276 |
Fluorouracil |
500 mg/m2 IV, d 1, q 21 days |
|
Epirubicin HCl |
100 mg/m2 IV, d 1, q 21 days |
||
|
Cyclophosphamide |
500 mg/m2 IV, d 1, q 21 days |
||
|
FEC-50 (total, 6 cycles) N=289 Tamoxifen 30 mg daily x 3 years, postmenopausal women, any receptor status |
Fluorouracil |
500 mg/m2 IV, d 1, q 21 days |
|
|
Epirubicin HCl |
50 mg/m2 IV, d 1, q 21 days |
||
|
Cyclophosphamide |
500 mg/m2 IV, d 1, q 21 days |
a In women who underwent lumpectomy, breast irradiation was to be administered after completion of study chemotherapy.
b Patients also received prophylactic antibiotic therapy with trimethoprim-sulfamethoxazole or fluoroquinolone for the duration of their chemotherapy.
c All women were to receive breast irradiation after the completion of chemotherapy.
In the MA-5 trial, the median age of the study population was 45 years. Approximately 60% of patients had 1 to 3 involved nodes and approximately 40% had ≥ 4 nodes involved with tumor. In the GFEA-05 study, the median age was 51 years and approximately half of the patients were postmenopausal. About 17% of the study population had 1 to 3 positive nodes and 80% of patients had ≥ 4 involved lymph nodes. Demographic and tumor characteristics were well-balanced between treatment arms in each study.
Relapse-free survival (RFS) and overall survival (OS) were analyzed using Kaplan-Meier methods in the intent-to-treat (ITT) patient populations in each study. Results were initially analyzed after up to 5 years of follow-up and these results are presented in the text below and in Table 3. Results after up to 10 years of follow-up are presented in Table 6. In Study MA-5, epirubicin hydrochloride-containing combination therapy (CEF-120) showed significantly longer RFS than CMF (5 year estimates were 62% versus 53%, stratified logrank for the overall RFS p=0.013). The estimated reduction in the risk of relapse was 24% at 5 years. The OS was also greater for the epirubicin hydrochloride-containing CEF-120 regimen than for the CMF regimen (5 year estimate 77% versus 70%; stratified logrank for overall survival p=0.043; non-stratified logrank p=0.13). The estimated reduction in the risk of death was 29% at 5 years.
In Study GFEA-05, patients treated with the higher-dose epirubicin hydrochloride regimen (FEC-100) had a significantly longer 5 year RFS (estimated 65% versus 52%, logrank for the overall RFS p=0.007) and OS (estimated 76% versus 65%, logrank for the overall survival p=0.007) than patients given the lower dose regimen (FEC-50). The estimated reduction in risk of relapse was 32% at 5 years. The estimated reduction in the risk of death was 31% at 5 years. Results of follow-up up to 10 years (median follow-up = 8.8 years and 8.3 years, respectively, for Study MA-5 and Study GFEA-05) are presented in Table 6.
Although the trials were not powered for subgroup analyses, in the MA-5 study, improvements in favor of CEF-120 vs. CMF were observed, in RFS and OS both in patients with 1 to 3 node positive and in those with ≥4 node positive tumor involvement. In the GFEA-05 study, improvements in RFS and OS were observed in both pre- and postmenopausal women treated with FEC-100 compared to FEC-50.
Table 6. Efficacy Results from Phase 3 Studies of Patients with Early Breast Cancer*
| MA-5 Study | GFEA-05 Study |
|||
| CEF-120 N=356 | CMF N=360 | FEC-100 N=276 | FEC-50 N=289 |
|
| RFS at 5 yrs (%) | 62 | 53 | 65 | 52 |
| Hazard ratio† | 0.76 | 0.68 |
||
| 2-sided 95% CI | (0.60, 0.96) | (0.52, 0.89) |
||
| Logrank Test stratified** | (p = 0.013) | (p = 0.007) |
||
| OS at 5 yrs (%) | 77 | 70 | 76 | 65 |
| Hazard ratio† | 0.71 | 0.69 |
||
| 2-sided 95% CI | (0.52, 0.98) | (0.51, 0.92) |
||
| Logrank Test stratified** | (p = 0.043) (unstratified p = 0.13) | (p = 0.007) |
||
| RFS at 10 yrs (%) | 51 | 44 | 49 | 43 |
| Hazard ratio† | 0.78 | 0.78 |
||
| 2-sided 95% CI | (0.63, 0.95) | (0.62, 0.99) |
||
| Logrank Test stratified** | (p = 0.017) (unstratified p = 0.023) | (p = 0.040) (unstratified p = 0.09) |
||
| OS at 10 yrs (%) | 61 | 57 | 56 | 50 |
| Hazard ratio† | 0.82 | 0.75 |
||
| 2-sided 95% CI | (0.65, 1.04) | (0.58, 0.96) |
||
| Logrank Test stratified** | (p = 0.100) (unstratified p = 0.18) | (p = 0.023) (unstratified p = 0.039) |
||
*Based on Kaplan-Meier estimates
**Patients in MA-5 were stratified by nodal status (1 to 3, 4 to 10, and >10 positive nodes), type of initial surgery (lumpectomy versus mastectomy), and by hormone receptor status (ER or PR positive (≥10 fmol), both negative (<10 fmol), or unknown status). Patients in GFEA-05 were stratified by nodal status (1 to 3, 4 to 10, and >10 positive nodes).
†Hazard ratio: CMF:CEF-120 in MA-5, FEC-50:FEC-100 in GFEA-05
The Kaplan-Meier curves for RFS and OS from Study MA-5 are shown in Figures 3 and 4 and those for Study GFEA-05 are shown in Figures 5 and 6.
Figure 3. Relapse-Free Survival in Study MA-5
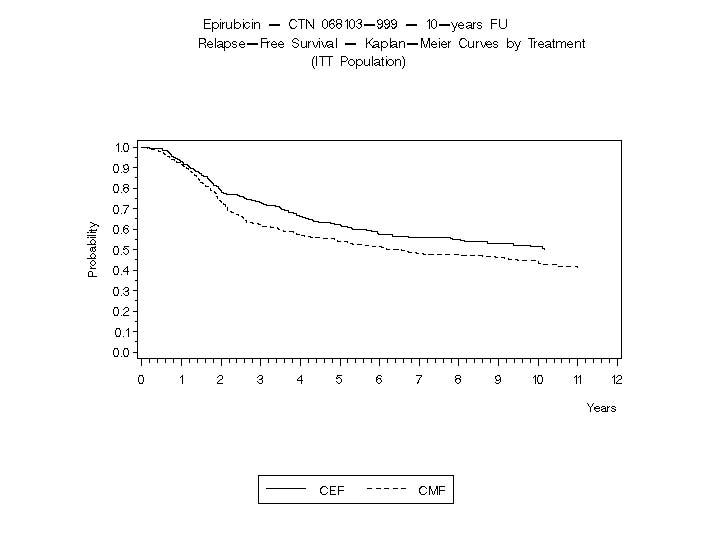
Figure 4. Overall Survival in Study MA-5
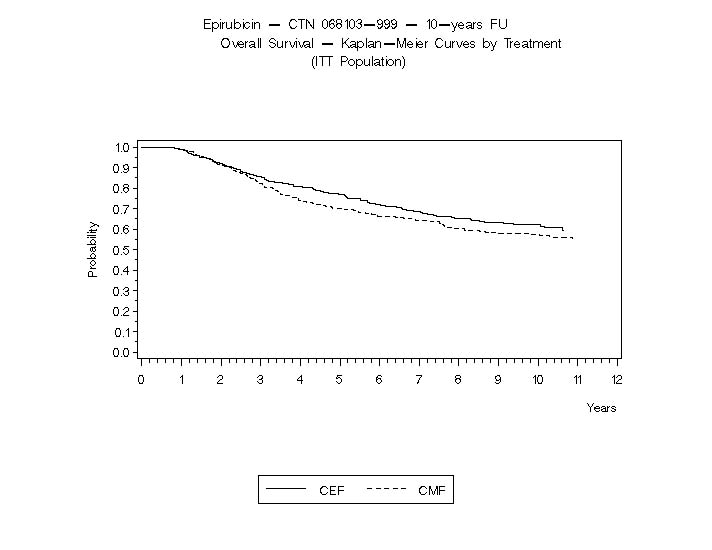
Figure 5. Relapse-Free Survival in Study GFEA-05
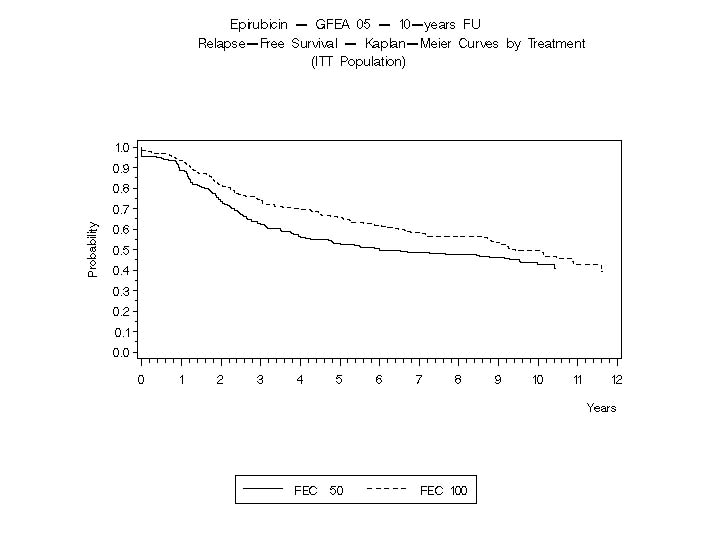
Figure 6. Overall Survival in Study GFEA-05
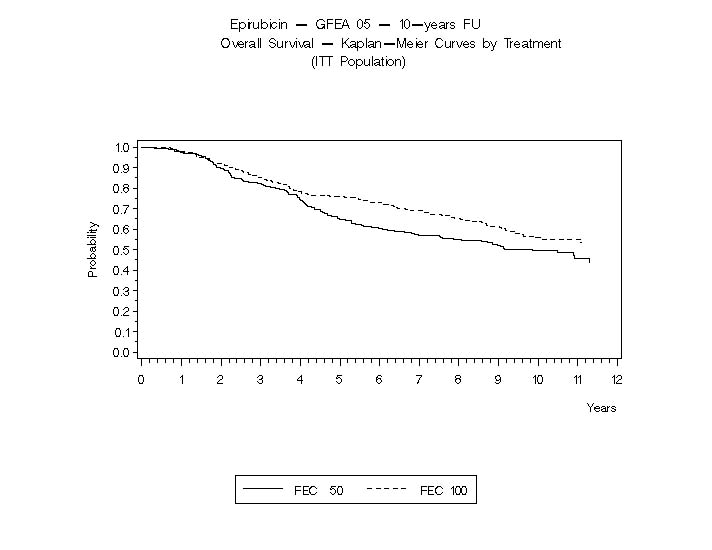
See Table 6 for statistics on 5 and 10 year analyses.
15. References
1. NIOSH Alert: Preventing occupational exposures to antineoplastic and other hazardous drugs in healthcare settings. 2004. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
2. OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html
3. American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am JHealth-Syst Pharm. 2006; 63:1172-1193.
4. Polovich, M., White, J. M., & Kelleher, L.O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd. ed.) Pittsburgh, PA: Oncology Nursing Society.
16. How is Epirubicin supplied
Epirubicin Hydrochloride Injection, USP is available in glass single-use vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use solution in the following strengths:
NDC 0143-9202-01; 50 mg/25 mL single use vial; individually boxed.
NDC 0143-9203-01; 200 mg/100 mL single use vial; individually boxed.
Store refrigerated between 2ºC and 8ºC (36ºF and 46ºF). Do not freeze. Protect from light.
Storage of the solution for injection at refrigerated conditions can result in the formation of a gelled product. This gelled product will return to a slightly viscous to mobile solution after 2 to a maximum of 4 hours equilibration at controlled room temperature (15 to 25ºC). Solution for injection should be used within 24 hours after removal from refrigeration.
Manufactured by:
THYMOORGAN PHARMAZIE GmbH,
Schiffgraben 23, 38690 Goslar, Germany
Distributed by:
Hikma Pharmaceuticals USA Inc.
Berkeley Heights, NJ 07922
Revised March 2020
127.207.013/01
17. Patient Counseling Information
Inform patients of the expected adverse effects of epirubicin hydrochloride, including gastrointestinal symptoms (nausea, vomiting, diarrhea, and stomatitis), alopecia and potential neutropenic complications.
Patients should understand that there is a risk of irreversible myocardial damage associated with treatment with epirubicin hydrochloride, as well as a risk of treatment-related leukemia.
Patients should consult their physician if vomiting, dehydration, fever, evidence of infection, symptoms of CHF, or injection-site pain occurs following therapy with epirubicin hydrochloride.
Advise patients that their urine may appear red for 1 to 2 days after administration of epirubicin hydrochloride and that they should not be alarmed.
Because epirubicin hydrochloride may induce chromosomal damage in sperm, advise men undergoing treatment with epirubicin hydrochloride to use effective contraceptive methods. Women treated with epirubicin hydrochloride may develop irreversible amenorrhea, or premature menopause.



| EPIRUBICIN HYDROCHLORIDE
epirubicin hydrochloride injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| EPIRUBICIN HYDROCHLORIDE
epirubicin hydrochloride injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Hikma Pharmaceuticals USA Inc. (001230762) |
Frequently asked questions
- How soon will my hair grow back after chemotherapy?
- How soon can you start chemo after port placement?
- Will I lose my hair during chemotherapy treatment?
More about epirubicin
- Check interactions
- Compare alternatives
- Side effects
- Dosage information
- During pregnancy
- Drug class: antibiotics/antineoplastics
- Breastfeeding
- En español