Lupron Depot Prescribing Information
Package insert / product label
Generic name: leuprolide acetate
Dosage form: injection
Drug classes: Gonadotropin releasing hormones, Hormones / antineoplastics
J Code (medical billing code): J9217 (7.5 mg, injection)
Medically reviewed by Drugs.com. Last updated on Apr 15, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
LUPRON DEPOT (leuprolide acetate for depot suspension)
Initial U.S. Approval: 1989
Recent Major Changes
Indications and Usage for Lupron Depot
LUPRON DEPOT is a gonadotropin releasing hormone (GnRH) agonist indicated for:
- treatment of advanced prostate cancer. (1)
Lupron Depot Dosage and Administration
LUPRON DEPOT must be administered under the supervision of a physician. Due to different release characteristics, the dosage strengths are not additive and must be selected based upon the desired dosing schedule. (2)
- LUPRON DEPOT 7.5 mg for 1-month administration, given as a single intramuscular injection every 4 weeks. (2.1)
- LUPRON DEPOT 22.5 mg for 3-month administration, given as a single intramuscular injection every 12 weeks. (2.2)
- LUPRON DEPOT 30 mg for 4-month administration, given as a single intramuscular injection every 16 weeks. (2.3)
- LUPRON DEPOT 45 mg for 6-month administration, given as a single intramuscular injection every 24 weeks. (2.4)
Dosage Forms and Strengths
7.5 mg, 22.5 mg, 30 mg, and 45 mg injections in a kit with prefilled dual chamber syringe. (3)
Contraindications
- Hypersensitivity to GnRH, GnRH agonist or any of the excipients in LUPRON DEPOT. (4)
Warnings and Precautions
- Tumor Flare: Increased serum testosterone (~ 50% above baseline) may occur when initiating treatment with LUPRON DEPOT. Monitor for worsening of prostate cancer symptoms during the first few weeks of treatment. Monitor patients for increased bone pain, neuropathy, hematuria, ureteral obstruction, and spinal cord compression. Spinal cord compressions may contribute to paralysis with or without fatal complications. (5.1)
- Metabolic Syndrome: The use of GnRH agonists may lead to an increased risk of metabolic changes such as hyperglycemia, diabetes, hyperlipidemia, and non-alcoholic fatty liver disease. Monitor for signs and symptoms of metabolic syndrome including lipids, blood glucose level and/or HbA1c and manage according to current treatment guidelines. (5.2)
- Cardiovascular Diseases: Increased risk of myocardial infarction, sudden cardiac death and stroke has been reported in association with use of GnRH analogs in men. Monitor for cardiovascular disease and manage according to institutional guidelines. (5.3)
- Effect on QT/QTc Interval: Androgen deprivation therapy may prolong the QT interval. Consider risks and benefits. (5.4)
- Convulsions have been observed in patients with or without a history of predisposing factors. Manage convulsions according to institutional guidelines. (5.5)
- Severe Cutaneous Adverse Reactions (SCARs), including Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN), occurred in patients treated with LUPRON DEPOT. Interrupt LUPRON DEPOT if signs or symptoms of SCARs develop. Permanently discontinue if SCARs are confirmed. (5.6)
- Embryo-Fetal Toxicity: LUPRON DEPOT may cause fetal harm. (5.8, 8.1)
Adverse Reactions/Side Effects
- LUPRON DEPOT 7.5 mg for 1-month administration: The most common adverse reactions (>10%) were general pain, hot flashes/sweats, GI disorders, edema, respiratory disorder, urinary disorder. (6.1)
- LUPRON DEPOT 22.5 mg for 3-month administration: The most common adverse reactions (>10%) were general pain, injection site reaction, hot flashes/sweats, GI disorders, joint disorders, testicular atrophy, urinary disorders. (6.2)
- LUPRON DEPOT 30 mg for 4-month administration: The most common adverse reactions (>10%) were asthenia, flu syndrome, general pain, headache, injection site reaction, hot flashes/sweats, GI disorders, edema, skin reaction, urinary disorders. (6.3)
- LUPRON DEPOT 45 mg for 6-month administration: The most common adverse reactions (>10%) were hot flush, injection site pain, upper respiratory infection, and fatigue. (6.4)
In postmarketing experience, mood swings, depression, rare reports of suicidal ideation and attempt, rare reports of pituitary apoplexy, and rare reports of serious drug-induced liver injury have been reported. (6.5)
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Use In Specific Populations
- Females and males of reproductive potential: LUPRON DEPOT may impair fertility. Counsel patients on pregnancy planning and prevention. (8.3)
- Pediatric: These LUPRON DEPOT formulations are not indicated for use in children. See the LUPRON DEPOT PED® prescribing information for the use of leuprolide acetate in children with central precocious puberty.
- Geriatric: This label reflects clinical trials for LUPRON DEPOT in prostate cancer in which the majority of the subjects studied were at least 65 years of age.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2024
Full Prescribing Information
1. Indications and Usage for Lupron Depot
LUPRON DEPOT 7.5 mg for 1-month administration, 22.5 mg for 3-month administration, 30 mg for 4-month administration, and 45 mg for 6-month administration (leuprolide acetate) are indicated for the treatment of advanced prostate cancer.
2. Lupron Depot Dosage and Administration
LUPRON DEPOT must be administered under the supervision of a physician.
In patients treated with GnRH analogues for prostate cancer, treatment is usually continued upon development of non-metastatic and metastatic castration-resistant prostate cancer.
| Dosage | 7.5 mg for 1-Month Administration | 22.5 mg for 3-Month Administration | 30 mg for 4-Month Administration | 45 mg for 6-Month Administration |
| Recommended dose | 1 injection every 4 weeks | 1 injection every 12 weeks | 1 injection every 16 weeks | 1 injection every 24 weeks |
2.1 LUPRON DEPOT 7.5 mg for 1-Month Administration
The recommended dose of LUPRON DEPOT 7.5 mg for 1-month administration is one injection every 4 weeks. Do not use concurrently a fractional dose, or a combination of doses of this or any depot formulation due to different release characteristics.
Incorporated in a depot formulation, the lyophilized microspheres must be reconstituted and should be administered every 4 weeks as a single intramuscular injection.
For optimal performance of the prefilled dual chamber syringe (PDS), read and follow the instructions in Section 2.5.
2.2 LUPRON DEPOT 22.5 mg for 3-Month Administration
The recommended dose of LUPRON DEPOT 22.5 mg for 3-month administration is one injection every 12 weeks. Do not use concurrently a fractional dose, or a combination of doses of this or any depot formulation due to different release characteristics.
Incorporated in a depot formulation, the lyophilized microspheres must be reconstituted and should be administered every 12 weeks as a single intramuscular injection.
For optimal performance of the prefilled dual chamber syringe (PDS), read and follow the instructions in Section 2.5.
2.3 LUPRON DEPOT 30 mg for 4-Month Administration
The recommended dose of LUPRON DEPOT 30 mg for 4-month administration is one injection every 16 weeks. Do not use concurrently a fractional dose, or a combination of doses of this or any depot formulation due to different release characteristics.
Incorporated in a depot formulation, the lyophilized microspheres must be reconstituted and should be administered every 16 weeks as a single intramuscular injection.
For optimal performance of the prefilled dual chamber syringe (PDS), read and follow the instructions in Section 2.5.
2.4 LUPRON DEPOT 45 mg for 6-Month Administration
The recommended dose of LUPRON DEPOT 45 mg for 6-month administration is one injection every 24 weeks. Do not use concurrently a fractional dose, or a combination of doses of this or any depot formulation due to different release characteristics.
Incorporated in a depot formulation, the lyophilized microspheres must be reconstituted and should be administered every 24 weeks as a single intramuscular injection.
For optimal performance of the prefilled dual chamber syringe (PDS), read and follow the instructions in Section 2.5.
2.5 Reconstitution and Administration for Injection of LUPRON DEPOT
- Reconstitute and administer the lyophilized microspheres as a single intramuscular injection.
- Inject the suspension immediately or discard if not used within two hours, because LUPRON DEPOT does not contain a preservative.
1. Visually inspect the LUPRON DEPOT powder. DO NOT USE the syringe if clumping or caking is evident. A thin layer of powder on the wall of the syringe is considered normal prior to mixing with the diluent. The diluent should appear clear and colorless.
2. To prepare for injection, screw the white plunger into the end stopper until the stopper begins to turn (see Figure 1 and Figure 2).
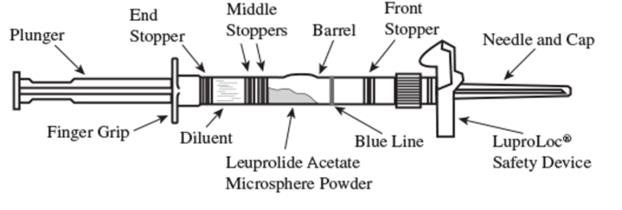
Figure 1
Figure 2
3. Hold the syringe UPRIGHT. Release the diluent by SLOWLY PUSHING (6 to 8 seconds) the plunger until the first middle stopper is at the blue line in the middle of the barrel (see Figure 3).
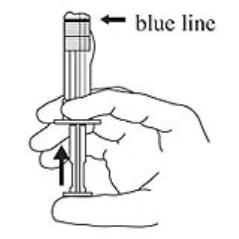
Figure 3
4. Keep the syringe UPRIGHT. Mix the microspheres (powder) thoroughly by gently shaking the syringe until the powder forms a uniform suspension. The suspension will appear milky. If the powder adheres to the stopper or caking/clumping is present, tap the syringe with your finger to disperse. DO NOT USE if any of the powder has not gone into suspension (see Figure 4).
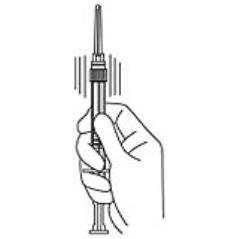
Figure 4
5. Keep the syringe UPRIGHT. With the opposite hand pull the needle cap upward without twisting.
6. Keep the syringe UPRIGHT. Advance the plunger to expel the air from the syringe. Now the syringe is ready for injection.
7. After cleaning the injection site with an alcohol swab, administer the intramuscular injection by inserting the needle at a 90 degree angle into the gluteal area, anterior thigh, or deltoid; injection sites should be alternated (see Figure 5).
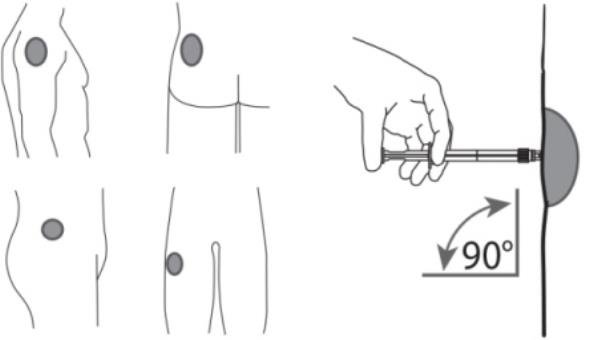
Figure 5
NOTE: If a blood vessel is accidentally penetrated, aspirated blood will be visible just below the luer lock (see Figure 6) and can be seen through the transparent LuproLoc® safety device. If blood is present, remove the needle immediately. Do not inject the medication.
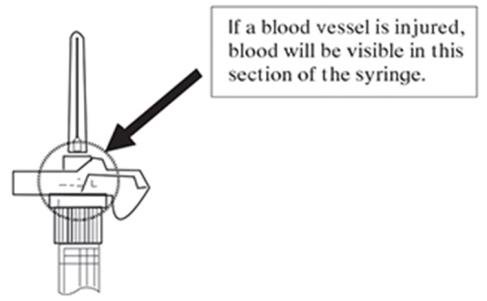
Figure 6
8. Inject the entire contents of the syringe intramuscularly.
9. Withdraw the needle. Once the syringe has been withdrawn, immediately activate the LuproLoc® safety device by pushing the arrow on the lock upward towards the needle tip with the thumb or finger, as illustrated, until the needle cover of the safety device over the needle is fully extended and a CLICK is heard or felt (see Figure 7).
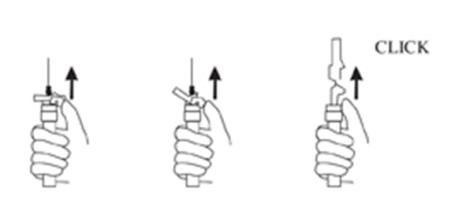
Figure 7
10. Dispose of the syringe according to local regulations/procedures.
3. Dosage Forms and Strengths
LUPRON DEPOT 7.5 mg for 1-month administration, 22.5 mg for 3-month administration, 30 mg for 4-month administration, and 45 mg for 6-month administration are each supplied as a kit with prefilled dual chamber syringe.
4. Contraindications
LUPRON DEPOT is contraindicated in:
- Hypersensitivity
LUPRON DEPOT is contraindicated in individuals with known hypersensitivity to GnRH agonists or any of the excipients in LUPRON DEPOT. Reports of anaphylactic reactions to GnRH agonists have been reported in the medical literature.
5. Warnings and Precautions
5.1 Tumor Flare
Initially, LUPRON DEPOT, like other GnRH agonists, causes increases in serum levels of testosterone to approximately 50% above baseline during the first weeks of treatment. Patients may experience worsening of symptoms or onset of new signs and symptoms during the first few weeks of treatment, including bone pain, neuropathy, hematuria, or bladder outlet obstruction. Spinal cord compression may contribute to paralysis with or without fatal complications.
Monitor patients for tumor flare symptoms during the first few weeks of treatment with LUPRON DEPOT. Closely monitor patients with metastatic vertebral lesions and/or with urinary tract obstruction for new or worsening symptoms.
5.2 Metabolic Syndrome
The use of GnRH agonists may lead to metabolic changes such as hyperglycemia, diabetes mellitus, and hyperlipidemia. Non-alcoholic fatty liver disease, including cirrhosis, occurred in the post-marketing setting. Hyperglycemia may represent new-onset diabetes mellitus or worsening of glycemic control in patients with pre-existing diabetes. Monitor for changes in serum lipids, blood glucose and/or glycosylated hemoglobin (HbA1c) in patients receiving a GnRH agonist, and manage according to current treatment guidelines.
5.3 Cardiovascular Diseases
Increased risk of developing myocardial infarction, sudden cardiac death and stroke has been reported in association with use of GnRH agonists in men. The risk appears low based on the reported odds ratios, and should be evaluated carefully along with cardiovascular risk factors when determining a treatment for patients with prostate cancer. Patients receiving a GnRH agonist should be monitored for symptoms and signs suggestive of development of cardiovascular disease and be managed according to institutional guidelines.
5.4 Effect on QT/QTc Interval
Androgen deprivation therapy may prolong the QT/QTc interval. Providers should consider whether the benefits of androgen deprivation therapy outweigh the potential risks in patients with congenital long QT syndrome, congestive heart failure, frequent electrolyte abnormalities, and in patients taking drugs known to prolong the QT interval. Electrolyte abnormalities should be corrected. Consider periodic monitoring of electrocardiograms and electrolytes.
5.5 Convulsions
Postmarketing reports of convulsions have been observed in patients on leuprolide acetate therapy. These included patients with a history of seizures, epilepsy, cerebrovascular disorders, central nervous system anomalies or tumors, and in patients on concomitant medications that have been associated with convulsions such as bupropion and SSRIs. Convulsions have also been reported in patients in the absence of any of the conditions mentioned above. Patients receiving a GnRH agonist who experience convulsion should be managed according to institutional guidelines.
5.6 Severe Cutaneous Adverse Reactions
Severe cutaneous adverse reactions (SCARs), including Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN), drug reaction with eosinophilia and systemic symptoms (DRESS), and acute generalized exanthematous pustulosis (AGEP), occurred in patients receiving LUPRON DEPOT; including cases with visceral involvement and/or requiring skin grafts [see Adverse Reactions (6.5)].
Monitor patients for the development of SCARs. Advise patients of the signs and symptoms of SCARs (e.g., a prodrome of fever, flu-like symptoms, mucosal lesions, progressive skin rash, or lymphadenopathy).
If a SCAR is suspected, interrupt LUPRON DEPOT until the etiology of the reaction has been determined. Consultation with a dermatologist is recommended. If a SCAR is confirmed, or for other grade 4 skin reactions, permanently discontinue LUPRON DEPOT.
5.7 Laboratory Tests
Monitor serum levels of testosterone following injection of LUPRON DEPOT 7.5 mg for 1-month administration, 22.5 mg for 3-month administration, 30 mg for 4-month administration, or 45 mg for 6-month administration. In the majority of patients, testosterone levels increased above baseline, and then declined thereafter to castrate levels (< 50 ng/dL) within four weeks. [see Clinical Studies (14) and Adverse Reactions (6)].
5.8 Embryo-Fetal Toxicity
Based on findings in animal studies, LUPRON DEPOT may cause fetal harm when administered to a pregnant woman. In animal developmental and reproductive toxicology studies, administration of the monthly formulation of leuprolide acetate on day 6 of pregnancy (sustained exposure was expected throughout the period of organogenesis) caused adverse embryo-fetal toxicity in animals at doses less than the human dose, based on body surface area, using an estimated daily dose. Advise pregnant patients and females of reproductive potential of the potential risk to the fetus [see Use in Specific Populations (8.1)].
6. Adverse Reactions/Side Effects
The following is discussed in more detail in other sections of the labeling:
- Tumor Flare [see Warnings and Precautions (5.1)]
- Metabolic Syndrome [see Warnings and Precautions (5.2)]
- Cardiovascular Disease [see Warnings and Precautions (5.3)]
- Effect on QT/QTc Interval [see Warnings and Precautions (5.4)]
- Convulsions [see Warnings and Precautions (5.5)]
- Severe Cutaneous Adverse Reactions [see Warnings and Precautions (5.6)]
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1 LUPRON DEPOT 7.5 mg for 1-Month Administration
In the majority of patients testosterone levels increased above baseline during the first week, declining thereafter to baseline levels or below by the end of the second week of treatment.
Potential exacerbations of signs and symptoms during the first few weeks of treatment is a concern in patients with vertebral metastases and/or urinary obstruction or hematuria which, if aggravated, may lead to neurological problems such as temporary weakness and/or paresthesia of the lower limbs or worsening of urinary symptoms [see Warnings and Precautions (5.1)].
In a clinical trial of LUPRON DEPOT 7.5 mg for 1-month administration, the following adverse reactions were reported in 5% or more of the patients during the initial 24-week treatment period.
| Table 2. Adverse Reactions Reported in ≥ 5% of Patients | ||
| LUPRON DEPOT 7.5 mg for 1-Month Administration (N=56) | ||
| N | (%) | |
| Body As A Whole | ||
| General pain | 13 | (23.2) |
| Infection | 3 | (5.4) |
| Cardiovascular System | ||
| Hot flashes/sweats* | 32 | (57.1) |
| Digestive System | ||
| GI disorders | 8 | (14.3) |
| Metabolic and Nutritional Disorders | ||
| Edema | 8 | (14.3) |
| Nervous System | ||
| Libido decreased* | 3 | (5.4) |
| Respiratory System | ||
| Respiratory disorder | 6 | (10.7) |
| Urogenital System | ||
| Urinary disorder | 7 | (12.5) |
| Impotence* | 3 | (5.4) |
| Testicular atrophy* | 3 | (5.4) |
| * Due to the expected physiologic effect of decreased testosterone levels. | ||
In this same study, the following adverse reactions were reported in less than 5% of the patients on LUPRON DEPOT 7.5 mg for 1-month administration.
Body As A Whole - asthenia, cellulitis, fever, headache, injection site reaction, neoplasm
Cardiovascular System - angina, congestive heart failure
Digestive System - anorexia, dysphagia, eructation, peptic ulcer
Blood and Lymphatic System - ecchymosis
Musculoskeletal System - myalgia
Nervous System - agitation, insomnia/sleep disorders, neuromuscular disorders
Respiratory System - emphysema, hemoptysis, lung edema, sputum increased
Skin and Appendages - hair disorder, skin reaction
Urogenital System - balanitis, breast enlargement, urinary tract infection
Laboratory Abnormalities
Abnormalities of certain parameters were observed, but their relationship to drug treatment are difficult to assess in this population. The following were recorded in ≥5% of patients at final visit: Decreased albumin, decreased hemoglobin/hematocrit, decreased prostatic acid phosphatase, decreased total protein, decreased urine specific gravity, hyperglycemia, hyperuricemia, increased BUN, increased creatinine, increased liver function tests (AST, LDH), increased phosphorus, increased platelets, increased prostatic acid phosphatase, increased total cholesterol, increased urine specific gravity, leukopenia.
6.2 LUPRON DEPOT 22.5 mg for 3-Month Administration
In two clinical trials of LUPRON DEPOT 22.5 mg for 3-month administration, the following adverse reactions were reported to have a possible or probable relationship to drug as ascribed by the treating physician in 5% or more of the patients receiving the drug.
| Table 3. Adverse Reactions Reported in ≥ 5% of Patients | ||
| LUPRON DEPOT 22.5 mg for 3-Month Administration | ||
| Body System/Reaction | N=94 | (%) |
| Body As A Whole | ||
| Asthenia | 7 | (7.4) |
| General pain | 25 | (26.6) |
| Headache | 6 | (6.4) |
| Injection site reaction | 13 | (13.8) |
| Cardiovascular System | ||
| Hot flashes/sweats | 55 | (58.5) |
| Digestive System | ||
| GI disorders | 15 | (16.0) |
| Musculoskeletal System | ||
| Joint disorders | 11 | (11.7) |
| Central/Peripheral Nervous System | ||
| Dizziness/vertigo | 6 | (6.4) |
| Insomnia/sleep disorders | 8 | (8.5) |
| Neuromuscular disorders | 9 | (9.6) |
| Respiratory System | ||
| Respiratory disorders | 6 | (6.4) |
| Skin and Appendages | ||
| Skin reaction | 8 | (8.5) |
| Urogenital System | ||
| Testicular atrophy | 19 | (20.2) |
| Urinary disorders | 14 | (14.9) |
In these same studies, the following adverse reactions were reported in less than 5% of the patients on LUPRON DEPOT 22.5 mg for 3-month administration.
Body As A Whole - enlarged abdomen, fever
Cardiovascular System - arrhythmia, bradycardia, heart failure, hypertension, hypotension, varicose vein
Digestive System - anorexia, duodenal ulcer, increased appetite, thirst/dry mouth
Blood and Lymphatic System - anemia, lymphedema
Metabolic and Nutritional Disorders - dehydration, edema
Central/Peripheral Nervous System - anxiety, delusions, depression, hypesthesia, libido decreased*, nervousness, paresthesia
Respiratory System - epistaxis, pharyngitis, pleural effusion, pneumonia
Special Senses - abnormal vision, amblyopia, dry eyes, tinnitus
Urogenital System - gynecomastia, impotence*, penis disorders, testis disorders.
* Physiologic effect of decreased testosterone.
Laboratory Abnormalities
Abnormalities of certain parameters were observed, but are difficult to assess in this population. The following were recorded in ≥5% of patients: increased BUN, hyperglycemia, hyperlipidemia (total cholesterol, LDL-cholesterol, triglycerides), hyperphosphatemia, abnormal liver function tests, increased PT, increased PTT. Additional laboratory abnormalities reported were: decreased platelets, decreased potassium and increased WBC.
6.3 LUPRON DEPOT 30 mg for 4-Month Administration
The 4-month formulation of LUPRON DEPOT 30 mg was utilized in clinical trials that studied the drug in 49 nonorchiectomized prostate cancer patients for 32 weeks or longer and in 24 orchiectomized prostate cancer patients for 20 weeks.
In the above described clinical trials, the following adverse reactions were reported in ≥ 5% of the patients during the treatment period.
| Table 4. Adverse Reactions Reported in ≥ 5% of Patients | ||||
| LUPRON DEPOT 30 mg for 4-Month Administration | ||||
| Body System/Events | Nonorchiectomized | Orchiectomized | ||
| Study 013 | Study 012 | |||
| N=49 | (%) | N=24 | (%) | |
| Body As A Whole | ||||
| Asthenia | 6 | (12.2) | 1 | (4.2) |
| Flu syndrome | 6 | (12.2) | 0 | (0.0) |
| General pain | 16 | (32.7) | 1 | (4.2) |
| Headache | 5 | (10.2) | 1 | (4.2) |
| Injection site reaction | 4 | (8.2) | 9 | (37.5) |
| Cardiovascular System | ||||
| Hot flashes/sweats | 23 | (46.9) | 2 | (8.3) |
| Digestive System | ||||
| GI disorders | 5 | (10.2) | 3 | (12.5) |
| Metabolic and Nutritional Disorders | ||||
| Dehydration | 4 | (8.2) | 0 | (0.0) |
| Edema | 4 | (8.2) | 5 | (20.8) |
| Musculoskeletal System | ||||
| Joint disorder | 8 | (16.3) | 1 | (4.2) |
| Myalgia | 4 | (8.2) | 0 | (0.0) |
| Nervous System | ||||
| Dizziness/vertigo | 3 | (6.1) | 2 | (8.3) |
| Neuromuscular disorders | 3 | (6.1) | 1 | (4.2) |
| Paresthesia | 4 | (8.2) | 1 | (4.2) |
| Respiratory System | ||||
| Respiratory disorder | 4 | (8.2) | 1 | (4.2) |
| Skin and Appendages | ||||
| Skin reaction | 6 | (12.2) | 0 | (0.0) |
| Urogenital System | ||||
| Urinary disorders | 5 | (10.2) | 4 | (16.7) |
In these same studies, the following adverse reactions were reported in less than 5% of the patients on LUPRON DEPOT 30 mg for 4-month administration.
Body As A Whole - abscess, accidental injury, allergic reaction, cyst, fever, generalized edema, hernia, neck pain, neoplasm
Cardiovascular System - atrial fibrillation, deep thrombophlebitis, hypertension
Digestive System - anorexia, eructation, gastrointestinal hemorrhage, gingivitis, gum hemorrhage, hepatomegaly, increased appetite, intestinal obstruction, periodontal abscess
Blood and Lymphatic System - lymphadenopathy
Metabolic and Nutritional Disorders - healing abnormal, hypoxia, weight loss
Musculoskeletal System - leg cramps, pathological fracture, ptosis
Nervous System - abnormal thinking, amnesia, confusion, convulsion, dementia, depression, insomnia/sleep disorders, libido decreased*, neuropathy, paralysis
Respiratory System - asthma, bronchitis, hiccup, lung disorder, sinusitis, voice alteration
Skin and Appendages - herpes zoster, melanosis
Urogenital System - bladder carcinoma, epididymitis, impotence*, prostate disorder, testicular atrophy*, urinary incontinence, urinary tract infection.
* Physiologic effect of decreased testosterone.
Laboratory Abnormalities
Abnormalities of certain parameters were observed, but their relationship to drug treatment is difficult to assess in this population. The following were recorded in ≥ 5% of patients: decreased bicarbonate, decreased hemoglobin/hematocrit/RBC, hyperlipidemia (total cholesterol, LDL-cholesterol, triglycerides), decreased HDL-cholesterol, eosinophilia, increased glucose, increased liver function tests (ALT, AST, GGTP, LDH), increased phosphorus. Additional laboratory abnormalities were reported: increased BUN and PT, leukopenia, thrombocytopenia, uricaciduria.
6.4 LUPRON DEPOT 45 mg for 6-Month Administration
One open label, multicenter study was conducted with LUPRON DEPOT 45 mg for 6-month administration in 151 prostate cancer patients. Patients were treated for 48 weeks, with 139/151 receiving two injections 24 weeks apart.
In the above described clinical trial, the following adverse reactions were reported in ≥ 5% of the patients during the treatment period. The Table 5 includes all adverse reactions reported in ≥ 5% of patients as well as the incidences of these adverse reactions that were considered, by the treating physician, to have a definite or possible relationship to LUPRON DEPOT.
| Table 5. Adverse Reactions in ≥ 5% of Patients | ||||
| LUPRON DEPOT 45 mg for 6-Month Administration | ||||
| Treatment Emergent | Treatment Related | |||
| Adverse Event | N = 151 | (%) | N = 151 | (%) |
| Hot flush/flushing | 89 | 58.9 | 88 | 58.3 |
| Injection site pain/discomfort | 29 | 19.2 | 16 | 10.6 |
| Upper respiratory tract infection/influenza-like illness1 | 32 | 21.2 | 0 | 0 |
| Fatigue/lethargy | 20 | 13.2 | 18 | 11.9 |
| Constipation | 15 | 9.9 | 5 | 3.3 |
| Arthralgia | 14 | 9.3 | 2 | 1.3 |
| Insomnia/sleep disorder | 13 | 8.6 | 5 | 3.3 |
| Headache/sinus headache | 12 | 7.9 | 3 | 2.0 |
| Musculoskeletal pain/myalgia | 12 | 7.9 | 3 | 2.0 |
| Second primary neoplasm2 | 11 | 7.3 | 0 | 0 |
| Cough | 10 | 6.6 | 2 | 1.3 |
| Hematuria/hemorrhagic cystitis | 10 | 6.6 | 0 | 0 |
| Hypertension/BP increased | 10 | 6.6 | 3 | 2.0 |
| Rash | 9 | 6.0 | 3 | 2.0 |
| Dysuria | 9 | 6.0 | 1 | 0.7 |
| Urinary tract infection/cystitis | 9 | 6.0 | 0 | 0 |
| Anemia/hemoglobin decreased | 10 | 6.6 | 2 | 1.3 |
| Back pain | 8 | 5.3 | 0 | 0 |
| COPD | 8 | 5.3 | 0 | 0 |
| Dizziness | 8 | 5.3 | 3 | 2.0 |
| Dyspnea/dyspnea on exertion | 8 | 5.3 | 2 | 1.3 |
| Nocturia | 8 | 5.3 | 2 | 1.3 |
| Peripheral/pitting edema | 8 | 5.3 | 2 | 1.3 |
| Coronary artery disease/angina | 8 | 5.3 | 1 | 0.7 |
1Includes influenza, nasal congestion, nasopharyngitis, rhinorrhea, upper respiratory tract infection, and viral upper respiratory tract infection
2Includes basal cell carcinoma, bladder transitional cell carcinoma, lung neoplasm, malignant melanoma, non-Hodgkin’s lymphoma, and squamous cell carcinoma
The following adverse reactions led to discontinuation; fatigue, hot flush, second primary neoplasm, asthenia, coronary artery disease, constipation, hyperkalemia, and sleep disorder. Serious adverse reactions in ≥ 2% of patients, regardless of causality, included chronic obstructive pulmonary disease, coronary artery disease/angina, cerebrovascular accident/transient ischemic attack, pneumonia, and second primary neoplasms.
Laboratory Abnormalities
At baseline, 13.9% of patients had a CTCAE v4.0 grade 1 or 2 decreased hemoglobin. During the study, 42.4% of subjects had grade 1 decreased hemoglobin (10 - <12-5 g/dL), 2.0% had grade 2 (8 - <10 g/dL) and 1.3% of subjects had grade 3 or 4 (<8 g/dL). Likewise, 28.5% of patients had a grade 1 or 2 increased cholesterol at baseline while 55.0% had grade 1 increased cholesterol (>199- 300 mg/dL), 3.3% had a grade 2 increase (>300-400 mg/dL), and 0.7% of subjects had grade 3 (>400 mg/dL) during the study.
6.5 Postmarketing
The following adverse reactions have been identified during post-approval use of LUPRON DEPOT. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
During postmarketing surveillance, which includes other dosage forms and other patient populations, the following adverse reactions were reported.
Mood swings, including depression, have been reported. There have been very rare reports of suicidal ideation and attempt. Many, but not all, of these patients had a history of depression or other psychiatric illness. Patients should be counseled on the possibility of development or worsening of depression during treatment with LUPRON DEPOT.
Symptoms consistent with an anaphylactoid or asthmatic process have occurred (incidence rate of about 0.002%).
Changes in Bone Density - Decreased bone density has been reported in the medical literature in men who have had orchiectomy or who have been treated with a GnRH agonist analog. In a clinical trial, 25 men with prostate cancer, 12 of whom had been treated previously with leuprolide acetate for at least six months, underwent bone density studies as a result of pain. The leuprolide-treated group had lower bone density scores than the nontreated control group. It can be anticipated that long periods of medical castration in men will have effects on bone density.
Pituitary Apoplexy - During post-marketing surveillance, rare cases of pituitary apoplexy (a clinical syndrome secondary to infarction of the pituitary gland) have been reported after the administration of gonadotropin-releasing hormone agonists. In a majority of these cases, a pituitary adenoma was diagnosed, with a majority of pituitary apoplexy cases occurring within 2 weeks of the first dose, and some within the first hour. In these cases, pituitary apoplexy has presented as sudden headache, vomiting, visual changes, ophthalmoplegia, altered mental status, and sometimes cardiovascular collapse. Immediate medical attention has been required.
Localized reactions including induration and abscess have been reported at the site of injection.
Symptoms consistent with fibromyalgia (e.g., joint and muscle pain, headaches, sleep disorders, gastrointestinal distress, and shortness of breath) have been reported individually and collectively.
Cardiovascular System - hypotension, myocardial infarction, pulmonary embolism
Respiratory, Thoracic and Mediastinal disorder - interstitial lung disease
Hepato-biliary disorder - serious drug-induced liver injury, non-alcoholic fatty liver disease
Skin reactions - rash, urticaria, photosensitivity, erythema multiforme, bullous dermatitis, dermatitis exfoliative, DRESS, SJS/ TEN, and AGEP
Blood and Lymphatic System - decreased WBC
Central/Peripheral Nervous System - convulsion, peripheral neuropathy, spinal fracture/paralysis
Endocrine System - diabetes mellitus
Musculoskeletal System - tenosynovitis-like symptoms
Urogenital System - prostate pain
See other LUPRON DEPOT and LUPRON Injection prescribing information for other reactions reported in women and pediatric populations.
7. Drug Interactions
7.1 Drug/Laboratory Test Interactions
Administration of LUPRON DEPOT in therapeutic doses results in suppression of the pituitary-gonadal system. Normal function is usually restored within three months after treatment is discontinued. Due to the suppression of the pituitary-gonadal system by LUPRON DEPOT, diagnostic tests of pituitary gonadotropic and gonadal functions conducted during treatment and up to three months after discontinuation of LUPRON DEPOT may be affected.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings in animal studies and mechanism of action, LUPRON DEPOT may cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data in pregnant women to inform the drug-associated risk. In animal developmental and reproductive toxicology studies, administration of a monthly formulation of leuprolide acetate on day 6 of pregnancy (sustained exposure was expected throughout the period of organogenesis) caused adverse embryo-fetal toxicity in animals at doses less than the human dose based on body surface area using an estimated daily dose (see data). Advise pregnant patients and females of reproductive potential of the potential risk to the fetus.
Data
Animal Data
Major fetal malformations were observed in developmental and reproductive toxicology studies in rabbits after a single administration of the monthly formulation of leuprolide acetate on day 6 of pregnancy at doses of 0.00024, 0.0024, and 0.024 mg/kg (approximately 1/1600 to 1/16 the human dose based on body surface area using an estimated daily dose in animals and humans). Since a depot formulation was utilized in the study, a sustained exposure to leuprolide was expected throughout the period of organogenesis and to the end of gestation. Similar studies in rats did not demonstrate an increase in fetal malformations, however, there was increased fetal mortality and decreased fetal weights with the two higher doses of the monthly formulation of leuprolide acetate in rabbits and with the highest dose (0.024 mg/kg) in rats.
8.2 Lactation
The safety and efficacy of LUPRON DEPOT have not been established in females. There is no information regarding the presence of LUPRON DEPOT in human milk, the effects on the breastfed child, or the effects on milk production. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in a breastfed child from LUPRON DEPOT, a decision should be made to discontinue breastfeeding or discontinue the drug, taking into account the importance of the drug to the mother.
8.3 Females and Males of Reproductive Potential
Infertility
Males
Based on findings in animals and mechanism of action, LUPRON DEPOT may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
10. Overdosage
There is no experience of overdosage in clinical trials. In rats, a single subcutaneous dose of 100 mg/kg (approximately 4,000 times the estimated daily human dose based on body surface area), resulted in dyspnea, decreased activity, and excessive scratching. In early clinical trials with daily subcutaneous leuprolide acetate, doses as high as 20 mg/day for up to two years caused no adverse effects differing from those observed with the 1 mg/day dose.
11. Lupron Depot Description
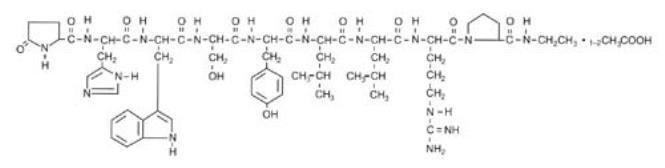
Leuprolide acetate is a synthetic nonapeptide analog of naturally occurring gonadotropin-releasing hormone (GnRH). The analog possesses greater potency than the natural hormone. The chemical name is 5-oxo-L-prolyl-L-histidyl-L-tryptophyl-L-seryl-L-tyrosyl-D-leucyl-L-leucyl-L-arginyl-N-ethyl-L-prolinamide acetate (salt) with the following structural formula:
LUPRON DEPOT 7.5 mg for 1-month administration is available in a prefilled dual-chamber syringe containing sterile lyophilized microspheres which, when mixed with diluent, becomes a suspension intended as a monthly intramuscular injection.
The front chamber of LUPRON DEPOT 7.5 mg for 1-month administration prefilled dual-chamber syringe contains leuprolide acetate (7.5 mg), purified gelatin (1.3 mg), DL-lactic and glycolic acids copolymer (66.2 mg), and D-mannitol (13.2 mg). The second chamber of diluent contains carboxymethylcellulose sodium (5 mg), D-mannitol (50 mg), polysorbate 80 (1 mg), water for injection, USP, and glacial acetic acid, USP to control pH.
LUPRON DEPOT 22.5 mg for 3-month administration is available in a prefilled dual-chamber syringe containing sterile lyophilized microspheres which, when mixed with diluent, become a suspension intended as an intramuscular injection to be given ONCE EVERY 12 WEEKS.
The front chamber of LUPRON DEPOT 22.5 mg for 3-month administration prefilled dual-chamber syringe contains leuprolide acetate (22.5 mg), polylactic acid (198.6 mg) and D-mannitol (38.9 mg). The second chamber of diluent contains carboxymethylcellulose sodium (7.5 mg), D-mannitol (75.0 mg), polysorbate 80 (1.5 mg), water for injection, USP, and glacial acetic acid, USP to control pH.
LUPRON DEPOT 30 mg for 4-month administration is available in a prefilled dual-chamber syringe containing sterile lyophilized microspheres which, when mixed with diluent, become a suspension intended as an intramuscular injection to be given ONCE EVERY 16 WEEKS.
The front chamber of LUPRON DEPOT 30 mg for 4-month administration prefilled dual-chamber syringe contains leuprolide acetate (30 mg), polylactic acid (264.8 mg) and D-mannitol (51.9 mg). The second chamber of diluent contains carboxymethylcellulose sodium (7.5 mg), D-mannitol (75.0 mg), polysorbate 80 (1.5 mg), water for injection, USP, and glacial acetic acid, USP to control pH.
LUPRON DEPOT 45 mg for 6-month administration is available in a prefilled dual-chamber syringe containing sterile lyophilized microspheres which, when mixed with diluent, become a suspension intended as an intramuscular injection to be given ONCE EVERY 24 WEEKS.
The front chamber of LUPRON DEPOT 45 mg for 6-month administration prefilled dual-chamber syringe contains leuprolide acetate (45 mg), polylactic acid (169.9 mg), D-mannitol (39.7 mg), and stearic acid (10.1 mg). The second chamber of diluent contains carboxymethylcellulose sodium (7.5 mg), D-mannitol (75.0 mg), polysorbate 80 (1.5 mg), water for injection, USP, and glacial acetic acid, USP to control pH.
12. Lupron Depot - Clinical Pharmacology
12.1 Mechanism of Action
Leuprolide acetate, a GnRH agonist, acts as an inhibitor of gonadotropin secretion. Animal studies indicate that following an initial stimulation, continuous administration of leuprolide acetate results in suppression of ovarian and testicular steroidogenesis. This effect was reversible upon discontinuation of drug therapy.
Administration of leuprolide acetate has resulted in inhibition of the growth of certain hormone dependent tumors (prostatic tumors in Noble and Dunning male rats and DMBA-induced mammary tumors in female rats) as well as atrophy of the reproductive organs.
12.2 Pharmacodynamics
In humans, administration of leuprolide acetate results in an initial increase in circulating concentrations of luteinizing hormone (LH) and follicle stimulating hormone (FSH), leading to a transient increase in concentrations of the gonadal steroids (testosterone and dihydrotestosterone in males, and estrone and estradiol in premenopausal females). However, continuous administration of leuprolide acetate results in decreased concentrations of LH and FSH. In males, testosterone is reduced to castrate concentrations. In premenopausal females, estrogens are reduced to postmenopausal concentrations. These decreases occur within two to four weeks after initiation of treatment, and castrate concentrations of testosterone in prostatic cancer patients have been demonstrated for more than five years.
Leuprolide acetate is not active when given orally.
12.3 Pharmacokinetics
Absorption
LUPRON DEPOT 7.5 mg for 1-Month Administration
Following a single injection of LUPRON DEPOT 7.5 mg for 1-month administration to patients, mean plasma measured concentrations were 20 ng/mL at 4 hours and 0.36 ng/mL at 4 weeks. However, intact leuprolide and an inactive major metabolite could not be distinguished by the assay which was employed in the study.
LUPRON DEPOT 22.5 mg for 3-Month Administration
Following a single injection of LUPRON DEPOT 22.5 mg for 3-month administration in patients, mean peak plasma concentrations were 48.9 ng/mL at 4 hours and then declined to 0.67 ng/mL at 12 weeks. Leuprolide appeared to be released at a constant rate following the onset of steady-state concentrations during the third week after dosing, providing steady plasma concentrations through the 12-week dosing interval. However, intact leuprolide and an inactive major metabolite could not be distinguished by the assay which was employed in the study. The initial burst, followed by a decline to a steady-state concentration, was similar to the release pattern seen with the monthly formulation.
LUPRON DEPOT 30 mg for 4-Month Administration
Following a single injection of LUPRON DEPOT 30 mg for 4-month administration in sixteen orchiectomized prostate cancer patients, mean plasma concentrations were 59.3 ng/mL at 4 hours and then declined to 0.30 ng/mL at 16 weeks. Mean plasma concentrations from weeks 3.5 to 16 was 0.44 ± 0.20 ng/mL (range: 0.20-1.06). Leuprolide appeared to be released at a constant rate following the onset of steady-state concentrations during the fourth week after dosing, providing steady plasma concentrations throughout the 16-week dosing interval. However, intact leuprolide and an inactive major metabolite could not be distinguished by the assay which was employed in the study. The initial burst, followed by a decline to a steady-state concentration, was similar to the release pattern seen with the other depot formulations.
LUPRON DEPOT 45 mg for 6-Month Administration
Following a single injection of LUPRON DEPOT 45 mg for 6-month administration in 26 prostate cancer patients, mean peak plasma concentration of 6.7 ng/mL was observed at 2 hours and then declined to 0.07 ng/mL at 24 weeks. Leuprolide appeared to be released continuously following the onset of steady-state concentrations during the third week after dosing providing steady plasma concentrations through the 24-week dosing interval. The initial burst, followed by a decline to a steady-state concentration, was similar to the release pattern seen with the other depot formulations. In this study, mean plasma concentration-time profiles were similar after the first and second dose.
Distribution
The mean steady-state volume of distribution of leuprolide following intravenous bolus administration to healthy male volunteers was 27 L. In vitro binding to human plasma proteins ranged from 43% to 49%.
Elimination
The mean systemic clearance of leuprolide following intravenous bolus administration to healthy male volunteers was 7.6 L/h, and terminal elimination half-life was approximately 3 hours based on a two compartment model.
Following administration of LUPRON DEPOT 3.75 mg to 3 patients, less than 5% of the dose was recovered as parent and M-I metabolite in the urine.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenicity studies were conducted with leuprolide acetate in rats and mice. In rats, a dose-related increase of benign pituitary hyperplasia and benign pituitary adenomas was noted at 24 months when the drug was administered subcutaneously at daily doses (0.6 to 4 mg/kg). There was a significant but not dose-related increase of pancreatic islet-cell adenomas in females and of testicular interstitial cell adenomas in males (highest incidence in the low dose group). In mice, no leuprolide acetate-induced tumors or pituitary abnormalities were observed at a dose as high as 60 mg/kg for two years. Patients have been treated with leuprolide acetate for up to three years with doses as high as 10 mg/day and for two years with doses as high as 20 mg/day without demonstrable pituitary abnormalities.
Genotoxicity studies were conducted with leuprolide acetate using bacterial and mammalian systems. These studies provided no evidence of mutagenic effects or chromosomal aberrations.
Leuprolide may reduce male and female fertility. Administration of leuprolide acetate to male and female rats at dose of 0.024, 0.24, and 2.4 mg/kg as monthly depot formulation for up to 3 months (approximately as low as 1/30 of the human dose based on body surface area using an estimated daily dose in animals and humans) caused atrophy of the reproductive organs, and suppression of reproductive function. These changes were reversible upon cessation of treatment.
14. Clinical Studies
14.1 LUPRON DEPOT 7.5 mg for 1-Month Administration
In an open-label, non-comparative, multicenter clinical study of LUPRON DEPOT 7.5 mg for 1-month administration, 56 patients with stage D2 prostatic adenocarcinoma and no prior systemic treatment were enrolled. The objectives were to determine if a 7.5 mg depot formulation of leuprolide injected once every 4 weeks would reduce and maintain serum testosterone to castrate range (≤50 ng/dL), to evaluate objective clinical response, and to assess the safety of the formulation. During the initial 24 weeks, serum testosterone was measured weekly, biweekly, or every four weeks and objective tumor response assessments were performed at Weeks 12 and 24. Once the patient completed the initial 24-week treatment phase, treatment continued at the investigator’s discretion. Data from the initial 24-week treatment phase are summarized in this section.
In the majority of patients, serum testosterone increased by 50% or more above baseline during the first week of treatment. Serum testosterone suppressed to the castrate range within 30 days of the initial depot injection in 94% (51/54) of patients for whom testosterone suppression was achieved (2 patients withdrew prior to onset of suppression) and within 66 days in all 54 patients. Mean serum testosterone suppressed to castrate level by Week 3. The median dosing interval between injections was 28 days. One escape from suppression (2 consecutive testosterone values greater than 50 ng/dL after achieving castrate level) was noted at Week 18, associated with a substantial dosing delay. In this patient, serum testosterone returned to the castrate range at the next monthly measurement. Serum testosterone was minimally above the castrate range on a single occasion for 4 other patients. No clinical significance was attributed to these rises in testosterone.
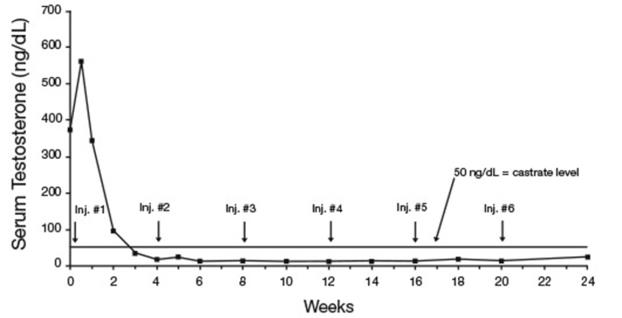
Figure 8. LUPRON DEPOT 7.5 mg for 1-Month Administration Mean Serum Testosterone Concentrations
Secondary efficacy endpoints evaluated included objective tumor response, assessed by clinical evaluations of tumor burden (complete response, partial response, objectively stable, and progression), as well as changes in local disease status, assessed by digital rectal examination, and changes in prostatic acid phosphatase (PAP). These evaluations were performed at Weeks 12 and 24. The objective tumor response analysis showed a “no progression” (i.e. Complete or partial response, or stable disease) in 77% (40/52) of patients at Week 12, and in 84% (42/50) of patients at Week 24. Local disease improved or remained stable in all (42) patients evaluated at Week 12 and in 98% (41/42) of patients elevated at Week 24. PAP normalized or decreased at Week 12 and/or 24 in the majority of patients with elevated baseline PAP.
Periodic monitoring of serum testosterone and PSA levels is recommended, especially if the anticipated clinical or biochemical response to treatment has not been achieved. It should be noted that results of testosterone determinations are dependent on assay methodology. It is advisable to be aware of the type and precision of the assay methodology to make appropriate clinical and therapeutic decisions.
14.2 LUPRON DEPOT 22.5 mg for 3-Month Administration
In clinical studies, serum testosterone was suppressed to castrate within 30 days in 87 of 92 (95%) patients and within an additional two weeks in three patients. Two patients did not suppress for 15 and 28 weeks, respectively. Suppression was maintained in all of these patients with the exception of transient minimal testosterone elevations in one of them, and in another an increase in serum testosterone to above the castrate range was recorded during the 12 hour observation period after a subsequent injection. This represents stimulation of gonadotropin secretion.
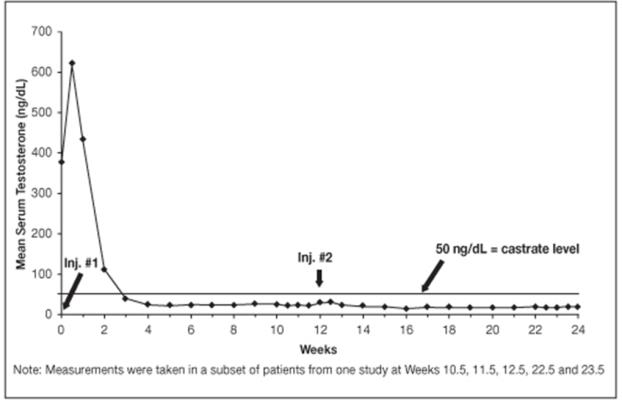
Figure 9. LUPRON DEPOT 22.5 mg for 3-Month Administration Mean Serum Testosterone Concentrations
An 85% rate of “no progression” was achieved during the initial 24 weeks of treatment. A decrease from baseline in serum PSA of ≥90% was reported in 71% of the patients and a change to within the normal range (≤3.99 ng/mL) in 63% of the patients.
Periodic monitoring of serum testosterone and PSA levels is recommended, especially if the anticipated clinical or biochemical response to treatment has not been achieved. It should be noted that results of testosterone determinations are dependent on assay methodology. It is advisable to be aware of the type and precision of the assay methodology to make appropriate clinical and therapeutic decisions.
14.3 LUPRON DEPOT 30 mg for 4-Month Administration
In an open-label, noncomparative, multicenter clinical study of LUPRON DEPOT 30 mg for 4-month administration, 49 patients with stage D2 prostatic adenocarcinoma (with no prior treatment) were enrolled. The objectives were to determine whether a 30 mg depot formulation of leuprolide injected once every 16 weeks would reduce and maintain serum testosterone levels at castrate levels (≤ 50 ng/dL), and to assess the safety of the formulation. The study was divided into an initial 32-week treatment phase and a long-term treatment phase. Serum testosterone levels were determined biweekly or weekly during the first 32 weeks of treatment. Once the patient completed the initial 32-week treatment period, treatment continued at the investigator’s discretion with serum testosterone levels being done every 4 months prior to the injection.
In the majority of patients, testosterone levels increased 50% or more above the baseline during the first week of treatment. Mean serum testosterone subsequently suppressed to castrate levels within 30 days of the first injection in 94% of patients and within 43 days in all 49 patients during the initial 32-week treatment period. The median dosing interval between injections was 112 days. One escape from suppression (two consecutive testosterone values greater than 50 ng/dL after castrate levels achieved) was noted at Week 16. In this patient, serum testosterone increased to above the castrate range following the second depot injection (Week 16) but returned to the castrate level by Week 18. No adverse reactions were associated with this rise in serum testosterone. A second patient had a rise in testosterone at Week 17, then returned to the castrate level by Week 18 and remained there through Week 32. In the long-term treatment phase two patients experienced testosterone elevations, both at Week 48. Testosterone for one patient returned to the castrate range at Week 52, and one patient discontinued the study at Week 48 due to disease progression.
Secondary efficacy endpoints evaluated in the study were the objective tumor response as assessed by clinical evaluations of tumor burden (complete response, partial response, objectively stable and progression) and evaluations of changes in prostatic involvement and prostate-specific antigen (PSA). These evaluations were performed at Weeks 16 and 32 of the treatment phase. The long-term treatment phase monitored PSA at each visit (every 16 weeks). The objective tumor response analysis showed “no progression” (i.e. complete or partial response, or stable disease) in 86% (37/43) of patients at Week 16, and in 77% (37/48) of patients at Week 32. Local disease improved or remained stable in all patients evaluated at Week 16 and/or 32. For patients with elevated baseline PSA, 50% (23/46) had a normal PSA (less than 4.0 ng/mL) at Week 16, and 51% (19/37) had a normal PSA at Week 32.
Periodic monitoring of serum testosterone and PSA levels is recommended, especially if the anticipated clinical or biochemical response to treatment has not been achieved. It should be noted that results of testosterone determinations are dependent on assay methodology. It is advisable to be aware of the type and precision of the assay methodology to make appropriate clinical and therapeutic decisions.
Using historical comparisons, the safety and efficacy of LUPRON DEPOT 30 mg for 4-month administration appear similar to the other LUPRON DEPOT formulations.
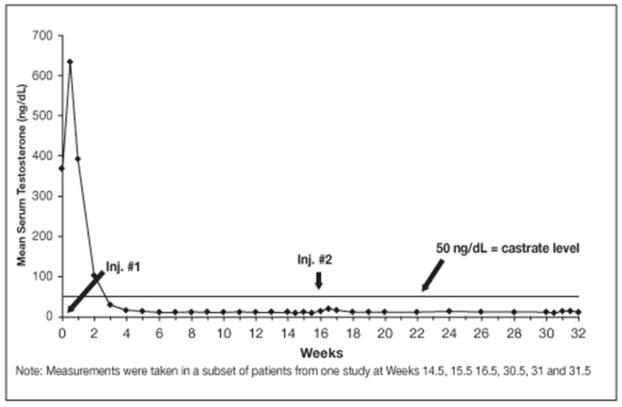
Figure 10. LUPRON DEPOT 30 mg for 4-Month Administration Mean Serum Testosterone Concentrations
14.4 LUPRON DEPOT 45 mg for 6-Month Administration
An open-label, non-comparative, multicenter clinical study of LUPRON DEPOT 45 mg for 6-month administration enrolled 151 patients with prostate cancer. The study drug was administered as two intramuscular injections of LUPRON DEPOT 45 mg at 24 week intervals (139/151 received 2 injections), and patients were followed for a total of 48 weeks.
Among 148 patients who had testosterone value at Week 4, serum testosterone was suppressed to castrate levels (< 50 ng/dL) from Week 4 through Week 48 in an estimated 93.4% (two-sided 95% CI: 89.2%, 97.6%) of patients. One patient failed to achieve testosterone suppression by Week 4, and eight patients had escapes from suppression (any testosterone value > 50 ng/dL after castrate levels were achieved). Mean testosterone levels increased to 608 ng/dL from a baseline of 435 ng/dL during the first week of treatment. By Week 4, the mean testosterone concentration had decreased to below castrate levels (16 ng/dL).
Periodic monitoring of serum testosterone levels is recommended, especially if the anticipated clinical or biochemical response to treatment has not been achieved. Testosterone determinations are dependent on assay methodology and it is advisable to be aware of the type and precision of the assay methodology to make appropriate clinical and therapeutic decisions.
Figure 11 below shows the mean testosterone concentration at various time points.
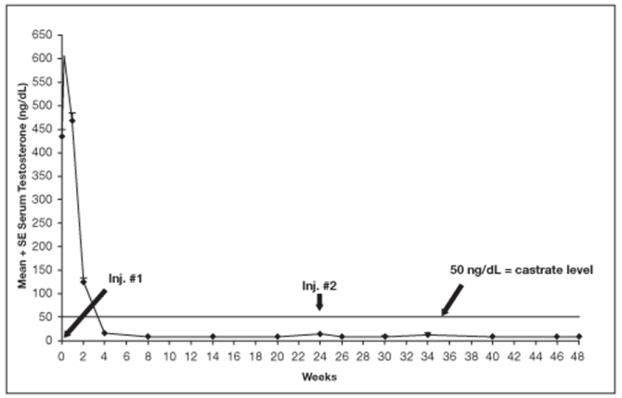
Figure 11. LUPRON DEPOT 45 mg for 6-Month Administration Serum Testosterone Concentrations (Mean + SE)
16. How is Lupron Depot supplied
Each LUPRON DEPOT 7.5 mg for 1-month administration kit (NDC 0074-3642-03), 22.5 mg for 3-month administration kit (NDC 0074-3346-03), 30 mg for 4-month administration kit (NDC 0074-3683-03), 45 mg for 6-month administration kit (NDC 0074-3473-03) contains:
- one prefilled dual-chamber syringe containing needle with LuproLoc® safety device
- one plunger
- two alcohol swabs
- a complete prescribing information enclosure
The prefilled dual-chamber syringe of LUPRON DEPOT 7.5 mg for 1-month administration contains sterile lyophilized microspheres of leuprolide acetate incorporated in a biodegradable lactic acid/glycolic acid copolymer.
The prefilled dual-chamber syringe of LUPRON DEPOT 22.5 mg for 3-month administration, 30 mg for 4-month administration, 45 mg for 6-month administration contains sterile lyophilized microspheres of leuprolide acetate incorporated in a biodegradable lactic acid polymer.
When mixed with 1 mL of accompanying diluent, LUPRON DEPOT 7.5 mg for 1-month administration is administered as a single monthly intramuscular injection.
When mixed with 1.5 mL of accompanying diluent, LUPRON DEPOT 22.5 mg for 3-month administration is administered as a single intramuscular injection EVERY 12 WEEKS.
When mixed with 1.5 mL of accompanying diluent, LUPRON DEPOT 30 mg for 4-month administration is administered as a single intramuscular injection EVERY 16 WEEKS.
When mixed with 1.5 mL of accompanying diluent, LUPRON DEPOT 45 mg for 6-month administration is administered as a single intramuscular injection EVERY 24 WEEKS.
Store between 20° to 25°C (68° to 77°F); excursions permitted to 15°C–30°C (59°F–86°F) [See USP Controlled Room Temperature].
17. Patient Counseling Information
Hypersensitivity Reactions
- Inform patients that if they have experienced hypersensitivity with other GnRH agonist drugs like LUPRON DEPOT, LUPRON DEPOT is contraindicated [see Contraindications (4)].
Tumor Flare
- Inform patients that LUPRON DEPOT can cause tumor flare during the first weeks of treatment. Inform patients that the increase in testosterone can cause an increase in urinary symptoms or pain. Advise patients to contact their healthcare provider if bone pain, neuropathy, hematuria, bladder outlet obstruction, spinal cord compression, or new or worsened symptoms occur after beginning LUPRON DEPOT treatment [see Warnings and Precautions (5.1)].
Metabolic Syndrome
- Advise patients that there is an increased risk of metabolic changes such as hyperglycemia, diabetes, hyperlipidemia, and non-alcoholic fatty liver disease with LUPRON DEPOT therapy. Inform patients that periodic monitoring for metabolic changes is required when being treated with LUPRON DEPOT [see Warnings and Precautions (5.2)].
Cardiovascular Disease
- Inform patients that there is an increased risk of myocardial infarction, sudden cardiac death, and stroke with LUPRON DEPOT treatment. Advise patients to immediately report signs and symptoms associated with these events to their healthcare provider for evaluation [see Warnings and Precautions (5.3)].
Severe Cutaneous Adverse Reactions
- Inform patients that severe cutaneous adverse reactions (SCARs), including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), drug reaction with eosinophilia and systemic symptoms (DRESS), and acute generalized exanthematous pustulosis (AGEP), which may be life threatening or fatal, may occur during treatment with LUPRON DEPOT. Advise patients to contact their healthcare provider or seek medical attention right away if they experience signs or symptoms of SCARs [see Warnings and Precautions (5.6)].
Urogenital Disorders
- Advise patients that LUPRON DEPOT may cause impotence [see Adverse Reactions (6)].
Infertility
- Inform patients that LUPRON DEPOT may cause infertility [see Use in Specific Populations (8.3)].
Continuation of LUPRON DEPOT Treatment
- Inform patients that LUPRON DEPOT is usually continued, often with additional medication, after the development of non-metastatic and metastatic castration-resistant prostate cancer [see Dosage and Administration (2.1)].
Manufactured for
AbbVie Inc.
North Chicago, IL 60064
by Takeda Pharmaceutical Company Limited
Osaka, Japan 540-8645
20082648
NDC 0074-3473-03
FOR ADULT USE
45 mg for 6-month administration
Single Dose Administration Kit with prefilled dual-chamber syringe.
LupronDepot®
(Leuprolide Acetate for Depot Suspension)
45 mg for 6-month administration
FOR INTRAMUSCULAR INJECTION
The front chamber contains: leuprolide acetate 45 mg
• polylactic acid 169.9 mg • D-mannitol 39.7 mg • stearic acid 10.1 mg
The second chamber contains: carboxymethylcellulose sodium 7.5 mg
• D-mannitol 75.0 mg • polysorbate 80 1.5 mg • water for injection, USP,
and glacial acetic acid, USP to control pH
Rx only
NDC 0074-3346-03
FOR ADULT USE
22.5 mg for 3-month administration
Single Dose Administration Kit with prefilled dual-chamber syringe.
LupronDepot®
(Leuprolide Acetate for Depot Suspension)
22.5 mg for 3-month administration
FOR INTRAMUSCULAR INJECTION
The front chamber contains: leuprolide acetate 22.5 mg
• polylactic acid 198.6 mg • D-mannitol 38.9 mg
The second chamber contains: carboxymethylcellulose sodium 7.5 mg
• D-mannitol 75.0 mg • polysorbate 80 1.5 mg • water for injection, USP,
and glacial acetic acid, USP to control pH
Rx only

NDC 0074-3683-03
FOR ADULT USE
30 mg for 4-month administration
Single Dose Administration Kit with prefilled dual-chamber syringe.
LupronDepot®
(Leuprolide Acetate for Depot Suspension)
30 mg for 4-month administration
FOR INTRAMUSCULAR INJECTION
The front chamber contains: leuprolide acetate 30 mg
• polylactic acid 264.8 mg • D-mannitol 51.9 mg
The second chamber contains: carboxymethylcellulose sodium 7.5 mg
• D-mannitol 75.0 mg • polysorbate 80 1.5 mg • water for injection, USP,
and glacial acetic acid, USP to control pH
Rx only
NDC 0074–3642–03
FOR ADULT USE 7.5 mg for 1–month administration
Single Dose Administration Kit with prefilled dual-chamber syringe.
LupronDepot®
(Leuprolide Acetate for Depot Suspension)
7.5 mg for 1–month administration
FOR INTRAMUSCULAR INJECTION
The front chamber contains: leuprolide acetate 7.5 mg • purified gelatin 1.3 mg • DL-lactic and glycolic acids copolymer 66.2 mg • D-mannitol 13.2 mg
The second chamber contains: carboxymethylcellulose sodium 5 mg • D-mannitol 50 mg • polysorbate 80 1 mg • water for injection, USP, and glacial acetic acid, USP to control pH
Rx only

| LUPRON DEPOT
leuprolide acetate kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| LUPRON DEPOT
leuprolide acetate kit |
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
| LUPRON DEPOT
leuprolide acetate kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| LUPRON DEPOT
leuprolide acetate kit |
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
| Labeler - AbbVie Inc. (078458370) |
Frequently asked questions
- How long should you take it for prostate cancer?
- Are Lupron Depot and Eligard the same drug?
- Can you get pregnant on Lupron Depot?
- Is this a chemotherapy treatment?
- Does it need to be refrigerated?
- How do you inject it for prostate cancer?
- Will I get my period while on Lupron?
- What does this drug do for IVF?
More about Lupron Depot (leuprolide)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (215)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- Support group
- Drug class: gonadotropin releasing hormones
- En español
Patient resources
Professional resources
Other brands
Eligard, Camcevi, Fensolvi, Lupron Depot-Gyn