Methotrexate Injection: Package Insert / Prescribing Info
Package insert / product label
Dosage form: injection, solution
Drug classes: Antimetabolites, Antipsoriatics, Antirheumatics, Other immunosuppressants
Medically reviewed by Drugs.com. Last updated on Feb 20, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- References
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
METHOTREXATE injection, for intravenous, intramuscular, subcutaneous, or intrathecal use
Initial U.S. Approval: 1953
WARNING: EMBRYO-FETAL TOXICITY, HYPERSENSITIVITY
REACTIONS, BENZYL ALCOHOL TOXICITY, and OTHER
SERIOUS ADVERSE REACTIONS
See full prescribing information for complete boxed warning.
- Methotrexate Injection can cause embryo-fetal toxicity, including fetal death. Use in non-neoplastic diseases is contraindicated during pregnancy. Advise females and males of reproductive potential to use effective contraception during and after treatment with Methotrexate Injection. (4, 5.1, 8.1, 8.3)
- Methotrexate Injection is contraindicated in patients with a history of severe hypersensitivity reactions to methotrexate, including anaphylaxis. (4, 5.2).
- Formulations with benzyl alcohol can cause severe central nervous toxicity or metabolic acidosis. Use only preservative-free Methotrexate Injection for treatment of neonates or low-birth weight infants, and for intrathecal use. Do not use benzyl alcohol-containing formulations for high-dose regimens unless immediate treatment is required and preservative-free formulations are not available. (2.1, 5.3)
- Other serious adverse reactions, including death, have been reported with methotrexate. Closely monitor for infections and adverse reactions of the bone marrow, kidneys, liver, nervous system, gastrointestinal tract, lungs, and skin. Withhold or discontinue Methotrexate Injection as appropriate. (5.4, 5.5, 5.6, 5.7, 5.8, 5.9, 5.10, 5.11)
Indications and Usage for Methotrexate Injection
Methotrexate Injection is a folate analog metabolic inhibitor indicated for:
- The following neoplastic diseases for the:
◦ Treatment of adult and pediatric patients with acute lymphoblastic leukemia as part of a combination chemotherapy regimen (1.1)
◦ Prophylaxis and treatment of adult and pediatric patients with meningeal leukemia (1.2)
◦ Treatment of adult and pediatric patients with non-Hodgkin lymphoma (1.3)
◦ Treatment of adult and pediatric patients with osteosarcoma as part of a combination chemotherapy regimen (1.4)
◦ Treatment of adults with breast cancer as part of a combination chemotherapy regimen (1.5)
◦ Treatment of adults with squamous cell carcinoma of the head and neck as single-agent (1.6)
◦ Treatment of adults with gestational trophoblastic neoplasia as part of a combination chemotherapy regimen (1.7) - Treatment of adults with rheumatoid arthritis (RA). (1.8)
- Treatment of pediatric patients with polyarticular juvenile idiopathic arthritis (pJIA). (1.9)
- Treatment of adults with severe psoriasis. (1.10)
Methotrexate Injection Dosage and Administration
- Verify pregnancy status in females of reproductive potential before starting Methotrexate Injection. (2.1, 4, 5.1)
- Neoplastic diseases: Refer to the prescribing information for disease specific dosing recommendations. Follow guidelines for high-dose regimens. (2.2, 2.3, 2.4, 2.5, 2.6, 2.7, 2.8, 2.9)
- RA: Recommended starting dosage of 7.5 mg once weekly intramuscularly; adjust dose to achieve an optimal response. (2.10)
- pJIA: Recommended starting dosage of 10 mg/m2 once weekly subcutaneously or intramuscularly; adjust dose to achieve an optimal response. (2.11)
- Psoriasis: Recommended dosage of 10 mg to 25 mg once weekly intramuscularly or intravenously; adjust dose to achieve optimal response. Once achieved, reduce to lowest possible dosage. (2.12)
Dosage Forms and Strengths
Injection: (3)
- Preservative-free (single-dose vials): 50 mg/2 mL, 100 mg/4 mL, 200 mg/8 mL and 250 mg/10 mL (25 mg/mL)
Contraindications
Warnings and Precautions
- Secondary malignancies can occur. (5.13)
- Tumor lysis syndrome can occur in patients with rapidly growing tumors. (5.14)
- Immunizations and Risks associated with Live Vaccines: Immunizations may be ineffective. Live vaccines are not recommended due to risk of disseminated infection. (5.15)
- Infertility: Can cause impairment of fertility, oligospermia, and menstrual dysfunction. (5.16, 8.3)
Adverse Reactions/Side Effects
Common adverse reactions include ulcerative stomatitis, leukopenia, nausea, and abdominal distress. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eugia US LLC at 1-866-850-2876 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Drug Interactions
Refer to full prescribing information for drug interactions with Methotrexate Injection. (7)
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2025
Full Prescribing Information
WARNING: EMBRYO-FETAL TOXICITY, HYPERSENSITIVITY REACTIONS, BENZYL ALCOHOL TOXICITY, and OTHER SERIOUS ADVERSE REACTIONS
• Methotrexate Injection can cause embryo-fetal toxicity, including fetal death. For non-neoplastic diseases, Methotrexate Injection is contraindicated in pregnancy. Advise females and males of reproductive potential to use effective contraception [see Contraindications (4), Warnings and Precautions (5.1), and Use in Specific Populations (8.1, 8.3)].
• Methotrexate Injection is contraindicated in patients with a history of severe hypersensitivity reactions to methotrexate, including anaphylaxis [see Contraindications (4) and Warnings and Precautions (5.2)].
• Formulations with benzyl alcohol can cause severe central nervous toxicity or metabolic acidosis. Use only preservative-free Methotrexate Injection for treatment of neonates or low-birth weight infants and for intrathecal use. Do not use benzyl alcohol-containing formulations for high-dose regimens unless immediate treatment is required and preservative-free formulations are not available [see Dosage and Administration (2.1) and Warnings and Precautions (5.3)].
• Other serious adverse reactions, including death, have been reported with methotrexate. Closely monitor for infections and adverse reactions of the bone marrow, kidneys, liver, nervous system, gastrointestinal tract, lungs, and skin. Withhold or discontinue Methotrexate Injection as appropriate [see Warnings and Precautions (5.4, 5.5, 5.6, 5.7, 5.8, 5.9, 5.10, 5.11)].
1. Indications and Usage for Methotrexate Injection
1.1 Acute Lymphoblastic Leukemia
Methotrexate Injection is indicated for the treatment of adult and pediatric patients with acute lymphoblastic leukemia (ALL) as part of a combination chemotherapy regimen.
1.2 Meningeal Leukemia: Prophylaxis and Treatment
Methotrexate Injection is indicated for the prophylaxis and treatment of meningeal leukemia in adult and pediatric patients.
1.3 Non-Hodgkin Lymphoma
Methotrexate Injection is indicated for the treatment of adults and pediatric patients with Non-Hodgkin lymphoma.
1.4 Osteosarcoma
Methotrexate Injection is indicated for the treatment of adults and pediatric patients with osteosarcoma as part of a combination chemotherapy regimen.
1.5 Breast Cancer
Methotrexate Injection is indicated for the treatment of adults with breast cancer as part of a combination chemotherapy regimen.
1.6 Squamous Cell Carcinoma of the Head and Neck
Methotrexate Injection is indicated for the treatment of adults with squamous cell carcinoma of the head and neck as a single-agent.
1.7 Gestational Trophoblastic Neoplasia
Methotrexate Injection is indicated for the treatment of adults with gestational trophoblastic neoplasia (GTN) as part of a combination chemotherapy regimen.
1.8 Rheumatoid Arthritis
Methotrexate Injection is indicated for the treatment of adults with rheumatoid arthritis (RA).
2. Methotrexate Injection Dosage and Administration
2.1 Important Dosage and Safety Information
- Use only preservative-free Methotrexate Injection for treatment of neonates or low-birth weight infants and for intrathecal use. Do not use benzyl alcohol-containing formulations for high-dose regimens unless immediate treatment is required and preservative-free formulations are not available [see Warnings and Precautions (5.3) and Use in Specific Populations (8.4)].
- Verify pregnancy status in females of reproductive potential before starting Methotrexate Injection [see Contraindications (4) and Warnings and Precautions (5.1)].
- For patients switching between a methotrexate product administered orally and Methotrexate Injection, consider potential differences in bioavailability.
2.2 Recommended Monitoring and Concomitant Therapies for Intermediate- and High-Dose Regimens
To decrease the risk of severe adverse reactions [see Warnings and Precautions (5)]:
- Administer leucovorin rescue in patients receiving Methotrexate Injection doses of 500 mg/m2 or greater (e.g., high-dose).
- Consider leucovorin rescue for patients receiving Methotrexate Injection doses between 100 mg/m2 to less than 500 mg/m2 (e.g., intermediate-dose).
Refer to the leucovorin Prescribing Information for additional information.
- For high-dose Methotrexate Injection regimens, follow the supportive care and monitoring instructions below. Also consider for patients receiving intermediate-dose Methotrexate Injection regimens.
- Monitor serum creatinine, electrolytes, at baseline and at least daily during therapy
- Administer intravenous fluids starting before the first dose and continuing throughout treatment to maintain adequate hydration and urine output
- Alkalinize urine starting before the first dose and continuing throughout treatment to maintain a urinary pH of 7 or higher
- Monitor methotrexate concentrations at least daily and adjust hydration and leucovorin dosing as needed
- Administer glucarpidase in patients who have toxic plasma methotrexate concentrations (>1 micromole per liter) and delayed methotrexate clearance due to impaired renal function (refer to the glucarpidase Prescribing Information for additional information)
2.3 Recommended Dosage for Acute Lymphoblastic Leukemia
Methotrexate Injection is used as part of a multi-drug regimen. The recommended dosage varies from 10 to 5,000 mg/m2 intravenously. For high dose Methotrexate Injection regimens, use leucovorin rescue in accordance with high-dose methotrexate regimen guidelines [see Dosage and Administration (2.2)]. Lower doses (e.g., 20 to 30 mg/m2/week) may be used intramuscularly. Individualize the dose and schedule of Methotrexate Injection based on disease state, patient risk category, concurrent drugs used, phase of treatment, and response to treatment.
2.4 Recommended Dosage for Meningeal Leukemia: Prophylaxis and Treatment
Use only preservative-free Methotrexate Injection for intrathecal use.
Prior to administration, dilute preservative-free Methotrexate Injection to a concentration of 1 mg/mL in preservative-free 0.9% Sodium Chloride Injection, USP.
The recommended intrathecal dose of Methotrexate Injection (preservative-free) is based on age:
- less than 1 year: 6 mg
- 1 to less than 2 years: 8 mg
- 2 to less than 3 years: 10 mg
- 3 to less than 9 years: 12 mg
- greater than or equal to 9 years: 12 to 15 mg
For treatment of meningeal leukemia, intrathecal methotrexate may be given at intervals of 2 or more days up to twice weekly; however, administration at intervals of less than 1 week may result in increased subacute toxicity. For meningeal leukemia prophylaxis, Methotrexate Injection is administered no more than once weekly.
For patients with Down Syndrome, administer leucovorin rescue with intrathecal Methotrexate Injection.
2.5 Recommended Dosage for Non-Hodgkin Lymphoma
The recommended dosage of Methotrexate Injection varies. When used in combination, recommended dosages range from 10 mg/m2 to 8,000 mg/m2 intravenously. When used as a single agent, recommended dosages include 8,000 mg/m2 intravenously for central nervous system-directed therapy or 5 to 75 mg intravenously for cutaneous forms of Non-Hodgkin lymphoma.
As part of a combination chemotherapy regimen, a recommended dosage of Methotrexate Injection is 1,000 mg/m2 or 3,000 mg/m2 as an intravenous infusion over 24 hours followed by leucovorin rescue in accordance with high-dose methotrexate regimen guidelines [see Dosage and Administration (2.2)].
For central nervous system-directed therapy, a recommended dosage of Methotrexate Injection is 8,000 mg/m2 as an intravenous infusion over 4 hours as a single agent or in combination with immunochemotherapy at doses ranging from 3,000 mg/m2 to 8,000 mg/m2 followed by leucovorin rescue in accordance with high-dose methotrexate regimen guidelines [see Dosage and Administration (2.2)].
For intrathecal Methotrexate Injection (preservative-free), the recommended dose is based on age [see Dosage and Administration (2.4)]. The frequency of administration varies based on whether it is being used for treatment or prophylaxis, and other factors.
2.6 Recommended Dosage for Osteosarcoma
The recommended dosage of Methotrexate Injection is typically 12 g/m2 (maximum 20 g/dose) as an intravenous infusion over 4 hours administered as a component of a combination chemotherapy regimen. Administer leucovorin rescue in accordance with high-dose methotrexate regimen guidelines [see Dosage and Administration (2.2)]. Subsequent doses may need to be adjusted based on observed peak serum methotrexate concentrations. Dosage and schedule may vary based upon factors such as patient comorbidities, disease state, and prior treatments.
2.7 Recommended Dosage for Breast Cancer
A recommended dosage of Methotrexate Injection is 40 mg/m2 intravenously as a component of a cyclophosphamide- and fluorouracil-based multi-drug regimen.
2.8 Recommended Dosage for Squamous Cell Carcinoma of Head and Neck
The recommended dosage of Methotrexate Injection ranges from 40 to 60 mg/m2 intravenously once weekly.
2.9 Recommended Dosage for Gestational Trophoblastic Neoplasia
For patients with low-risk gestational trophoblastic neoplasia (GTN) a recommended dosage for Methotrexate Injection is 30 mg/m2 to 200 mg/m2 or 0.4 mg/kg to 1 mg/kg intravenously or intramuscularly.
For patients with high-risk GTN, a recommended dosage for Methotrexate Injection is 300 mg/m2 over 12 hours as an intravenous infusion as a component of a multi-drug regimen.
2.10 Recommended Dosage for Rheumatoid Arthritis
The recommended starting dosage of Methotrexate Injection is 7.5 mg once weekly, administered intramuscularly with escalation to achieve optimal response. Dosages of more than 20 mg once weekly result in an increased risk of serious adverse reactions, including myelosuppression.
When responses are observed, the majority occurred between 3 and 6 weeks from initiation of treatment; however, responses have occurred up to 12 weeks after treatment initiation.
Administer folic acid or folinic acid to reduce the risk of methotrexate adverse reactions [see Warnings and Precautions (5.12)].
2.11 Recommended Dosage for Polyarticular Juvenile Idiopathic Arthritis
The recommended starting dosage of Methotrexate Injection is 10 mg/m2 once weekly administered subcutaneously or intramuscularly, with escalation to achieve optimal response. Dosages over 30 mg/m2 per week may result in an increased risk of serious adverse reactions, including myelosuppression. When responses are observed, the majority occurred between 3 and 6 weeks from initiation of treatment; however, responses have occurred up to 12 weeks after treatment initiation.
Administer folic acid or folinic acid to reduce the risk of methotrexate adverse reactions [see Warnings and Precautions (5.12)].
2.12 Recommended Dosage for Psoriasis
The recommended dosage of Methotrexate Injection is 10 mg to 25 mg intramuscularly or intravenously once weekly until adequate response is achieved.
Adjust the dose gradually to achieve optimal clinical response; do not exceed 25 mg per week. Once optimal clinical response has been achieved, reduce the dosage to the lowest possible dosing regimen.
Administer folic acid or folinic acid to reduce the risk of methotrexate adverse reactions [see Warnings and Precautions (5.12)].
2.13 Dosage Modifications for Adverse Reactions
Discontinue Methotrexate Injection for:
- Anaphylaxis or other severe hypersensitivity reactions [see Warnings and Precautions (5.2)]
- Lymphoproliferative disease [see Warnings and Precautions (5.13)]
Withhold, dose reduce or discontinue Methotrexate Injection as appropriate for:
- Myelosuppression [see Warnings and Precautions (5.4)]
Withhold or discontinue Methotrexate Injection as appropriate for:
- Serious infections [see Warnings and Precautions (5.5)]
- Renal toxicity [see Warnings and Precautions (5.6)]
- Hepatotoxicity [see Warnings and Precautions (5.7)]
- Neurotoxicity [see Warnings and Precautions (5.8)]
- Gastrointestinal toxicity [see Warnings and Precautions (5.9)]
- Pulmonary toxicity [see Warnings and Precautions (5.10)]
- Dermatologic reactions [see Warnings and Precautions (5.11)]
2.14 Administration and Handling Information
Methotrexate Injection is a hazardous drug. Follow applicable special handling and disposable procedures.1
Preservative-free (single-dose vial)
Methotrexate Injection preservative-free may be administered by intramuscular, intravenous, subcutaneous, or intrathecal injection.
• Use only preservative-free Methotrexate Injection for treatment of neonates or low-birth weight infants and for intrathecal use[see Warning and Precautions (5.3) and Use in Specific Populations (8.4)].
• Use preservative-free Methotrexate Injection for high-dose regimens unless immediate treatment is required, and preservative-free formulations are not available [see Warning and Precautions (5.3) and Use in Specific Populations (8.4)].
• Preservative-free Methotrexate Injection may be further diluted before use with preservative-free 0.9% Sodium Chloride Injection, USP. Diluted product should be used within 4 hours when stored at room temperature (20°C to 25°C) or 24 hours when stored under refrigeration (2°C to 8°C).
• Visually inspect for particulate matter and discoloration prior to administration. Discard if particulate matter or discoloration is observed.
3. Dosage Forms and Strengths
Injection: Methotrexate Injection, USP is a clear, yellowish solution and is supplied in single-dose vials (preservative-free) in the following strengths:
Preservative-free (single-dose vial)
- 50 mg/2 mL, 100 mg/4 mL, 200 mg/8 mL and 250 mg/10 mL (25 mg/mL)
4. Contraindications
Methotrexate Injection is contraindicated in:
- Patients with history of severe hypersensitivity to methotrexate [see Warnings and Precautions (5.2)].
- Pregnancy in patients with non-neoplastic diseases [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1)].
5. Warnings and Precautions
5.1 Embryo-Fetal Toxicity
Based on published reports and its mechanism of action, methotrexate can cause embryo-fetal toxicity, including fetal death when administered to a pregnant woman.
Methotrexate Injection is contraindicated for use in pregnant women with non-neoplastic diseases. Advise pregnant women with neoplastic diseases of the potential risk to a fetus. The preservative benzyl alcohol can cross the placenta; when possible, use the preservative-free formulation when Methotrexate Injection is needed during pregnancy to treat a neoplastic disease [see Warnings and Precautions (5.3)].
Advise females of reproductive potential to use effective contraception during Methotrexate Injection treatment and for 6 months after the final dose. Advise males with female partners of reproductive potential to use effective contraception during Methotrexate Injection treatment and for 3 months after the final dose [see Contraindications (4) and Use in Specific Populations (8.1, 8.3, 8.4)].
5.2 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis, can occur with methotrexate [see Adverse Reactions (6.1)]. If signs or symptoms of anaphylaxis or any other serious hypersensitivity reaction occurs, immediately discontinue Methotrexate Injection and institute appropriate therapy [see Contraindications (4)].
5.3 Risks of Serious Adverse Reactions due to Benzyl Alcohol-Preservative
Formulations with benzyl alcohol can cause severe central nervous toxicity or metabolic acidosis, if used in neonates or low-birth weight infants, intrathecally, or in high-dose regimens. Use only preservative-free Methotrexate Injection for treatment of neonates or low-birth weight infants and for intrathecal use. Do not use benzyl alcohol-containing formulations for high-dose regimens unless immediate treatment is required, and preservative-free formulations are not available. The preservative benzyl alcohol can cross the placenta; when possible, use the preservative-free formulation when Methotrexate Injection is needed during pregnancy to treat a neoplastic disease [see Use in Specific Populations (8.1)].
Serious and Fatal Adverse Reactions Including Gasping Syndrome in Neonates and Low-Birth Weight Infants
Serious and fatal adverse reactions including “gasping syndrome” can occur in neonates and low birth weight infants treated with drugs containing benzyl alcohol, including Methotrexate Injection with preservative. The “gasping syndrome” is characterized by central nervous system (CNS) depression, metabolic acidosis, and gasping respirations.
When prescribing in infants (non-neonate, non-low-birth weight), if a preservative-free formulation of Methotrexate Injection is not available and use of a benzyl alcohol-containing formulation is necessary, consider the combined daily metabolic load of benzyl alcohol from all sources including Methotrexate Injection (Methotrexate Injection contains 9.4 mg of benzyl alcohol/per mL) and other drugs containing benzyl alcohol. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known [see Use in Specific Populations (8.4)].
Neurotoxicity Due to Intrathecal Administration
Serious neurotoxicity can occur following the intrathecal administration of Methotrexate Injection containing the preservative benzyl alcohol.
Metabolic Acidosis with High-Dose Therapy
Severe metabolic acidosis can occur with Methotrexate Injection that contains the preservative benzyl alcohol.
5.4 Myelosuppression
Methotrexate suppresses hematopoiesis and can cause severe and life-threatening pancytopenia, anemia, aplastic anemia, leukopenia, neutropenia, and thrombocytopenia [see Adverse Reactions (6.1)].
Obtain blood counts at baseline and periodically during treatment. Monitor patients for possible clinical complications of myelosuppression. Provide supportive care and withhold, reduce dose, or discontinue Methotrexate Injection as needed.
5.5 Serious Infections
Patients treated with methotrexate are at increased risk for developing life-threatening or fatal bacterial, fungal, or viral infections including opportunistic infections such as Pneumocystis jiroveci pneumonia, invasive fungal infections, hepatitis B reactivation, tuberculosis primary infection or reactivation, and disseminated Herpes zoster and cytomegalovirus infections.
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with Methotrexate Injection. Withhold or discontinue Methotrexate Injection in patients who develop serious infections.
5.6 Renal Toxicity
Methotrexate can cause renal toxicity including irreversible acute renal failure. Monitor renal function and withhold or discontinue methotrexate as needed for severe renal toxicity.
For patients receiving high-dose regimens, follow recommendations to decrease the risk of renal injury and mitigate renal toxicity [see Dosage and Administration (2.2)].
Patients with impaired renal function are at increased risk for methotrexate toxicity [see Use in Specific Populations (8.6)].
Consider administration of glucarpidase in patients with toxic plasma methotrexate concentrations (>1 micromole per liter) and delayed clearance due to impaired renal function. [see Dosage and Administration (2.2)].
5.7 Hepatotoxicity
Methotrexate can cause severe and potentially irreversible hepatotoxicity including fibrosis, cirrhosis, and fatal liver failure [see Adverse Reactions (6.1, 6.2)].
In patients with psoriasis, fibrosis or cirrhosis may occur in the absence of symptoms or abnormal liver function tests. In patients with psoriasis, the risk of hepatotoxicity appears to increase with total cumulative dose and generally occurs after receipt of a total cumulative dose of 1.5 g or more.
The safety of Methotrexate Injection in patients with liver disease is unknown. Avoid use of Methotrexate Injection in patients with chronic liver disease, unless benefits clearly outweigh the risks. The risk of hepatotoxicity is increased with heavy alcohol consumption.
Assess liver function prior to initiating Methotrexate Injection and monitor liver function tests during treatment. Withhold or discontinue Methotrexate Injection as appropriate.
5.8 Neurotoxicity
Methotrexate can cause severe acute and chronic neurotoxicity which can be progressive, irreversible, and fatal. Serious neurotoxicity, including generalized and focal seizures, have occurred in pediatric patients [see Use in Specific Populations (8.4)]. Monitor patients for signs of neurotoxicity and withhold or discontinue Methotrexate Injection when appropriate.
Leukoencephalopathy
Leukoencephalopathy can occur with intermediate and high-dose intravenous regimens, intrathecal methotrexate, and low-dose methotrexate therapy. The risk of leukoencephalopathy is increased with prior cranial radiation.
Transient Acute Neurologic Syndrome
A transient acute stroke-like syndrome can occur with high-dose methotrexate. Clinical manifestations include confusion, hemiparesis, transient blindness, seizures, and coma.
Neurologic Adverse Reactions Associated with Intrathecal Administration
Intrathecal methotrexate can cause the following additional neurologic adverse reactions:
- Acute chemical arachnoiditis manifested by symptoms such as headache, back pain, nuchal rigidity, and fever.
- Subacute myelopathy characterized by paraparesis or paraplegia.
Avoid the intrathecal use of Methotrexate Injection that contains the preservative benzyl alcohol because of the risk of serious neurotoxicity [see Warnings and Precautions (5.3)].
5.9 Gastrointestinal Toxicity
Methotrexate can cause diarrhea, vomiting, stomatitis, hemorrhagic enteritis and fatal intestinal perforation [see Adverse Reactions (6.1)]. Patients with peptic ulcer disease or ulcerative colitis are at a greater risk of developing severe gastrointestinal adverse reactions.
Withhold or discontinue Methotrexate Injection for severe gastrointestinal toxicity, and institute appropriate supportive care as needed.
5.10 Pulmonary Toxicity
Methotrexate-induced pulmonary toxicity including acute or chronic interstitial pneumonitis and irreversible or fatal cases can occur at all dose levels. Monitor patients for signs of pulmonary toxicity and withhold or discontinue Methotrexate Injection as appropriate.
5.11 Dermatologic Reactions
Severe, including fatal, dermatologic reactions, such as toxic epidermal necrolysis, Stevens-Johnson syndrome, exfoliative dermatitis, skin necrosis, and erythema multiforme, can occur with methotrexate [see Adverse Reactions (6.1, 6.2)].
Psoriasis may be aggravated by concomitant exposure to ultraviolet radiation.
Methotrexate can also cause radiation recall dermatitis and photodermatitis (sunburn) reactivation.
Monitor patients for signs of dermatologic toxicity and withhold or permanently discontinue Methotrexate Injection for severe dermatologic adverse reactions. Counsel patients to avoid excessive sun exposure and use sun protection measures.
5.12 Folic Acid Supplementation
Neoplastic Diseases
Products containing folic acid or its derivatives may decrease the clinical effectiveness of methotrexate. Avoid use of products containing folic acid or folinic acid unless clinically indicated [see Drug Interactions (7.1)].
Non-neoplastic Diseases
Folate deficiency may increase methotrexate adverse reactions. Administer folic acid or folinic acid to patients with rheumatoid arthritis, pJIA, and psoriasis [see Dosage and Administration (2.10, 2.11, 2.12)].
5.13 Secondary Malignancies
Secondary malignancies can occur at all dose levels of methotrexate. In some cases, lymphoproliferative disease that occurred during therapy with low-dose methotrexate regressed completely following withdrawal of methotrexate. If lymphoproliferative disease occurs, discontinue Methotrexate Injection and institute appropriate treatment if lymphoma does not regress.
5.14 Tumor Lysis Syndrome
Methotrexate can induce tumor lysis syndrome in patients with rapidly growing tumors. Institute appropriate treatment for prevention and management of tumor lysis syndrome.
5.15 Immunization and Risks Associated with Live Vaccines
Immunization during Methotrexate Injection treatment may be ineffective.
Disseminated infections following administration of live vaccines have been reported.
Update immunizations according to immunization guidelines prior to initiating Methotrexate Injection. Immunization with live vaccines is not recommended during treatment. The interval between live vaccinations and initiation of Methotrexate Injection should be in accordance with current vaccination guidelines for patients on immunosuppressive therapies.
5.16 Infertility
Based on published reports, methotrexate can cause impairment of fertility, oligospermia, and menstrual dysfunction. It is not known if the infertility may be reversible in affected patients. Discuss the risk of effects on reproduction with female and male patients of reproductive potential [see Use in Specific Populations (8.3)].
5.17 Increased Risk of Adverse Reactions Due to Third Space Accumulation
Methotrexate can exit slowly from third space accumulations resulting in prolonged terminal plasma half-life and toxicity. Evacuate significant third-space accumulations prior to Methotrexate Injection administration [see Clinical Pharmacology (12.3)].
5.18 Increased Risk of Soft Tissue and Bone Toxicity with Concomitant Radiotherapy
Concomitant radiation therapy increases the risk of soft tissue necrosis and osteonecrosis associated with methotrexate.
5.19 Risk of Serious Adverse Reactions with Medication Errors
Serious adverse reactions, including death, have occurred due to medication errors. Most commonly, these errors occurred in patients who were taking methotrexate daily when a weekly dosing regimen was prescribed. Ensure that patients receive the recommended dosage, because medication errors have led to death.
6. Adverse Reactions/Side Effects
The following adverse reactions are described, or described in greater detail, in other sections:
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
- Myelosuppression [see Warnings and Precautions (5.4)]
- Serious Infections [see Warnings and Precautions (5.5)]
- Renal Toxicity [see Warnings and Precautions (5.6)]
- Hepatotoxicity [see Warnings and Precautions (5.7)]
- Neurotoxicity [see Warnings and Precautions (5.8)]
- Gastrointestinal Toxicity [see Warnings and Precautions (5.9)]
- Pulmonary Toxicity [see Warnings and Precautions (5.10)]
- Dermatologic Reactions [see Warnings and Precautions (5.11)]
- Secondary Malignancies [see Warnings and Precautions (5.13)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.14)]
- Increased Risk of Adverse Reactions due to Third Space Accumulation [see Warnings and Precautions (5.17)]
6.1 Clinical Trials Experience
Because clinical trials and other studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Commonly reported adverse reactions include ulcerative stomatitis, leukopenia, nausea, and abdominal distress. Other frequently reported adverse reactions are infection, malaise, fatigue, chills, fever, and dizziness.
Rheumatoid Arthritis
The approximate incidences of methotrexate-attributed (i.e., placebo rate subtracted) adverse reactions in 12- to 18-week double-blind studies in patients (n=128) with RA treated with low-dose oral (7.5 mg per week to 15 mg per week) pulse methotrexate are listed below. Most patients were on concomitant NSAIDs and some received corticosteroids. Hepatic histology was not examined in these short-term studies.
Incidence ≥10%: Elevated liver function tests 15%, nausea/vomiting 10%.
Incidence 3% to <10%: Stomatitis, thrombocytopenia (platelet count less than 100,000/mm3).
Incidence 1% to <3%: Rash/pruritus/dermatitis, diarrhea, alopecia, leukopenia (white blood cell count less than 3,000/mm3), pancytopenia, dizziness.
Two other controlled trials of patients (n=680) with RA on 7.5 mg per week to 15 mg per week oral doses showed the following adverse reactions:
Incidence 1%: Interstitial pneumonitis.
Other less common adverse reactions: Decreased hematocrit, headache, upper respiratory infection, anorexia, arthralgias, chest pain, coughing, dysuria, eye discomfort, epistaxis, fever, infection, sweating, tinnitus, vaginal discharge.
Polyarticular Juvenile Idiopathic Arthritis (pJIA)
The approximate incidences of adverse reactions reported in patients 2 to 18 years of age with pJIA treated with oral, weekly doses of methotrexate (5 mg/m2 per week to 20 mg/m2 per week or 0.1 mg/kg per week to 0.65 mg/kg per week) were as follows (most patients were receiving concomitant NSAIDs, and some received corticosteroids): elevated liver function tests, 14%; gastrointestinal reactions (e.g., nausea, vomiting, diarrhea), 11%; stomatitis, 2%; leukopenia, 2%; headache, 1.2%; alopecia, 0.5%; dizziness, 0.2%; rash, 0.2%.
Psoriasis
In two published series of adult psoriasis patients (n=204, 248) treated with methotrexate doses up to 25 mg per week for up to 4 years, adverse reaction rates were similar to those in patients with RA, except for alopecia, photosensitivity, and “burning of skin lesions” (each 3% to 10%). Painful plaque erosions have been reported.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of methotrexate. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: Aplastic anemia, lymphadenopathy, hypogammaglobulinemia
Cardiovascular disorders: Thromboembolic events (including arterial thrombosis, cerebral thrombosis, deep vein thrombosis, retinal vein thrombosis, thrombophlebitis, and pulmonary embolus), pericarditis, pericardial effusion, hypotension, sudden death
Endocrine: Diabetes
Eye disorders: Optic neuropathy, blurred vision, ocular irritation, conjunctivitis, xerophthalmia
Gastrointestinal disorders: Hemorrhagic enteritis, intestinal perforation, gingivitis, pancreatitis, pharyngitis, hematemesis, melena, gastrointestinal ulceration and bleeding
Hepatobiliary disorders: Acute hepatitis, decreased serum albumin, fibrosis, cirrhosis, liver failure
Immune system disorders: Anaphylaxis, anaphylactoid reactions, vasculitis
Metabolism: Hyperglycemia
Musculoskeletal disorders: Stress fracture, soft tissue necrosis, arthralgia, myalgia, osteoporosis
Nervous system disorders: Headaches, drowsiness, blurred vision, speech impairment (including dysarthria and aphasia), transient cognitive dysfunction, mood alteration, unusual cranial sensations, paresis, encephalopathy, leukoencephalopathy, and convulsions. Also, spinal radiculopathy with intrathecal use
Renal disorders: Severe renal toxicity including renal failure, azotemia, hematuria, proteinuria, cystitis
Reproductive disorders: Defective oogenesis or spermatogenesis, loss of libido, impotence, gynecomastia, menstrual dysfunction
Respiratory disorders: Pulmonary fibrosis, respiratory failure, chronic interstitial obstructive pulmonary disease, pleuritic pain and thickening, alveolitis
Skin disorders: Toxic epidermal necrolysis, Stevens-Johnson syndrome, exfoliative dermatitis, skin necrosis, and erythema multiforme, erythematous rashes, pruritus, alopecia, skin ulceration, accelerated nodulosis, urticaria, pigmentary changes, ecchymosis, telangiectasia, photosensitivity, acne, furunculosis
General disorders and administration site conditions: Injection site necrosis, injection site reaction
7. Drug Interactions
7.1 Effects of Other Drugs on Methotrexate
Drugs that Increase Methotrexate Exposure
Coadministration of methotrexate with the following products may increase methotrexate plasma concentrations, which may increase the risk of methotrexate severe adverse reactions.
Increased organ specific adverse reactions may also occur when methotrexate is coadministered with hepatotoxic or nephrotoxic products. If coadministration cannot be avoided, monitor closely for methotrexate adverse reactions when coadministered with:
- Penicillin or sulfonamide antibiotics
- Highly protein-bound drugs (e.g., oral anticoagulants, phenytoin, salicylates, sulfonamides, sulfonylureas, and tetracyclines)
- Proton pump inhibitors
- Probenecid
- Antifolate drugs (e.g., dapsone, pemetrexed, pyrimethamine and sulfonamides)
- Aspirin and other nonsteroidal anti-inflammatory drugs
Unexpectedly severe and fatal gastrointestinal toxicity can occur with concomitant administration of methotrexate (primarily at high dose) and nonsteroidal anti-inflammatory drugs (NSAIDs)
- Mercaptopurine
- Hepatotoxic products
- Weak acids (e.g., salicylates)
- Nephrotoxic products
Nitrous Oxide
Coadministration of methotrexate with nitrous oxide anesthesia potentiates the effect of methotrexate on folate-dependent metabolic pathways, which may increase the risk of severe methotrexate adverse reactions. Avoid nitrous oxide anesthesia in patients receiving methotrexate. Consider alternative therapies in patients who have received prior nitrous oxide anesthesia.
Folic Acid
Coadministration of methotrexate with folic acid or its derivatives decreases the clinical effectiveness of methotrexate in patients with neoplastic diseases. Methotrexate competes with reduced folates for active transport across cell membranes. Instruct patients to take folic or folinic acid only as directed by their healthcare provider [see Warnings and Precautions (5.12)].
7.2 Effects of Methotrexate on Other Drugs
Theophylline
Coadministration of methotrexate with theophylline increases theophylline plasma concentrations which may increase the risk of theophylline adverse reactions. Monitor theophylline levels and adjust the theophylline dosage in accordance with approved product labeling.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Methotrexate Injection is contraindicated in pregnant women with non-neoplastic diseases. Based on published reports and its mechanism of action, methotrexate can cause embryo-fetal toxicity and fetal death when administered to a pregnant woman [see Data and Clinical Pharmacology (12.1)]. There are no animal data that meet current standards for nonclinical developmental toxicity studies. Advise pregnant women with neoplastic diseases of the potential risk to a fetus. The preservative benzyl alcohol can cross the placenta; when possible, use the preservative-free formulation when Methotrexate Injection is needed during pregnancy to treat a neoplastic disease [see Warnings and Precautions (5.3) and Use in Specific Populations (8.4)].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
Published data from case reports, literature reviews, and observational studies report that methotrexate exposure during pregnancy is associated with an increased risk of embryo-fetal toxicity and fetal death. Methotrexate exposure during the first trimester of pregnancy is associated with an increased incidence of spontaneous abortions and multiple adverse developmental outcomes, including skull anomalies, facial dysmorphism, CNS abnormalities, limb abnormalities, and sometimes cardiac anomalies and intellectual impairment. Adverse outcomes associated with exposure during second and third trimesters of pregnancy include intrauterine growth restriction and functional abnormalities. Because methotrexate is widely distributed and persists in the body for a prolonged period, there is a potential risk to the fetus from preconception methotrexate exposure.
A prospective multicenter study evaluated pregnancy outcomes in women taking methotrexate less than or equal to 30 mg/week after conception. The rate of spontaneous abortion/miscarriage in pregnant women exposed to methotrexate was 42.5% (95% confidence interval [95% CI] 29.2 to 58.7), which was higher than in unexposed patients with autoimmune disease (22.5%, 95% CI 16.8 to 29.7) and unexposed patients with non-autoimmune disease (17.3%, 95% CI 13 to 22.8). Of the live births, the rate of major birth defects in pregnant women exposed to methotrexate after conception was higher than in unexposed patients with autoimmune disease (adjusted odds ratio (OR) 1.8 [95% CI 0.6 to 5.7]) and unexposed patients with non-autoimmune disease (adjusted OR 3.1 [95% CI 1.03 to 9.5]) (2.9%). Major birth defects associated with pregnancies exposed to methotrexate after conception were not always consistent with methotrexate-associated adverse developmental outcomes.
8.2 Lactation
Risk Summary
Limited published literature reports the presence of methotrexate in human milk in low amounts, with the highest breast milk to plasma concentration ration reported to be 0.08:1. No information is available on the effects of methotrexate on a breastfed infant or on milk production. Because of the potential for serious adverse reactions from methotrexate in breastfed infants, advise women not to breastfeed during treatment with Methotrexate Injection and for 1 week after the final dose.
8.3 Females and Males of Reproductive Potential
Methotrexate can cause malformations and fetal death at doses less than or equal to the recommended clinical doses [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating Methotrexate Injection [see Contraindications (4) and Use in Specific Populations (8.1)].
Contraception
Females
Advise females of reproductive potential to use effective contraception during and for 6 months after the final dose of Methotrexate Injection therapy.
Males
Methotrexate can cause chromosomal damage to sperm cells. Advise males with female partners of reproductive potential to use effective contraception during and for 3 months after the final dose of Methotrexate Injection therapy.
Infertility
Females
Based on published reports of female infertility after therapy with methotrexate, advise females of reproductive potential that Methotrexate Injection can cause impairment of fertility and menstrual dysfunction during and after cessation of therapy. It is not known if the infertility may be reversed in all affected females.
Males
Based on published reports of male infertility after therapy with methotrexate, advise males that Methotrexate Injection can cause oligospermia or infertility during and after cessation of therapy. It is not known if the infertility may be reversed in all affected males.
8.4 Pediatric Use
The safety and effectiveness of Methotrexate Injection in pediatric patients have been established for ALL, meningeal leukemia prophylaxis and treatment, non-Hodgkin lymphoma, osteosarcoma and in pJIA. Clinical studies evaluating the use of methotrexate in pediatric patients with pJIA demonstrated safety comparable to that observed in adults with RA [see Adverse Reactions (6.1)]. The safety and effectiveness of Methotrexate Injection have not been established in pediatric patients for the treatment of breast cancer, squamous cell carcinoma of the head and neck, gestational trophoblastic neoplasia, rheumatoid arthritis, and psoriasis. Additional risk information is described below.
Risks of Serious Adverse Reactions due to Benzyl Alcohol-Preservative
Due to the risk of serious adverse reactions and fatal gasping syndrome following administration of intravenous solutions containing the preservative benzyl alcohol in neonates, use only preservative-free Methotrexate Injection in neonates and low-birth weight infants. The “gasping syndrome” is characterized by CNS depression, metabolic acidosis, and gasping respirations.
Serious adverse reactions including fatal reactions and the “gasping syndrome” occurred in premature neonates and low-birth weight infants in the neonatal intensive care unit who received drugs containing benzyl alcohol as a preservative. In these cases, benzyl alcohol dosages of 99 to 234 mg/kg/day produced high levels of benzyl alcohol and its metabolites in the blood and urine (blood levels of benzyl alcohol were 0.61 to 1.378 mmol/L). Additional adverse reactions include gradual neurological deterioration, seizures, intracranial hemorrhage, hematological abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and cardiovascular collapse. Preterm, low-birth weight infants may be more likely to develop these reactions because they may be less able to metabolize benzyl alcohol.
When prescribing in infants (non-neonate, non-low-birth weight), if a preservative-free formulation of Methotrexate Injection is not available and use of a benzyl alcohol-containing formulation is necessary, consider the combined daily metabolic load of benzyl alcohol from all sources including Methotrexate Injection (Methotrexate Injection contains 9.4 mg of benzyl alcohol/per mL) and other drugs containing benzyl alcohol. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known.
Do not administer methotrexate formulations containing benzyl alcohol intrathecally due to the risk of severe neurotoxicity [see Warnings and Precautions (5.3)].
Leukemia/Lymphoma
Serious neurotoxicity, frequently manifested as generalized or focal seizures, has been reported with unexpectedly increased frequency among pediatric patients with acute lymphoblastic leukemia who were treated with intermediate-dose intravenous methotrexate (1 g/m2) [see Warnings and Precautions (5.8)].
8.5 Geriatric Use
Clinical studies of methotrexate did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Renal Impairment
Methotrexate elimination is reduced in patients with renal impairment [creatinine clearance (CLcr) less than 90 mL/min, calculated using Cockcroft-Gault] [see Clinical Pharmacology (12.3)]. Patients with renal impairment are at increased risk for methotrexate adverse reactions.
Follow recommendations to promote methotrexate elimination and decrease risk of acute kidney injury and other methotrexate toxicities in patients who are receiving intermediate- or high-dose regimens [see Dosage and Administration (2.2) and Warnings and Precautions (5.6)]. Consider reducing the dose or discontinuing Methotrexate Injection in patients with renal impairment as appropriate.
8.7 Hepatic Impairment
The pharmacokinetics and safety of methotrexate in patients with hepatic impairment is unknown Patients with hepatic impairment may be at increased risk for methotrexate adverse reaction based on elimination characteristics of methotrexate [see Clinical Pharmacology (12.3)]. Consider reducing the dose or discontinuing Methotrexate Injection in patients with hepatic impairment as appropriate [see Warnings and Precautions (5.7)].
10. Overdosage
Manifestations
Overdosage, including fatal overdosage, has occurred with methotrexate [see Warnings and Precautions (5.19)].
Manifestations of overdosage include adverse reactions reported at pharmacologic doses, particularly hematologic and gastrointestinal reactions (e.g., leukopenia, thrombocytopenia, anemia, pancytopenia, myelosuppression, mucositis, stomatitis, oral ulceration, nausea, vomiting, gastrointestinal ulceration, or gastrointestinal bleeding). In some cases, no symptoms were reported; however, sepsis or septic shock, renal failure, and aplastic anemia were also reported.
Manifestations of intrathecal overdosage include CNS symptoms (e.g., headache, nausea and vomiting, seizure or convulsion, and acute toxic encephalopathy). In some cases, no symptoms were reported; however, cerebellar herniation associated with increased intracranial pressure and acute toxic encephalopathy have also been reported.
Management
Leucovorin and levoleucovorin are indicated to diminish the toxicity and counteract the effect of inadvertently administered overdosages of methotrexate. Administer leucovorin or levoleucovorin as soon as possible after overdosage (refer to the leucovorin or levoleucovorin prescribing information). Monitor serum methotrexate concentrations closely to guide leucovorin or levoleucovorin therapy. Monitor serum creatinine concentrations closely because high serum methotrexate concentrations may cause renal damage leading to acute renal failure.
Glucarpidase is indicated for the treatment of toxic methotrexate concentrations in patients with delayed methotrexate clearance due to impaired renal function (refer to the glucarpidase prescribing information). If glucarpidase is used, do not administer leucovorin within 2 hours before or after a dose of glucarpidase because leucovorin is a substrate for glucarpidase.
Hydration and urinary alkalinization may be necessary to prevent the precipitation of methotrexate and/or its metabolites in the renal tubules. Neither hemodialysis nor peritoneal dialysis has been shown to improve methotrexate elimination. However, effective clearance of methotrexate has been reported with acute, intermittent hemodialysis using a high-flux dialyzer.
11. Methotrexate Injection Description
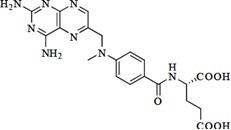
Methotrexate, USP is a folate analog metabolic inhibitor with the chemical name of N-[4-[[(2,4-diamino-6-pteridinyl) methyl]methylamino]benzoyl]-L-glutamic acid and a molecular· weight of 454.44. The molecular formula is C20H22N8O5, and the structural formula is shown below:
Preservative-free Methotrexate Injection, USP is supplied in sterile single-dose vials for intravenous, intramuscular, subcutaneous, or intrathecal use.
- Each 25 mg/mL, 2 mL, 4 mL, 8 mL and 10 mL vial contains 50 mg, 100 mg, 200 mg, and 250 mg methotrexate, USP equivalent to 54.8 mg, 109.6 mg, 219.3 mg and 274.18 mg of methotrexate sodium respectively, and the following inactive ingredients: Sodium chloride 9.8 mg, 19.6 mg, 39.2 mg and 49 mg respectively. May contain sodium hydroxide and/or hydrochloric acid to adjust pH between 7.0 to 9.0.
12. Methotrexate Injection - Clinical Pharmacology
12.1 Mechanism of Action
Methotrexate inhibits dihydrofolic acid reductase. Dihydrofolates must be reduced to tetrahydrofolates by this enzyme before they can be utilized as carriers of one-carbon groups in the synthesis of purine nucleotides and thymidylate. Therefore, methotrexate interferes with DNA synthesis, repair, and cellular replication. Actively proliferating tissues such as malignant cells, bone marrow, fetal cells, buccal and intestinal mucosa, and cells of the urinary bladder are in general more sensitive to this effect of methotrexate.
The mechanism of action in rheumatoid arthritis, pJIA, and in psoriasis is unknown.
12.3 Pharmacokinetics
Distribution
After intravenous administration, the initial volume of distribution is approximately 0.18 L/kg (18% of body weight) and steady-state volume of distribution is approximately 0.4 L/to 0.8 L/kg (40% to 80% of body weight).
Methotrexate competes with reduced folates for active transport across cell membranes by means of a single carrier-mediated active transport process. At serum concentrations greater than 100 micromolar, passive diffusion becomes a major pathway by which effective intracellular concentrations can be achieved.
Methotrexate in serum is approximately 50% protein bound.
Methotrexate may be displaced from plasma albumin by various compounds, including sulfonamides, salicylates, tetracyclines, chloramphenicol, and phenytoin.
Methotrexate does not penetrate the blood-cerebrospinal fluid barrier in therapeutic amounts when given intravenously, intramuscularly, or subcutaneously.
Elimination
The terminal half-life reported for methotrexate is approximately 3 to 10 hours for patients receiving treatment for psoriasis, or rheumatoid arthritis or low-dose antineoplastic therapy (less than 30 mg/m2).
Following intravenous administration of high-dose methotrexate, the terminal half-life is 8 hours to 15 hours.
Metabolism
Methotrexate undergoes hepatic and intracellular metabolism to polyglutamated forms that can be converted back to methotrexate by hydrolase enzymes. These polyglutamates act as inhibitors of dihydrofolate reductase and thymidylate synthetase. Small amounts of methotrexate polyglutamates may remain in tissues for extended periods. The retention and prolonged drug action of these active metabolites vary among different cells, tissues, and tumors. Methotrexate undergoes minor metabolism to 7-hydroxymethotrexate, and accumulation may become significant following high dosages. The aqueous solubility of 7-hydroxymethotrexate is 3- to 5-fold lower than the solubility of methotrexate.
Excretion
Renal excretion is the primary route of elimination and is dependent upon dosage and route of administration. With intravenous administration, 80% to 90% of the administered dose is excreted unchanged in the urine within 24 hours. There is limited biliary excretion amounting to 10% or less of the administered dose. Enterohepatic recirculation of methotrexate has been proposed.
Renal excretion occurs by glomerular filtration and active tubular secretion. Nonlinear elimination due to saturation of renal tubular reabsorption has been observed in psoriatic patients at doses between 7.5 mg and 30 mg.
Specific Populations
Pediatric Patients
In pediatric patients receiving methotrexate for acute lymphoblastic leukemia (6.3 mg/m2 to 30 mg/m2), or for JIA (3.75 mg/m2 to 26.2 mg/m2), the terminal half-life has been reported to range from 0.7 to 5.8 hours or from 0.9 to 2.3 hours, respectively [see Use in Specific Populations (8.4)]. Patients with Renal impairment
The elimination half-life of methotrexate increases with the severity of renal impairment, with high inter-individual variability [see Use in Specific Populations (8.6)]
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Methotrexate has been evaluated in a number of animal studies for carcinogenic potential with inconclusive results. There is evidence that methotrexate causes chromosomal damage to animal somatic cells and human bone marrow cells [see Use in Specific Populations (8.1, 8.2, 8.3)].
16. How is Methotrexate Injection supplied
How Supplied
Methotrexate Injection, USP is a sterile, clear, yellowish solution available with preservative-free (single-dose vials) as follows:
| Strength/Fill volume
| NDC number
| Pack style
|
| Preservative-free |
||
| 50 mg/2 mL | 55150-510-01 | Single-Dose Vial packaged individually |
| 55150-510-05 | Single-Dose Vials in a carton of 5 |
|
| 100 mg/4 mL | 55150-511-01 | Single-Dose Vial packaged individually |
| 55150-511-10 | Single-Dose Vials in a carton of 10 |
|
| 200 mg/8 mL | 55150-512-01 | Single-Dose Vial packaged individually |
| 55150-512-10 | Single-Dose Vials in a carton of 10 |
|
| 250 mg/10 mL | 55150-513-01 | Single-Dose Vial packaged individually |
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Protect from light.
Methotrexate Injection, USP is a hazardous drug. Follow applicable special handling and disposal procedures.1
The vial stopper is not made with natural rubber latex.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information). Embryo-Fetal Toxicity
- Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Contraindications (4), Warnings and Precautions (5.1), and Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during methotrexate therapy and for 6 months after the final dose [see Use in Specific Populations (8.3)].
- Advise males of reproductive potential to use effective contraception during methotrexate therapy and for 3 months after the final dose [see Use in Specific Populations (8.3)].
Hypersensitivity Reactions
Advise patients of the potential risk of hypersensitivity and that Methotrexate Injection is contraindicated in patients with a history of severe hypersensitivity to methotrexate. Advise patients to seek immediate medical attention if signs or symptoms of a hypersensitivity reaction occur [see Warnings and Precautions (5.2)].
Myelosuppression and Serious Infections
Advise patient to contact their healthcare provider immediately for new onset fever, symptoms of infection, easy bruising or persistent bleeding [see Warnings and Precautions (5.4, 5.5)].
Renal Toxicity
Advise patients that methotrexate can cause renal toxicity. Advise patients to immediately contact their healthcare provider for signs or symptoms of renal toxicity, such as marked increases or decreases in urinary output [see Warnings and Precautions (5.6)].
Hepatotoxicity
Advise patients to report signs or symptoms of hepatic toxicity and avoidance of alcohol during methotrexate treatment [see Warnings and Precautions (5.7)].
Neurotoxicity
Advise patient to contact their healthcare provider immediately if they develop new neurological symptoms [see Warnings and Precautions (5.8)].
Gastrointestinal Toxicity
Advise patients to contact their healthcare provider if they develop diarrhea, vomiting, or stomatitis. Advise patients to immediately contact their healthcare provider for high fever, rigors, persistent or severe abdominal pain, severe constipation, hematemesis, or melena [see Warnings and Precautions (5.9)].
Pulmonary Toxicity
Advise patients to contact their healthcare provider for symptoms of cough, fever, and dyspnea [see Warnings and Precautions (5.10)].
Dermatologic Toxicity
Advise patients that Methotrexate Injection can cause serious skin rash and to immediately contact their healthcare provider for new or worsening skin rash. Advise patients to avoid excessive sun exposure and to use sun protection measures [see Warnings and Precautions (5.11)].
Secondary Malignancies
Advise patients on the risk of second primary malignancies during treatment with Methotrexate Injection [see Warnings and Precautions (5.13)].
Lactation
Advise women not to breastfeed during treatment with methotrexate and for 1 week after the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise females and males of reproductive potential that methotrexate may cause impairment of fertility [see Use in Specific Populations (8.3)].
Drug Interactions
- Advise patients and caregivers to inform their healthcare provider of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7)].
- Instruct patients being treated for neoplastic indication to not take products containing folic acid or folinic acid unless directed to do so by their healthcare provider [see Warnings and Precautions (5.12)].
Distributed by:
Eugia US LLC
279 Princeton-Hightstown Rd.
E. Windsor, NJ 08520
Manufactured by:
Eugia Pharma Specialities Limited
Hyderabad - 500032
India
|
Patient Information Methotrexate (Meth-oh-trex-ate) Injection for intravenous, intramuscular, subcutaneous, or intrathecal use |
| What is the most important information I should know about Methotrexate Injection?
Methotrexate Injection can cause serious side effects that may be severe and lead to death, including: Harm to an unborn baby, including birth defects or death of an unborn baby. Females who can become pregnant: • Your healthcare provider should do a pregnancy test before you start taking Methotrexate Injection to see if you are pregnant. • If you are being treated for a medical condition other than cancer, do not receive or take Methotrexate Injection if you are pregnant. See “Do not receive Methotrexate Injection if”. • If you are taking Methotrexate Injection to treat your cancer, you and your healthcare provider will decide if you will receive or take Methotrexate Injection if you are pregnant. • Use effective birth control (contraception) during treatment and for 6 months after your final dose of Methotrexate Injection. Ask your healthcare provider what forms of birth control you can use during this time. Tell your healthcare provider right away if you become pregnant or think you are pregnant during treatment with Methotrexate Injection. Males with female partners who are able to become pregnant: • Use effective birth control during treatment and for 3 months after your final dose of Methotrexate Injection. Tell your healthcare provider right away if your female partner becomes pregnant during treatment with Methotrexate Injection. Severe allergic reactions. Severe allergic reactions can happen with Methotrexate Injection. • Do not receive Methotrexate Injection if you have had a severe allergic reaction to methotrexate in the past. Get medical help right away if you develop any of the signs or symptoms of a severe allergic reaction to Methotrexate Injection, including:
Call your healthcare provider right away if you develop: • a new fever (temperature of 100.4°F or higher) • symptoms of infection • easy bruising or bleeding that will not stop Your healthcare provider may give you medicines to support your blood counts or give you transfusions if needed, and change your dose or stop your treatment with Methotrexate Injection if needed. Serious infections. People who receive treatment with Methotrexate Injection have an increased risk of developing serious infections that can be life-threatening or lead to death. These infections include: • bacterial infections • fungal infections • viral infections • certain infections that happen because your immune system is weakened • hepatitis B infection that comes back (reactivation) • tuberculosis (TB) infection that is new or that comes back (reactivation) • shingles (herpes zoster) • cytomegalovirus infections Your healthcare provider will closely watch you for signs and symptoms of infection during treatment with Methotrexate Injection. Your healthcare provider may hold or stop your treatment with Methotrexate Injection if you develop a serious infection. Kidney problems. Methotrexate Injection can cause kidney damage including sudden kidney failure that may not go away (irreversible). People who already have kidney problems have an increased risk of kidney problems with Methotrexate Injection. Your healthcare provider will check your kidney function during treatment, and will hold or stop Methotrexate Injection treatment as needed for severe kidney damage. Call your healthcare provider right away if you have signs or symptoms of kidney problems such as a big change in the amount of urine that you make, either increased or decreased. Liver problems. Methotrexate Injection can cause severe liver problems including liver scarring (fibrosis), cirrhosis, and liver failure that may not get better (possibly irreversible) and can cause death. • In people with psoriasis who receive Methotrexate Injection, liver fibrosis or cirrhosis may happen without any symptoms or abnormal liver tests. The risk for liver problems in people with psoriasis increases with the amount of Methotrexate Injection that you receive over time. • Your healthcare provider will do tests to monitor your liver function before you start and during treatment with Methotrexate Injection, and may hold or stop your treatment with Methotrexate Injection, if needed. • The risk of liver problems is increased with heavy use of alcohol. Avoid drinking alcohol during Methotrexate Injection treatment. Tell your healthcare provider if you develop any signs or symptoms of liver problems during treatment with Methotrexate Injection, including: • tiredness • easy bleeding or bruising • loss of appetite • nausea • difficulty thinking clearly • swelling in your legs, feet, or ankles • weight loss • itchy skin • yellowing of your skin or the white part of your eyes • weaknees Brain and spinal cord (nervous system) problems. Methotrexate Injection can cause nervous system problems that can be severe and last for a short time or last for a long time. These nervous system problems can get progressively worse, may not get better (possibly irreversible), and can cause death. • Serious nervous system problems can happen in children who receive Methotrexate Injection, including seizures that can begin on one side of the brain (focal seizures) or on both sides of the brain (generalized seizures). • The risk for a nervous system problem called leukoencephalopathy is increased in people who have had radiation treatment to their head and spine (craniospinal irradiation) in the past. Call your healthcare provider if you develop any new neurological symptoms. • People who receive high-dose Methotrexate Injection can develop sudden symptoms that are like the symptoms of a stroke, but they last a short time and may go away (transient). • People who receive injections of Methotrexate Injection into their spine (intrathecal methotrexate) can develop inflammation of the lining around the spinal nerves. Call your healthcare provider right away if you or your child develop any new signs or symptoms of a nervous system problem during treatment with Methotrexate Injection, including: • confusion • weakness on one side of your body • sudden blindness that goes away • seizures • coma • headache • back pain • stiff neck • fever Severe stomach and intestine (gastrointestinal) problems. Methotrexate Injection can cause diarrhea, vomiting, mouth sores, stomach and intestinal inflammation with severe bleeding, and tears in the intestinal wall (perforation), and can lead to death.
Call your healthcare provider if you develop symptoms of a lung problem, including: cough, fever, and trouble breathing. Skin reactions. Severe skin reactions can happen with Methotrexate Injection, that can be serious and can lead to death. • In people with psoriasis: Your psoriasis may get worse if you are exposed to sunlight or other types of ultraviolet light. • Methotrexate Injection can cause reactivation of skin reactions that can happen after radiation therapy (radiation recall) and can cause sunburn to come back (photodermatitis). Limit sunlight exposure during treatment with Methotrexate Injection. Use sunscreen and wear protective clothing when you will be exposed to sunlight during treatment with Methotrexate Injection. Call your healthcare provider right away if you develop a new or worsening skin rash during treatment with Methotrexate Injection. See “What are the possible side effects of Methotrexate Injection?” for more information about side effects. |
| What is Methotrexate Injection? Methotrexate Injection is a prescription medicine used: in adults and children: • in combination with other chemotherapy medicines to treat acute lymphoblastic leukemia (ALL) to help prevent (prophylaxis) and to treat leukemia that spreads to the covering of the brain and spinal cord (meninges). • to treat non-Hodgkin lymphoma • in combination with other chemotherapy medicines to treat osteosarcoma in adults: • in combination with other chemotherapy medicines to treat breast cancer • alone to treat squamous cell carcinoma of the head and neck • in combination with other chemotherapy medicines to treat gestational trophoblastic neoplasia Methotrexate Injection is a prescription medicine used: • in adults to treat rheumatoid arthritis (RA) • in children to treat polyarticular juvenile idiopathic arthritis (pJIA) • in adults to treat severe psoriasis |
| Do not receive Methotrexate Injection if you: • have had a severe allergic reaction to Methotrexate Injection. See “What is the most important information I should know about Methotrexate Injection?” • you are pregnant and are being treated, or will be treated with Methotrexate Injection for rheumatoid arthritis, pJIA, or severe psoriasis, or for any disease other than cancer. Methotrexate Injection can cause harm to an unborn baby including birth defects or death of an unborn baby. See “What is the most important information I should know about Methotrexate Injection?” Before you receive Methotrexate Injection, tell your healthcare provider about all of your medical conditions, including if you: • have kidney problems or are receiving dialysis treatments • have liver problems • have a history of neurologic problems, including seizures • drink-alcohol containing beverages during treatment with Methotrexate Injection, or if there are any changes in the amount of alcoholic beverages you drink • have fluid in your stomach-area (ascites) • have lung problems or fluid in your lungs (pleural effusion) • plan to have any surgeries with general anesthesia, including dental surgery • have stomach ulcers (peptic ulcer disease) • have ulcerative colitis • have recently received or are scheduled to receive a vaccine. You should not receive live vaccines during treatment with Methotrexate Injection. • are breastfeeding or plan to breastfeed. Methotrexate may pass into your breast milk. Do not breastfeed during treatment and for 1 week after your last dose of Methotrexate Injection. Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Taking certain medicines can affect the amount of methotrexate in your blood and can increase your risk for serious side effects. |
| How will I receive or take Methotrexate Injection?
• Depending on your medical condition and the dose of Methotrexate Injection that is prescribed by your healthcare provider, Methotrexate Injection can be given to you:
• Your healthcare provider will decide your dose, how you will receive Methotrexate Injection, and how often you need to receive it, depending on your medical condition that is being treated. • If you are receiving high-dose Methotrexate Injection to treat your cancer, you will receive the medicine leucovorin to help prevent severe side effects (“rescue”) to your bone marrow and other normal cells in your body. You will also receive intravenous (IV) fluids and other medicines to help prevent and treat side effects. • If you are receiving a “moderate-dose” of Methotrexate Injection to treat your cancer, you may also receive leucovorin. • Do not take folic acid or folinic acid during treatment with Methotrexate Injection to treat your cancer, unless your healthcare provider tells you to. Taking folic acid or folinic acid with Methotrexate Injection may make your treatment less effective. • Your healthcare provider will do blood tests to check for side effects during treatment with Methotrexate Injection. • Your healthcare provider may stop your treatment, change when you receive your treatment, or change the dose of your treatment if you have certain side effects while receiving Methotrexate Injection. If you are receiving Methotrexate Injection for treatment of severe psoriasis, rheumatoid arthritis, or polyarticular juvenile idiopathic arthritis: • You should receive your Methotrexate Injection dose 1 time each week, not every day. Serious side effects and death have happened in people who mistakenly have taken Methotrexate every day instead of 1 time each week. • Take folic acid or folinic acid every day during treatment with Methotrexate Injection, as instructed by your healthcare provider, to help reduce the chance of developing certain side effects, such as mouth sores. • If you receive too much Methotrexate Injection call your healthcare provider or go to your nearest hospital emergency room right way. You will need to receive a medicine as soon as possible to help reduce side effects that could be severe and could cause death. In all patients receiving Methotrexate Injection: • If you miss receiving a dose of Methotrexate Injection, call your healthcare provider for instructions about when to receive your next dose of Methotrexate Injection. |
| What are the possible side effects of Methotrexate Injection?
Methotrexate Injection can cause serious side effects, including: • See “What is the most important information I should know about Methotrexate Injection?” • Tumor lysis syndrome (TLS). TLS is caused by the fast breakdown of cancer cells. TLS can cause kidney failure and the need for dialysis treatment, abnormal heart rhythm, seizure, and sometimes death. Your healthcare provider may do blood tests to check you for TLS if you are receiving Methotrexate Injection as a cancer treatment. Your healthcare provider will treat you as needed to prevent or manage TLS if you develop it during treatment with Methotrexate Injection. • New (secondary) cancers. New (secondary) cancers can happen in people who take or receive Methotrexate Injection at any dose.
The most common side effects of Methotrexate Injection include: • mouth sores or ulcers • nausea • decreased white blood cell count. See “What is the most important information I should know about Methotrexate Injection?” • upset stomach Possible fertility problems (infertility) in males and females. Methotrexate Injection can cause fertility problems in males and females, and cause sperm production to stop in males, and menstrual problems in females. In females, your periods (menstrual cycle) may be irregular or completely stop when you receive Methotrexate Injection. Your periods may or may not return to normal following treatment. It is not known if your fertility will return after treatment. Talk to your healthcare provider about your risk for infertility if this is a concern for you. These are not all of the possible side effects of Methotrexate Injection. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. General information about the safe and effective use of Methotrexate Injection. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about Methotrexate Injection that is written for health professionals. |
| What are the ingredients in Methotrexate Injection?
Active ingredient: Methotrexate, USP. Inactive ingredients for Methotrexate Injection, USP Preservative-free: sodium chloride. May contain sodium hydroxide and/or hydrochloric acid to adjust pH between 7.0 to 9.0. Distributed by: Eugia US LLC 279 Princeton-Hightstown Rd. E. Windsor, NJ 08520 Manufactured by: Eugia Pharma Specialities Limited Hyderabad - 500032 India For more information, go to eugiaus.com or call 1-866-850-2876. |
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: February 2025
PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 50 mg per 2 mL - Container Label
Rx only NDC 55150-510-01
Methotrexate
Injection, USP
50 mg per 2 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous, Intramuscular,
Subcutaneous and Intrathecal Use Only
Sterile Isotonic Solution
2 mL Single-Dose Vial

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 50 mg per 2 mL - Container-Carton (1's Pack)
Rx only NDC 55150-510-01
Methotrexate
Injection, USP
50 mg per 2 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous,
Intramuscular, Subcutaneous
and Intrathecal Use Only
Sterile Isotonic Solution
1 x 2 mL Single-Dose Vial
eugia

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 50 mg per 2 mL - Container-Carton (5's Pack)
Rx only NDC 55150-510-05
Methotrexate
Injection, USP
50 mg per 2 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous, Intramuscular, Subcutaneous and
Intrathecal Use Only
Sterile Isotonic Solution
5 x 2 mL Single-Dose Vials
eugia

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 100 mg per 4 mL - Container Label
Rx only NDC 55150-511-01
Methotrexate
Injection, USP
100 mg per 4 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous, Intramuscular,
Subcutaneous and Intrathecal Use Only
Sterile Isotonic Solution
4 mL Single-Dose Vial

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 100 mg per 4 mL - Container-Carton (1's Pack)
Rx only NDC 55150-511-01
Methotrexate
Injection, USP
100 mg per 4 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous,
Intramuscular, Subcutaneous
and Intrathecal Use Only
Sterile Isotonic Solution
1 x 4 mL Single-Dose Vial
eugia

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 100 mg per 4 mL - Container-Carton (10's Pack)
Rx only NDC 55150-511-10
Methotrexate
Injection, USP
100 mg per 4 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous, Intramuscular, Subcutaneous and
Intrathecal Use Only
Sterile Isotonic Solution
10 x 4 mL Single-Dose Vials
eugia

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 200 mg per 8 mL - Container Label
Rx only NDC 55150-512-01
Methotrexate
Injection, USP
200 mg per 8 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous, Intramuscular,
Subcutaneous and
Intrathecal Use Only
Sterile Isotonic Solution
8 mL Single-Dose Vial

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 200 mg per 8 mL - Container-Carton (1's Pack)
Rx only NDC 55150-512-01
Methotrexate
Injection, USP
200 mg per 8 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous,
Intramuscular,
Subcutaneous and
Intrathecal Use Only
Sterile Isotonic Solution
1 x 8 mL Single-Dose Vial
eugia

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 200 mg per 8 mL - Container-Carton (10's Pack)
Rx only NDC 55150-512-10
Methotrexate
Injection, USP
200 mg per 8 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous, Intramuscular, Subcutaneous and
Intrathecal Use Only
Sterile Isotonic Solution
10 x 8 mL Single-Dose Vials
eugia

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 250 mg per 10 mL - Container Label
Rx only NDC 55150-513-01
Methotrexate
Injection, USP
250 mg per 10 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous, Intramuscular,
Subcutaneous and
Intrathecal Use Only
Sterile Isotonic Solution
10 mL Single-Dose Vial

PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 250 mg per 10 mL- Container-Carton (1's Pack)
Rx only NDC 55150-513-01
Methotrexate
Injection, USP
250 mg per 10 mL
(25 mg/mL)
Preservative free
Hazardous Drug
For Intravenous,
Intramuscular,
Subcutaneous and
Intrathecal Use Only
Sterile Isotonic Solution
1 x 10 mL Single-Dose Vial
eugia

| METHOTREXATE
methotrexate injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| METHOTREXATE
methotrexate injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| METHOTREXATE
methotrexate injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| METHOTREXATE
methotrexate injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Eugia US LLC (968961354) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Mylan Laboratories Limited | 676159830 | ANALYSIS(55150-510, 55150-511, 55150-512, 55150-513) , STERILIZE(55150-510, 55150-511, 55150-512, 55150-513) , LABEL(55150-510, 55150-511, 55150-512, 55150-513) , MANUFACTURE(55150-510, 55150-511, 55150-512, 55150-513) , PACK(55150-510, 55150-511, 55150-512, 55150-513) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| EUGIA Pharma Specialities Limited | 872201704 | ANALYSIS(55150-510, 55150-511, 55150-512, 55150-513) , MANUFACTURE(55150-510, 55150-511, 55150-512, 55150-513) , PACK(55150-510, 55150-511, 55150-512, 55150-513) | |
Frequently asked questions
- Why should I take folic acid with methotrexate?
- What causes Plaque Psoriasis?
- How long does it take for methotrexate to work?
- Does methotrexate cause weight gain?
- How do I know if methotrexate is working for rheumatoid arthritis?
- What are the different brands of methotrexate?
- How long does methotrexate stay in your system?
- How does methotrexate work for ectopic pregnancy?
More about methotrexate
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (335)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- Support group
- Drug class: antimetabolites
- Breastfeeding
Patient resources
Professional resources
Other brands
Otrexup, Trexall, Rasuvo, Xatmep, ... +3 more