Optimark Prescribing Information
Package insert / product label
Generic name: gadoversetamide
Dosage form: injection, solution
Drug class: Magnetic resonance imaging contrast media
Medically reviewed by Drugs.com. Last updated on Jun 17, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
OPTIMARK (gadoversetamide) injection, for intravenous use
Initial U.S. Approval: 1999
WARNING: NEPHROGENIC SYSTEMIC FIBROSIS (NSF)
See full prescribing information for complete boxed warning.
Gadolinium-based contrast agents (GBCAs) increase the risk for NSF among patients with impaired elimination of the drugs. Avoid use of GBCAs in these patients unless the diagnostic information is essential and not available with non-contrasted MRI or other modalities. NSF may result in fatal or debilitating fibrosis affecting the skin, muscle and internal organs (5.1).
-
Do not administer Optimark to patients with:
- chronic, severe kidney disease (GFR <30 mL/min/1.73m2), or
- acute kidney injury (4).
- Screen patients for acute kidney injury and other conditions that may reduce renal function. For patients at risk for chronically reduced renal function (e.g. age >60 years, hypertension or diabetes), estimate the glomerular filtration rate (GFR) through laboratory testing (5.1).
Recent Major Changes
Warnings and Precautions, Gadolinium Retention (5.3) 4/2018
Indications and Usage for Optimark
Optimark is a gadolinium-based paramagnetic contrast agent for diagnostic magnetic resonance imaging (MRI) indicated for intravenous use:
- In patients with abnormal blood-brain barrier or abnormal vascularity of the brain, spine and associated tissues (1)
- To provide contrast enhancement and facilitate visualization of lesions with abnormal vascularity in the liver in patients who are highly suspect for liver structural abnormalities on computed tomography (1)
Optimark Dosage and Administration
Dosage Forms and Strengths
Injection: Each ml of Optimark contains 330.9 mg gadoversetamide (3)
Contraindications
Warnings and Precautions
- Nephrogenic Systemic Fibrosis (NSF) has occurred in patients with impaired elimination of Gadolinium Based Contrast Agents (GBCAs). Higher than recommended dosing or repeat dosing appears to increase the risk. (5.1)
- Hypersensitivity reactions including fatal reactions have occurred, particularly in patients with history of allergy, drug reactions, or other hypersensitivity-like disorders. Monitor these patients closely during and after administration of Optimark. (5.2)
- Gadolinium is retained for months or years in brain, bone, and other organs. (5.3)
- Acute Kidney Injury (AKI) has occurred in patients with preexisting renal insufficiency. Use the lowest necessary dose of Optimark in these patients. (5.4)
- Interference with laboratory measurements of serum iron, copper and zinc and in the measurement of serum calcium using the ortho-cresophthalin complexone (OCP) colorimetric method has occurred. (5.5)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence >2%) are: headache, vasodilatation, taste perversion, dizziness, nausea and paresthesia. (6)
To report SUSPECTED ADVERSE REACTIONS contact Liebel-Flarsheim Company LLC at 1-855-266-5037 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2018
Full Prescribing Information
WARNING: NEPHROGENIC SYSTEMIC FIBROSIS
Gadolinium-based contrast agents (GBCAs) increase the risk for NSF among patients with impaired elimination of the drugs. Avoid use of GBCAs in these patients unless the diagnostic information is essential and not available with non-contrasted MRI or other modalities. NSF may result in fatal or debilitating fibrosis affecting the skin, muscle and internal organs.
- Do not administer Optimark to patients with:
- chronic, severe kidney disease (GFR <30 mL/min/1.73m2), or
- acute kidney injury [see Contraindications (4)].
- Screen patients for acute kidney injury and other conditions that may reduce renal function. For patients at risk for chronically reduced renal function (e.g. age >60 years, hypertension or diabetes), estimate the glomerular filtration rate (GFR) through laboratory testing.
- Do not exceed the recommended Optimark dose and allow a sufficient period of time for elimination of the drug from the body prior to any re-administration [see Warnings and Precautions (5.1)].
1. Indications and Usage for Optimark
2. Optimark Dosage and Administration
2.1 Dosing Guidelines
- Administer Optimark as a bolus peripheral intravenous injection at a dose of 0.2 mL/kg (0.1 mmol/kg) and at a rate of 1 to 2 mL/sec delivered by manual or by power injection (see Table 1).
- Use sterile technique to withdraw and administer Optimark.
- Follow injection with a 5 mL normal saline flush to ensure complete administration of the contrast.
- Discard unused portions of the drug.
|
Table 1 Dosage Chart for Optimark Injection |
|
|
Body Weight |
0.1 mmol/kg |
|
40 |
8 |
|
50 |
10 |
|
60 |
12 |
|
70 |
14 |
|
80 |
16 |
|
90 |
18 |
|
100 |
20 |
|
110 |
22 |
|
120 |
24 |
|
130 |
26 |
|
140 |
28 |
|
150 |
30 |
2.2 Drug Handling
- Visually inspect Optimark for particulate matter and discoloration prior to administration. Do not use the solution if it is discolored or particulate matter is present.
- Do not mix Optimark with other medications or parenteral nutrition and do not administer Optimark in the same intravenous line as other medications because of the potential for chemical incompatibility.
2.3 Imaging
- Complete the imaging procedure within 1 hour of the injection of Optimark.
- Paramagnetic contrast agents may impair the visualization of lesions seen on non-contrast MRI. Interpret Optimark MR images with companion non-contrast MR images [see Clinical Pharmacology (12.2)].
3. Dosage Forms and Strengths
Optimark is supplied as a clear, colorless to slightly yellow solution for injection containing 330.9 mg gadoversetamide per mL (equivalent to 0.5 mmol/mL).
4. Contraindications
Optimark is contraindicated in patients with:
- chronic, severe kidney disease (glomerular filtration rate, GFR <30 mL/min/1.73m2)
- acute kidney injury
- known hypersensitivity reactions to gadolinium, gadoversetamide or any of the inert ingredients.
5. Warnings and Precautions
5.1 Nephrogenic Systemic Fibrosis (NSF)
Gadolinium-based contrast agents (GBCAs) increase the risk for NSF among patients with impaired elimination of the drugs. Avoid use of GBCAs among these patients unless the diagnostic information is essential and not available with non-contrast enhanced MRI or other modalities. The GBCA-associated NSF risk appears highest for patients with chronic, severe kidney disease (GFR <30 mL/min/1.73m2) as well as patients with acute kidney injury. Do not administer Optimark to these patients. The risk appears lower for patients with chronic, moderate kidney disease (GFR 30 to 59 mL/min/1.73m2) and little, if any, for patients with chronic, mild kidney disease (GFR 60 to 89 mL/min/1.73m2). NSF may result in fatal or debilitating fibrosis affecting the skin, muscle and internal organs. Report any diagnosis of NSF following Optimark administration to Liebel-Flarsheim Company LLC (1‑855‑266‑5037) or FDA (1-800-FDA-1088 or www.fda.gov/medwatch).
Screen patients for acute kidney injury and other conditions that may reduce renal function. Features of acute kidney injury consist of rapid (over hours to days) and usually reversible decrease in kidney function, commonly in the setting of surgery, severe infection, injury, or drug-induced kidney toxicity. Serum creatinine levels and estimated GFR may not reliably assess renal function in the setting of acute kidney injury. For patients at risk for chronic kidney disease (e.g., age >60 years, diabetes mellitus or chronic hypertension), estimate the GFR through laboratory testing.
Among the factors that may increase the risk for NSF are repeated or higher than recommended doses of a GBCA and the degree of renal impairment at the time of exposure. Record the specific GBCA and the dose administered to a patient. When administering Optimark, do not exceed the recommended dose and allow a sufficient period of time for elimination of the drug prior to any re-administration [see Dosage and Administration (2.1)].
5.2 Hypersensitivity Reactions
Severe hypersensitivity reactions including anaphylaxis have been observed with administration of gadolinium products including Optimark. Before administering Optimark ensure the availability of resuscitation equipment and personnel trained in resuscitation techniques. Patients with a history of allergy, drug reactions or other hypersensitivity-like disorders may be at greater risk and should be closely observed during the procedure and for several hours after drug administration. If a reaction occurs, stop Optimark and immediately begin appropriate therapy including resuscitation.
5.3 Gadolinium Retention
Gadolinium is retained for months or years in several organs. The highest concentrations (nanomoles per gram of tissue) have been identified in the bone, followed by other organs (e.g. brain, skin, kidney, liver, and spleen). The duration of retention also varies by tissue and is longest in bone. Linear GBCAs cause more retention than macrocyclic GBCAs. At equivalent doses, gadolinium retention varies among the linear agents with Omniscan (gadodiamide) and Optimark (gadoversetamide) causing greater retention than other linear agents [Eovist (gadoxetate disodium), Magnevist (gadopentetate dimeglumine), MultiHance (gadobenate dimeglumine)]. Retention is lowest and similar among the macrocyclic GBCAs [Dotarem (gadoterate meglumine), Gadavist (gadobutrol), ProHance (gadoteridol)].
Consequences of gadolinium retention in the brain have not been established. Pathologic and clinical consequences of GBCA administration and retention in skin and other organs have been established in patients with impaired renal function [see Warnings and Precautions (5.1)]. There are rare reports of pathologic skin changes in patients with normal renal function. Adverse events involving multiple organ systems have been reported in patients with normal renal function without an established causal link to gadolinium retention [see Adverse Reactions (6.2)].
While clinical consequences of gadolinium retention have not been established in patients with normal renal function, certain patients might be at higher risk. These include patients requiring multiple lifetime doses, pregnant and pediatric patients, and patients with inflammatory conditions. Consider the retention characteristics of the agent when choosing a GBCA for these patients. Minimize repetitive GBCA imaging studies, particularly closely spaced studies when possible.
5.4 Acute Kidney Injury (AKI)
In patients with chronically reduced renal function, acute kidney injury requiring dialysis has occurred with the use of GBCAs. The risk of acute kidney injury may increase with increasing dose of the contrast agent; administer the lowest dose necessary for adequate imaging.
5.5 Interference with Laboratory Testing
Interference by Optimark in the measurements of serum iron, copper and zinc has been observed. Optimark causes interference in the measurement of serum calcium using the ortho-cresophthalin complexone (OCP) colorimetric method. In the presence of Optimark, OCP produces an erroneous, low value for serum calcium. The magnitude of this artifact is proportional to the concentration of Optimark in the blood, and accurate values can be obtained approximately 90 minutes following injection. In patients with renal insufficiency, clearance of Optimark is slowed and the interference with calcium determination by OCP is prolonged. Neither the arsenazo III dye system nor the inductively coupled plasma mass spectroscopy methods for calcium assay are affected by Optimark.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in other sections of the prescribing information:
- Nephrogenic Systemic Fibrosis [see Contraindications (4), Boxed Warning and Warnings and Precautions (5.1)]
- Hypersensitivity reactions [see Contraindications (4) and Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The adverse reactions described in this section were observed in a total of 1,309 subjects (24 healthy volunteers and 1,285 patients in clinical trials). Patients ranged in age from 12 to 85 years (mean age of 50 years) and 680 subjects (52%) were men. The ethnic distribution was 84% White, 9% Black, 3% Asian, and 4% other.
Overall, 460 subjects (35%) reported at least one adverse reaction. Most adverse reactions were mild or moderate in severity. The most commonly noted adverse reactions were: injection associated discomfort (26%), headache (9.4%), vasodilatation (6.4%), taste perversion (6.2%), dizziness (3.7%), nausea (3.2%), and paresthesia (2.2%). Table 2 lists adverse reactions reported in 1% or greater of patients.
|
Table 2 Adverse Reactions Experienced by ≥1% of Patients |
|
|
Body System or Event |
Optimark (N = 1309) |
|
Injection associated discomfort |
26.4% |
|
Headache |
9.4% |
|
Vasodilatation |
6.4% |
|
Taste Perversion |
6.2% |
|
Dizziness |
3.7% |
|
Nausea |
3.2% |
|
Paresthesia |
2.2% |
|
Diarrhea |
1.9% |
|
Pain Abdomen |
1.8% |
|
Asthenia |
1.5% |
|
Injection Site Reaction |
1.5% |
|
Rhinitis |
1.5% |
|
Dyspepsia |
1.2% |
|
Pain Back |
1.2% |
|
Pain |
1.0% |
The following adverse reactions occurred in less than 1% of the patients:
Body as a Whole: allergic reaction, facial edema, fever, malaise, neck rigidity, neck pain, pelvic pain, increased sweating
Cardiovascular: arrhythmia, chest pain, hypertension, hypotension, pallor, palpitation, syncope, tachycardia, vasospasm
Digestive: anorexia, constipation, dry mouth, dysphagia, eructation, increased salivation, thirst, vomiting
Metabolic and Nutritional: increased creatinine, edema, hypercalcemia
Musculoskeletal: arthralgia, leg cramps, myalgia, spasm
Nervous System: agitation, anxiety, confusion, diplopia, dystonia, hypertonia, hypesthesia, somnolence, tremor, vertigo
Respiratory System: cough, dyspnea, laryngismus, pharyngitis, sinusitis, voice alteration
Skin and Appendages: erythema multiforme, pruritus, rash, thrombophlebitis, urticaria
Special Senses: parosmia, tinnitus
Urogenital: oliguria
6.2 Post-marketing Experience
The following adverse reactions have been identified during post-approval use of Optimark. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to Optimark.
- Nephrogenic Systemic Fibrosis (NSF)
- Hypersensitivity reactions including bronchospasm and laryngeal/pharyngeal edema
- Seizures
General Disorders and Administration Site Conditions: Adverse events with variable onset and duration have been reported after GBCA administration [see Warnings and Precautions (5.3)]. These include heterogeneous clusters of symptoms in the neurological, cutaneous, and musculoskeletal systems, fatigue, asthenia, pain syndromes, and gadolinium retention.
Skin: Gadolinium associated plaques
7. Drug Interactions
Drug interactions with other contrast agents and other drugs have not been studied.
8. Use In Specific Populations
8.1 Pregnancy
GBCAs cross the placenta and result in fetal exposure and gadolinium retention. The human data on the association between GBCAs and adverse fetal outcomes are limited and inconclusive. Because of the potential risks of gadolinium to the fetus, use Optimark only if imaging is essential during pregnancy and cannot be delayed.
Contrast enhancement is visualized in the human placenta and fetal tissues after maternal GBCA administration.
Cohort studies and case reports on exposure to GBCAs during pregnancy have not reported a clear association between GBCAs and adverse effects in the exposed neonates. However, a retrospective cohort study, comparing pregnant women who had a GBCA MRI to pregnant women who did not have an MRI, reported a higher occurrence of stillbirths and neonatal deaths in the group receiving GBCA MRI. Limitations of this study include a lack of comparison with non-contrast MRI and lack of information about the maternal indication for MRI. Overall, these data preclude a reliable evaluation of the potential risk of adverse fetal outcomes with the use of GBCAs in pregnancy.
GBCAs administered to pregnant non-human primates (0.1 mmol/kg on gestational days 85 and 135) result in measurable gadolinium concentration in the offspring in bone, brain, skin, liver, kidney, and spleen for at least 7 months. GBCAs administered to pregnant mice (2 mmol/kg daily on gestational days 16 through 19) result in measurable gadolinium concentrations in the pups in bone, brain, kidney, liver, blood, muscle, and spleen at one month postnatal age.
Gadoversetamide administered to rats reduced neonatal weights from birth through weaning at maternal doses of 0.5 mmol/kg/day (1 times the human dose based on body surface area) for 5 weeks (including gestation) and paternal doses of 0.5 mmol/kg/day for 12 weeks. This effect was not observed at 0.1 mmol/kg (0.2 times the human dose based on body surface area). Maternal toxicity was not observed at any dose.
Gadoversetamide injection caused a reduced mean fetal weight, abnormal liver lobation, delayed ossification of sternebrae, and delayed behavioral development (startle reflex and air righting reflex) in fetuses from female rats dosed with 4.9 mmol/kg/day (10 times the human dose based on body surface area) on days 7 through 17 of gestation. These effects were not observed at 0.7 mmol/kg/day (1 times the human dose based on body surface area). Maternal toxicity was observed at 4.9 mmol/kg/day.
Gadoversetamide injection caused forelimb flexures and cardiovascular changes in fetuses from female rabbits dosed with 0.4 and 1.6 mmol/kg/day (respectively, 1 and 4 times the human dose based on body surface area) on gestation days 6 through 18. The cardiovascular changes were malformed thoracic arteries, a septal defect, and abnormal ventricle. These effects were not observed at 0.1 mmol/kg/day (0.3 times the human dose based on body surface area). Maternal toxicity was not observed at any dose.
8.3 Nursing Mothers
Radiolabeled gadoversetamide (153Gd) was excreted in the milk of lactating rats receiving a single intravenous dose of 0.1 mmol/kg. Women should discontinue nursing and discard breast milk up to 72 hours after Optimark administration [see Clinical Pharmacology (12.3)].
8.4 Pediatric Use
The safety and effectiveness of Optimark in pediatric patients have not been established. Pediatric patients may be particularly vulnerable to adverse GBCA reactions due to renal immaturity or unrecognized renal insufficiency.
8.5 Geriatric Use
Since gadoversetamide is cleared from the body by glomerular filtration, the risk of adverse reactions may be greater in patients with impaired renal function (GFR ≥30 and <90 mL/min/1.73m2). Due to the risk for NSF, estimate the GFR through laboratory testing for patients >60 years of age [see Warnings and Precautions (5.1)].
8.6 Renal Impairment
A single intravenous dose of 0.1 mmol/kg of Optimark was administered to 28 patients (17 men and 11 women) with impaired renal function (mean serum creatinine of 2.4 mg/dL). Sixteen patients had concurrent central nervous system or liver pathology. Renal impairment was shown to delay the elimination of gadoversetamide (see Table 3). The mean cumulative urinary excretion of gadoversetamide at 72 hours was approximately 93.5% for renally impaired patients and 95.8% for subjects with normal renal function. Dose adjustments in renal impairment have not been studied. Optimark has been shown to be removed from the body by hemodialysis [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
A single intravenous dose of 0.1 mmol/kg of Optimark was administered to 4 patients (2 men and 2 women) with impaired hepatic function. Hepatically impaired patients with normal renal function had plasma kinetics similar to normal subjects (see Table 3).
|
Table 3 Elimination Profiles of Normal, Renally Impaired and Hepatically Impaired Men and Women (mean ± SD) |
||
|
Population |
Elimination t1/2 (hours) |
|
|
Men (N = 52) |
Women (N = 48) |
|
|
Healthy Volunteers |
1.73 ± 0.31 (N = 8) |
1.73 ± 0.40 (N = 4) |
|
Normal Patients |
1.90 ± 0.50 (N = 25) |
1.94 ± 0.57 (N = 31) |
|
Renally Impaired |
8.74 ± 5.14 (N = 17) |
6.91 ± 2.46 (N = 11) |
|
Hepatically Impaired |
2.09 ± 0.03 (N = 2) |
2.35 ± 1.09 (N = 2) |
10. Overdosage
Clinical consequences of overdosage with Optimark have not been reported. Treatment of overdose is directed toward supporting vital functions and prompt institution of symptomatic therapy. Optimark has been shown to be dialyzable [see Clinical Pharmacology (12.3)].
11. Optimark Description
Optimark (gadoversetamide) injection is a nonionic gadolinium chelate of diethylenetriamine pentaacetic acid bismethoxyethylamide (gadoversetamide), for intravenous injection.
Optimark injection is provided as a sterile, preservative-free, nonpyrogenic, clear, and colorless to pale yellow, aqueous solution of gadoversetamide. Each mL of Optimark contains 330.9 mg of gadoversetamide (0.5 millimole), 28.4 mg of calcium versetamide sodium (0.05 millimole), 0.7 mg calcium chloride dihydrate (0.005 millimole), and water for injection. Sodium hydroxide and/or hydrochloric acid may have been added for pH adjustment.
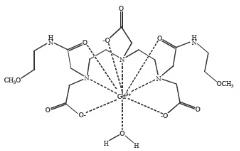
Gadoversetamide is designated chemically as [8, 11-bis(carboxymethyl)-14-[2-[(2-methoxyethyl)amino]-2-oxoethyl]-6-oxo-2-oxa-5,8,11,14-tetraazahexadecan-16-oato(3-)] gadolinium with a formula weight of 661.77 g/mol and empirical formula of C20H34N5O10Gd. The structural formula of gadoversetamide in aqueous solution is:
Optimark has a pH of 5.5 to 7.5. Pertinent physiochemical data are provided below (Table 4).
|
Table 4 Physiochemical Properties of Optimark |
|
|
Osmolality (mOsmol/kg water) @ 37°C |
1110 |
|
Viscosity (cP) @ 20°C |
3.1 |
|
@ 37°C |
2.0 |
|
Density (g/mL) @ 25°C |
1.160 |
Optimark has an osmolality of approximately 3.9 times that of plasma (285 mOsm/kg water) and is hypertonic under conditions of use.
12. Optimark - Clinical Pharmacology
12.1 Mechanism of Action
Gadoversetamide is a paramagnetic agent that develops a magnetic moment when placed in a magnetic field. The relatively large magnetic moment can enhance the relaxation rates of water protons in its vicinity, leading to an increase in signal intensity (brightness) of tissues.
12.2 Pharmacodynamics
In MRI, visualization of normal and pathological brain, spinal and hepatic tissue depends in part on variations in the radiofrequency signal intensity that occurs with: 1) changes in proton density; 2) alterations of the spin-lattice or longitudinal relaxation time (T1); and 3) variation of the spin-spin or transverse relaxation time (T2). When placed in a magnetic field, gadoversetamide decreases T1 and T2 relaxation times in tissues where it accumulates. At the recommended dose, the effect is primarily on T1 relaxation time, and produces an increase in signal intensity (brightness).
12.3 Pharmacokinetics
The pharmacokinetics of intravenously administered gadoversetamide in normal subjects conforms to a two-compartment open-model with mean distribution and elimination half-lives (reported as mean ± SD) of about 13.3 ± 6.8 and 103.6 ± 19.5 minutes.
Distribution
Gadoversetamide does not undergo protein binding in vitro. In pregnant and lactating rats which received 153Gd-labeled gadoversetamide, radioactivity was detected in the placenta, fetus, and maternal milk. The volume of distribution at steady state of gadoversetamide in normal subjects is 162 ± 25 mL/kg, roughly equivalent to that of extracellular water.
Disruption of the blood-brain barrier or abnormal vascularity allows accumulation of gadoversetamide in the extravascular spaces of lesions.
Following GBCA administration, gadolinium is present for months or years in brain, bone, skin, and other organs [see Warnings and Precautions (5.3)].
Metabolism
Gadoversetamide is not metabolized.
Elimination
Gadoversetamide (0.1 mmol/kg) is eliminated primarily in the urine with 95.5 ± 17.4% (mean ± SD) of the administered dose eliminated by 24 hours. Animal data demonstrated that insignificant levels of 153Gd-labeled gadoversetamide are eliminated via the feces. In experimentally induced anephria in the rat, hepatobiliary excretion did not significantly compensate for the absence of urinary elimination. The renal and plasma clearance rates of gadoversetamide in normal subjects are similar (69 ± 15.4 and 72 ± 16.3 mL/hr/kg, respectively) indicating that the drug is cleared through the kidneys via glomerular filtration. Within the studied dose range (0.1 to 0.7 mmol/kg), the kinetics of gadoversetamide appear to be linear.
Gadoversetamide is removed from the body by hemodialysis. Approximately 98% of the administered dose (0.1 mmol/kg) was cleared from the circulation over the three dialysis sessions that occurred 2 hours, 48 hours, and 120 hours after injection. After each of three dialysis sessions, respectively, 70%, 93%, and 98% of the administered dose was cleared from the plasma. The mean dialysis clearance of gadoversetamide was 93.2 ± 17.1 mL/min, or 48% of the creatinine clearance (194 ± 18.6 mL/min), using a high flux PMMA membrane.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to evaluate the carcinogenic potential of gadoversetamide. The results of the following genotoxicity assays were negative: Salmonella/E. Coli reverse mutation (Ames) assay, mouse lymphoma mutagenesis assay, and the in vivo mammalian micronucleus assay. The in vitro CHO chromosome aberration assay without metabolic activation was positive.
Gadoversetamide administered to rats in a fertility study was shown to have irreversible reduction and degeneration of spermatocytes in testes and epididymides, and impaired male fertility following intravenous doses of 2.0 mmol/kg/day (4 times the human dose based on body surface area) for 7 weeks. These effects were not observed at dose of 0.5 mmol/kg/day (1 times the human dose based on body surface area).
In a separate 28-day repeat dose study in rats, gadoversetamide was shown to have irreversible reduction of male reproductive organ weights, degeneration of the germinal epithelium of the testes, presence of germ cells in the epididymides, and reduced sperm count following daily intravenous doses of 3.0 mmol/kg/day (6 times the human dose based on body surface area). These effects were not observed at 0.6 mmol/kg/day (1 times the human dose based on surface area). These effects were not observed in similar studies conducted in dogs.
In a single dose study in rats, gadoversetamide did not produce adverse effects on the male reproductive system 24 hours and 14 days after intravenous administration of 0.5 to 15 mmol/kg (1 to 25 times the human dose based on body surface area).
14. Clinical Studies
Optimark was evaluated in 4 controlled clinical trials (two liver and two CNS studies). Out of 461 patients who received Optimark, there were 252 men and 209 women with a mean age of 49 years (range 12 to 82 years); 83% were Caucasian, 9% Black, 3% Asian, and 5% other racial or ethnic groups. The trials were designed to compare combined non-contrast and Optimark 0.1 mmol/kg contrast MR images to non-contrast MR images, based on pre-specified imaging characteristics (endpoints).
In the two CNS studies, MR images were analyzed from 262 patients who were highly suspect for CNS disorders and received Optimark. Pre-contrast and pre-plus-post-contrast (combined) images were independently evaluated by three blinded readers (each reader examined approximately 1/3 of the images). The images were evaluated by the blinded readers for the following endpoints using a scale from 1 to 10: the level of conspicuity of all lesions, the ability to delineate lesion borders from parenchyma/structures, the number of lesions, and the confidence in the number of lesions. As shown in Table 5, the first row of each endpoint group represents the difference in the mean score of the combined pre- and post-contrast MRI from the mean score of the pre-contrast MRI alone. Also, the table shows the number of patients whose paired MRI images were better, worse or the same as the pre-contrast MRI. Results from the contrast image alone were not evaluated. In Table 5 for these endpoints, when read in combination with the non-contrast images, Optimark provided a statistically significant improvement over baseline. In addition to these measures, the images were evaluated for the blinded reader’s confidence in the diagnosis. Although improvement over baseline was noted, the diagnosis was not rigorously confirmed.
|
Table 5 Results of MRI Central Nervous System Studies with 0.1 mmol/kg Optimark |
||
|
Endpoints |
Study A |
Study B |
|
Optimark |
Optimark |
|
|
Conspicuity: |
0.39* |
0.66* |
|
Worse |
24 (18%) |
24 (19%) |
|
Same |
69 (52%) |
52 (40%) |
|
Better |
39 (30%) |
53 (41%) |
|
Border Delineation: |
0.70* |
0.86* |
|
Worse |
23 (17%) |
25 (19%) |
|
Same |
55 (42%) |
51 (40%) |
|
Better |
54 (41%) |
53 (41%) |
|
Number of Lesions: Pre Pair (b) |
1.8 2.0◊ |
3.0 3.3* |
|
Worse |
9 (7%) |
16 (12%) |
|
Same |
101 (77%) |
86 (67%) |
|
Better |
22 (16%) |
27 (21%) |
|
Confidence in Number of Lesions: |
0.11* |
0.56* |
|
Worse |
19 (14%) |
18 (14%) |
|
Same |
86 (65%) |
60 (47%) |
|
Better |
27 (20%) |
51 (40%) |
|
(a) Difference of means = (Side-by-side pre- and post-Optimark mean) - (pre-mean) |
||
|
(b) Pair = Side-by-side pre- and post-Optimark |
||
|
* Statistically significant for both the median (Wilcoxon test) and mean (paired t test) |
||
|
◊ Statistically significant for median (Wilcoxon test) |
||
|
† 1 patient was excluded from this analysis because a non-contrast image was not obtained for that patient |
||
In the two liver studies, MR images were analyzed from 199 patients with a suspected liver abnormality on a contrast CT who received Optimark. Patients had both pre-contrast and post-contrast MRI scans covering the entire liver. In each study, the images were read by 3 blinded readers (each reader examined approximately 1/3 of the images). Using a scale of 1 to 10, the images were evaluated by the blinded readers for the level of conspicuity of all lesions, the ability to delineate lesion borders from parenchyma/structures, the number of lesions and confidence in the number of lesions. The results are shown in Table 6. The first row of each endpoint group represents the difference in the mean score of the combined pre- and post-contrast MRI from the mean score of the pre-contrast MRI alone. Also, the table shows the number of patients whose paired MRI images were better, worse or the same as the pre-contrast MRI. Results from the contrast image alone were not evaluated. As shown in Table 6 for these endpoints, when read in combination with the non-contrast image, Optimark provided a statistically significant improvement over non-contrast images. In addition to these measures, the images were evaluated for the blinded reader’s confidence in the diagnosis. Although improvement over baseline was noted, the trial was not designed to rigorously confirm the diagnosis.
|
Table 6 Results of MRI Liver Studies with 0.1 mmol/kg Optimark |
||
|
Endpoints |
Study C |
Study D |
|
Optimark |
Optimark |
|
|
Conspicuity: |
0.77* |
0.75* |
|
Worse |
21 (21%) |
14 (14%) |
|
Same |
37 (37%) |
50 (50%) |
|
Better |
41 (41%) |
36 (36%) |
|
Border Delineation: |
0.77* |
0.69* |
|
Worse |
21 (21%) |
15 (15%) |
|
Same |
38 (38%) |
45 (45%) |
|
Better |
40 (40%) |
40 (40%) |
|
Number of Lesions: Pre Pair (b) |
2.4 3.0* |
3.5 3.8† |
|
Worse |
13 (13%) |
16 (16%) |
|
Same |
50 (51%) |
58 (58%) |
|
Better |
36 (36%) |
26 (26%) |
|
Confidence in Number of Lesions: Difference of Means |
1.6* |
1.0* |
|
Worse |
39 (39%) |
38 (38%) |
|
Same |
2 (2%) |
8 (8%) |
|
Better |
58 (59%) |
54 (54%) |
|
(a) Difference of means = (Side-by-side pre- and post-Optimark mean) - (pre-mean) (b) Pair = Side-by-side pre- and post-Optimark * Statistically significant for both the median (Wilcoxon test) and mean (paired t test) † Borderline statistical significance in paired t test |
||
A subsequent study of 140 normal volunteers evaluated the safety of Optimark 0.1 mmol/kg delivered by power injector. Imaging results were not studied. The normal volunteers were randomized to receive Optimark injected manually, or Optimark or saline injected at 3 different power injector rates. At 2 mL/sec, the adverse event rates were comparable in the Optimark and saline controls when delivered manually and by power injector. In these small sample sizes, there was a trend towards increasing adverse events with increasing rates of power injection. Patients with abnormal vascularity were not evaluated. The safety and efficacy of power injector rates higher than 2 mL/sec has not been established.
16. How is Optimark supplied
How Supplied
Optimark is a clear, colorless to slightly yellow solution containing 330.9 mg/mL (equivalent to 0.5 mmol/mL) of gadoversetamide for injection. Optimark is supplied in 10 mL vials containing 5 mL or 10 mL of solution and is also provided in 20 mL vials containing 15 mL or 20 mL of solution. Each single dose vial is rubber stoppered with an aluminum seal and the contents are sterile. Optimark is supplied in 10 mL, 15 mL, 20 mL or 30 mL syringes containing 10 mL, 15 mL, 20 mL or 30 mL of solution respectively. Each syringe is sealed with rubber closures and the contents are sterile. Vials and syringes are contained in shipping cartons with the following configurations:
5 mL in glass vials in cartons of 10 vials (NDC Code 0019-1177-02)
10 mL in glass vials in cartons of 10 vials (NDC Code 0019-1177-04)
15 mL in glass vials in cartons of 10 vials (NDC Code 0019-1177-06)
20 mL in glass vials in cartons of 10 vials (NDC Code 0019-1177-08)
10 mL in plastic syringes in cartons of 10 syringes (NDC Code 0019-1177-11)
15 mL in plastic syringes in cartons of 10 syringes (NDC Code 0019-1177-16)
20 mL in plastic syringes in cartons of 10 syringes (NDC Code 0019-1177-21)
30 mL in plastic syringes in cartons of 10 syringes (NDC Code 0019-1177-31)
Storage
Optimark should be stored at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature] and protected from light and freezing. Optimark may be stored at 37°C for up to one month in a contrast media warmer utilizing circulating warm air. For periods longer than one month, store at 20°C to 25°C (68°F to 77°F).
17. Patient Counseling Information
• Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Nephrogenic Systemic Fibrosis
Instruct patients to inform their physician if they:
- have a history of kidney disease
- have recently received a GBCA
GBCAs increase the risk for NSF in patients with impaired elimination of the drugs. To counsel patients at risk for NSF:
- describe the clinical manifestations of NSF
- describe procedures to screen for the detection of renal impairment
Instruct the patients to contact their physician if they develop signs or symptoms of NSF following Optimark administration, such as burning, itching, swelling, scaling, hardening and tightening of the skin; red or dark patches on the skin; stiffness in joints with trouble moving, bending or straightening the arms, hands, legs or feet; pain in the hip bones or ribs; or muscle weakness.
Gadolinium Retention
Advise patients that gadolinium is retained for months or years in brain, bone, skin, and other organs in patients with normal renal function. The clinical consequences of retention are unknown. Retention depends on multiple factors and is greater following administration of linear GBCAs than following administration of macrocyclic GBCAs [See Warnings and Precautions (5.3)].
Other
Instruct patients to inform their physician if they:
- are pregnant or breast feeding
- have a history of renal disease or heart disease, seizure, asthma or allergic respiratory diseases
This product is covered by U.S. Patent No. 5130120, 5137711, 5508388. The use of this product is covered by U.S. Patent No. 5130120 and 5137711.
Manufactured and Distributed by:
Liebel-Flarsheim Company LLC
Raleigh, NC 27616
Made in USA
GBT 1177Cb0418
Rx only
Medication Guide
| What is OPTIMARK?
• OPTIMARK is a prescription medicine called a gadolinium-based contrast agent (GBCA). OPTIMARK, like other GBCAs is injected into your vein and used with a magnetic resonance imaging (MRI) scanner. • An MRI exam with a GBCA, including OPTIMARK, helps your doctor to see problems better than an MRI exam without a GBCA. • Your doctor has reviewed your medical records and has determined that you would benefit from using a GBCA with your MRI exam |
| What is the most important information I should know about OPTIMARK?
• OPTIMARK contains a metal called gadolinium. Small amounts of gadolinium can stay in your body including the brain, bones, skin and other parts of your body for a long time (several months to years). • It is not known how gadolinium may affect you, but so far, studies have not found harmful effects in patients with normal kidneys. • Rarely, patients report pains, tiredness, and skin, muscle or bone ailments for a long time, but these symptoms have not been directly linked to gadolinium. • There are different GBCAs that can be used for your MRI exam. The amount of gadolinium that stays in the body is different for different gadolinium medicines. Gadolinium stays in the body more after Omniscan or Optimark than after Eovist, Magnevist, or MultiHance. Gadolinium stays in the body the least after Dotarem, Gadavist, or ProHance. • People who get many doses of gadolinium medicines, women who are pregnant and young children may be at increased risk from gadolinium staying in the body. • Some people with kidney problems who get gadolinium medicines can develop a condition with severe thickening of the skin, muscles and other organs in the body (nephrogenic systemic fibrosis). Your healthcare provider should screen you to see how well your kidneys are working before you receive OPTIMARK. |
| Do not receive OPTIMARK if you have had a severe allergic reaction to GBCAs including gadoversetamide, or any of the ingredients in OPTIMARK. |
| Before receiving OPTIMARK, tell your healthcare provider about all your medical conditions, including if you:
• have had any MRI procedures in the past where you received a GBCA. Your healthcare provider may ask you for more information including the dates of these MRI procedures. • are pregnant or plan to become pregnant. It is not known if OPTIMARK can harm your unborn baby. Talk to your healthcare provider about the possible risks to an unborn baby if a GBCA such as OPTIMARK is received during pregnancy • have kidney problems, diabetes, or high blood pressure. • have had an allergic reaction to dyes (contrast agents) including GBCAs |
| What are the possible side effects of OPTIMARK?
• See “What is the most important information I should know about OPTIMARK?” • Allergic reactions. OPTIMARK can cause allergic reactions that can sometimes be serious. Your healthcare provider will monitor you closely for symptoms of an allergic reaction. The most common side effects of OPTIMARK include: headache, vasodilatation, taste perversion, dizziness, nausea and paresthesia. These are not all the possible side effects of OPTIMARK. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
| General information about the safe and effective use of OPTIMARK.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your healthcare provider for information about OPTIMARK® that is written for health professionals. |
| What are the ingredients in OPTIMARK?
Active ingredient: Gadoversetamide Inactive ingredients:Calcium Versetamide Sodium, Calcium Chloride Dihydrate, Water, Sodium Hydroxide, Hydrochloric Acid Manufactured by: Liebel-Flarsheim Company LLC Raleigh, NC 27616 This Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: 4/2018 |
PACKAGE LABEL - PRINCIPAL DISPLAY PANEL - 30 mL syringe label
Sterile Solution
30 mL NDC 0019-1177-31
Optimark® 0.5mmol/mL
(gadoversetamide injection)
For Intravenous Injection Only
Dosage: See Package Insert.
Rx Only
MEDICATION AND FLUID PATHWAY ARE STERILE
OUTSIDE OF SYRINGE IS NOT STERILE
SINGLE DOSE UNIT, DISCARD UNUSED PORTION
PROTECT FROM LIGHT • PROTECT FROM FREEZING
Store at 20 to 25°C (68 to 77°F) [See USP Controlled Room Temperature]
Manufactured by:
Liebel-Flarsheim Company LLC
8800 Durant Road
Raleigh, NC 27616
Made in USA
11630916

| OPTIMARK
gadoversetamide injection, solution |
||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||
| Labeler - Liebel-Flarsheim Company LLC (057880002) |
More about OptiMARK (gadoversetamide)
- Check interactions
- Compare alternatives
- Side effects
- Dosage information
- During pregnancy
- Drug class: magnetic resonance imaging contrast media
- Breastfeeding