Lotensin Prescribing Information
Package insert / product label
Generic name: benazepril hydrochloride
Dosage form: tablet
Drug class: Angiotensin Converting Enzyme Inhibitors
Medically reviewed by Drugs.com. Last updated on Feb 22, 2023.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
LOTENSIN® (benazepril hydrochloride, USP) tablets, for oral use
Initial U.S. Approval: 1991
Indications and Usage for Lotensin
Lotensin is an angiotensin-converting enzyme (ACE) inhibitor indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. (1)
Lotensin Dosage and Administration
- Adult Patients: Initiate with 10 mg once daily (or 5 mg if patients is on diuretic). Titrate to 40 mg daily based on blood pressure response. (2.1)
- Pediatric patients age 6 years and above with glomerular filtration rate (GFR) >30 mL/min/1.73 m2: Initiate with 0.2 mg/kg once daily. Maximum dose is 0.6 mg/kg once daily.
- Renal Impairment: Initiate with 5 mg once daily in patients with GFR <30 mL/min/1.73 m2 (serum creatinine >3 mg/dL) (2.2)
Dosage Forms and Strengths
- Tablets: 10 mg, 20 mg, 40 mg
Contraindications
Warnings and Precautions
Adverse Reactions/Side Effects
The most common adverse reactions leading to discontinuation were headache (0.6%) and cough (0.5%) (6)
To report SUSPECTED ADVERSE REACTIONS, contact Validus Pharmaceuticals LLC at 1-866-982-5438 (1-866-9VALIDUS) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Diuretics: Excessive drop in blood pressure (7.1)
- Antidiabetics: Increased risk of hypoglycemia (7.2)
- NSAIDS: Increased risk of renal impairment and loss of antihypertensive efficacy (7.3)
- Dual inhibition of the renin-angiotensin system: Increased risk of renal impairment, hypotension and hyperkalemia (7.4)
- Lithium: Symptoms of lithium toxicity (7.6)
- Neprilysin Inhibitor: Increased risk of angioedema (7.7)
- Gold: Nitritoid reactions (7.8)
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2019
Related/similar drugs
amlodipine, lisinopril, metoprolol, losartan, furosemide, carvedilol, hydrochlorothiazide
Full Prescribing Information
WARNING: FETAL TOXICITY
When pregnancy is detected, discontinue Lotensin as soon as possible.
Drugs that act directly on the renin-angiotensin system can cause injury and death to the developing fetus [see Warnings and Precautions (5.1)].
1. Indications and Usage for Lotensin
Lotensin® is indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes including the class to which this drug principally belongs.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program’s Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mm Hg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in Black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
It may be used alone or in combination with thiazide diuretics.
2. Lotensin Dosage and Administration
2.1 Recommended Dosage
ADULTS
The recommended initial dose for patients not receiving a diuretic is 10 mg once a day. The usual maintenance dosage range is 20 to 40 mg per day administered as a single dose or in two equally divided doses. A dose of 80 mg gives an increased response, but experience with this dose is limited. The divided regimen was more effective in controlling trough (pre-dosing) blood pressure than the same dose given as a once-daily regimen.
Use with diuretics in adults
The recommended starting dose of Lotensin in a patient on a diuretic is 5 mg once daily. If blood pressure is not controlled with Lotensin alone, a low dose of diuretic may be added.
PEDIATRIC PATIENTS 6 YEARS OF AGE AND OLDER
The recommended starting dose for pediatric patients is 0.2 mg/kg once per day. Titrate as needed to 0.6 mg/kg once per day. Doses above 0.6 mg/kg (or in excess of 40 mg daily) have not been studied in pediatric patients.
Lotensin is not recommended in pediatric patients less than 6 years of age or in pediatric patients with GFR less than 30 mL/min/1.73m² [see Use in Specific Populations (8.3)].
2.2 Dose Adjustment for Renal Impairment
For adults with a GFR <30 mL/min/1.73 m² (serum creatinine >3 mg/dL), the recommended initial dose is 5 mg Lotensin once daily. Dosage may be titrated upward until blood pressure is controlled or to a maximum total daily dose of 40 mg. Lotensin can also worsen renal function [see Warnings and Precautions (5.3)].
2.3 Preparation of Suspension (for 150 mL of a 2 mg/mL Suspension)
Add 75 mL of Ora-Plus®* oral suspending vehicle to an amber polyethylene terephthalate (PET) bottle containing fifteen Lotensin 20 mg tablets, and shake for at least two minutes. Allow the suspension to stand for a minimum of 1 hour. After the standing time, shake the suspension for a minimum of one additional minute. Add 75 mL of Ora-Sweet®* oral syrup vehicle to the bottle and shake the suspension to disperse the ingredients. The suspension should be refrigerated at 2º to 8°C (36º to 46°F) and can be stored for up to 30 days in the PET bottle with a child-resistant screw- cap closure. Shake the suspension before each use.
*Ora-Plus® and Ora-Sweet® are registered trademarks of Paddock Laboratories, Inc. Ora Plus® contains carrageenan, citric acid, methylparaben, microcrystalline cellulose, carboxymethylcellulose sodium, potassium sorbate, simethicone, sodium phosphate monobasic, xanthan gum, and water. Ora-Sweet® contains citric acid, berry citrus flavorant, glycerin, methylparaben, potassium sorbate, sodium phosphate monobasic, sorbitol, sucrose, and water.
3. Dosage Forms and Strengths
Tablets: 10 mg, 20 mg, and 40 mg
- Each 10 mg tablet is dark yellow with “10” on one side and “LOTENSIN” on the other
- Each 20 mg tablet is pink with “20” on one side and “LOTENSIN” on the other
- Each 40 mg tablet is dark rose with “40” on one side and “LOTENSIN” on the other
4. Contraindications
Lotensin is contraindicated in patients:
- who are hypersensitive to benazepril or to any other ACE inhibitor
- with a history of angioedema with or without previous ACE inhibitor treatment
Lotensin is contraindicated in combination with a neprilysin inhibitor (e.g., sacubitril). Do not administer Lotensin within 36 hours of switching to or from sacubitril/valsartan, a neprilysin inhibitor [see Warnings and Precautions (5.2)].
Do not coadminister aliskiren with angiotensin receptor blockers, ACE inhibitors, including Lotensin in patients with diabetes [see Drug Interactions (7.4)].
5. Warnings and Precautions
5.1 Fetal Toxicity
Lotensin can cause fetal harm when administered to a pregnant woman. Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death.
Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue Lotensin as soon as possible [see Use in Specific Populations (8.1)].
5.2 Angioedema and Anaphylactoid Reactions
Angioedema
Head and Neck Angioedema
Angioedema of the face, extremities, lips, tongue, glottis, and/or larynx including some fatal reactions, have occured in patients treated with Lotensin. Patients with involvement of the tongue, glottis or larynx are likely to experience airway obstruction, especially those with a history of airway surgery. Lotensin should be promptly discontinued and appropriate therapy and monitoring should be provided until complete and sustained resolution of signs and symptoms of angioedema has occurred.
Patients with a history of angioedema unrelated to ACE inhibitor therapy may be at increased risk of angioedema while receiving an ACE inhibitor [see Contraindications (4)]. ACE inhibitors have been associated with a higher rate of angioedema in Black than in non-Black patients.
Patients receiving coadministration of ACE inhibitor and mTOR (mammalian target of rapamycin) inhibitor (e.g., temsirolimus, sirolimus, everolimus) therapy or a neprilysin inhibitor may be at increased risk for angioedema [see Drug Interactions (7.7)].
Intestinal Angioedema
Intestinal angioedema has occurred in patients treated with ACE inhibitors. These patients presented with abdominal pain (with or without nausea or vomiting); in some cases there was no prior history of facial angioedema and C-1 esterase levels were normal. In some cases, the angioedema was diagnosed by procedures including abdominal CT scan or ultrasound, or at surgery, and symptoms resolved after stopping the ACE inhibitor.
Anaphylactoid Reactions
Anaphylactoid Reactions During Desensitization
Two patients undergoing desensitizing treatment with hymenoptera venom while receiving ACE inhibitors sustained life-threatening anaphylactoid reactions.
Anaphylactoid Reactions During Dialysis
Sudden and potentially life threatening anaphylactoid reactions have occurred in some patients dialyzed with high-flux membranes and treated concomitantly with an ACE inhibitor. In such patients, dialysis must be stopped immediately, and aggressive therapy for anaphylactoid reactions must be initiated. Symptoms have not been relieved by antihistamines in these situations. In these patients, consideration should be given to using a different type of dialysis membrane or a different class of antihypertensive agent. Anaphylactoid reactions have also been reported in patients undergoing low-density lipoprotein apheresis with dextran sulfate absorption.
5.3 Impaired Renal Function
Monitor renal function periodically in patients treated with Lotensin. Changes in renal function, including acute renal failure, can be caused by drugs that inhibit the renin-angiotensin system.
Patients whose renal function may depend on the activity of the renin-angiotensin system (e.g., patients with renal artery stenosis, chronic kidney disease, severe congestive heart failure, post- myocardial infarction, or volume depletion) may be at particular risk of developing acute renal failure on Lotensin. Consider withholding or discontinuing therapy in patients who develop a clinically significant decrease in renal function on Lotensin.
5.4 Hypotension
Lotensin can cause symptomatic hypotension, sometimes complicated by oliguria, progressive azotemia, acute renal failure, or death. Patients at risk of excessive hypotension include those with the following conditions or characteristics: heart failure with systolic blood pressure below 100 mm Hg, ischemic heart disease, cerebrovascular disease, hyponatremia, high dose diuretic therapy, renal dialysis, or severe volume and/or salt depletion of any etiology.
In such patients, follow closely for the first 2 weeks of treatment and whenever the dose of benazepril or diuretic is increased. Avoid use of Lotensin in patients who are hemodynamically unstable after acute MI.
Surgery/Anesthesia
In patients undergoing major surgery or during anesthesia with agents that produce hypotension, Lotensin may block angiotensin II formation secondary to compensatory renin release. If hypotension occurs, correct by volume expansion.
5.5 Hyperkalemia
Serum potassium should be monitored periodically in patients receiving Lotensin. Drugs that inhibit the renin-angiotensin system can cause hyperkalemia. Risk factors for the development of hyperkalemia include renal insufficiency, diabetes mellitus, and the concomitant use of potassium-sparing diuretics, potassium supplements and/or potassium-containing salt substitutes [see Drug Interactions (7.1)].
5.6 Hepatic Failure
ACE inhibitors have been associated with a syndrome that starts with cholestatic jaundice and progresses to fulminant hepatic necrosis and (sometimes) death. The mechanism of this syndrome is not understood. Patients receiving ACE inhibitors who develop jaundice or marked elevations of hepatic enzymes should discontinue the ACE inhibitor and receive appropriate medical follow-up.
6. Adverse Reactions/Side Effects
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
Lotensin has been evaluated for safety in over 6000 patients with hypertension; over 700 of these patients were treated for at least one year. The overall incidence of reported adverse events was similar in Lotensin and placebo patients.
The reported side effects were generally mild and transient, and there was no relation between side effects and age, duration of therapy, or total dosage within the range of 2 to 80 mg.
Discontinuation of therapy because of a side effect was required in approximately 5% of U.S. patients treated with Lotensin and in 3% of patients treated with placebo. The most common reasons for discontinuation were headache (0.6%) and cough (0.5%).
Adverse reactions seen in at least 1% greater frequency in patients treated with Lotensin than placebo were headache (6% vs. 4%), dizziness (4% vs. 2%), somnolence (2% vs. 0%) and postural dizziness (2% vs. 0%).
Adverse reactions reported in controlled clinical trials (less than 1% more on benazepril than on placebo), and rarer events seen in post-marketing experience, include the following (in some, a causal relationship to drug use is uncertain):
Dermatologic: Stevens-Johnson syndrome, pemphigus, apparent hypersensitivity reactions (manifested by dermatitis, pruritus, or rash), photosensitivity, and flushing.
Gastrointestinal: Nausea, pancreatitis, constipation, gastritis, vomiting, and melena.
Hematologic: Thrombocytopenia and hemolytic anemia.
Neurologic/Psychiatric: Anxiety, decreased libido, hypertonia, insomnia, nervousness, and paresthesia.
Other: Fatigue, asthma, bronchitis, dyspnea, sinusitis, urinary tract infection, frequent urination, infection, arthritis, impotence, alopecia, arthralgia, myalgia, asthenia, sweating.
Laboratory Abnormalities:
Elevations of uric acid, blood glucose, serum bilirubin, and liver enzymes [see Warnings and Precautions (5)] have been reported, as have incidents of hyponatremia, electrocardiographic changes, eosinophilia, and proteinuria.
7. Drug Interactions
7.1 Diuretics
Hypotension
Patients on diuretics, especially those in whom diuretic therapy was recently instituted, may occasionally experience an excessive reduction of blood pressure after initiation of therapy with Lotensin. The possibility of hypotensive effects with Lotensin can be minimized by either discontinuing or decreasing the dose of diuretic prior to initiation of treatment with Lotensin [see Dosage and Administration (2.1)].
Hyperkalemia
Potassium-sparing diuretics (spironolactone, amiloride, triamterene, and others) can increase the risk of hyperkalemia. Therefore, if concomitant use of such agents is indicated, monitor the patient’s serum potassium frequently. Lotensin attenuates potassium loss caused by thiazide-type diuretics.
7.2 Antidiabetics
Concomitant administration of Lotensin and antidiabetic medicines (insulins, oral hypoglycemic agents) may increase the risk of hypoglycemia.
7.3 Non-Steroidal Anti-Inflammatory Agents including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, coadministration of NSAIDs, including selective COX-2 inhibitors, with ACE inhibitors, including benazepril, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving benazepril and NSAID therapy.
The antihypertensive effect of ACE inhibitors, including benazepril, may be attenuated by NSAIDs.
7.4 Dual Blockade of the Renin-Angiotensin System (RAS)
Dual Blockade of the RAS with angiotensin receptor blockers, ACE inhibitors, or aliskiren is associated with increased risks of hypotension, hyperkalemia, and changes in renal function (including acute renal failure) compared to monotherapy. Most patients receiving the combination of two RAS inhibitors do not obtain any additional benefit compared to monotherapy. In general, avoid combined use of RAS inhibitors. Closely monitor blood pressure, renal function and electrolytes in patients on Lotensin and other agents that affect the RAS.
Do not coadminister aliskiren with Lotensin in patients with diabetes. Avoid use of aliskiren with Lotensin in patients with renal impairment (GFR < 60 mL/min).
7.5 Mammalian Target of Rapamycin (mTOR) Inhibitors
Patients receiving coadministration of ACE inhibitor and mTOR inhibitor (e.g., temsirolimus, sirolimus, everolimus) therapy may be at increased risk for angioedema. Monitor for signs of angioedema [see Warnings and Precautions (5.2)].
7.6 Lithium
Lithium toxicity has been reported in patients receiving lithium concomitantly with Lotensin. Lithium toxicity was usually reversible upon discontinuation of lithium or Lotensin. Monitor serum lithium levels during concurrent use.
7.7 Neprilysin Inhibitor
Patients taking concomitant neprilysin inhibitors may be at increased risk for angioedema [see Warnings and Precautions].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Lotensin can cause fetal harm when administered to a pregnant woman. Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin angiotensin system from other antihypertensive agents. When pregnancy is detected, discontinue Lotensin as soon as possible.
The estimated background risk of major birth defects and miscarriage for the indicated population are unknown. All pregnancies have a background risk of birth defects, loss, or other adverse outcomes. In the general U.S. population, the estimated background risk of major defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Hypertension in pregnancy increases the maternal rush for pre-eclampsia, gestational diabetes, premature delivery, and delivery complications (e.g., need for cesarean section, and post-partum hemorrhage). Hypertension increases the fetal risk for intrauterine growth restriction and intrauterine death. Pregnant women with hypertension should be carefully monitored and managed accordingly.
Fetal/Neonatal Adverse Reactions
Oligohydramnios in pregnant women who use drugs affecting the renin-angiotensin system in the second and third trimesters of pregnancy can result in the following: reduced fetal renal function leading to anuria and renal failure, fetal lung hypoplasia and skeletal deformations, including skull hypoplasia, hypotension, and death. In the unusual case that there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system for a particular patient, apprise the mother of the potential risk to the fetus.
Perform serial ultrasound examinations to assess the intra-amniotic environment.
Fetal testing may be appropriate, based on the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. Closely observe infants with histories of in utero exposure to Lotensin for hypotension, oliguria, and hyperkalemia. If oliguria or hypotension occur in neonates with a history of in utero exposure to Lotensin, support blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and substituting for disordered renal function.
8.2 Lactation
Minimal amounts of unchanged benazepril and of benazeprilat are excreted into the breast milk of lactating women treated with benazepril. A newborn child ingesting entirely breast milk would receive less than 0.1% of the mg/kg maternal dose of benazepril and benazeprilat.
8.3 Pediatric Use
The antihypertensive effects of Lotensin have been evaluated in a double-blind study in pediatric patients 7 to 16 years of age [see Clinical Pharmacology (12.3)]. The pharmacokinetics of Lotensin have been evaluated in pediatric patients 6 to 16 years of age [see Clinical Pharmacology (12.3)].
Infants below the age of 1 year should not be given Lotensin because of the risk of effects on kidney development.
Safety and effectiveness of Lotensin have not been established in pediatric patients less than 6 years of age or in children with glomerular filtration rate < 30 mL/min/1.73m² [see Dosage and Administration (2.1) and Clinical Pharmacology 12.3)].
8.4 Geriatric Use
Of the total number of patients who received benazepril in U.S. clinical studies of Lotensin, 18% were 65 or older while 2% were 75 or older. No overall differences in effectiveness or safety were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Benazepril and benazeprilat are substantially excreted by the kidney. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function [see Dosage and Administration (2.2)].
8.5 Race
ACE inhibitors, including Lotensin, as monotherapy, have an effect on blood pressure that is less in Black patients than in non-Blacks.
8.6 Renal Impairment
Dose adjustment of Lotensin is required in patients undergoing hemodialysis or whose creatinine clearance is ≤ 30 mL/min. No dose adjustment of Lotensin is required in patients with creatinine clearance > 30 mL/min [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
10. Overdosage
Single oral doses of 3 g/kg benazepril were associated with significant lethality in mice. Rats, however, tolerated single oral doses of up to 6 g/kg. Reduced activity was seen at 1 g/kg in mice and at 5 g/kg in rats. Human overdoses of benazepril have not been reported, but the most common manifestation of human benazepril overdosage is likely to be hypotension, for which the usual treatment would be intravenous infusion of normal saline solution. Hypotension can be associated with electrolyte disturbances and renal failure.
Benazepril is only slightly dialyzable, but consider dialysis to support patients with severely impaired renal function [see Warnings and Precautions (5.3)].
If ingestion is recent, consider activated charcoal. Consider gastric decontamination (e.g., vomiting, gastric lavage) in the early period after ingestion.
Monitor for blood pressure and clinical symptoms. Supportive management should be employed to ensure adequate hydration and to maintain systemic blood pressure.
In the case of marked hypotension, infuse physiological saline solution; as needed, consider vasopressors (e.g., catecholamines i.v.).
11. Lotensin Description
Benazepril hydrochloride, USP is a white to off-white crystalline powder, soluble (> 100 mg/mL) in water, in ethanol, and in methanol. Its chemical name is benazepril 3-[[1-(ethoxy-carbonyl)-3 phenyl-(1S)-propyl]amino]-2,3,4,5-tetrahydro-2-oxo-1H-1-(3S)-benzazepine-1-acetic acid monohydrochloride; its structural formula is
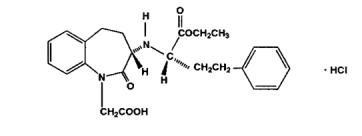
Its empirical formula is C24H28N2O5•HCl, and its molecular weight is 460.96.
Benazeprilat, the active metabolite of benazepril, is a non-sulfhydryl angiotensin-converting enzyme inhibitor.
Lotensin is supplied as tablets containing 10 mg, 20 mg, and 40 mg of benazepril hydrochloride for oral administration. The inactive ingredients are colloidal silicon dioxide, crospovidone, hydrogenated castor oil (10 mg and 20 mg tablets), hypromellose, iron oxides, lactose, magnesium stearate (40 mg tablets), microcrystalline cellulose, polysorbate 80, propylene glycol (40 mg tablets), starch, talc, and titanium dioxide.
12. Lotensin - Clinical Pharmacology
12.1 Mechanism of Action
Benazepril and benazeprilat inhibit angiotensin-converting enzyme (ACE) in human subjects and animals. Benazeprilat has much greater ACE inhibitory activity than does benazepril.
ACE is a peptidyl dipeptidase that catalyzes the conversion of angiotensin I to the vasoconstrictor substance, angiotensin II. Angiotensin II also stimulates aldosterone secretion by the adrenal cortex.
Inhibition of ACE results in decreased plasma angiotensin II, which leads to decreased vasopressor activity and to decreased aldosterone secretion. The latter decrease may result in a small increase of serum potassium.
Removal of angiotensin II negative feedback on renin secretion leads to increased plasma renin activity. In animal studies, benazepril had no inhibitory effect on the vasopressor response to angiotensin II and did not interfere with the hemodynamic effects of the autonomic neurotransmitters acetylcholine, epinephrine, and norepinephrine.
ACE is identical to kininase, an enzyme that degrades bradykinin. Whether increased levels of bradykinin, a potent vasodepressor peptide, play a role in the therapeutic effects of Lotensin remains to be elucidated. While the mechanism through which benazepril lowers blood pressure is believed to be primarily suppression of the renin-angiotensin-aldosterone system, benazepril has an antihypertensive effect even in patients with low-renin hypertension.
12.2 Pharmacodynamics
Single and multiple doses of 10 mg or more of Lotensin cause inhibition of plasma ACE activity by at least 80% to 90% for at least 24 hours after dosing. Pressor responses to exogenous angiotensin I were inhibited by 60% to 90% (up to 4 hours post-dose) at the 10 mg dose.
Drug Interactions
Lotensin has been used concomitantly with beta-adrenergic-blocking agents, calcium- channel-blocking agents, diuretics, digoxin, and hydralazine, without evidence of clinically important adverse interactions. Benazepril, like other ACE inhibitors, has had less than additive effects with beta-adrenergic blockers, presumably because both drugs lower blood pressure by inhibiting parts of the renin-angiotensin system.
12.3 Pharmacokinetics
The pharmacokinetics of benazepril are approximately dose-proportional within the dosage range of 10 to 80 mg.
Following oral administration of Lotensin, peak plasma concentrations of benazepril, and its active metabolite benazeprilat are reached within 0.5 to1.0 hour and 1 to 2 hours, respectively. While the bioavailability of benazepril is not affected by food, time to peak plasma concentrations of benazeprilat is delayed to 2 to 4 hours.
The serum protein binding of benazepril is about 96.7% and that of benazeprilat about 95.3%, as measured by equilibrium dialysis; on the basis of in vitro studies, the degree of protein binding should be unaffected by age, hepatic dysfunction, or concentration (over the concentration range of 0.24 - 23.6 µmol/L).
Benazepril is almost completely metabolized to benazeprilat by cleavage of the ester group (primarily in liver). Both benazepril and benazeprilat undergo glucuronidation.
Benazepril and benazeprilat are cleared predominantly by renal excretion. About 37% of an orally administered dose was recovered in urine as benazeprilat (20%), benazeprilat glucuronide (8%), benazepril glucuronide (4%) and as trace amounts of benazepril. Nonrenal (i.e., biliary) excretion accounts for approximately 11% to 12% of benazeprilat excretion. The effective half-life of benazeprilat following once daily repeat oral administration of benazepril hydrochloride is 10 to 11 hours. Thus, steady-state concentrations of benazeprilat should be reached after 2 or 3 doses of benazepril hydrochloride given once daily.
Accumulation ratio based on AUC of benazeprilat was 1.19 following once daily administration.
Specific Populations
Renal impairment
The pharmacokinetics of systemic exposure to benazepril and benazeprilat in patients with mild-to-moderate renal insufficiency (creatinine clearance > 30 mL/min) is similar to that in patients with normal renal function. In patients with creatinine clearance ≤ 30 mL/min, peak benazeprilat levels and the initial (alpha phase) half-life increase, and time to steady-state may be delayed [see Dosage and Administration (2)].
When dialysis was started 2 hours after ingestion of 10 mg of benazepril, approximately 6% of benazeprilat was removed in 4 hours of dialysis. The parent compound, benazepril, was not detected in the dialysate.
Hepatic impairment
In patients with hepatic insufficiency (due to cirrhosis), the pharmacokinetics of benazeprilat are essentially unaltered.
Drug Interactions
The pharmacokinetics of benazepril are not affected by the following drugs: hydrochlorothiazide, furosemide, chlorthalidone, digoxin, propranolol, atenolol, nifedipine, amlodipine, naproxen, acetylsalicylic acid, or cimetidine. Likewise the administration of benazepril does not substantially affect the pharmacokinetics of these medications (cimetidine kinetics were not studied).
Pediatrics
The pharmacokinetics of benazaprilat, evaluated in pediatric patients with hypertension following oral administration of a single dose is presented in table below.
| Age group | Cmax
(ng/mL) | Tmax*
(h) | AUC0-inf (ng/mL*h) | CL/F/wt (L/h/Kg) | T1/2
(h) |
| >1 to ≤ 24 months | 277 | 1 | 1328 | 0.26 | 5.0 |
| n=5 | (192, 391) | (0.6, 2) | (773, 2117) | (0.18, 0.4) | (4, 5.8) |
| >2 to ≤ 6 years | 200 | 2 | 978 | 0.36 | 5.5 |
| n=7 | (168, 244) | (1.4, 2.4) | (842, 1152) | (0.31, 0.42) | (4.7, 6.5) |
| >6 to ≤ 12 years | 221 | 2 | 1041 | 0.25 | 5.5 |
| n=7 | (194, 258) | (1.2, 2.2) | (855, 1313) | (0.21, 0.31) | (4.7, 6.5) |
| >12 to ≤ 17 years | 287 | 2 | 1794 | 0.16 | 5.1 |
| n=8 | (217, 420) | (1.3, 2.3) | (1478, 2340) | (0.13, 0.21) | (4.2, 5.7) |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No evidence of carcinogenicity was found when benazepril was administered to rats and mice for up to two years at doses of up to 150 mg/kg/day. When compared on the basis of body weights, this dose is 110 times the maximum recommended human dose. When compared on the basis of body surface areas, this dose is 18 and 9 times (rats and mice, respectively) the maximum recommended human dose (calculations assume a patient weight of 60 kg). No mutagenic activity was detected in the Ames test in bacteria (with or without metabolic activation), in an in vitro test for forward mutations in cultured mammalian cells, or in a nucleus anomaly test. In doses of 50 to 500 mg/kg/day (6 to 60 times the maximum recommended human dose based on mg/m² comparison and 37 to 375 times the maximum recommended human dose based on a mg/kg comparison), Lotensin had no adverse effect on the reproductive performance of male and female rats.
14. Clinical Studies
Hypertension
Adult Patients
In single-dose studies, Lotensin lowered blood pressure within 1 hour, with peak reductions achieved between 2 and 4 hours after dosing. The antihypertensive effect of a single dose persisted for 24 hours. In multiple-dose studies, once-daily doses of between 20 mg and 80 mg decreased seated pressure 24 hours after dosing by about 6 to 12 mm Hg systolic and 4 to 7 mm Hg diastolic. The trough values represent reductions of about 50% of that seen at peak.
Four dose-response studies using once-daily dosing were conducted in 470 mild-to-moderate hypertensive patients not using diuretics. The minimal effective once-daily dose of Lotensin was 10 mg; but further falls in blood pressure, especially at morning trough, were seen with higher doses in the studied dosing range (10 to 80 mg). In studies comparing the same daily dose of Lotensin given as a single morning dose or as a twice-daily dose, blood pressure reductions at the time of morning trough blood levels were greater with the divided regimen.
The antihypertensive effects of Lotensin were not appreciably different in patients receiving high- or low-sodium diets.
In normal human volunteers, single doses of benazepril caused an increase in renal blood flow but had no effect on glomerular filtration rate.
Use of Lotensin in combination with thiazide diuretics gives a blood-pressure-lowering effect greater than that seen with either agent alone. By blocking the renin-angiotensin-aldosterone axis, administration of Lotensin tends to reduce the potassium loss associated with the diuretic.
Pediatric Patients
In a clinical study of 107 pediatric patients, 7 to 16 years of age, with either systolic or diastolic pressure above the 95th percentile, patients were given 0.1 or 0.2 mg/kg then titrated up to 0.3 or 0.6 mg/kg with a maximum dose of 40 mg once daily. After four weeks of treatment, the 85 patients whose blood pressure was reduced on therapy were then randomized to either placebo or benazepril and were followed up for an additional two weeks. At the end of two weeks, blood pressure (both systolic and diastolic) in children withdrawn to placebo rose by 4 to 6 mm Hg more than in children on benazepril. No dose-response was observed.
16. How is Lotensin supplied
Lotensin is available as:
| Dose | Color | Engraving | Bottle of 100 |
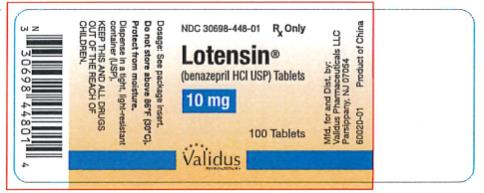
| 10 mg | Dark Yellow | Lotensin 10 | NDC 30698-448-01 |
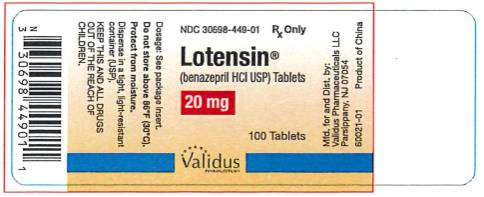
| 20 mg | Pink | Lotensin 20 | NDC 30698-449-01 |
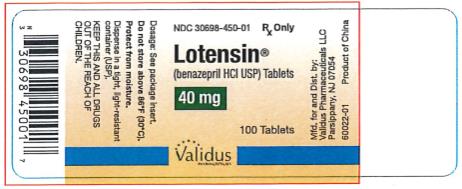
| 40 mg | Dark Rose | Lotensin 40 | NDC 30698-450-01 |
Storage: Do not store above 86°F (30ºC). Protect from moisture. Dispense in a tight container (USP).
17. Patient Counseling Information
Pregnancy: Tell female patients of childbearing age about the consequences of exposure to Lotensin during pregnancy. Discuss treatment options with women planning to become pregnant. Instruct patients to report pregnancies to their physicians as soon as possible.
Angioedema: Angioedema, including laryngeal edema, can occur at any time with treatment with ACE inhibitors. Tell patients to report immediately any signs or symptoms suggesting angioedema (swelling of face, eyes, lips, or tongue, or difficulty in breathing) and to take no more drugs until they have consulted with the prescribing physician.
Symptomatic Hypotension: Tell patients to report light-headedness especially during the first few days of therapy. If actual syncope occurs, tell the patients to discontinue the drug until they have consulted with the prescribing physician.
Tell patients that excessive perspiration and dehydration may lead to an excessive fall in blood pressure because of a reduction in fluid volume. Other causes of volume depletion such as vomiting or diarrhea may also lead to a fall in blood pressure; advise patients accordingly.
Hyperkalemia: Tell patients not to use potassium supplements or salt substitutes containing potassium without consulting the prescribing physician.
Hypoglycemia: Tell diabetic patients treated with oral antidiabetic agents or insulin starting an ACE inhibitor to monitor for hypoglycemia closely, especially during the first month of combined use.
Manufactured for and Distributed by:
Validus Pharmaceuticals LLC
119 Cherry Hill Road, Suite 310
Parsippany, NJ 07054
info@validuspharma.com
www.validuspharma.com
1-866-982-5438 (1-866-9VALIDUS)
Product of China
© 2018 Validus Pharmaceuticals LLC
60023-05 January 2019
| LOTENSIN
benazepril hydrochloride tablet |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| LOTENSIN
benazepril hydrochloride tablet |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| LOTENSIN
benazepril hydrochloride tablet |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| Labeler - Validus Pharmaceuticals LLC (801194619) |
More about Lotensin (benazepril)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (5)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- Generic availability
- Drug class: Angiotensin Converting Enzyme Inhibitors
- Breastfeeding
- En español
