Nuedexta: Package Insert / Prescribing Info
Package insert / product label
Generic name: dextromethorphan hydrobromide and quinidine sulfate
Dosage form: capsule, gelatin coated
Drug class: Miscellaneous central nervous system agents
Medically reviewed by Drugs.com. Last updated on Dec 5, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Clinical Studies
- Drug Interactions
- Use In Specific Populations
- Drug Abuse and Dependence
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
NUEDEXTA (dextromethorphan hydrobromide and quinidine sulfate) capsules, for oral use
Initial U.S. Approval: 2010
Indications and Usage for Nuedexta
NUEDEXTA is a combination product containing dextromethorphan hydrobromide (an uncompetitive NMDA receptor antagonist and sigma-1 agonist) and quinidine sulfate (a CYP450 2D6 inhibitor) indicated for the treatment of pseudobulbar affect (PBA). (1)
Nuedexta Dosage and Administration
Dosage Forms and Strengths
Capsules: Dextromethorphan hydrobromide 20 mg/quinidine sulfate 10 mg. (3)
Contraindications
- Concomitant use with quinidine, quinine, or mefloquine. (4.1)
- Patients with a history of quinidine, quinine or mefloquine-induced thrombocytopenia, hepatitis, or other hypersensitivity reactions. (4.2)
- Patients with known hypersensitivity to dextromethorphan. (4.2)
- Use with an MAOI or within 14 days of stopping an MAOI. Allow 14 days after stopping NUEDEXTA before starting an MAOI. (4.3)
- Prolonged QT interval, congenital long QT syndrome, history suggestive of torsades de pointes, or heart failure. (4.4)
- Complete atrioventricular (AV) block without implanted pacemaker, or patients at high risk of complete AV block. (4.4)
- Concomitant use with drugs that both prolong QT interval and are metabolized by CYP2D6 (e.g., thioridazine or pimozide). (4.4)
Warnings and Precautions
- Thrombocytopenia or other hypersensitivity reactions: Discontinue if occurs. (5.1)
- Hepatitis: Discontinue if occurs. (5.2)
- QT Prolongation: Monitor ECG if concomitant use of drugs that prolong QT interval cannot be avoided or if concomitant CYP3A4 inhibitors used. (5.3)
- Left ventricular hypertrophy (LVH) or left ventricular dysfunction (LVD): Monitor ECG in patients with LVH or LVD. (5.3)
- CYP2D6 substrate: Nuedexta inhibits CYP2D6. Accumulation of parent drug and/or failure of metabolite formation may decrease safety and/or efficacy of concomitant CYP2D6 metabolized drugs. Adjust dose of CYP2D6 substrate or use alternative treatment when clinically indicated. (5.4, 12.4)
- Dizziness: Take precautions to reduce falls. (5.5)
- Serotonin syndrome: Use of NUEDEXTA with selective serotonin reuptake inhibitor (SSRIs) or tricyclic antidepressants increases the risk. Discontinue if occurs. (5.6, 7.4)
- Anticholinergic effects of quinidine: Monitor for worsening in myasthenia gravis and other sensitive conditions. (5.7)
Adverse Reactions/Side Effects
The most common adverse reactions (incidence of ≥3% and two-fold greater than placebo) in patients taking NUEDEXTA are diarrhea, dizziness, cough, vomiting, asthenia, peripheral edema, urinary tract infection, influenza, increased gamma-glutamyltransferase, and flatulence. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Otsuka America Pharmaceutical, Inc. at 1-800-438-9927 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Desipramine: Exposure increases 8-fold. Reduce desipramine dose and adjust based on clinical response. (7.5, 12.4)
- Paroxetine: Exposure increases 2-fold. Reduce paroxetine dose and adjust based on clinical response. (7.5, 12.4)
- Digoxin: Increased digoxin substrate plasma concentration may occur. (7.6)
Use In Specific Populations
- Pregnancy: Based on animal data, may cause fetal harm. (8.1)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2022
Full Prescribing Information
1. Indications and Usage for Nuedexta
NUEDEXTA is indicated for the treatment of pseudobulbar affect (PBA).
PBA occurs secondary to a variety of otherwise unrelated neurologic conditions, and is characterized by involuntary, sudden, and frequent episodes of laughing and/or crying. PBA episodes typically occur out of proportion or incongruent to the underlying emotional state. PBA is a specific condition, distinct from other types of emotional lability that may occur in patients with neurological disease or injury.
2. Nuedexta Dosage and Administration
2.1 Recommended Dose
The recommended starting dose of NUEDEXTA is one capsule daily by mouth for the initial seven days of therapy. On the eighth day of therapy and thereafter, the daily dose should be a total of two capsules a day, given as one capsule every 12 hours.
The need for continued treatment should be reassessed periodically, as spontaneous improvement of PBA occurs in some patients.
3. Dosage Forms and Strengths
NUEDEXTA capsules contain 20 mg dextromethorphan hydrobromide and 10 mg quinidine sulfate in a brick red gelatin capsule with "DMQ 20-10" printed in white ink on the capsule.
4. Contraindications
4.1 Quinidine and Related Drugs
NUEDEXTA contains quinidine and should not be used concomitantly with other drugs containing quinidine, quinine, or mefloquine.
4.2 Hypersensitivity
NUEDEXTA is contraindicated in patients with a history of NUEDEXTA, quinine, mefloquine or quinidine-induced thrombocytopenia, hepatitis, bone marrow depression or lupus-like syndrome. NUEDEXTA is also contraindicated in patients with a known hypersensitivity to dextromethorphan (e.g., rash, hives) [see Warnings and Precautions (5.1)].
4.3 MAOIs
NUEDEXTA is contraindicated in patients taking monoamine oxidase inhibitors (MAOIs) or in patients who have taken MAOIs within the preceding 14 days, due to the risk of serious and possibly fatal drug interactions, including serotonin syndrome. Allow at least 14 days after stopping NUEDEXTA before starting an MAOI [see Drug Interactions (7.1)].
4.4 Cardiovascular
NUEDEXTA is contraindicated in patients with a prolonged QT interval, congenital long QT syndrome or a history suggestive of torsades de pointes, and in patients with heart failure [see Warnings and Precautions (5.3)].
NUEDEXTA is contraindicated in patients receiving drugs that both prolong QT interval and are metabolized by CYP2D6 (e.g., thioridazine and pimozide), as effects on QT interval may be increased [see Drug Interactions (7.2)].
NUEDEXTA is contraindicated in patients with complete atrioventricular (AV) block without implanted pacemakers, or in patients who are at high risk of complete AV block.
5. Warnings and Precautions
5.1 Thrombocytopenia and Other Hypersensitivity Reactions
Quinidine can cause immune-mediated thrombocytopenia that can be severe or fatal. Non-specific symptoms, such as lightheadedness, chills, fever, nausea, and vomiting, can precede or occur with thrombocytopenia. NUEDEXTA should be discontinued immediately if thrombocytopenia occurs, unless the thrombocytopenia is clearly not drug-related, as continued use increases the risk for fatal hemorrhage. Likewise, NUEDEXTA should not be restarted in sensitized patients, because more rapid and more severe thrombocytopenia than the original episode can occur. NUEDEXTA should not be used if immune-mediated thrombocytopenia from structurally related drugs, including quinine and mefloquine is suspected, as cross-sensitivity can occur. Quinidine-associated thrombocytopenia usually, but not always, resolves within a few days of discontinuation of the sensitizing drug.
Quinidine has also been associated with a lupus-like syndrome involving polyarthritis, sometimes with a positive antinuclear antibody test. Other associations include rash, bronchospasm, lymphadenopathy, hemolytic anemia, vasculitis, uveitis, angioedema, agranulocytosis, the sicca syndrome, myalgia, elevation in serum levels of skeletal-muscle enzymes, and pneumonitis.
5.2 Hepatotoxicity
Hepatitis, including granulomatous hepatitis, has been reported in patients receiving quinidine, generally during the first few weeks of therapy. Fever may be a presenting symptom, and thrombocytopenia or other signs of hypersensitivity may also occur. Most cases remit when quinidine is withdrawn.
5.3 Cardiac Effects
NUEDEXTA causes dose-dependent QTc prolongation [see Clinical Pharmacology (12.2)]. QT prolongation can cause torsades de pointes-type ventricular tachycardia, with the risk increasing as the degree of prolongation increases. When initiating NUEDEXTA in patients at risk of QT prolongation and torsades de pointes, electrocardiographic (ECG) evaluation of QT interval should be conducted at baseline and 3 to 4 hours after the first dose. This includes patients concomitantly taking/initiating drugs that prolong the QT interval or that are strong or moderate CYP3A4 inhibitors, and patients with left ventricular hypertrophy (LVH) or left ventricular dysfunction (LVD). LVH and LVD are more likely to be present in patients with chronic hypertension, known coronary artery disease, or history of stroke. LVH and LVD can be diagnosed utilizing echocardiography or another suitable cardiac imaging modality.
Strong and moderate CYP3A inhibitors include, but are not limited to, atazanavir, clarithromycin, indinavir, itraconazole, ketoconazole, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, amprenavir, aprepitant, diltiazem, erythromycin, fluconazole, fosamprenavir, grapefruit juice, and verapamil.
Reevaluate ECG if risk factors for arrhythmia change during the course of treatment with NUEDEXTA. Risk factors include concomitant use of drugs associated with QT prolongation, electrolyte abnormality (hypokalemia, hypomagnesemia), bradycardia, and family history of QT abnormality. Hypokalemia and hypomagnesemia should be corrected prior to initiation of therapy with NUEDEXTA, and should be monitored during treatment.
If patients taking NUEDEXTA experience symptoms that could indicate the occurrence of cardiac arrhythmias, e.g., syncope or palpitations, NUEDEXTA should be discontinued and the patient further evaluated.
5.4 Concomitant use of CYP2D6 Substrates
The quinidine in NUEDEXTA inhibits CYP2D6 in patients in whom CYP2D6 is not otherwise genetically absent or its activity otherwise pharmacologically inhibited [see Warnings and Precautions (5.8) and Clinical Pharmacology (12.3, 12.5)]. Because of this effect on CYP2D6, accumulation of parent drug and/or failure of active metabolite formation may decrease the safety and/or the efficacy of drugs used concomitantly with NUEDEXTA that are metabolized by CYP2D6 [see Drug Interactions (7.5)].
5.5 Dizziness
NUEDEXTA may cause dizziness [see Adverse Reactions (6.1)]. Precautions to reduce the risk of falls should be taken, particularly for patients with motor impairment affecting gait or a history of falls. In a controlled trial of NUEDEXTA, 10% of patients on NUEDEXTA and 5% on placebo experienced dizziness.
5.6 Serotonin Syndrome
When used with SSRIs (such as fluoxetine) or tricyclic antidepressants (such as clomipramine and imipramine), NUEDEXTA may cause "serotonin syndrome", with changes including altered mental status, hypertension, restlessness, myoclonus, hyperthermia, hyperreflexia, diaphoresis, shivering, and tremor [see Drug Interactions (7.4), Overdosage (10)].
5.7 Anticholinergic Effects of Quinidine
Monitor for worsening clinical condition in myasthenia gravis and other conditions that may be adversely affected by anticholinergic effects.
5.8 CYP2D6 Poor Metabolizers
The quinidine component of NUEDEXTA is intended to inhibit CYP2D6 so that higher exposure to dextromethorphan can be achieved compared to when dextromethorphan is given alone [see Warnings and Precautions (5.4) and Clinical Pharmacology (12.3, 12.5)]. Approximately 7 to 10% of Caucasians and 3 to 8% of African Americans lack the capacity to metabolize CYP2D6 substrates and are classified as poor metabolizers (PMs). The quinidine component of NUEDEXTA is not expected to contribute to the effectiveness of NUEDEXTA in PMs, but adverse events of the quinidine are still possible. In those patients who may be at risk of significant toxicity due to quinidine, genotyping to determine if they are PMs should be considered prior to making the decision to treat with NUEDEXTA.
6. Adverse Reactions/Side Effects
A total of 946 patients participated in four Phase 3 controlled and uncontrolled PBA studies and received at least one dose of the combination product of dextromethorphan/quinidine in various strengths at the recommended or higher than the recommended dose. Of those patients, 393 patients were exposed for at least 180 days and 294 patients were exposed for at least one year. Median exposure was 168 days.
Controlled trials enrolled only patients with either ALS or MS. Uncontrolled studies enrolled 136 patients with PBA secondary to a wide variety of underlying neurological conditions including stroke (45 patients) and traumatic brain injury (23 patients). Consequently, patients with other underlying neurologic diseases may experience other adverse reactions not described below.
6.1 Clinical Trials Experience
A 12-week, placebo-controlled study evaluated NUEDEXTA (dextromethorphan 20 mg/quinidine 10 mg) (N=107) and a 30 mg dextromethorphan/10 mg quinidine combination (N=110) compared to placebo (N=109). Approximately 60% of patients had ALS and 40% had MS. Patients were 25 to 80 years of age, with a mean age of approximately 51 years. Three (3) ALS patients in each drug treatment arm and 1 ALS patient in the placebo arm died during the 12-week placebo-control period. All deaths were consistent with the natural progression of ALS.
Adverse Reactions Leading to Discontinuation
The most commonly reported adverse reactions (incidence ≥2% and greater than placebo) that led to discontinuation with the 20 mg dextromethorphan/10 mg quinidine twice daily dose were muscle spasticity (3%), respiratory failure (1%), abdominal pain (2%), asthenia (2%), dizziness (2%), fall (1%), and muscle spasms (2%).
Most Common Adverse Reactions
Adverse drug reactions that occurred in ≥3% of patients receiving the 20 mg dextromethorphan/10 mg quinidine twice daily dose, and at an incidence of ≥2 times placebo in short-term clinical trials in ALS and MS are provided in Table 1. Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to the rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
| NUEDEXTA N=107 % | Placebo N=109 % |
|
|---|---|---|
| Diarrhea | 13 | 6 |
| Dizziness | 10 | 5 |
| Cough | 5 | 2 |
| Vomiting | 5 | 1 |
| Asthenia | 5 | 2 |
| Peripheral edema | 5 | 1 |
| Urinary tract infection | 4 | 1 |
| Influenza | 4 | 1 |
| Increased gamma-glutamyltransferase | 3 | 0 |
| Flatulence | 3 | 1 |
6.2 Long-Term Exposure with NUEDEXTA
The experience in open-label clinical trials is consistent with the safety profile observed in the placebo-controlled clinical trials.
6.3 Safety Experience of Individual Components
The following adverse reactions have been reported with the use of the individual components of NUEDEXTA, dextromethorphan and quinidine, from post-marketing experience. Because these reactions are reported voluntarily from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Dextromethorphan
Drowsiness, dizziness, nervousness or restlessness, nausea, vomiting, and stomach pain.
Quinidine
Cinchonism is most often a sign of chronic quinidine toxicity, but it may appear in sensitive patients after a single moderate dose of several hundred milligrams. Cinchonism is characterized by nausea, vomiting, diarrhea, headache tinnitus, hearing loss, vertigo, blurred vision, diplopia, photophobia, confusion, and delirium.
Convulsions, apprehension, and ataxia have been reported with quinidine therapy, but it is not clear that these were not simply the results of hypotension and consequent cerebral hypoperfusion in patients being treated for cardiovascular indications. Acute psychotic reactions have been reported to follow the first dose of quinidine, but these reactions appear to be extremely rare. Other adverse reactions occasionally reported with quinidine therapy include depression, mydriasis, disturbed color perception, night blindness, scotomata, optic neuritis, visual field loss, photosensitivity, keratopathy, and abnormalities of skin pigmentation.
Related/similar drugs
7. Drug Interactions
7.1 MAOIs
Do not use NUEDEXTA with monoamine oxidase inhibitors (MAOIs) or in patients who have taken MAOIs within the preceding 14 days [see Contraindications (4.3)].
7.2 Drugs that Prolong QT and are Metabolized by CYP2D6
Do not use with drugs that both prolong QT interval and are metabolized by CYP2D6 (e.g., thioridazine or pimozide) [see Contraindications (4.4)].
7.3 Drugs that Prolong QT and Concomitant CYP3A4 Inhibitors
Recommend ECG in patients taking drugs with NUEDEXTA that prolong the QT interval and in patients taking concomitant moderate or strong CYP3A4 inhibitors [see Warnings and Precautions (5.3)].
7.4 SSRIs and Tricyclic Antidepressants
Use of NUEDEXTA with SSRIs or tricyclic antidepressants increases the risk of 'serotonin syndrome' [see Warnings and Precautions (5.6)].
7.5 CYP2D6 Substrate
The co-administration of NUEDEXTA with drugs that undergo extensive CYP2D6 metabolism may result in altered drug effects, due to accumulation of parent drug and/or failure of metabolite formation [see Warnings and Precautions (5.4)]. Therapy with medications that are primarily metabolized by CYP2D6 and that have a relatively narrow therapeutic index should be initiated at a low dose if a patient is receiving NUEDEXTA concurrently. If NUEDEXTA is added to the treatment regimen of a patient already receiving a drug primarily metabolized by CYP2D6, the need for a dose modification of the original medication should be considered. The extent to which CYP2D6 interactions may pose clinical problems will depend on the pharmacokinetics of the substrate involved.
In cases of prodrugs whose actions are mediated by the CYP2D6-produced metabolites (for example, codeine and hydrocodone, whose analgesic and antitussive effects appear to be mediated by morphine and hydromorphone, respectively), it may not be possible to achieve the desired clinical benefits in the presence of NUEDEXTA due to quinidine-mediated inhibition of CYP2D6. Consider use of alternative treatment with NUEDEXTA when clinically indicated.
Drug interactions with desipramine and paroxetine have been studied in controlled clinical trials with a higher dose combination of dextromethorphan/quinidine (dextromethorphan 30 mg/quinidine 30 mg) than NUEDEXTA; study results are described below. No other drug interactions with CYP2D6 substrates have been systematically investigated, although concomitant use of such drugs was allowed in clinical trials with NUEDEXTA and in clinical trials with higher dose formulations of dextromethorphan/quinidine.
Desipramine (CYP2D6 substrate):
The tricyclic antidepressant desipramine is metabolized primarily by CYP2D6. A drug interaction study was conducted between a higher combination dose of dextromethorphan (dextromethorphan 30 mg/quinidine 30 mg) and desipramine 25 mg. The combination dose of dextromethorphan/quinidine increased steady state desipramine levels approximately 8-fold. If NUEDEXTA and desipramine are prescribed concomitantly, the initial dose of desipramine should be markedly reduced. The dose of desipramine can then be adjusted based on clinical response; however, a dose above 40 mg/day is not recommended.
Paroxetine (CYP2D6 inhibitor and substrate):
When the combination dose of dextromethorphan 30 mg/quinidine 30 mg was added to paroxetine at steady state, paroxetine exposure (AUC0-24) increased by 1.7 fold and Cmax increased by 1.5 fold. Consideration should be given to initiating treatment with a lower dose of paroxetine if given with NUEDEXTA. The dose of paroxetine can then be adjusted based on clinical response; however, dosage above 35 mg/day is not recommended.
7.6 Digoxin
Quinidine is an inhibitor of P-glycoprotein. Concomitant administration of quinidine with digoxin, a P-glycoprotein substrate, results in serum digoxin levels that may be as much as doubled. Plasma digoxin concentrations should be closely monitored in patients taking NUEDEXTA concomitantly, and the digoxin dose reduced, as necessary.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of NUEDEXTA in pregnant women. In oral studies conducted in rats and rabbits, a combination of dextromethorphan/quinidine demonstrated developmental toxicity, including teratogenicity (rabbits) and embryo lethality, when given to pregnant animals (see Data). In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
When dextromethorphan/quinidine was administered orally (0/0, 5/100, 15/100, and 50/100 mg/kg/day) to pregnant rats during the period of organogenesis, embryo-fetal deaths were observed at the highest dose tested and reduced skeletal ossification was observed at all doses. The lowest dose tested (5/100 mg/kg/day) is approximately 1/50 times the recommended human dose (RHD) of 40/20 mg/day on a mg/m2 basis. Oral administration to pregnant rabbits during organogenesis in two separate studies (0/0, 5/60, 15/60, and 30/60 mg/kg/day; 0/0, 5/100, 15/100, and 50/100 mg/kg/day) resulted in an increased incidence of fetal malformations at all but the lowest dose tested. The no-effect dose (5/100 mg/kg/day) is approximately 2/100 times the RHD on a mg/m2 basis.
When dextromethorphan/quinidine was orally administered to female rats during pregnancy and lactation in two separate studies (0/0, 5/100, 15/100, and 30/100 mg/kg/day; 0/0, 5/100, 15/100, and 50/100 mg/kg/day), pup survival and pup weight were decreased at all doses, and developmental delay was observed in offspring at the mid and high doses. A no-effect dose for adverse developmental effects was not identified. The lowest dose tested (5/100 mg/kg/day) is approximately 1/50 times the RHD on a mg/m2 basis.
When dextromethorphan/quinidine was orally administered (0/0, 5/50, 15/50, 25/50 mg/kg) to male and female rats on Postnatal Day 7, the highest dose resulted in neuronal death in brain (thalamus and medulla oblongata). Postnatal Day 7 in rat corresponds to the third trimester of the gestation through the first several months of life but may extend to approximately three years of age in humans.
8.2 Lactation
Risk Summary
Quinidine is excreted in human milk. It is not known whether dextromethorphan is excreted in human milk. There are no data on the effects of quinidine or dextromethorphan on the breastfed infant or the effects on milk production. The development and health benefits of breastfeeding should be considered along with the mother's clinical need for NUEDEXTA and any potential adverse effects on the breastfed infant from NUEDEXTA or from the underlying material condition.
8.4 Pediatric Use
The safety and effectiveness in pediatric patients below the age of 18 have not been established.
8.5 Geriatric Use
Of the total number of patients with PBA in clinical studies of NUEDEXTA, 14% were 65 years old and over, while 2% were 75 and over. Clinical studies of NUEDEXTA did not include sufficient number of patients aged 65 and over to determine whether they respond differently than younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Dose adjustment of NUEDEXTA is not required in patients with mild to moderate renal impairment [see Clinical Pharmacology (12.3)]. The pharmacokinetics of NUEDEXTA have not been evaluated in patients with severe renal impairment; however, increases in dextromethorphan and/or quinidine levels are likely to be observed.
8.7 Hepatic Impairment
Dose adjustment of NUEDEXTA is not required in patients with mild to moderate hepatic impairment. The pharmacokinetics of NUEDEXTA have not been evaluated in patients with severe hepatic impairment; however, increases in dextromethorphan and/or quinidine levels are likely to be observed.
9. Drug Abuse and Dependence
NUEDEXTA is a low-affinity uncompetitive NMDA antagonist and sigma-1 receptor agonist that has not been systematically studied in animals or humans for its potential for abuse, tolerance, or physical dependence. However, NUEDEXTA is a combination product containing dextromethorphan and quinidine, and cases of dextromethorphan abuse have been reported, predominantly in adolescents.
While clinical trials did not reveal drug-seeking behavior, these observations were not systematic and it is not possible to predict on the basis of this experience the extent to which NUEDEXTA will be misused, diverted, and/or abused once marketed. Therefore, patients with a history of drug abuse should be observed closely for signs of NUEDEXTA misuse or abuse (e.g., development of tolerance, increases in dose, drug-seeking behavior).
10. Overdosage
Evaluation and treatment of NUEDEXTA overdose is based on experience with the individual components, dextromethorphan and quinidine. Metabolism of the dextromethorphan component of NUEDEXTA is inhibited by the quinidine component, such that adverse effects of overdose due to NUEDEXTA might be more severe or more persistent compared to overdose of dextromethorphan alone.
During development of NUEDEXTA, dose combinations of dextromethorphan/quinidine containing up to 6-times higher dextromethorphan dose and 12-times higher quinidine dose were studied. The most common adverse events were mild to moderate nausea, dizziness, and headache.
The most important adverse effects of acute quinidine overdose are ventricular arrhythmias and hypotension. Other signs and symptoms of overdose may include vomiting, diarrhea, tinnitus, high-frequency hearing loss, vertigo, blurred vision, diplopia, photophobia, headache, confusion, and delirium.
While therapeutic doses of quinidine for treatment of cardiac arrhythmia or malaria are generally 10-fold, or more, higher than the dose of quinidine in NUEDEXTA, potentially fatal cardiac arrhythmia, including torsades de pointes, can occur at quinidine exposures that are possible from NUEDEXTA overdose.
Adverse effects of dextromethorphan overdose include nausea, vomiting, stupor, coma, respiratory depression, seizures, tachycardia, hyperexcitability, and toxic psychosis. Other adverse effects include ataxia, nystagmus, dystonia, blurred vision, and changes in muscle reflexes. Dextromethorphan may cause serotonin syndrome, and this risk is increased by overdose, particularly if taken with other serotonergic agents, SSRIs or tricyclic antidepressants.
10.1 Treatment of Overdose
While serum quinidine levels can be measured, electrocardiographic monitoring of the QTc interval is a better predictor of quinidine-induced arrhythmia. Treatment of hemodynamically unstable polymorphic ventricular tachycardia (including torsades de pointes) is either immediate cardioversion or, if a cardiac pacemaker is in place or immediately available, immediate overdrive pacing. After pacing or cardioversion, further management must be guided by the length of the QTc interval. Factors contributing to QTc prolongation (especially hypokalemia and hypomagnesemia) should be sought out and (if possible) aggressively corrected. Prevention of recurrent torsades de pointes may require sustained overdrive pacing or the cautious administration of isoproterenol (30 to 150 ng/kg/min).
Because of the theoretical possibility of QT-prolonging effects that might be additive to those of quinidine, other antiarrhythmics with Class I (procainamide) or Class III activities should (if possible) be avoided.
If the post-cardioversion QTc interval is prolonged, then the pre-cardioversion polymorphic ventricular tachyarrhythmia was (by definition) torsades de pointes. In this case, class Ib antiarrhythmics like lidocaine are unlikely to be of value, and other Class I and Class III antiarrhythmics are likely to exacerbate the situation.
Quinidine-induced hypotension that is not due to an arrhythmia is likely to be a consequence of quinidine-related α-blockade and vasorelaxation. Treatment of hypotension should be directed at symptomatic and supportive measures. Repletion of central volume (Trendelenburg positioning, saline infusion) may be sufficient therapy; other interventions reported to have been beneficial in this setting are those that increase peripheral vascular resistance, including α-agonist catecholamines (norepinephrine).
Quinidine:
Adequate studies of orally administered activated charcoal in human overdoses of quinidine have not been reported, but there are animal data showing significant enhancement of systemic elimination following this intervention, and there is at least one human case report in which the elimination half-life of quinidine in the serum was apparently shortened by repeated gastric lavage. Activated charcoal should be avoided if an ileus is present; the conventional dose is 1 g/kg, administered every 2 to 6 hours as a slurry with 8 mL/kg of tap water. Although renal elimination of quinidine might theoretically be accelerated by maneuvers to acidify the urine, such maneuvers are potentially hazardous and of no demonstrated benefit. Quinidine is not usefully removed from the circulation by dialysis. Following quinidine overdose, drugs that delay elimination of quinidine (cimetidine, carbonic anhydrase inhibitors, thiazide diuretics) should be withdrawn unless absolutely required.
Dextromethorphan:
Treatment of dextromethorphan overdosage should be directed at symptomatic and supportive measures.
11. Nuedexta Description
NUEDEXTA is an oral formulation of dextromethorphan hydrobromide USP and quinidine sulfate USP in a fixed dose combination.
Dextromethorphan hydrobromide is the pharmacologically active ingredient of NUEDEXTA that acts on the central nervous system (CNS). The chemical name is dextromethorphan hydrobromide: morphinan, 3-methoxy-17-methyl-, (9α, 13α, 14α), hydrobromide monohydrate. Dextromethorphan hydrobromide has the empirical formula C18H25NO•HBr•H2O with a molecular weight of 370.33. The structural formula is:

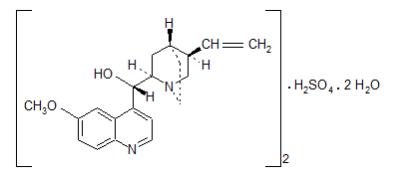
Quinidine sulfate is a specific inhibitor of CYP2D6-dependent oxidative metabolism used in NUEDEXTA to increase the systemic bioavailability of dextromethorphan. The chemical name is quinidine sulfate: cinchonan-9-o1, 6'-methoxy-, (9S) sulfate (2:1), (salt), dihydrate. Quinidine sulfate dihydrate has the empirical formula of (C20H24N2O2)2•H2SO4•2H2O with a molecular weight of 782.96. The structural formula is:
The combination product, NUEDEXTA, is a white to off-white powder. NUEDEXTA is available for oral use as NUEDEXTA which contains 20 mg dextromethorphan hydrobromide and 10 mg quinidine sulfate. The active ingredients are dextromethorphan hydrobromide monohydrate USP and quinidine sulfate dihydrate USP. Inactive ingredients in the capsule are croscarmellose sodium NF, microcrystalline cellulose NF, colloidal silicon dioxide NF, lactose monohydrate NF, and magnesium stearate NF.
12. Nuedexta - Clinical Pharmacology
12.1 Mechanism of Action
Dextromethorphan (DM) is a sigma-1 receptor agonist and an uncompetitive NMDA receptor antagonist. Quinidine increases plasma levels of dextromethorphan by competitively inhibiting cytochrome P450 2D6, which catalyzes a major biotransformation pathway for dextromethorphan. The mechanism by which dextromethorphan exerts therapeutic effects in patients with pseudobulbar affect is unknown.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of dextromethorphan 30 mg/quinidine 10 mg (for 7 doses) on QTc prolongation was evaluated in a randomized, double-blind (except for moxifloxacin), placebo- and positive-controlled (400 mg moxifloxacin) crossover thorough QT study in 50 fasted normal healthy men and women with CYP2D6 extensive metabolizer (EM) genotype. Mean changes in QTcF were 6.8 ms for dextromethorphan 30 mg/quinidine 10 mg and 9.1 ms for the reference positive control (moxifloxacin). The maximum mean (95% upper confidence bound) difference from placebo after baseline correction was 10.2 (12.6) ms. This test dose is adequate to represent the steady state exposure in patients with CYP2D6 extensive metabolizer phenotype.
The effects of supratherapeutic doses of dextromethorphan/quinidine (30 mg/30 mg and 60 mg/60 mg, for 7 doses) on QTc prolongation was evaluated in a randomized, placebo-controlled, double-blind, crossover design with an additional open-label positive control (400 mg moxifloxacin) arm in 36 healthy volunteers. The maximum mean (95% upper confidence bound) differences from placebo after baseline-correction were 10.2 (14.6) and 18.4 (22.7) ms following dextromethorphan/quinidine doses of 30 mg/30 mg and 60/60 mg, respectively. The supratherapeutic doses are adequate to represent exposure increases due to drug-drug interactions and organ dysfunctions.
12.3 Pharmacokinetics
NUEDEXTA contains dextromethorphan and quinidine, both of which are metabolized primarily by liver enzymes. Quinidine's primary pharmacological action in NUEDEXTA is to competitively inhibit the metabolism of dextromethorphan catalyzed by CYP2D6 in order to increase and prolong plasma concentrations of dextromethorphan [see Warnings and Precautions (5.4, 5.8), and Clinical Pharmacology (12.5)]. Studies were conducted with the individual components of NUEDEXTA in healthy subjects to determine single-dose and multiple-dose kinetics of orally administered dextromethorphan in combination with quinidine. The increase in dextromethorphan levels appeared approximately dose proportional when the dextromethorphan dose was increased from 20 mg to 30 mg in the presence of 10 mg of quinidine.
Absorption
Following single and repeated combination doses of dextromethorphan 30 mg/quinidine 10 mg, dextromethorphan/quinidine-treated subjects had an approximately 20-fold increase in dextromethorphan exposure compared to dextromethorphan given without quinidine.
Following repeated doses of dextromethorphan 30 mg/quinidine 10 mg and dextromethorphan 20 mg/quinidine 10 mg (NUEDEXTA), maximal plasma concentrations (Cmax) of dextromethorphan are reached approximately 3 to 4 hours after dosing and maximal plasma concentrations of quinidine are reached approximately 1 to 2 hours after dosing.
In extensive metabolizers, mean Cmax and AUC0-12 values of dextromethorphan and dextrorphan increased as doses of dextromethorphan increased from 20 to 30 mg; mean Cmax and AUC0-12 values of quinidine appeared similar.
The mean plasma Cmax of quinidine following twice daily co-administration of dextromethorphan 30 mg/quinidine 10 mg in patients with PBA was within 1 to 3% of the concentrations required for antiarrhythmic efficacy (2 to 5 mcg/mL).
NUEDEXTA may be taken without regard to meals as food does not affect the exposure of dextromethorphan and quinidine significantly.
Distribution
After NUEDEXTA administration, protein binding remains essentially the same as that after administration of the individual components; dextromethorphan is approximately 60 to 70% protein bound and quinidine is approximately 80 to 89% protein bound.
Metabolism and Excretion
NUEDEXTA is a combination product containing dextromethorphan and quinidine. Dextromethorphan is metabolized by CYP2D6 and quinidine is metabolized by CYP3A4. After dextromethorphan 30 mg/quinidine 30 mg administration in extensive metabolizers, the elimination half-life of dextromethorphan was approximately 13 hours and the elimination half-life of quinidine was approximately 7 hours.
There are several hydroxylated metabolites of quinidine. The major metabolite of quinidine is 3-hydroxyquinidine. The 3-hydroxymetabolite is considered to be at least half as pharmacologically active as quinidine with respect to cardiac effects such as QT prolongation.
When the urine pH is less than 7, about 20% of administered quinidine appears unchanged in the urine, but this fraction drops to as little as 5% when the urine is more alkaline. Renal clearance involves both glomerular filtration and active tubular secretion, moderated by (pH-dependent) tubular reabsorption.
Specific Populations
Geriatric Use
The pharmacokinetics of dextromethorphan/quinidine have not been investigated systematically in elderly subjects (aged >65 years), although such subjects were included in the clinical program. A population pharmacokinetic analysis of 170 subjects (148 subjects <65 years old and 22 subjects ≥65 years old) administered dextromethorphan 30 mg/quinidine 30 mg revealed similar pharmacokinetics in subjects <65 years and those ≥65 years of age.
Pediatric Use
The pharmacokinetics of NUEDEXTA in pediatric patients have not been studied.
Gender
A population pharmacokinetic analysis based on data from 109 subjects (75 male; 34 female) showed no apparent gender differences in the pharmacokinetics of NUEDEXTA.
Race
A population pharmacokinetic analysis of race with 109 subjects (20 Caucasian; 71 Hispanic; 18 Black) revealed no apparent racial differences in the pharmacokinetics of NUEDEXTA.
Renal Impairment
In a study of a combination dose of dextromethorphan 30 mg/quinidine 30 mg TWICE DAILY in 12 subjects with mild (CLCR 50 to 80 mL/min) or moderate (CLCR 30 to 50 mL/min) renal impairment (6 each) compared to 9 healthy subjects (matched in gender, age, and weight range to impaired subjects), subjects showed little difference in quinidine or dextromethorphan pharmacokinetics compared to healthy subjects. Dose adjustment is, therefore, not required in mild or moderate renal impairment. NUEDEXTA has not been studied in patients with severe renal impairment.
Hepatic Impairment
In a study of a combination dose of dextromethorphan 30 mg/quinidine 30 mg TWICE DAILY in 12 subjects with mild or moderate hepatic impairment (as indicated by the Child-Pugh method; 6 each) compared to 9 healthy subjects (matched in gender, age, and weight range to impaired subjects), subjects with moderate hepatic impairment showed similar dextromethorphan AUC and Cmax and clearance compared to healthy subjects. Mild to moderate hepatic impairment had little effect on quinidine pharmacokinetics. Patients with moderate impairment showed an increased frequency of adverse events. Therefore, dosage adjustment is not required in patients with mild and moderate hepatic impairment, although additional monitoring for adverse reactions should be considered. Quinidine clearance is unaffected by hepatic cirrhosis, although there is an increased volume of distribution that leads to an increase in the elimination half-life. Neither dextromethorphan alone nor NUEDEXTA has been evaluated in patients with severe hepatic impairment.
12.4 Drug-Drug Interactions
The potential for dextromethorphan and quinidine to inhibit or induce cytochrome P450 in vitro were evaluated in human microsomes. Dextromethorphan did not inhibit (<20% inhibition) any of the tested isoenzymes: CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 in human liver microsomes at concentrations up to 5 microM. Quinidine did not inhibit (<30% inhibition) CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2E1, or CYP3A4 in human microsomes at concentrations up to 5 microM. Quinidine inhibited CYP2D6 with a half maximal inhibitory concentration (IC50) of less than 0.05 microM. Neither dextromethorphan nor quinidine induced CYP1A2, CYP2B6 or CYP3A4 in human hepatocytes at concentrations up to 4.8 microM.
Desipramine (CYP2D6 substrate)
Co-administration of dextromethorphan 30 mg/quinidine 30 mg with the tricyclic antidepressant desipramine, a CYP2D6 substrate, when desipramine was given at a dose of 25 mg once daily in 13 healthy volunteers resulted in an approximately 8-fold increase in steady state desipramine exposure (Cmin) compared to desipramine given alone. Therefore, concomitant administration of NUEDEXTA and drugs undergoing CYP2D6 metabolism should be evaluated for appropriate dose adjustment or alternative medication if the concomitant medication depends primarily on CYP2D6 metabolism and has a narrow therapeutic index, or if it relies on CYP2D6 for conversion to an active species [see Warnings and Precautions (5.4)].
Paroxetine (CYP2D6 inhibitor and substrate)
Co-administration of the selective serotonin reuptake inhibitor paroxetine and a higher combination dose of dextromethorphan/quinidine (dextromethorphan 30 mg/quinidine 30 mg) was studied in 27 healthy volunteers. Group 1 (N = 14) received paroxetine 20 mg once daily for 12 days followed by the addition of dextromethorphan 30 mg/quinidine 30 mg twice daily for 8 days. Group 2 (N = 13) received dextromethorphan 30 mg/quinidine 30 mg twice daily for 8 days followed by the addition of paroxetine 20 mg once daily for 12 days. Dextromethorphan exposure (AUC0-12) and Cmax increased by 1.5 fold and 1.4 fold, respectively, and quinidine exposure (AUC0-12) and Cmax increased by 1.4 fold and 1.3 fold, respectively, and dextrorphan exposure (AUC0-12) and Cmax decreased by 14% and 18%, respectively, and paroxetine exposure (AUC0-24) and Cmax increased by 2.3 fold and 2.0 fold, respectively, when paroxetine was added to the combination dose of dextromethorphan/quinidine at steady state (Group 2).
When the combination dose of dextromethorphan/quinidine was added to paroxetine at steady state (Group 1), paroxetine exposure (AUC0-24) and Cmax increased by 1.7 fold and 1.5 fold, respectively, while dextromethorphan and quinidine exposure did not change significantly and dextrorphan exposure (AUC0-12) and Cmax decreased by 34% and 33%, respectively.
Based on these results, when NUEDEXTA is prescribed with drugs such as paroxetine that inhibit or are extensively metabolized by CYP2D6, consideration should be given to initiating treatment with a lower dose. The dose of paroxetine can then be adjusted based on clinical response; however, dosage above 35 mg/day is not recommended [see Warnings and Precautions (5.4)].
NMDA receptor antagonists (memantine)
A drug interaction study was conducted between a higher combination dose of dextromethorphan/quinidine (dextromethorphan 30 mg/quinidine 30 mg) and memantine 20 mg/day to investigate the pharmacokinetic and pharmacodynamic interactions in 52 healthy subjects. Both dextromethorphan and memantine are antagonists of the N-methyl-D-aspartate (NMDA) receptor, which could theoretically result in an additive effect at NMDA receptors and potentially an increased incidence of adverse events. There was no significant difference in the plasma concentrations of dextromethorphan and dextrorphan before and after the administration of memantine. Plasma concentrations of quinidine increased 20 to 30% when memantine was added to dextromethorphan 30 mg/quinidine 30 mg.
12.5 Pharmacogenomics
The quinidine component of NUEDEXTA is intended to inhibit CYP2D6 so that higher exposure to dextromethorphan can be achieved compared to when dextromethorphan is given alone. Approximately 7 to 10% of Caucasians and 3 to 8% of African Americans generally lack the capacity to metabolize CYP2D6 substrates and are classified as PMs. The quinidine component of NUEDEXTA is not expected to contribute to the effectiveness of NUEDEXTA in PMs, but adverse events of the quinidine are still possible. In those patients who may be at risk of significant toxicity due to quinidine, genotyping to determine if they are PMs should be considered prior to making the decision to treat with NUEDEXTA [see Warnings and Precautions (5.4, 5.8), and Clinical Pharmacology (12.3)].
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 26-week carcinogenicity study in the Tg.rasH2 transgenic mouse, dextromethorphan and quinidine, alone and in combination, at oral doses up to 100/100 mg/kg/day did not show any evidence of carcinogenic potential.
In a two-year carcinogenicity study in rats, dextromethorphan/quinidine were administered at oral doses of 0/0, 5/100, 20/100, 50/100, 50/0, 0/100 mg/kg/day. No biologically significant tumor findings were observed. The highest dose tested (50/100 mg/kg/day) is approximately 12/50 times the maximum recommended human dose (MRHD) of 40/20 mg/day on a mg/m2 basis.
Mutagenesis
Dextromethorphan/quinidine was negative in an in vitro chromosomal aberration assay in human lymphocytes.
Dextromethorphan was negative in in vitro (bacterial reverse mutation, chromosomal aberration in human lymphocytes) and in vivo (mouse micronucleus) assays.
Quinidine was negative in an in vitro bacterial reverse mutation assay and in an in vivo mouse micronucleus assay. Quinidine induced chromosomal aberrations in an in vitro chromosomal aberration assay in the presence of metabolic activation.
Impairment of fertility
When dextromethorphan/quinidine was administered orally (0/0, 5/100, 15/100, and 50/100 mg/kg/day) to male and female rats prior to and during mating, and continuing to Day 7 of gestation in females, no effect on fertility was observed up to the highest dose tested, which is approximately 12/50 times the MRHD on a mg/m2 basis.
14. Clinical Studies
The efficacy of NUEDEXTA was demonstrated in one trial in patients with pseudobulbar affect (PBA). These patients had underlying amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS). Other trials at higher doses (dextromethorphan 30 mg/quinidine 30 mg) provided supportive evidence.
In the NUEDEXTA trial, patients with PBA were randomized to receive NUEDEXTA dextromethorphan 20 mg/quinidine 10 mg, (N=107), dextromethorphan 30 mg/quinidine 10 mg (N=110), or placebo (N=109) for 12 weeks.
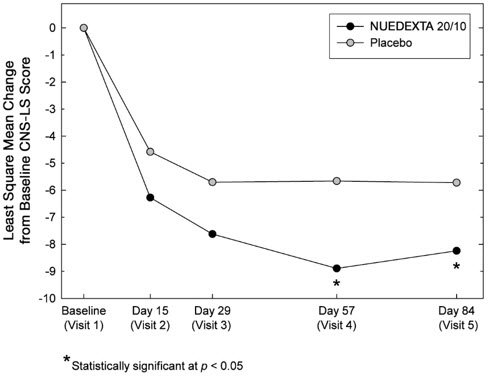
The primary outcome measure, laughing and crying episodes (Figure 1), was statistically significantly lower in each dextromethorphan/quinidine arm compared to placebo, based on an analysis of the sums of the episode counts over the double-blind phase. The secondary endpoint was the Center for Neurologic Studies Lability Scale (CNS-LS), a seven-item self-report questionnaire with 3 items assessing crying and 4 assessing laughter. CNS-LS was analyzed based on the difference between the mean scores on Day 84 and baseline, and was also statistically significantly lower in each dextromethorphan/quinidine arm compared to placebo (Figure 2). There were no clinically important differences between NUEDEXTA and the dextromethorphan 30 mg/quinidine 10 mg arm.
Figure 1: Mean PBA Episode Rates by Visit
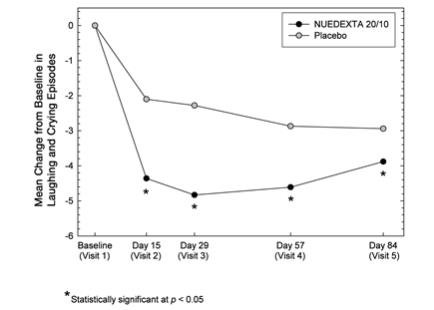
Figure 2: Least Square Mean CNS-LS Scores by Visit
Two additional studies conducted using a higher dose combination of dextromethorphan/quinidine (dextromethorphan 30 mg/quinidine 30 mg) provided supportive evidence of NUEDEXTA efficacy. The first was a 4-week study in PBA patients with underlying ALS, and the second was a 12-week study in patients with underlying MS. In both studies, the primary outcome measure, CNS-LS, and the secondary outcome measure, laughing and crying episodes, were statistically significantly decreased by the dextromethorphan/quinidine combination.
16. How is Nuedexta supplied
NUEDEXTA is supplied as brick red gelatin capsules imprinted with "DMQ 20-10". NUEDEXTA is supplied in the following package configuration:
| Package Configuration
| Capsule Strength (mg) | NDC Code |
| Bottles of 60 (30 day supply) | dextromethorphan 20 mg/quinidine 10 mg | 59148-053-16 |
Storage
Store NUEDEXTA capsules at controlled room temperature, 25°C (77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17. Patient Counseling Information
Hypersensitivity
Patients should be advised a hypersensitivity reaction to NUEDEXTA could occur. Patients should be instructed to seek medical attention immediately if they experience symptoms indicative of hypersensitivity after taking NUEDEXTA [see Contraindications (4.2), Warnings and Precautions (5.1)].
Cardiac effects
Patients should be advised to consult their healthcare provider immediately if they feel faint or lose consciousness. Patients should be counseled to inform their healthcare provider if they have any personal or family history of QTc prolongation [see Contraindications (4.4), Warnings and Precautions (5.3), Drug Interactions (7)].
Dizziness
Patients should be advised that NUEDEXTA may cause dizziness. Precautions to reduce the risk of falls should be taken, particularly for patients with motor impairment affecting gait or a history of falls [see Warnings and Precautions (5.5), Adverse Reactions (6.1)].
Drug Interactions
Inform patients that NUEDEXTA increases the risk of adverse drug interactions. Instruct patients to inform their healthcare provider about all the medications that they are taking before taking NUEDEXTA. Before taking any new medications, patients should tell their healthcare provider that they are taking NUEDEXTA [see Drug Interactions (7)].
Dosing Instructions
Instruct patients to take NUEDEXTA exactly as prescribed. Instruct patients not to take more than 2 capsules in a 24-hour period and to make sure that there is an approximate 12-hour interval between doses, and not to take a double dose after they miss a dose [see Dosage and Administration (2.1)].
General
Patients should not share or give NUEDEXTA to others, even if they have the same symptoms, because it may harm them.
Advise patients to contact their healthcare provider if their PBA symptoms persist or worsen.
Advise patients to keep this and all medications out of reach of children and pets.
Distributed and Marketed by Otsuka America Pharmaceutical, Inc., Rockville, MD 20850 USA
NUEDEXTA is a registered trademark of Avanir Pharmaceuticals, Inc. used under license by Otsuka America Pharmaceutical, Inc.

| NUEDEXTA
dextromethorphan hydrobromide and quinidine sulfate capsule, gelatin coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Otsuka America Pharmaceutical, Inc (008314390) |
More about Nuedexta (dextromethorphan / quinidine)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (23)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- FDA approval history
- Drug class: miscellaneous central nervous system agents
- En español
