Keytruda Prescribing Information
Package insert / product label
Generic name: pembrolizumab
Dosage form: injection, powder, lyophilized, for solution
Drug class: Anti-PD-1 and PD-L1 monoclonal antibodies (immune checkpoint inhibitors)
J Code (medical billing code): J9271 (1 mg, injection)
Medically reviewed by Drugs.com. Last updated on Aug 18, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
KEYTRUDA® (pembrolizumab) injection, for intravenous use
Initial U.S. Approval: 2014
Recent Major Changes
Indications and Usage for Keytruda
KEYTRUDA is a programmed death receptor-1 (PD-1)-blocking antibody indicated:
Melanoma
- for the treatment of patients with unresectable or metastatic melanoma. (1.1)
- for the adjuvant treatment of adult and pediatric (12 years and older) patients with Stage IIB, IIC, or III melanoma following complete resection. (1.1)
Non-Small Cell Lung Cancer (NSCLC)
- in combination with pemetrexed and platinum chemotherapy, as first-line treatment of patients with metastatic nonsquamous NSCLC, with no EGFR or ALK genomic tumor aberrations. (1.2)
- in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, as first-line treatment of patients with metastatic squamous NSCLC. (1.2)
- as a single agent for the first-line treatment of patients with NSCLC expressing PD-L1 [Tumor Proportion Score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
- as a single agent for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA. (1.2, 2.1)
- for the treatment of patients with resectable (tumors ≥4 cm or node positive) NSCLC in combination with platinum-containing chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery. (1.2)
- as a single agent, for adjuvant treatment following resection and platinum-based chemotherapy for adult patients with Stage IB (T2a ≥4 cm), II, or IIIA NSCLC. (1.2)
Head and Neck Squamous Cell Cancer (HNSCC)
- in combination with platinum and FU for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC. (1.3)
- as a single agent for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [Combined Positive Score (CPS) ≥1] as determined by an FDA-approved test. (1.3, 2.1)
- as a single agent for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy. (1.3)
Classical Hodgkin Lymphoma (cHL)
- for the treatment of adult patients with relapsed or refractory cHL. (1.4)
- for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy. (1.4)
Primary Mediastinal Large B-Cell Lymphoma (PMBCL)
- for the treatment of adult and pediatric patients with refractory PMBCL, or who have relapsed after 2 or more prior lines of therapy. (1.5)
- Limitations of Use: KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Cancer
- in combination with enfortumab vedotin, for the treatment of adult patients with locally advanced or metastatic urothelial cancer. (1.6)
- as a single agent for the treatment of patients with locally advanced or metastatic urothelial carcinoma who:
- are not eligible for any platinum-containing chemotherapy, or
- who have disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy. (1.6)
- as a single agent for the treatment of patients with Bacillus Calmette-Guerin (BCG)-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS) with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy. (1.6)
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
- for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. (1.7, 2.1)
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer (CRC)
- for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC) as determined by an FDA-approved test. (1.8, 2.1)
Gastric Cancer
- in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, for the first-line treatment of adults with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.1 (1.9)
- in combination with fluoropyrimidine- and platinum-containing chemotherapy, for the first-line treatment of adults with locally advanced unresectable or metastatic HER2-negative gastric or gastroesophageal junction (GEJ) adenocarcinoma. (1.9)
Esophageal Cancer
- for the treatment of patients with locally advanced or metastatic esophageal or gastroesophageal junction (GEJ) (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
- in combination with chemoradiotherapy, for the treatment of patients with FIGO 2014 Stage III-IVA cervical cancer. (1.11)
- in combination with chemotherapy, with or without bevacizumab, for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test. (1.11, 2.1)
- as a single agent for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test. (1.11, 2.1)
Hepatocellular Carcinoma (HCC)
- for the treatment of patients with HCC secondary to hepatitis B who have received prior systemic therapy other than a PD-1/PD-L1-containing regimen. (1.12)
Biliary Tract Cancer (BTC)
- in combination with gemcitabine and cisplatin, for the treatment of patients with locally advanced unresectable or metastatic biliary tract cancer. (1.13)
Merkel Cell Carcinoma (MCC)
- for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma. (1.14)
Renal Cell Carcinoma (RCC)
- in combination with axitinib, for the first-line treatment of adult patients with advanced RCC. (1.15)
- in combination with lenvatinib, for the first-line treatment of adult patients with advanced RCC. (1.15)
- for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions. (1.15)
Endometrial Carcinoma
- in combination with carboplatin and paclitaxel, followed by KEYTRUDA as a single agent, for the treatment of adult patients with primary advanced or recurrent endometrial carcinoma. (1.16)
- in combination with lenvatinib, for the treatment of adult patients with advanced endometrial carcinoma that is mismatch repair proficient (pMMR) as determined by an FDA-approved test or not MSI-H, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation. (1.16, 2.1)
- as a single agent, for the treatment of adult patients with advanced endometrial carcinoma that is MSI-H or dMMR, as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation. (1.16, 2.1)
Tumor Mutational Burden-High (TMB-H) Cancer
- for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase (mut/Mb)] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options.1 (1.17, 2.1)
- Limitations of Use: The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma (cSCC)
- for the treatment of patients with recurrent or metastatic cSCC or locally advanced cSCC that is not curable by surgery or radiation. (1.18)
Triple-Negative Breast Cancer (TNBC)
- for the treatment of patients with high-risk early-stage TNBC in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery. (1.19)
- in combination with chemotherapy, for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA approved test. (1.19, 2.1)
Adult Classical Hodgkin Lymphoma and Adult Primary Mediastinal Large B-Cell Lymphoma: Additional Dosing Regimen of 400 mg Every 6 Weeks
- for use at an additional recommended dosage of 400 mg every 6 weeks for Classical Hodgkin Lymphoma and Primary Mediastinal Large B-Cell Lymphoma in adults.2 (1.20, 2.2)
1 This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
2 This indication is approved under accelerated approval based on pharmacokinetic data, the relationship of exposure to efficacy, and the relationship of exposure to safety. Continued approval for this dosing may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Keytruda Dosage and Administration
- Melanoma: 200 mg every 3 weeks or 400 mg every 6 weeks; 2 mg/kg (up to 200 mg) every 3 weeks for pediatrics. (2.2)
- NSCLC: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- HNSCC: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- cHL or PMBCL: 200 mg every 3 weeks or 400 mg every 6 weeks for adults; 2 mg/kg (up to 200 mg) every 3 weeks for pediatrics. (2.2)
- Urothelial Cancer: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- MSI-H or dMMR Cancer: 200 mg every 3 weeks or 400 mg every 6 weeks for adults; 2 mg/kg (up to 200 mg) every 3 weeks for pediatrics. (2.2)
- MSI-H or dMMR CRC: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- Gastric Cancer: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- Esophageal Cancer: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- Cervical Cancer: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- HCC: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- BTC: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- MCC: 200 mg every 3 weeks or 400 mg every 6 weeks for adults; 2 mg/kg (up to 200 mg) every 3 weeks for pediatrics. (2.2)
- RCC: 200 mg every 3 weeks or 400 mg every 6 weeks as a single agent in the adjuvant setting, or in the advanced setting with either:
- axitinib 5 mg orally twice daily or
- lenvatinib 20 mg orally once daily. (2.2)
- Endometrial Carcinoma: 200 mg every 3 weeks or 400 mg every 6 weeks
- in combination with carboplatin and paclitaxel regardless of MMR or MSI status, or
- in combination with lenvatinib 20 mg orally once daily for pMMR or not MSI-H tumors, or
- as a single agent for MSI-H or dMMR tumors. (2.2)
- TMB-H Cancer: 200 mg every 3 weeks or 400 mg every 6 weeks for adults; 2 mg/kg (up to 200 mg) every 3 weeks for pediatrics. (2.2)
- cSCC: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- TNBC: 200 mg every 3 weeks or 400 mg every 6 weeks. (2.2)
- Administer KEYTRUDA as an intravenous infusion over 30 minutes after dilution. (2.4)
- See Full Prescribing Information for dosage modifications for adverse reactions and preparation and administration instructions. (2.3, 2.4)
Dosage Forms and Strengths
- Injection: 100 mg/4 mL (25 mg/mL) solution in a single-dose vial (3)
Contraindications
None. (4)
Warnings and Precautions
- Immune-Mediated Adverse Reactions (5.1)
- Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue, including the following: immune-mediated pneumonitis, immune-mediated colitis, immune-mediated hepatitis, immune-mediated endocrinopathies, immune-mediated nephritis with renal dysfunction, immune-mediated dermatologic adverse reactions, and solid organ transplant rejection.
- Monitor for early identification and management. Evaluate liver enzymes, creatinine, and thyroid function at baseline and periodically during treatment.
- Withhold or permanently discontinue based on severity and type of reaction.
- Infusion-related reactions: Interrupt, slow the rate of infusion, or permanently discontinue KEYTRUDA based on the severity of reaction. (5.2)
- Complications of allogeneic HSCT: Fatal and other serious complications can occur in patients who receive allogeneic HSCT before or after being treated with a PD-1/PD-L1 blocking antibody. (5.3)
- Treatment of patients with multiple myeloma with a PD-1 or PD-L1 blocking antibody in combination with a thalidomide analogue plus dexamethasone is not recommended outside of controlled clinical trials. (5.4)
- Embryo-Fetal toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective method of contraception. (5.5, 8.1, 8.3)
Adverse Reactions/Side Effects
Most common adverse reactions (reported in ≥20% of patients) were:
- KEYTRUDA as a single agent: fatigue, musculoskeletal pain, rash, diarrhea, pyrexia, cough, decreased appetite, pruritus, dyspnea, constipation, pain, abdominal pain, nausea, and hypothyroidism. (6.1)
- KEYTRUDA in combination with chemotherapy or chemoradiotherapy: fatigue/asthenia, nausea, constipation, diarrhea, decreased appetite, rash, vomiting, cough, dyspnea, pyrexia, alopecia, peripheral neuropathy, mucosal inflammation, stomatitis, headache, weight loss, abdominal pain, arthralgia, myalgia, insomnia, palmar-plantar erythrodysesthesia, urinary tract infection, and hypothyroidism. (6.1)
- KEYTRUDA in combination with chemotherapy and bevacizumab: peripheral neuropathy, alopecia, anemia, fatigue/asthenia, nausea, neutropenia, diarrhea, hypertension, thrombocytopenia, constipation, arthralgia, vomiting, urinary tract infection, rash, leukopenia, hypothyroidism, and decreased appetite. (6.1)
- KEYTRUDA in combination with axitinib: diarrhea, fatigue/asthenia, hypertension, hepatotoxicity, hypothyroidism, decreased appetite, palmar-plantar erythrodysesthesia, nausea, stomatitis/mucosal inflammation, dysphonia, rash, cough, and constipation. (6.1)
- KEYTRUDA in combination with lenvatinib: hypothyroidism, hypertension, fatigue, diarrhea, musculoskeletal disorders, nausea, decreased appetite, vomiting, stomatitis, weight loss, abdominal pain, urinary tract infection, proteinuria, constipation, headache, hemorrhagic events, palmar-plantar erythrodysesthesia, dysphonia, rash, hepatotoxicity, and acute kidney injury. (6.1)
- KEYTRUDA in combination with enfortumab vedotin: rash, peripheral neuropathy, fatigue, pruritus, diarrhea, alopecia, weight loss, decreased appetite, dry eye, nausea, constipation, dysgeusia, and urinary tract infection. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme LLC at 1-877-888-4231 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 8/2024
Full Prescribing Information
1. Indications and Usage for Keytruda
1.1 Melanoma
KEYTRUDA® is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with Stage IIB, IIC, or III melanoma following complete resection.
1.2 Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [Tumor Proportion Score (TPS) ≥1%] as determined by an FDA-approved test [see Dosage and Administration (2.1)], with no EGFR or ALK genomic tumor aberrations, and is:
- Stage III where patients are not candidates for surgical resection or definitive chemoradiation, or
- metastatic.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test [see Dosage and Administration (2.1)], with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
KEYTRUDA is indicated for the treatment of patients with resectable (tumors ≥4 cm or node positive) NSCLC in combination with platinum-containing chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, as a single agent, is indicated as adjuvant treatment following resection and platinum-based chemotherapy for adult patients with Stage IB (T2a ≥4 cm), II, or IIIA NSCLC.
1.3 Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [Combined Positive Score (CPS) ≥1] as determined by an FDA-approved test [see Dosage and Administration (2.1)].
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
1.4 Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
1.5 Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy.
Limitations of Use: KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
1.6 Urothelial Cancer
KEYTRUDA, in combination with enfortumab vedotin, is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma:
- who are not eligible for any platinum-containing chemotherapy, or
- who have disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with Bacillus Calmette-Guerin (BCG)-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS) with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
1.7 Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options [see Dosage and Administration (2.1)].
1.8 Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC) as determined by an FDA-approved test [see Dosage and Administration (2.1)].
1.9 Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of adults with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test [see Dosage and Administration (2.1)].
This indication is approved under accelerated approval based on tumor response rate and durability of response [see Clinical Studies (14.9)]. Continued approval of this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
KEYTRUDA, in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of adults with locally advanced unresectable or metastatic HER2-negative gastric or gastroesophageal junction (GEJ) adenocarcinoma.
1.10 Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or gastroesophageal junction (GEJ) (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
- in combination with platinum- and fluoropyrimidine-based chemotherapy, or
- as a single agent after one or more prior lines of systemic therapy for patients with tumors of squamous cell histology that express PD-L1 (CPS ≥10) as determined by an FDA-approved test [see Dosage and Administration (2.1)].
1.11 Cervical Cancer
KEYTRUDA, in combination with chemoradiotherapy (CRT), is indicated for the treatment of patients with FIGO 2014 Stage III-IVA cervical cancer.
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test [see Dosage and Administration (2.1)].
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test [see Dosage and Administration (2.1)].
1.12 Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) secondary to hepatitis B who have received prior systemic therapy other than a PD-1/PD-L1-containing regimen.
1.13 Biliary Tract Cancer
KEYTRUDA, in combination with gemcitabine and cisplatin, is indicated for the treatment of patients with locally advanced unresectable or metastatic biliary tract cancer (BTC).
1.14 Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC).
1.15 Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA, in combination with lenvatinib, is indicated for the first-line treatment of adult patients with advanced RCC.
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions [see Clinical Studies (14.15)].
1.16 Endometrial Carcinoma
KEYTRUDA, in combination with carboplatin and paclitaxel, followed by KEYTRUDA as a single agent, is indicated for the treatment of adult patients with primary advanced or recurrent endometrial carcinoma.
KEYTRUDA, in combination with lenvatinib, is indicated for the treatment of adult patients with advanced endometrial carcinoma that is mismatch repair proficient (pMMR) as determined by an FDA-approved test or not MSI-H, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation [see Dosage and Administration (2.1)].
KEYTRUDA, as a single agent, is indicated for the treatment of adult patients with advanced endometrial carcinoma that is MSI-H or dMMR, as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation [see Dosage and Administration (2.1)].
1.17 Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase (mut/Mb)] solid tumors, as determined by an FDA-approved test [see Dosage and Administration (2.1)], that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response [see Clinical Studies (14.17)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Limitations of Use: The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
1.18 Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
1.19 Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test [see Dosage and Administration (2.1)].
1.20 Adult Classical Hodgkin Lymphoma and Adult Primary Mediastinal Large B-Cell Lymphoma: Additional Dosing Regimen of 400 mg Every 6 Weeks
KEYTRUDA is indicated for use at an additional recommended dosage of 400 mg every 6 weeks for classical Hodgkin lymphoma and primary mediastinal large B-cell lymphoma in adults [see Indications and Usage (1.4, 1.5), Dosage and Administration (2.2)]. This indication is approved under accelerated approval based on pharmacokinetic data, the relationship of exposure to efficacy, and the relationship of exposure to safety [see Clinical Pharmacology (12.2), Clinical Studies (14.20)]. Continued approval for this dosage may be contingent upon verification and description of clinical benefit in the confirmatory trials.
2. Keytruda Dosage and Administration
2.1 Patient Selection
Information on FDA-approved tests for patient selection is available at:
http://www.fda.gov/CompanionDiagnostics.
Patient Selection for Single-Agent Treatment
Select patients for treatment with KEYTRUDA as a single agent based on the presence of positive PD-L1 expression in:
- Stage III NSCLC who are not candidates for surgical resection or definitive chemoradiation [see Clinical Studies (14.2)].
- metastatic NSCLC [see Clinical Studies (14.2)].
- first-line treatment of metastatic or unresectable, recurrent HNSCC [see Clinical Studies (14.3)].
- previously treated recurrent locally advanced or metastatic esophageal cancer [see Clinical Studies (14.10)].
- recurrent or metastatic cervical cancer with disease progression on or after chemotherapy [see Clinical Studies (14.11)].
For the MSI-H/dMMR indications, select patients for treatment with KEYTRUDA as a single agent based on MSI-H/dMMR status in tumor specimens [see Clinical Studies (14.7, 14.8)].
For the TMB-H indication, select patients for treatment with KEYTRUDA as a single agent based on TMB-H status in tumor specimens [see Clinical Studies (14.17)].
Because subclonal dMMR mutations and microsatellite instability may arise in high-grade gliomas during temozolomide therapy, it is recommended to test for TMB-H, MSI-H, and dMMR in the primary tumor specimens obtained prior to initiation of temozolomide chemotherapy in patients with high-grade gliomas.
Additional Patient Selection Information for MSI-H or dMMR in Patients with non-CRC Solid Tumors
Due to discordance between local tests and FDA-approved tests, confirmation of MSI-H or dMMR status is recommended by an FDA-approved test in patients with MSI-H or dMMR solid tumors, if feasible. If unable to perform confirmatory MSI-H/dMMR testing, the presence of TMB ≥10 mut/Mb, as determined by an FDA-approved test, may be used to select patients for treatment [see Clinical Studies (14.7)].
Patient Selection for Combination Therapy
For use of KEYTRUDA in combination with chemotherapy and trastuzumab, select patients based on the presence of positive PD-L1 expression (CPS ≥1) in locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma [see Clinical Studies (14.9)].
For use of KEYTRUDA in combination with chemotherapy, with or without bevacizumab, select patients based on the presence of positive PD-L1 expression in persistent, recurrent, or metastatic cervical cancer [see Clinical Studies (14.11)].
For the pMMR/not MSI-H advanced endometrial carcinoma indication, select patients for treatment with KEYTRUDA in combination with lenvatinib based on MSI or MMR status in tumor specimens [see Clinical Studies (14.16)].
For use of KEYTRUDA in combination with chemotherapy, select patients based on the presence of positive PD-L1 expression in locally recurrent unresectable or metastatic TNBC [see Clinical Studies (14.19)].
Additional Patient Selection Information
- An FDA-approved test for the detection of not MSI-H is currently unavailable for the selection of patients with not MSI-H endometrial carcinoma for treatment with KEYTRUDA in combination with lenvatinib [see Clinical Studies (14.16)].
2.2 Recommended Dosage
| Indication | Recommended Dosage of KEYTRUDA | Duration/Timing of Treatment |
|---|---|---|
|
||
| Monotherapy | ||
| Adult patients with unresectable or metastatic melanoma | 200 mg every 3 weeks*
or 400 mg every 6 weeks* | Until disease progression or unacceptable toxicity |
| Adjuvant treatment of adult patients with melanoma, NSCLC, or RCC | 200 mg every 3 weeks*
or 400 mg every 6 weeks* | Until disease recurrence, unacceptable toxicity, or up to 12 months |
| Adult patients with NSCLC, HNSCC, cHL, PMBCL, locally advanced or metastatic Urothelial Carcinoma, MSI-H or dMMR Cancer, MSI-H or dMMR CRC, MSI-H or dMMR Endometrial Carcinoma, Esophageal Cancer, Cervical Cancer, HCC, MCC, TMB-H Cancer, or cSCC | 200 mg every 3 weeks*
or 400 mg every 6 weeks* | Until disease progression, unacceptable toxicity, or up to 24 months |
| Adult patients with high-risk BCG- unresponsive NMIBC | 200 mg every 3 weeks*
or 400 mg every 6 weeks* | Until persistent or recurrent high-risk NMIBC, disease progression, unacceptable toxicity, or up to 24 months |
| Pediatric patients with cHL, PMBCL, MSI-H or dMMR Cancer, MCC, or TMB- H Cancer | 2 mg/kg every 3 weeks (up to a maximum of 200 mg)* | Until disease progression, unacceptable toxicity, or up to 24 months |
| Pediatric patients (12 years and older) for adjuvant treatment of melanoma | 2 mg/kg every 3 weeks (up to a maximum of 200 mg)* | Until disease recurrence, unacceptable toxicity, or up to 12 months |
| Combination Therapy† | ||
| Adult patients with resectable NSCLC | 200 mg every 3 weeks*
or 400 mg every 6 weeks* Administer KEYTRUDA prior to chemotherapy when given on the same day. | Neoadjuvant treatment in combination with chemotherapy for 12 weeks or until disease progression that precludes definitive surgery or unacceptable toxicity, followed by adjuvant treatment with KEYTRUDA as a single agent after surgery for 39 weeks or until disease recurrence or unacceptable toxicity |
| Adult patients with NSCLC, HNSCC, HER2-negative Gastric Cancer, Esophageal Cancer, or BTC | 200 mg every 3 weeks*
or 400 mg every 6 weeks* Administer KEYTRUDA prior to chemotherapy when given on the same day. | Until disease progression, unacceptable toxicity, or up to 24 months |
| Adult patients with locally advanced or metastatic urothelial cancer | 200 mg every 3 weeks*
or 400 mg every 6 weeks* Administer KEYTRUDA after enfortumab vedotin when given on the same day. | Until disease progression, unacceptable toxicity, or up to 24 months |
| Adult patients with HER2-positive Gastric Cancer | 200 mg every 3 weeks*
or 400 mg every 6 weeks* Administer KEYTRUDA prior to trastuzumab and chemotherapy when given on the same day. | Until disease progression, unacceptable toxicity, or up to 24 months |
| Adult patients with Cervical Cancer | 200 mg every 3 weeks*
or 400 mg every 6 weeks* Administer KEYTRUDA prior to chemoradiotherapy or prior to chemotherapy with or without bevacizumab when given on the same day. | Until disease progression, unacceptable toxicity, or for KEYTRUDA, up to 24 months |
| Adult patients with RCC | 200 mg every 3 weeks*
or 400 mg every 6 weeks* Administer KEYTRUDA in combination with axitinib 5 mg orally twice daily‡ or Administer KEYTRUDA in combination with lenvatinib 20 mg orally once daily. | Until disease progression, unacceptable toxicity, or for KEYTRUDA, up to 24 months |
| Adult patients with Endometrial Carcinoma | 200 mg every 3 weeks*
or 400 mg every 6 weeks* Administer KEYTRUDA prior to carboplatin and paclitaxel when given on the same day. or Administer KEYTRUDA in combination with lenvatinib 20 mg orally once daily. | Until disease progression, unacceptable toxicity, or for KEYTRUDA, up to 24 months |
| Adult patients with high-risk early-stage TNBC | 200 mg every 3 weeks*
or 400 mg every 6 weeks* Administer KEYTRUDA prior to chemotherapy when given on the same day. | Neoadjuvant treatment in combination with chemotherapy for 24 weeks (8 doses of 200 mg every 3 weeks or 4 doses of 400 mg every 6 weeks) or until disease progression or unacceptable toxicity, followed by adjuvant treatment with KEYTRUDA as a single agent for up to 27 weeks (9 doses of 200 mg every 3 weeks or 5 doses of 400 mg every 6 weeks) or until disease recurrence or unacceptable toxicity§ |
| Adult patients with locally recurrent unresectable or metastatic TNBC | 200 mg every 3 weeks*
or 400 mg every 6 weeks* Administer KEYTRUDA prior to chemotherapy when given on the same day. | Until disease progression, unacceptable toxicity, or up to 24 months |
2.3 Dose Modifications
No dose reduction for KEYTRUDA is recommended. In general, withhold KEYTRUDA for severe (Grade 3) immune-mediated adverse reactions. Permanently discontinue KEYTRUDA for Life-threatening (Grade 4) immune-mediated adverse reactions, recurrent severe (Grade 3) immune-mediated reactions that require systemic immunosuppressive treatment, or an inability to reduce corticosteroid dose to 10 mg or less of prednisone or equivalent per day within 12 weeks of initiating steroids.
Dosage modifications for KEYTRUDA for adverse reactions that require management different from these general guidelines are summarized in Table 2.
| Adverse Reaction | Severity* | Dosage Modification |
|---|---|---|
| ALT = alanine aminotransferase, AST = aspartate aminotransferase, DRESS = Drug Rash with Eosinophilia and Systemic Symptoms, SJS = Stevens Johnson Syndrome, TEN = toxic epidermal necrolysis, ULN = upper limit normal | ||
|
||
| Immune-Mediated Adverse Reactions [see Warnings and Precautions (5.1)] | ||
| Pneumonitis | Grade 2 | Withhold† |
| Grade 3 or 4 | Permanently discontinue | |
| Colitis | Grade 2 or 3 | Withhold† |
| Grade 4 | Permanently discontinue | |
|
Hepatitis with no tumor involvement of the liver | AST or ALT increases to more than 3 and up to 8 times ULN or Total bilirubin increases to more than 1.5 and up to 3 times ULN | Withhold† |
| For liver enzyme elevations in patients treated with combination therapy with axitinib, see Table 3. | AST or ALT increases to more than 8 times ULN or Total bilirubin increases to more than 3 times ULN | Permanently discontinue |
| Hepatitis with tumor involvement of the liver‡ | Baseline AST or ALT is more than 1 and up to 3 times ULN and increases to more than 5 and up to 10 times ULN or Baseline AST or ALT is more than 3 and up to 5 times ULN and increases to more than 8 and up to 10 times ULN | Withhold† |
| ALT or AST increases to more than 10 times ULN or Total bilirubin increases to more than 3 times ULN | Permanently discontinue | |
| Endocrinopathies | Grade 3 or 4 | Withhold until clinically stable or permanently discontinue depending on severity |
| Nephritis with Renal Dysfunction | Grade 2 or 3 increased blood creatinine | Withhold† |
| Grade 4 increased blood creatinine | Permanently discontinue | |
| Exfoliative Dermatologic Conditions | Suspected SJS, TEN, or DRESS | Withhold† |
| Confirmed SJS, TEN, or DRESS | Permanently discontinue | |
| Myocarditis | Grade 2, 3, or 4 | Permanently discontinue |
| Neurological Toxicities | Grade 2 | Withhold† |
| Grade 3 or 4 | Permanently discontinue | |
| Hematologic toxicity in patients with cHL or PMBCL | Grade 4 | Withhold until resolution to Grades 0 or 1 |
| Other Adverse Reactions | ||
| Infusion-related reactions [see Warnings and Precautions (5.2)] | Grade 1 or 2 | Interrupt or slow the rate of infusion |
| Grade 3 or 4 | Permanently discontinue | |
The following table represents dosage modifications that are different from those described above for KEYTRUDA or in the Full Prescribing Information for the drug administered in combination.
| Treatment | Adverse Reaction | Severity | Dosage Modification |
|---|---|---|---|
| ALT = alanine aminotransferase, AST = aspartate aminotransferase, ULN = upper limit normal | |||
|
|||
| KEYTRUDA in combination with axitinib | Liver enzyme elevations* | ALT or AST increases to at least 3 times but less than 10 times ULN without concurrent total bilirubin at least 2 times ULN | Withhold both KEYTRUDA and axitinib until resolution to Grades 0 or 1† |
| ALT or AST increases to more than 3 times ULN with concurrent total bilirubin at least 2 times ULN or ALT or AST ≥10 times ULN | Permanently discontinue both KEYTRUDA and axitinib |
||
Recommended Dose Modifications for Adverse Reactions for KEYTRUDA in Combination with Lenvatinib
When administering KEYTRUDA in combination with lenvatinib, modify the dosage of one or both drugs. Withhold or discontinue KEYTRUDA as shown in Table 2. Refer to lenvatinib prescribing information for additional dose modification information.
2.4 Preparation and Administration
Preparation for Intravenous Infusion
- Visually inspect the solution for particulate matter and discoloration. The solution is clear to slightly opalescent, colorless to slightly yellow. Discard the vial if visible particles are observed.
- Dilute KEYTRUDA injection (solution) prior to intravenous administration.
- Withdraw the required volume from the vial(s) of KEYTRUDA and transfer into an intravenous (IV) bag containing 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP. Mix diluted solution by gentle inversion. Do not shake. The final concentration of the diluted solution should be between 1 mg/mL to 10 mg/mL.
- Discard any unused portion left in the vial.
Storage of Diluted Solution
The product does not contain a preservative.
Store the diluted solution from the KEYTRUDA 100 mg/4 mL vial either:
- At room temperature for no more than 6 hours from the time of dilution. This includes room temperature storage of the diluted solution, and the duration of infusion.
- Under refrigeration at 2°C to 8°C (36°F to 46°F) for no more than 96 hours from the time of dilution. If refrigerated, allow the diluted solution to come to room temperature prior to administration. Do not shake.
Discard after 6 hours at room temperature or after 96 hours under refrigeration.
Do not freeze.
3. Dosage Forms and Strengths
- Injection: 100 mg/4 mL (25 mg/mL) clear to slightly opalescent, colorless to slightly yellow solution in a single-dose vial
5. Warnings and Precautions
5.1 Severe and Fatal Immune-Mediated Adverse Reactions
KEYTRUDA is a monoclonal antibody that belongs to a class of drugs that bind to either the programmed death-receptor 1 (PD-1) or the PD-ligand 1 (PD-L1), blocking the PD-1/PD-L1 pathway, thereby removing inhibition of the immune response, potentially breaking peripheral tolerance and inducing immune-mediated adverse reactions. Important immune-mediated adverse reactions listed under WARNINGS AND PRECAUTIONS may not include all possible severe and fatal immune-mediated adverse reactions.
Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue and can affect more than one body system simultaneously. Immune-mediated adverse reactions can occur at any time after starting treatment with a PD-1/PD-L1 blocking antibody. While immune-mediated adverse reactions usually manifest during treatment with PD-1/PD-L1 blocking antibodies, immune-mediated adverse reactions can also manifest after discontinuation of PD-1/PD-L1 blocking antibodies.
Early identification and management of immune-mediated adverse reactions are essential to ensure safe use of PD-1/PD-L1 blocking antibodies. Monitor patients closely for symptoms and signs that may be clinical manifestations of underlying immune-mediated adverse reactions. Evaluate liver enzymes, creatinine, and thyroid function at baseline and periodically during treatment. For patients with TNBC treated with KEYTRUDA in the neoadjuvant setting, monitor blood cortisol at baseline, prior to surgery, and as clinically indicated. In cases of suspected immune-mediated adverse reactions, initiate appropriate workup to exclude alternative etiologies, including infection. Institute medical management promptly, including specialty consultation as appropriate.
Withhold or permanently discontinue KEYTRUDA depending on severity [see Dosage and Administration (2.3)]. In general, if KEYTRUDA requires interruption or discontinuation, administer systemic corticosteroid therapy (1 to 2 mg/kg/day prednisone or equivalent) until improvement to Grade 1 or less. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Consider administration of other systemic immunosuppressants in patients whose immune-mediated adverse reactions are not controlled with corticosteroid therapy.
Toxicity management guidelines for adverse reactions that do not necessarily require systemic steroids (e.g., endocrinopathies and dermatologic reactions) are discussed below.
Immune-Mediated Pneumonitis
KEYTRUDA can cause immune-mediated pneumonitis. The incidence of pneumonitis is higher in patients who have received prior thoracic radiation. Immune-mediated pneumonitis occurred in 3.4% (94/2799) of patients receiving KEYTRUDA, including fatal (0.1%), Grade 4 (0.3%), Grade 3 (0.9%), and Grade 2 (1.3%) adverse reactions. Systemic corticosteroids were required in 67% (63/94) of patients with pneumonitis. Pneumonitis led to permanent discontinuation of KEYTRUDA in 1.3% (36) of patients and withholding of KEYTRUDA in 0.9% (26) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement; of these, 23% had recurrence of pneumonitis. Pneumonitis resolved in 59% of the 94 patients.
In clinical studies enrolling 389 adult patients with cHL who received KEYTRUDA as a single agent, pneumonitis occurred in 31 (8%) patients, including Grades 3-4 pneumonitis in 2.3% of patients. Patients received high-dose corticosteroids for a median duration of 10 days (range: 2 days to 53 months). Pneumonitis rates were similar in patients with and without prior thoracic radiation. Pneumonitis led to discontinuation of KEYTRUDA in 21 (5.4%) patients. Of the patients who developed pneumonitis, 42% interrupted KEYTRUDA, 68% discontinued KEYTRUDA, and 77% had resolution.
In a clinical study enrolling 580 adult patients with resected NSCLC (KEYNOTE-091) who received KEYTRUDA as a single agent for adjuvant treatment, pneumonitis occurred in 41 (7%) patients, including fatal (0.2%), Grade 4 (0.3%), and Grade 3 (1%) adverse reactions. Patients received high-dose corticosteroids for a median duration of 10 days (range: 1 day to 2.3 months). Pneumonitis led to discontinuation of KEYTRUDA in 26 (4.5%) of patients. Of the patients who developed pneumonitis, 54% interrupted KEYTRUDA, 63% discontinued KEYTRUDA, and 71% had resolution.
Immune-Mediated Colitis
KEYTRUDA can cause immune-mediated colitis, which may present with diarrhea. Cytomegalovirus (CMV) infection/reactivation has been reported in patients with corticosteroid-refractory immune-mediated colitis. In cases of corticosteroid-refractory colitis, consider repeating infectious workup to exclude alternative etiologies. Immune-mediated colitis occurred in 1.7% (48/2799) of patients receiving KEYTRUDA, including Grade 4 (<0.1%), Grade 3 (1.1%), and Grade 2 (0.4%) adverse reactions. Systemic corticosteroids were required in 69% (33/48) of patients with colitis. Additional immunosuppressant therapy was required in 4.2% of patients. Colitis led to permanent discontinuation of KEYTRUDA in 0.5% (15) of patients and withholding of KEYTRUDA in 0.5% (13) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement; of these, 23% had recurrence of colitis. Colitis resolved in 85% of the 48 patients.
Hepatotoxicity and Immune-Mediated Hepatitis
KEYTRUDA as a Single Agent
KEYTRUDA can cause immune-mediated hepatitis. Immune-mediated hepatitis occurred in 0.7% (19/2799) of patients receiving KEYTRUDA, including Grade 4 (<0.1%), Grade 3 (0.4%), and Grade 2 (0.1%) adverse reactions. Systemic corticosteroids were required in 68% (13/19) of patients with hepatitis. Eleven percent of these patients required additional immunosuppressant therapy. Hepatitis led to permanent discontinuation of KEYTRUDA in 0.2% (6) of patients and withholding of KEYTRUDA in 0.3% (9) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement; of these, none had recurrence of hepatitis. Hepatitis resolved in 79% of the 19 patients.
KEYTRUDA with Axitinib
KEYTRUDA in combination with axitinib can cause hepatic toxicity with higher than expected frequencies of Grades 3 and 4 ALT and AST elevations compared to KEYTRUDA alone. Monitor liver enzymes before initiation of and periodically throughout treatment. Consider more frequent monitoring of liver enzymes as compared to when the drugs are administered as single agents. For elevated liver enzymes, interrupt KEYTRUDA and axitinib, and consider administering corticosteroids as needed [see Dosage and Administration (2.3)].
With the combination of KEYTRUDA and axitinib, Grades 3 and 4 increased ALT (20%) and increased AST (13%) were seen. Fifty-nine percent of the patients with increased ALT received systemic corticosteroids. In patients with ALT ≥3 times ULN (Grades 2-4, n=116), ALT resolved to Grades 0-1 in 94%. Among the 92 patients who were rechallenged with either KEYTRUDA (n=3) or axitinib (n=34) administered as a single agent or with both (n=55), recurrence of ALT ≥3 times ULN was observed in 1 patient receiving KEYTRUDA, 16 patients receiving axitinib, and 24 patients receiving both KEYTRUDA and axitinib. All patients with a recurrence of ALT ≥3 ULN subsequently recovered from the event.
Immune-Mediated Endocrinopathies
Adrenal Insufficiency
KEYTRUDA can cause primary or secondary adrenal insufficiency. For Grade 2 or higher adrenal insufficiency, initiate symptomatic treatment, including hormone replacement as clinically indicated. Withhold KEYTRUDA depending on severity [see Dosage and Administration (2.3)].
Adrenal insufficiency occurred in 0.8% (22/2799) of patients receiving KEYTRUDA, including Grade 4 (<0.1%), Grade 3 (0.3%), and Grade 2 (0.3%) adverse reactions. Systemic corticosteroids were required in 77% (17/22) of patients with adrenal insufficiency; of these, the majority remained on systemic corticosteroids. Adrenal insufficiency led to permanent discontinuation of KEYTRUDA in <0.1% (1) of patients and withholding of KEYTRUDA in 0.3% (8) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement.
Hypophysitis
KEYTRUDA can cause immune-mediated hypophysitis. Hypophysitis can present with acute symptoms associated with mass effect such as headache, photophobia, or visual field defects. Hypophysitis can cause hypopituitarism. Initiate hormone replacement as indicated. Withhold or permanently discontinue KEYTRUDA depending on severity [see Dosage and Administration (2.3)].
Hypophysitis occurred in 0.6% (17/2799) of patients receiving KEYTRUDA, including Grade 4 (<0.1%), Grade 3 (0.3%), and Grade 2 (0.2%) adverse reactions. Systemic corticosteroids were required in 94% (16/17) of patients with hypophysitis; of these, the majority remained on systemic corticosteroids. Hypophysitis led to permanent discontinuation of KEYTRUDA in 0.1% (4) of patients and withholding of KEYTRUDA in 0.3% (7) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement.
Thyroid Disorders
KEYTRUDA can cause immune-mediated thyroid disorders. Thyroiditis can present with or without endocrinopathy. Hypothyroidism can follow hyperthyroidism. Initiate hormone replacement for hypothyroidism or institute medical management of hyperthyroidism as clinically indicated. Withhold or permanently discontinue KEYTRUDA depending on severity [see Dosage and Administration (2.3)].
Thyroiditis occurred in 0.6% (16/2799) of patients receiving KEYTRUDA, including Grade 2 (0.3%). No patients discontinued KEYTRUDA due to thyroiditis. KEYTRUDA was withheld in <0.1% (1) of patients.
Hyperthyroidism occurred in 3.4% (96/2799) of patients receiving KEYTRUDA, including Grade 3 (0.1%) and Grade 2 (0.8%). Hyperthyroidism led to permanent discontinuation of KEYTRUDA in <0.1% (2) of patients and withholding of KEYTRUDA in 0.3% (7) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement.
The incidence of new or worsening hyperthyroidism was higher in 580 patients with resected NSCLC, occurring in 11% of patients receiving KEYTRUDA as a single agent as adjuvant treatment (KEYNOTE-091), including Grade 3 (0.2%) hyperthyroidism.
Hypothyroidism occurred in 8% (237/2799) of patients receiving KEYTRUDA, including Grade 3 (0.1%) and Grade 2 (6.2%). Hypothyroidism led to permanent discontinuation of KEYTRUDA in <0.1% (1) of patients and withholding of KEYTRUDA in 0.5% (14) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement. The majority of patients with hypothyroidism required long-term thyroid hormone replacement.
The incidence of new or worsening hypothyroidism was higher in 1185 patients with HNSCC, occurring in 16% of patients receiving KEYTRUDA as a single agent or in combination with platinum and FU, including Grade 3 (0.3%) hypothyroidism. The incidence of new or worsening hypothyroidism was higher in 389 patients with cHL (17%) receiving KEYTRUDA as a single agent, including Grade 1 (6.2%) and Grade 2 (10.8%) hypothyroidism.
The incidence of new or worsening hypothyroidism was higher in 580 patients with resected NSCLC, occurring in 22% of patients receiving KEYTRUDA as a single agent as adjuvant treatment (KEYNOTE-091), including Grade 3 (0.3%) hypothyroidism.
Type 1 Diabetes Mellitus, which can present with Diabetic Ketoacidosis
Monitor patients for hyperglycemia or other signs and symptoms of diabetes. Initiate treatment with insulin as clinically indicated. Withhold KEYTRUDA depending on severity [see Dosage and Administration (2.3)].
Type 1 diabetes mellitus occurred in 0.2% (6/2799) of patients receiving KEYTRUDA. Type 1 diabetes mellitus led to permanent discontinuation in <0.1% (1) of patients and withholding of KEYTRUDA in <0.1% (1) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement. All patients with Type 1 diabetes mellitus required long-term insulin therapy.
Immune-Mediated Nephritis with Renal Dysfunction
KEYTRUDA can cause immune-mediated nephritis. Immune-mediated nephritis occurred in 0.3% (9/2799) of patients receiving KEYTRUDA, including Grade 4 (<0.1%), Grade 3 (0.1%), and Grade 2 (0.1%) adverse reactions. Systemic corticosteroids were required in 89% (8/9) of patients with nephritis. Nephritis led to permanent discontinuation of KEYTRUDA in 0.1% (3) of patients and withholding of KEYTRUDA in 0.1% (3) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement; of these, none had recurrence of nephritis. Nephritis resolved in 56% of the 9 patients.
Immune-Mediated Dermatologic Adverse Reactions
KEYTRUDA can cause immune-mediated rash or dermatitis. Exfoliative dermatitis, including Stevens Johnson Syndrome, DRESS, and toxic epidermal necrolysis (TEN), has occurred with PD-1/PD-L1 blocking antibodies. Topical emollients and/or topical corticosteroids may be adequate to treat mild to moderate non-exfoliative rashes. Withhold or permanently discontinue KEYTRUDA depending on severity [see Dosage and Administration (2.3)].
Immune-mediated dermatologic adverse reactions occurred in 1.4% (38/2799) of patients receiving KEYTRUDA, including Grade 3 (1%) and Grade 2 (0.1%) adverse reactions. Systemic corticosteroids were required in 40% (15/38) of patients with immune-mediated dermatologic adverse reactions. Immune-mediated dermatologic adverse reactions led to permanent discontinuation of KEYTRUDA in 0.1% (2) of patients and withholding of KEYTRUDA in 0.6% (16) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement; of these, 6% had recurrence of immune-mediated dermatologic adverse reactions. Immune-mediated dermatologic adverse reactions resolved in 79% of the 38 patients.
Other Immune-Mediated Adverse Reactions
The following clinically significant immune-mediated adverse reactions occurred at an incidence of <1% (unless otherwise noted) in patients who received KEYTRUDA or were reported with the use of other PD-1/PD-L1 blocking antibodies. Severe or fatal cases have been reported for some of these adverse reactions.
Cardiac/Vascular: Myocarditis, pericarditis, vasculitis
Nervous System: Meningitis, encephalitis, myelitis and demyelination, myasthenic syndrome/myasthenia gravis (including exacerbation), Guillain-Barré syndrome, nerve paresis, autoimmune neuropathy
Ocular: Uveitis, iritis and other ocular inflammatory toxicities can occur. Some cases can be associated with retinal detachment. Various grades of visual impairment, including blindness, can occur. If uveitis occurs in combination with other immune-mediated adverse reactions, consider a Vogt-Koyanagi-Harada-like syndrome, as this may require treatment with systemic steroids to reduce the risk of permanent vision loss.
Gastrointestinal: Pancreatitis, to include increases in serum amylase and lipase levels, gastritis, duodenitis
Musculoskeletal and Connective Tissue: Myositis/polymyositis, rhabdomyolysis (and associated sequelae, including renal failure), arthritis (1.5%), polymyalgia rheumatica
Endocrine: Hypoparathyroidism
Hematologic/Immune: Hemolytic anemia, aplastic anemia, hemophagocytic lymphohistiocytosis, systemic inflammatory response syndrome, histiocytic necrotizing lymphadenitis (Kikuchi lymphadenitis), sarcoidosis, immune thrombocytopenic purpura, solid organ transplant rejection, other transplant (including corneal graft) rejection
5.2 Infusion-Related Reactions
KEYTRUDA can cause severe or life-threatening infusion-related reactions, including hypersensitivity and anaphylaxis, which have been reported in 0.2% of 2799 patients receiving KEYTRUDA. Monitor patients for signs and symptoms of infusion-related reactions including rigors, chills, wheezing, pruritus, flushing, rash, hypotension, hypoxemia, and fever. Interrupt or slow the rate of infusion for mild (Grade 1) or moderate (Grade 2) infusion-related reactions. For severe (Grade 3) or life-threatening (Grade 4) infusion-related reactions, stop infusion and permanently discontinue KEYTRUDA [see Dosage and Administration (2.3)].
5.3 Complications of Allogeneic HSCT
Fatal and other serious complications can occur in patients who receive allogeneic hematopoietic stem cell transplantation (HSCT) before or after being treated with a PD-1/PD-L1 blocking antibody. Transplant-related complications include hyperacute graft-versus-host-disease (GVHD), acute GVHD, chronic GVHD, hepatic veno-occlusive disease (VOD) after reduced intensity conditioning, and steroid-requiring febrile syndrome (without an identified infectious cause). These complications may occur despite intervening therapy between PD-1/PD-L1 blockade and allogeneic HSCT.
5.4 Increased Mortality in Patients with Multiple Myeloma when KEYTRUDA is Added to a Thalidomide Analogue and Dexamethasone
In two randomized trials in patients with multiple myeloma, the addition of KEYTRUDA to a thalidomide analogue plus dexamethasone, a use for which no PD-1 or PD-L1 blocking antibody is indicated, resulted in increased mortality. Treatment of patients with multiple myeloma with a PD-1 or PD-L1 blocking antibody in combination with a thalidomide analogue plus dexamethasone is not recommended outside of controlled trials.
5.5 Embryo-Fetal Toxicity
Based on its mechanism of action, KEYTRUDA can cause fetal harm when administered to a pregnant woman. Animal models link the PD-1/PD-L1 signaling pathway with maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue. Advise women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with KEYTRUDA and for 4 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling.
- Severe and fatal immune-mediated adverse reactions [see Warnings and Precautions (5.1)].
- Infusion-related reactions [see Warnings and Precautions (5.2)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS AND PRECAUTIONS reflect exposure to KEYTRUDA as a single agent in 2799 patients in three randomized, open-label, active-controlled trials (KEYNOTE-002, KEYNOTE-006, and KEYNOTE-010), which enrolled 912 patients with melanoma and 682 patients with NSCLC, and one single-arm trial (KEYNOTE-001), which enrolled 655 patients with melanoma and 550 patients with NSCLC. In addition to the 2799 patients, certain subsections in the WARNINGS AND PRECAUTIONS describe adverse reactions observed with exposure to KEYTRUDA as a single agent in a randomized, placebo-controlled trial (KEYNOTE-091), which enrolled 580 patients with resected NSCLC, a non-randomized, open-label, multi-cohort trial (KEYNOTE-012), a non-randomized, open-label, single-cohort trial (KEYNOTE-055), and two randomized, open-label, active-controlled trials (KEYNOTE-040 and KEYNOTE-048 single agent arms), which enrolled 909 patients with HNSCC; in two non-randomized, open-label trials (KEYNOTE-013 and KEYNOTE-087) and one randomized, open-label, active-controlled trial (KEYNOTE-204), which enrolled 389 patients with cHL; in a randomized, open-label, active-controlled trial (KEYNOTE-048 combination arm), which enrolled 276 patients with HNSCC; in combination with axitinib in a randomized, active-controlled trial (KEYNOTE-426), which enrolled 429 patients with RCC; and in post-marketing use. Across all trials, KEYTRUDA was administered at doses of 2 mg/kg intravenously every 3 weeks, 10 mg/kg intravenously every 2 weeks, 10 mg/kg intravenously every 3 weeks, or 200 mg intravenously every 3 weeks. Among the 2799 patients, 41% were exposed for 6 months or more and 21% were exposed for 12 months or more.
Melanoma
Ipilimumab-Naive Melanoma
The safety of KEYTRUDA for the treatment of patients with unresectable or metastatic melanoma who had not received prior ipilimumab and who had received no more than one prior systemic therapy was investigated in KEYNOTE-006. KEYNOTE-006 was a multicenter, open-label, active-controlled trial where patients were randomized (1:1:1) and received KEYTRUDA 10 mg/kg every 2 weeks (n=278) or KEYTRUDA 10 mg/kg every 3 weeks (n=277) until disease progression or unacceptable toxicity or ipilimumab 3 mg/kg every 3 weeks for 4 doses unless discontinued earlier for disease progression or unacceptable toxicity (n=256) [see Clinical Studies (14.1)]. Patients with autoimmune disease, a medical condition that required systemic corticosteroids or other immunosuppressive medication; a history of interstitial lung disease; or active infection requiring therapy, including HIV or hepatitis B or C, were ineligible.
The median duration of exposure was 5.6 months (range: 1 day to 11.0 months) for KEYTRUDA and similar in both treatment arms. Fifty-one and 46% of patients received KEYTRUDA 10 mg/kg every 2 or 3 weeks, respectively, for ≥6 months. No patients in either arm received treatment for more than one year.
The study population characteristics were: median age of 62 years (range: 18 to 89); 60% male; 98% White; 32% had an elevated lactate dehydrogenase (LDH) value at baseline; 65% had M1c stage disease; 9% with history of brain metastasis; and approximately 36% had been previously treated with systemic therapy which included a BRAF inhibitor (15%), chemotherapy (13%), and immunotherapy (6%).
In KEYNOTE-006, the adverse reaction profile was similar for the every 2 week and every 3 week schedule, therefore summary safety results are provided in a pooled analysis (n=555) of both KEYTRUDA arms. Adverse reactions leading to permanent discontinuation of KEYTRUDA occurred in 9% of patients. Adverse reactions leading to discontinuation of KEYTRUDA in more than one patient were colitis (1.4%), autoimmune hepatitis (0.7%), allergic reaction (0.4%), polyneuropathy (0.4%), and cardiac failure (0.4%). Adverse reactions leading to interruption of KEYTRUDA occurred in 21% of patients; the most common (≥1%) was diarrhea (2.5%). Tables 4 and 5 summarize selected adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-006.
| Adverse Reaction | KEYTRUDA 10 mg/kg every 2 or 3 weeks | Ipilimumab | ||
|---|---|---|---|---|
| n=555 | n=256 | |||
| All Grades†
(%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) |
|
|
||||
| General | ||||
| Fatigue | 28 | 0.9 | 28 | 3.1 |
| Skin and Subcutaneous Tissue | ||||
| Rash‡ | 24 | 0.2 | 23 | 1.2 |
| Vitiligo§ | 13 | 0 | 2 | 0 |
| Musculoskeletal and Connective Tissue | ||||
| Arthralgia | 18 | 0.4 | 10 | 1.2 |
| Back pain | 12 | 0.9 | 7 | 0.8 |
| Respiratory, Thoracic and Mediastinal | ||||
| Cough | 17 | 0 | 7 | 0.4 |
| Dyspnea | 11 | 0.9 | 7 | 0.8 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 16 | 0.5 | 14 | 0.8 |
| Nervous System | ||||
| Headache | 14 | 0.2 | 14 | 0.8 |
Other clinically important adverse reactions occurring in ≥10% of patients receiving KEYTRUDA were diarrhea (26%), nausea (21%), and pruritus (17%).
| Laboratory Test† | KEYTRUDA 10 mg/kg every 2 or 3 weeks | Ipilimumab | ||
|---|---|---|---|---|
| All Grades‡
% | Grades 3-4 % | All Grades % | Grades 3-4 % |
|
|
||||
| Chemistry | ||||
| Hyperglycemia | 45 | 4.2 | 45 | 3.8 |
| Hypertriglyceridemia | 43 | 2.6 | 31 | 1.1 |
| Hyponatremia | 28 | 4.6 | 26 | 7 |
| Increased AST | 27 | 2.6 | 25 | 2.5 |
| Hypercholesterolemia | 20 | 1.2 | 13 | 0 |
| Hematology | ||||
| Anemia | 35 | 3.8 | 33 | 4.0 |
| Lymphopenia | 33 | 7 | 25 | 6 |
Other laboratory abnormalities occurring in ≥20% of patients receiving KEYTRUDA were increased hypoalbuminemia (27% all Grades; 2.4% Grades 3-4), increased ALT (23% all Grades; 3.1% Grades 3-4), and increased alkaline phosphatase (21% all Grades, 2% Grades 3-4).
Ipilimumab-Refractory Melanoma
The safety of KEYTRUDA in patients with unresectable or metastatic melanoma with disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor, was investigated in KEYNOTE-002. KEYNOTE-002 was a multicenter, partially blinded (KEYTRUDA dose), randomized (1:1:1), active-controlled trial in which 528 patients received KEYTRUDA 2 mg/kg (n=178) or 10 mg/kg (n=179) every 3 weeks or investigator's choice of chemotherapy (n=171), consisting of dacarbazine (26%), temozolomide (25%), paclitaxel and carboplatin (25%), paclitaxel (16%), or carboplatin (8%) [see Clinical Studies (14.1)]. Patients with autoimmune disease, severe immune-related toxicity related to ipilimumab, defined as any Grade 4 toxicity or Grade 3 toxicity requiring corticosteroid treatment (greater than 10 mg/day prednisone or equivalent dose) for greater than 12 weeks; medical conditions that required systemic corticosteroids or other immunosuppressive medication; a history of interstitial lung disease; or an active infection requiring therapy, including HIV or hepatitis B or C, were ineligible.
The median duration of exposure to KEYTRUDA 2 mg/kg every 3 weeks was 3.7 months (range: 1 day to 16.6 months) and to KEYTRUDA 10 mg/kg every 3 weeks was 4.8 months (range: 1 day to 16.8 months). In the KEYTRUDA 2 mg/kg arm, 36% of patients were exposed to KEYTRUDA for ≥6 months and 4% were exposed for ≥12 months. In the KEYTRUDA 10 mg/kg arm, 41% of patients were exposed to KEYTRUDA for ≥6 months and 6% of patients were exposed to KEYTRUDA for ≥12 months.
The study population characteristics were: median age of 62 years (range: 15 to 89); 61% male; 98% White; 41% had an elevated LDH value at baseline; 83% had M1c stage disease; 73% received two or more prior therapies for advanced or metastatic disease (100% received ipilimumab and 25% a BRAF inhibitor); and 15% with history of brain metastasis.
In KEYNOTE-002, the adverse reaction profile was similar for the 2 mg/kg dose and 10 mg/kg dose, therefore summary safety results are provided in a pooled analysis (n=357) of both KEYTRUDA arms. Adverse reactions resulting in permanent discontinuation occurred in 12% of patients receiving KEYTRUDA; the most common (≥1%) were general physical health deterioration (1%), asthenia (1%), dyspnea (1%), pneumonitis (1%), and generalized edema (1%). Adverse reactions leading to interruption of KEYTRUDA occurred in 14% of patients; the most common (≥1%) were dyspnea (1%), diarrhea (1%), and maculo-papular rash (1%). Tables 6 and 7 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-002.
| Adverse Reaction | KEYTRUDA 2 mg/kg or 10 mg/kg every 3 weeks | Chemotherapy† | ||
|---|---|---|---|---|
| n=357 | n=171 | |||
| All Grades‡
(%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) |
|
|
||||
| Skin and Subcutaneous Tissue | ||||
| Pruritus | 28 | 0 | 8 | 0 |
| Rash§ | 24 | 0.6 | 8 | 0 |
| Gastrointestinal | ||||
| Constipation | 22 | 0.3 | 20 | 2.3 |
| Diarrhea | 20 | 0.8 | 20 | 2.3 |
| Abdominal pain | 13 | 1.7 | 8 | 1.2 |
| Respiratory, Thoracic and Mediastinal | ||||
| Cough | 18 | 0 | 16 | 0 |
| General | ||||
| Pyrexia | 14 | 0.3 | 9 | 0.6 |
| Asthenia | 10 | 2.0 | 9 | 1.8 |
| Musculoskeletal and Connective Tissue | ||||
| Arthralgia | 14 | 0.6 | 10 | 1.2 |
Other clinically important adverse reactions occurring in patients receiving KEYTRUDA were fatigue (43%), nausea (22%), decreased appetite (20%), vomiting (13%), and peripheral neuropathy (1.7%).
| Laboratory Test† | KEYTRUDA 2 mg/kg or 10 mg/kg every 3 weeks | Chemotherapy | ||
|---|---|---|---|---|
| All Grades‡
% | Grades 3-4 % | All Grades % | Grades 3-4 % |
|
|
||||
| Chemistry | ||||
| Hyperglycemia | 49 | 6 | 44 | 6 |
| Hypoalbuminemia | 37 | 1.9 | 33 | 0.6 |
| Hyponatremia | 37 | 7 | 24 | 3.8 |
| Hypertriglyceridemia | 33 | 0 | 32 | 0.9 |
| Increased alkaline phosphatase | 26 | 3.1 | 18 | 1.9 |
| Increased AST | 24 | 2.2 | 16 | 0.6 |
| Decreased bicarbonate | 22 | 0.4 | 13 | 0 |
| Hypocalcemia | 21 | 0.3 | 18 | 1.9 |
| Increased ALT | 21 | 1.8 | 16 | 0.6 |
Other laboratory abnormalities occurring in ≥20% of patients receiving KEYTRUDA were anemia (44% all Grades; 10% Grades 3-4) and lymphopenia (40% all Grades; 9% Grades 3-4).
Adjuvant Treatment of Resected Stage IIB or IIC Melanoma
Among the 969 patients with Stage IIB or IIC melanoma enrolled in KEYNOTE-716 [see Clinical Studies (14.1)] treated with KEYTRUDA, the median duration of exposure to KEYTRUDA was 9.9 months (range: 0 to 15.4 months). Patients with autoimmune disease or a medical condition that required immunosuppression or mucosal or ocular melanoma were ineligible. Adverse reactions occurring in patients with Stage IIB or IIC melanoma were similar to those occurring in 1011 patients with Stage III melanoma from KEYNOTE-054 or the 2799 patients with melanoma or NSCLC treated with KEYTRUDA as a single agent.
Adjuvant Treatment of Stage III Resected Melanoma
The safety of KEYTRUDA as a single agent was investigated in KEYNOTE-054, a randomized (1:1) double-blind trial in which 1019 patients with completely resected Stage IIIA (>1 mm lymph node metastasis), IIIB or IIIC melanoma received 200 mg of KEYTRUDA by intravenous infusion every 3 weeks (n=509) or placebo (n=502) for up to one year [see Clinical Studies (14.1)]. Patients with active autoimmune disease or a medical condition that required immunosuppression or mucosal or ocular melanoma were ineligible. Seventy-six percent of patients received KEYTRUDA for 6 months or longer.
The study population characteristics were: median age of 54 years (range: 19 to 88), 25% age 65 or older; 62% male; and 94% ECOG PS of 0 and 6% ECOG PS of 1. Sixteen percent had Stage IIIA, 46% had Stage IIIB, 18% had Stage IIIC (1-3 positive lymph nodes), and 20% had Stage IIIC (≥4 positive lymph nodes).
Two patients treated with KEYTRUDA died from causes other than disease progression; causes of death were drug reaction with eosinophilia and systemic symptoms and autoimmune myositis with respiratory failure. Serious adverse reactions occurred in 25% of patients receiving KEYTRUDA. Adverse reactions leading to permanent discontinuation occurred in 14% of patients receiving KEYTRUDA; the most common (≥1%) were pneumonitis (1.4%), colitis (1.2%), and diarrhea (1%). Adverse reactions leading to interruption of KEYTRUDA occurred in 19% of patients; the most common (≥1%) were diarrhea (2.4%), pneumonitis (2%), increased ALT (1.4%), arthralgia (1.4%), increased AST (1.4%), dyspnea (1%), and fatigue (1%). Tables 8 and 9 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-054.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks n=509 | Placebo n=502 |
||
|---|---|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) |
|
| Gastrointestinal | ||||
| Diarrhea | 28 | 1.2 | 26 | 1.2 |
| Nausea | 17 | 0.2 | 15 | 0 |
| Skin and Subcutaneous Tissue | ||||
| Pruritus | 19 | 0 | 12 | 0 |
| Rash | 13 | 0.2 | 9 | 0 |
| Musculoskeletal and Connective Tissue | ||||
| Arthralgia | 16 | 1.2 | 14 | 0 |
| Endocrine | ||||
| Hypothyroidism | 15 | 0 | 2.8 | 0 |
| Hyperthyroidism | 10 | 0.2 | 1.2 | 0 |
| Respiratory, Thoracic and Mediastinal | ||||
| Cough | 14 | 0 | 11 | 0 |
| General | ||||
| Asthenia | 11 | 0.2 | 8 | 0 |
| Influenza like illness | 11 | 0 | 8 | 0 |
| Investigations | ||||
| Weight loss | 11 | 0 | 8 | 0 |
| Laboratory Test† | KEYTRUDA 200 mg every 3 weeks | Placebo | ||
|---|---|---|---|---|
| All Grades‡
% | Grades 3-4 % | All Grades % | Grades 3-4 % |
|
|
||||
| Chemistry | ||||
| Increased ALT | 27 | 2.4 | 16 | 0.2 |
| Increased AST | 24 | 1.8 | 15 | 0.4 |
| Hematology | ||||
| Lymphopenia | 24 | 1 | 16 | 1.2 |
NSCLC
First-line treatment of metastatic nonsquamous NSCLC with pemetrexed and platinum chemotherapy
The safety of KEYTRUDA in combination with pemetrexed and investigator's choice of platinum (either carboplatin or cisplatin) was investigated in KEYNOTE-189, a multicenter, double-blind, randomized (2:1), active-controlled trial in patients with previously untreated, metastatic nonsquamous NSCLC with no EGFR or ALK genomic tumor aberrations [see Clinical Studies (14.2)]. A total of 607 patients received KEYTRUDA 200 mg, pemetrexed and platinum every 3 weeks for 4 cycles followed by KEYTRUDA and pemetrexed (n=405) or placebo, pemetrexed, and platinum every 3 weeks for 4 cycles followed by placebo and pemetrexed (n=202). Patients with autoimmune disease that required systemic therapy within 2 years of treatment; a medical condition that required immunosuppression; or who had received more than 30 Gy of thoracic radiation within the prior 26 weeks were ineligible.
The median duration of exposure to KEYTRUDA 200 mg every 3 weeks was 7.2 months (range: 1 day to 20.1 months). Sixty percent of patients in the KEYTRUDA arm were exposed to KEYTRUDA for ≥6 months. Seventy-two percent of patients received carboplatin.
The study population characteristics were: median age of 64 years (range: 34 to 84), 49% age 65 or older; 59% male; 94% White and 3% Asian; and 18% with history of brain metastases at baseline.
KEYTRUDA was discontinued for adverse reactions in 20% of patients. The most common adverse reactions resulting in permanent discontinuation of KEYTRUDA were pneumonitis (3%) and acute kidney injury (2%). Adverse reactions leading to the interruption of KEYTRUDA occurred in 53% of patients; the most common adverse reactions or laboratory abnormalities leading to interruption of KEYTRUDA (≥2%) were neutropenia (13%), asthenia/fatigue (7%), anemia (7%), thrombocytopenia (5%), diarrhea (4%), pneumonia (4%), increased blood creatinine (3%), dyspnea (2%), febrile neutropenia (2%), upper respiratory tract infection (2%), increased ALT (2%), and pyrexia (2%). Tables 10 and 11 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-189.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks Pemetrexed Platinum Chemotherapy n=405 | Placebo Pemetrexed Platinum Chemotherapy n=202 |
||
|---|---|---|---|---|
| All Grades*
(%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) |
|
| Gastrointestinal | ||||
| Nausea | 56 | 3.5 | 52 | 3.5 |
| Constipation | 35 | 1.0 | 32 | 0.5 |
| Diarrhea | 31 | 5 | 21 | 3.0 |
| Vomiting | 24 | 3.7 | 23 | 3.0 |
| General | ||||
| Fatigue† | 56 | 12 | 58 | 6 |
| Pyrexia | 20 | 0.2 | 15 | 0 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 28 | 1.5 | 30 | 0.5 |
| Skin and Subcutaneous Tissue | ||||
| Rash‡ | 25 | 2.0 | 17 | 2.5 |
| Respiratory, Thoracic and Mediastinal | ||||
| Cough | 21 | 0 | 28 | 0 |
| Dyspnea | 21 | 3.7 | 26 | 5 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks Pemetrexed Platinum Chemotherapy | Placebo Pemetrexed Platinum Chemotherapy |
||
|---|---|---|---|---|
| All Grades†
% | Grades 3-4 % | All Grades % | Grades 3-4 % |
|
|
||||
| Hematology | ||||
| Anemia | 85 | 17 | 81 | 18 |
| Lymphopenia | 64 | 22 | 64 | 25 |
| Neutropenia | 48 | 20 | 41 | 19 |
| Thrombocytopenia | 30 | 12 | 29 | 8 |
| Chemistry | ||||
| Hyperglycemia | 63 | 9 | 60 | 7 |
| Increased ALT | 47 | 3.8 | 42 | 2.6 |
| Increased AST | 47 | 2.8 | 40 | 1.0 |
| Hypoalbuminemia | 39 | 2.8 | 39 | 1.1 |
| Increased creatinine | 37 | 4.2 | 25 | 1.0 |
| Hyponatremia | 32 | 7 | 23 | 6 |
| Hypophosphatemia | 30 | 10 | 28 | 14 |
| Increased alkaline phosphatase | 26 | 1.8 | 29 | 2.1 |
| Hypocalcemia | 24 | 2.8 | 17 | 0.5 |
| Hyperkalemia | 24 | 2.8 | 19 | 3.1 |
| Hypokalemia | 21 | 5 | 20 | 5 |
First-line treatment of metastatic squamous NSCLC with carboplatin and either paclitaxel or paclitaxel protein-bound chemotherapy
The safety of KEYTRUDA in combination with carboplatin and investigator's choice of either paclitaxel or paclitaxel protein-bound was investigated in KEYNOTE-407, a multicenter, double-blind, randomized (1:1), placebo-controlled trial in 558 patients with previously untreated, metastatic squamous NSCLC [see Clinical Studies (14.2)]. Safety data are available for the first 203 patients who received KEYTRUDA and chemotherapy (n=101) or placebo and chemotherapy (n=102). Patients with autoimmune disease that required systemic therapy within 2 years of treatment; a medical condition that required immunosuppression; or who had received more than 30 Gy of thoracic radiation within the prior 26 weeks were ineligible.
The median duration of exposure to KEYTRUDA was 7 months (range: 1 day to 12 months). Sixty-one percent of patients in the KEYTRUDA arm were exposed to KEYTRUDA for ≥6 months. A total of 139 of 203 patients (68%) received paclitaxel and 64 patients (32%) received paclitaxel protein-bound in combination with carboplatin.
The study population characteristics were: median age of 65 years (range: 40 to 83), 52% age 65 or older; 78% male; 83% White; and 9% with history of brain metastases.
KEYTRUDA was discontinued for adverse reactions in 15% of patients, with no single type of adverse reaction accounting for the majority. Adverse reactions leading to interruption of KEYTRUDA occurred in 43% of patients; the most common (≥2%) were thrombocytopenia (20%), neutropenia (11%), anemia (6%), asthenia (2%), and diarrhea (2%). The most frequent (≥2%) serious adverse reactions were febrile neutropenia (6%), pneumonia (6%), and urinary tract infection (3%).
The adverse reactions observed in KEYNOTE-407 were similar to those observed in KEYNOTE-189 with the exception that increased incidences of alopecia (47% vs. 36%) and peripheral neuropathy (31% vs. 25%) were observed in the KEYTRUDA and chemotherapy arm compared to the placebo and chemotherapy arm in KEYNOTE-407.
Previously Untreated NSCLC
The safety of KEYTRUDA was investigated in KEYNOTE-042, a multicenter, open-label, randomized (1:1), active-controlled trial in 1251 patients with PD-L1 expressing, previously untreated Stage III NSCLC who were not candidates for surgical resection or definitive chemoradiation or metastatic NSCLC [see Clinical Studies (14.2)]. Patients received KEYTRUDA 200 mg every 3 weeks (n=636) or investigator's choice of chemotherapy (n=615), consisting of pemetrexed and carboplatin followed by optional pemetrexed (n=312) or paclitaxel and carboplatin followed by optional pemetrexed (n=303) every 3 weeks. Patients with EGFR or ALK genomic tumor aberrations; autoimmune disease that required systemic therapy within 2 years of treatment; a medical condition that required immunosuppression; or who had received more than 30 Gy of thoracic radiation within the prior 26 weeks were ineligible.
The median duration of exposure to KEYTRUDA was 5.6 months (range: 1 day to 27.3 months). Forty-eight percent of patients in the KEYTRUDA arm were exposed to KEYTRUDA 200 mg for ≥6 months.
The study population characteristics were: median age of 63 years (range: 25 to 90), 45% age 65 or older; 71% male; and 64% White, 30% Asian, and 2% Black. Nineteen percent were Hispanic or Latino. Eighty-seven percent had metastatic disease (Stage IV), 13% had Stage III disease (2% Stage IIIA and 11% Stage IIIB), and 5% had treated brain metastases at baseline.
KEYTRUDA was discontinued for adverse reactions in 19% of patients. The most common adverse reactions resulting in permanent discontinuation of KEYTRUDA were pneumonitis (3.0%), death due to unknown cause (1.6%), and pneumonia (1.4%). Adverse reactions leading to interruption of KEYTRUDA occurred in 33% of patients; the most common adverse reactions or laboratory abnormalities leading to interruption of KEYTRUDA (≥2%) were pneumonitis (3.1%), pneumonia (3.0%), hypothyroidism (2.2%), and increased ALT (2.0%). The most frequent (≥2%) serious adverse reactions were pneumonia (7%), pneumonitis (3.9%), pulmonary embolism (2.4%), and pleural effusion (2.2%).
Tables 12 and 13 summarize the adverse reactions and laboratory abnormalities, respectively, in patients treated with KEYTRUDA in KEYNOTE-042.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks n=636 | Chemotherapy n=615 |
||
|---|---|---|---|---|
| All Grades*
(%) | Grades 3-5 (%) | All Grades (%) | Grades 3-5 (%) |
|
| General | ||||
| Fatigue† | 25 | 3.1 | 33 | 3.9 |
| Pyrexia | 10 | 0.3 | 8 | 0 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 17 | 1.7 | 21 | 1.5 |
| Respiratory, Thoracic and Mediastinal | ||||
| Dyspnea | 17 | 2.0 | 11 | 0.8 |
| Cough | 16 | 0.2 | 11 | 0.3 |
| Skin and Subcutaneous Tissue | ||||
| Rash‡ | 15 | 1.3 | 8 | 0.2 |
| Gastrointestinal | ||||
| Constipation | 12 | 0 | 21 | 0.2 |
| Diarrhea | 12 | 0.8 | 12 | 0.5 |
| Nausea | 12 | 0.5 | 32 | 1.1 |
| Endocrine | ||||
| Hypothyroidism | 12 | 0.2 | 1.5 | 0 |
| Infections | ||||
| Pneumonia | 12 | 7 | 9 | 6 |
| Investigations | ||||
| Weight loss | 10 | 0.9 | 7 | 0.2 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks | Chemotherapy | ||
|---|---|---|---|---|
| All Grades†
% | Grades 3-4 % | All Grades % | Grades 3-4 % |
|
|
||||
| Chemistry | ||||
| Hyperglycemia | 52 | 4.7 | 51 | 5 |
| Increased ALT | 33 | 4.8 | 34 | 2.9 |
| Hypoalbuminemia | 33 | 2.2 | 29 | 1.0 |
| Increased AST | 31 | 3.6 | 32 | 1.7 |
| Hyponatremia | 31 | 9 | 32 | 8 |
| Increased alkaline phosphatase | 29 | 2.3 | 29 | 0.3 |
| Hypocalcemia | 25 | 2.5 | 19 | 0.7 |
| Hyperkalemia | 23 | 3.0 | 20 | 2.2 |
| Increased prothrombin INR | 21 | 2.0 | 15 | 2.9 |
| Hematology | ||||
| Anemia | 43 | 4.4 | 79 | 19 |
| Lymphopenia | 30 | 7 | 41 | 13 |
Previously Treated NSCLC
The safety of KEYTRUDA was investigated in KEYNOTE-010, a multicenter, open-label, randomized (1:1:1), active-controlled trial, in patients with advanced NSCLC who had documented disease progression following treatment with platinum-based chemotherapy and, if positive for EGFR or ALK genetic aberrations, appropriate therapy for these aberrations [see Clinical Studies (14.2)]. A total of 991 patients received KEYTRUDA 2 mg/kg (n=339) or 10 mg/kg (n=343) every 3 weeks or docetaxel (n=309) at 75 mg/m2 every 3 weeks. Patients with autoimmune disease, medical conditions that required systemic corticosteroids or other immunosuppressive medication, or who had received more than 30 Gy of thoracic radiation within the prior 26 weeks were ineligible.
The median duration of exposure to KEYTRUDA 2 mg/kg every 3 weeks was 3.5 months (range: 1 day to 22.4 months) and to KEYTRUDA 10 mg/kg every 3 weeks was 3.5 months (range 1 day to 20.8 months). The data described below reflect exposure to KEYTRUDA 2 mg/kg in 31% of patients exposed to KEYTRUDA for ≥6 months. In the KEYTRUDA 10 mg/kg arm, 34% of patients were exposed to KEYTRUDA for ≥6 months.
The study population characteristics were: median age of 63 years (range: 20 to 88), 42% age 65 or older; 61% male; 72% White and 21% Asian; and 8% with advanced localized disease, 91% with metastatic disease, and 15% with history of brain metastases. Twenty-nine percent received two or more prior systemic treatments for advanced or metastatic disease.
In KEYNOTE-010, the adverse reaction profile was similar for the 2 mg/kg and 10 mg/kg dose, therefore summary safety results are provided in a pooled analysis (n=682). Treatment was discontinued for adverse reactions in 8% of patients receiving KEYTRUDA. The most common adverse events resulting in permanent discontinuation of KEYTRUDA was pneumonitis (1.8%). Adverse reactions leading to interruption of KEYTRUDA occurred in 23% of patients; the most common (≥1%) were diarrhea (1%), fatigue (1.3%), pneumonia (1%), liver enzyme elevation (1.2%), decreased appetite (1.3%), and pneumonitis (1%). Tables 14 and 15 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-010.
| Adverse Reaction | KEYTRUDA 2 or 10 mg/kg every 3 weeks n=682 | Docetaxel 75 mg/m2 every 3 weeks n=309 |
||
|---|---|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) | All Grades†
(%) | Grades 3-4 (%) |
|
| Metabolism and Nutrition | ||||
| Decreased appetite | 25 | 1.5 | 23 | 2.6 |
| Respiratory, Thoracic and Mediastinal | ||||
| Dyspnea | 23 | 3.7 | 20 | 2.6 |
| Cough | 19 | 0.6 | 14 | 0 |
| Gastrointestinal | ||||
| Nausea | 20 | 1.3 | 18 | 0.6 |
| Constipation | 15 | 0.6 | 12 | 0.6 |
| Vomiting | 13 | 0.9 | 10 | 0.6 |
| Skin and Subcutaneous Tissue | ||||
| Rash‡ | 17 | 0.4 | 8 | 0 |
| Pruritus | 11 | 0 | 3 | 0.3 |
| Musculoskeletal and Connective Tissue | ||||
| Arthralgia | 11 | 1.0 | 9 | 0.3 |
| Back pain | 11 | 1.5 | 8 | 0.3 |
Other clinically important adverse reactions occurring in patients receiving KEYTRUDA were fatigue (25%), diarrhea (14%), asthenia (11%) and pyrexia (11%).
| Laboratory Test† | KEYTRUDA 2 or 10 mg/kg every 3 weeks | Docetaxel 75 mg/m2 every 3 weeks |
||
|---|---|---|---|---|
| All Grades‡
% | Grades 3-4 % | All Grades‡
% | Grades 3-4 % |
|
|
||||
| Chemistry | ||||
| Hyponatremia | 32 | 8 | 27 | 2.9 |
| Increased alkaline phosphatase | 28 | 3.0 | 16 | 0.7 |
| Increased AST | 26 | 1.6 | 12 | 0.7 |
| Increased ALT | 22 | 2.7 | 9 | 0.4 |
Other laboratory abnormalities occurring in ≥20% of patients receiving KEYTRUDA were hyperglycemia (44% all Grades; 4.1% Grades 3-4), anemia (37% all Grades; 3.8% Grades 3-4), hypertriglyceridemia (36% all Grades; 1.8% Grades 3-4), lymphopenia (35% all Grades; 9% Grades 3-4), hypoalbuminemia (34% all Grades; 1.6% Grades 3-4), and hypercholesterolemia (20% all Grades; 0.7% Grades 3-4).
Neoadjuvant and Adjuvant Treatment of Resectable NSCLC
The safety of KEYTRUDA in combination with neoadjuvant platinum-containing chemotherapy followed by surgery and continued adjuvant treatment with KEYTRUDA as a single agent after surgery was investigated in KEYNOTE-671, a multicenter, randomized (1:1), double-blind, placebo-controlled trial in patients with previously untreated and resectable Stage II, IIIA, or IIIB (N2) NSCLC by AJCC 8th edition [see Clinical Studies (14.2)]. Patients with active autoimmune disease that required systemic therapy within 2 years of treatment or a medical condition that required immunosuppression were ineligible.
The median duration of exposure to KEYTRUDA 200 mg every 3 weeks was 10.9 months (range: 1 day to 18.6 months). The study population characteristics were: median age of 64 years (range: 26 to 83), 45% age 65 or older, 7% age 75 or older; 71% male; 61% White, 31% Asian, 2% Black, 4% race not reported; 9% Hispanic or Latino.
Adverse reactions occurring in patients with resectable NSCLC receiving KEYTRUDA in combination with platinum containing chemotherapy, given as neoadjuvant treatment and continued as single agent adjuvant treatment, were generally similar to those occurring in patients in other clinical trials across tumor types receiving KEYTRUDA in combination with chemotherapy.
Neoadjuvant Phase of KEYNOTE-671
A total of 396 patients received at least 1 dose of KEYTRUDA in combination with platinum-containing chemotherapy as neoadjuvant treatment and 399 patients received at least 1 dose of placebo in combination with platinum-containing chemotherapy as neoadjuvant treatment.
Serious adverse reactions occurred in 34% of patients who received KEYTRUDA in combination with platinum-containing chemotherapy as neoadjuvant treatment; the most frequent (≥2%) serious adverse reactions were pneumonia (4.8%), venous thromboembolism (3.3%), and anemia (2%). Fatal adverse reactions occurred in 1.3% of patients, including death due to unknown cause (0.8%), sepsis (0.3%), and immune-mediated lung disease (0.3%).
Permanent discontinuation of any study drug due to an adverse reaction occurred in 18% of patients who received KEYTRUDA in combination with platinum-containing chemotherapy as neoadjuvant treatment; the most frequent (≥1%) adverse reactions that led to permanent discontinuation of any study drug were acute kidney injury (1.8%), interstitial lung disease (1.8%), anemia (1.5%), neutropenia (1.5%), and pneumonia (1.3%).
Of the 396 KEYTRUDA-treated patients and 399 placebo-treated patients who received neoadjuvant treatment, 6% (n=25) and 4.3% (n=17), respectively, did not receive surgery due to adverse reactions. The most frequent (≥1%) adverse reactions that led to cancellation of surgery in the KEYTRUDA arm was interstitial lung disease (1%).
Of the 325 KEYTRUDA-treated patients who received surgery, 3.1% (n=10) experienced delay of surgery (surgery more than 8 weeks from last neoadjuvant treatment if patient received less than 4 cycles of neoadjuvant therapy or more than 20 weeks after first dose of neoadjuvant treatment if patient received 4 cycles of neoadjuvant therapy) due to adverse reactions. Of the 317 placebo-treated patients who received surgery, 2.5% (n=8) experienced delay of surgery due to adverse reactions.
Of the 325 KEYTRUDA-treated patients who received surgery, 7% (n=22) did not receive adjuvant treatment due to adverse reactions. Of the 317 placebo-treated patients who received surgery, 3.2% (n=10) did not receive adjuvant treatment due to adverse reactions.
Adjuvant Phase of KEYNOTE-671
A total of 290 patients in the KEYTRUDA arm and 267 patients in the placebo arm received at least 1 dose of adjuvant treatment.
Of the patients who received single agent KEYTRUDA as adjuvant treatment, 14% experienced serious adverse reactions; the most frequent serious adverse reaction was pneumonia (3.4%). One fatal adverse reaction of pulmonary hemorrhage occurred. Permanent discontinuation of adjuvant KEYTRUDA due to an adverse reaction occurred in 12% of patients; the most frequent (≥1%) adverse reactions that led to permanent discontinuation of adjuvant KEYTRUDA were diarrhea (1.7%), interstitial lung disease (1.4%), AST increased (1%), and musculoskeletal pain (1%).
Adjuvant Treatment of Resected NSCLC
The safety of KEYTRUDA as a single agent was investigated in KEYNOTE-091, a multicenter, randomized (1:1), triple-blind, placebo-controlled trial in patients with completely resected Stage IB (T2a ≥4 cm), II, or IIIA NSCLC; adjuvant chemotherapy up to 4 cycles was optional [see Clinical Studies (14.2)]. A total of 1161 patients received KEYTRUDA 200 mg (n=580) or placebo (n=581) every 3 weeks. Patients were ineligible if they had active autoimmune disease, were on chronic immunosuppressive agents, or had a history of interstitial lung disease or pneumonitis.
The median duration of exposure to KEYTRUDA was 11.7 months (range: 1 day to 18.9 months). Sixty-eight percent of patients in the KEYTRUDA arm were exposed to KEYTRUDA for ≥6 months.
The adverse reactions observed in KEYNOTE-091 were generally similar to those occurring in other patients with NSCLC receiving KEYTRUDA as a single agent, with the exception of hypothyroidism (22%), hyperthyroidism (11%), and pneumonitis (7%). Two fatal adverse reactions of myocarditis occurred.
HNSCC
First-line treatment of metastatic or unresectable, recurrent HNSCC
The safety of KEYTRUDA, as a single agent and in combination with platinum (cisplatin or carboplatin) and FU chemotherapy, was investigated in KEYNOTE-048, a multicenter, open-label, randomized (1:1:1), active-controlled trial in patients with previously untreated, recurrent or metastatic HNSCC [see Clinical Studies (14.3)]. Patients with autoimmune disease that required systemic therapy within 2 years of treatment or a medical condition that required immunosuppression were ineligible. A total of 576 patients received KEYTRUDA 200 mg every 3 weeks either as a single agent (n=300) or in combination with platinum and FU (n=276) every 3 weeks for 6 cycles followed by KEYTRUDA, compared to 287 patients who received cetuximab weekly in combination with platinum and FU every 3 weeks for 6 cycles followed by cetuximab.
The median duration of exposure to KEYTRUDA was 3.5 months (range: 1 day to 24.2 months) in the KEYTRUDA single agent arm and was 5.8 months (range: 3 days to 24.2 months) in the combination arm. Seventeen percent of patients in the KEYTRUDA single agent arm and 18% of patients in the combination arm were exposed to KEYTRUDA for ≥12 months. Fifty-seven percent of patients receiving KEYTRUDA in combination with chemotherapy started treatment with carboplatin.
KEYTRUDA was discontinued for adverse reactions in 12% of patients in the KEYTRUDA single agent arm. The most common adverse reactions resulting in permanent discontinuation of KEYTRUDA were sepsis (1.7%) and pneumonia (1.3%). Adverse reactions leading to the interruption of KEYTRUDA occurred in 31% of patients; the most common adverse reactions leading to interruption of KEYTRUDA (≥2%) were pneumonia (2.3%), pneumonitis (2.3%), and hyponatremia (2%).
KEYTRUDA was discontinued for adverse reactions in 16% of patients in the combination arm. The most common adverse reactions resulting in permanent discontinuation of KEYTRUDA were pneumonia (2.5%), pneumonitis (1.8%), and septic shock (1.4%). Adverse reactions leading to the interruption of KEYTRUDA occurred in 45% of patients; the most common adverse reactions leading to interruption of KEYTRUDA (≥2%) were neutropenia (14%), thrombocytopenia (10%), anemia (6%), pneumonia (4.7%), and febrile neutropenia (2.9%).
Tables 16 and 17 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-048.
| KEYTRUDA 200 mg every 3 weeks | KEYTRUDA 200 mg every 3 weeks Platinum FU | Cetuximab Platinum FU |
||||
|---|---|---|---|---|---|---|
| Adverse Reaction | n=300 | n=276 | n=287 | |||
| All Grades*
(%) | Grades 3-4 (%) | All Grades*
(%) | Grades 3-4 (%) | All Grades*
(%) | Grades 3-4 (%) |
|
|
||||||
| General | ||||||
| Fatigue† | 33 | 4 | 49 | 11 | 48 | 8 |
| Pyrexia | 13 | 0.7 | 16 | 0.7 | 12 | 0 |
| Mucosal inflammation | 4.3 | 1.3 | 31 | 10 | 28 | 5 |
| Gastrointestinal | ||||||
| Constipation | 20 | 0.3 | 37 | 0 | 33 | 1.4 |
| Nausea | 17 | 0 | 51 | 6 | 51 | 6 |
| Diarrhea‡ | 16 | 0.7 | 29 | 3.3 | 35 | 3.1 |
| Vomiting | 11 | 0.3 | 32 | 3.6 | 28 | 2.8 |
| Dysphagia | 8 | 2.3 | 12 | 2.9 | 10 | 2.1 |
| Stomatitis | 3 | 0 | 26 | 8 | 28 | 3.5 |
| Skin | ||||||
| Rash§ | 20 | 2.3 | 17 | 0.7 | 70 | 8 |
| Pruritus | 11 | 0 | 8 | 0 | 10 | 0.3 |
| Respiratory, Thoracic and Mediastinal | ||||||
| Cough¶ | 18 | 0.3 | 22 | 0 | 15 | 0 |
| Dyspnea# | 14 | 2.0 | 10 | 1.8 | 8 | 1.0 |
| Endocrine | ||||||
| Hypothyroidism | 18 | 0 | 15 | 0 | 6 | 0 |
| Metabolism and Nutrition | ||||||
| Decreased appetite | 15 | 1.0 | 29 | 4.7 | 30 | 3.5 |
| Weight loss | 15 | 2 | 16 | 2.9 | 21 | 1.4 |
| Infections | ||||||
| PneumoniaÞ | 12 | 7 | 19 | 11 | 13 | 6 |
| Nervous System | ||||||
| Headache | 12 | 0.3 | 11 | 0.7 | 8 | 0.3 |
| Dizziness | 5 | 0.3 | 10 | 0.4 | 13 | 0.3 |
| Peripheral sensory neuropathyß | 1 | 0 | 14 | 1.1 | 7 | 1 |
| Musculoskeletal | ||||||
| Myalgiaà | 12 | 1.0 | 13 | 0.4 | 11 | 0.3 |
| Neck pain | 6 | 0.7 | 10 | 1.1 | 7 | 0.7 |
| Psychiatric | ||||||
| Insomnia | 7 | 0.7 | 10 | 0 | 8 | 0 |
| KEYTRUDA 200 mg every 3 weeks | KEYTRUDA 200 mg every 3 weeks Platinum FU | Cetuximab Platinum FU |
||||
|---|---|---|---|---|---|---|
| Laboratory Test* | All Grades†
(%) | Grades 3-4 (%) | All Grades†
(%) | Grades 3-4 (%) | All Grades†
(%) | Grades 3-4 (%) |
|
||||||
| Hematology | ||||||
| Lymphopenia | 54 | 25 | 69 | 35 | 74 | 45 |
| Anemia | 52 | 7 | 89 | 28 | 78 | 19 |
| Thrombocytopenia | 12 | 3.8 | 73 | 18 | 76 | 18 |
| Neutropenia | 7 | 1.4 | 67 | 35 | 71 | 42 |
| Chemistry | ||||||
| Hyperglycemia | 47 | 3.8 | 55 | 6 | 66 | 4.7 |
| Hyponatremia | 46 | 17 | 56 | 20 | 59 | 20 |
| Hypoalbuminemia | 44 | 3.2 | 47 | 4.0 | 49 | 1.1 |
| Increased AST | 28 | 3.1 | 24 | 2.0 | 37 | 3.6 |
| Increased ALT | 25 | 2.1 | 22 | 1.6 | 38 | 1.8 |
| Increased alkaline phosphatase | 25 | 2.1 | 27 | 1.2 | 33 | 1.1 |
| Hypercalcemia | 22 | 4.6 | 16 | 4.3 | 13 | 2.6 |
| Hypocalcemia | 22 | 1.1 | 32 | 4 | 58 | 7 |
| Hyperkalemia | 21 | 2.8 | 27 | 4.3 | 29 | 4.3 |
| Hypophosphatemia | 20 | 5 | 35 | 12 | 48 | 19 |
| Hypokalemia | 19 | 5 | 34 | 12 | 47 | 15 |
| Increased creatinine | 18 | 1.1 | 36 | 2.3 | 27 | 2.2 |
| Hypomagnesemia | 16 | 0.4 | 42 | 1.7 | 76 | 6 |
Previously treated recurrent or metastatic HNSCC
Among the 192 patients with HNSCC enrolled in KEYNOTE-012 [see Clinical Studies (14.3)], the median duration of exposure to KEYTRUDA was 3.3 months (range: 1 day to 27.9 months). Patients with autoimmune disease or a medical condition that required immunosuppression were ineligible for KEYNOTE-012.
The study population characteristics were: median age of 60 years (range: 20 to 84), 35% age 65 or older; 83% male; and 77% White, 15% Asian, and 5% Black. Sixty-one percent of patients had two or more lines of therapy in the recurrent or metastatic setting, and 95% had prior radiation therapy. Baseline ECOG PS was 0 (30%) or 1 (70%) and 86% had M1 disease.
KEYTRUDA was discontinued due to adverse reactions in 17% of patients. Serious adverse reactions occurred in 45% of patients receiving KEYTRUDA. The most frequent serious adverse reactions reported in at least 2% of patients were pneumonia, dyspnea, confusional state, vomiting, pleural effusion, and respiratory failure. The incidence of adverse reactions, including serious adverse reactions, was similar between dosage regimens (10 mg/kg every 2 weeks or 200 mg every 3 weeks); therefore, summary safety results are provided in a pooled analysis. The most common adverse reactions (occurring in ≥20% of patients) were fatigue, decreased appetite, and dyspnea. Adverse reactions occurring in patients with HNSCC were generally similar to those occurring in 2799 patients with melanoma or NSCLC treated with KEYTRUDA as a single agent, with the exception of increased incidences of facial edema (10% all Grades; 2.1% Grades 3-4) and new or worsening hypothyroidism [see Warnings and Precautions (5.1)].
Relapsed or Refractory cHL
KEYNOTE-204
The safety of KEYTRUDA was evaluated in KEYNOTE-204 [see Clinical Studies (14.4)]. Adults with relapsed or refractory cHL received KEYTRUDA 200 mg intravenously every 3 weeks (n=148) or brentuximab vedotin (BV) 1.8 mg/kg intravenously every 3 weeks (n=152). The trial required an ANC ≥1000/µL, platelet count ≥75,000/µL, hepatic transaminases ≤2.5 times the upper limit of normal (ULN), bilirubin ≤1.5 times ULN, and ECOG performance status of 0 or 1. The trial excluded patients with active non-infectious pneumonitis, prior pneumonitis requiring steroids, active autoimmune disease, a medical condition requiring immunosuppression, or allogeneic HSCT within the past 5 years. The median duration of exposure to KEYTRUDA was 10 months (range: 1 day to 2.2 years), with 68% receiving at least 6 months of treatment and 48% receiving at least 1 year of treatment.
Serious adverse reactions occurred in 30% of patients who received KEYTRUDA. Serious adverse reactions in ≥1% included pneumonitis, pneumonia, pyrexia, myocarditis, acute kidney injury, febrile neutropenia, and sepsis. Three patients (2%) died from causes other than disease progression: two from complications after allogeneic HSCT and one from unknown cause.
Permanent discontinuation of KEYTRUDA due to an adverse reaction occurred in 14% of patients; 7% of patients discontinued treatment due to pneumonitis. Dosage interruption of KEYTRUDA due to an adverse reaction occurred in 30% of patients. Adverse reactions which required dosage interruption in ≥3% of patients were upper respiratory tract infection, pneumonitis, transaminase increase, and pneumonia.
Thirty-eight percent of patients had an adverse reaction requiring systemic corticosteroid therapy.
Table 18 summarizes adverse reactions in KEYNOTE-204.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks N=148 | Brentuximab Vedotin 1.8 mg/kg every 3 weeks N=152 |
||
|---|---|---|---|---|
| All Grades*
(%) | Grades 3- 4 (%) | All Grades*
(%) | Grades 3- 4†
(%) |
|
|
||||
| Infections | ||||
| Upper respiratory tract infection‡ | 41 | 1.4 | 24 | 0 |
| Urinary tract infection | 11 | 0 | 3 | 0.7 |
| Musculoskeletal and Connective Tissue | ||||
| Musculoskeletal pain§ | 32 | 0 | 29 | 1.3 |
| Gastrointestinal | ||||
| Diarrhea¶ | 22 | 2.7 | 17 | 1.3 |
| Nausea | 14 | 0 | 24 | 0.7 |
| Vomiting | 14 | 1.4 | 20 | 0 |
| Abdominal pain# | 11 | 0.7 | 13 | 1.3 |
| General | ||||
| Pyrexia | 20 | 0.7 | 13 | 0.7 |
| FatigueÞ | 20 | 0 | 22 | 0.7 |
| Skin and Subcutaneous Tissue | ||||
| Rashß | 20 | 0 | 19 | 0.7 |
| Pruritus | 18 | 0 | 12 | 0 |
| Respiratory, Thoracic and Mediastinal | ||||
| Coughà | 20 | 0.7 | 14 | 0.7 |
| Pneumonitisè | 11 | 5 | 3 | 1.3 |
| Dyspneað | 11 | 0.7 | 7 | 0.7 |
| Endocrine | ||||
| Hypothyroidism | 19 | 0 | 3 | 0 |
| Nervous System | ||||
| Peripheral neuropathyø | 11 | 0.7 | 43 | 7 |
| Headacheý | 11 | 0 | 11 | 0 |
Clinically relevant adverse reactions in <10% of patients who received KEYTRUDA included herpes virus infection (9%), pneumonia (8%), oropharyngeal pain (8%), hyperthyroidism (5%), hypersensitivity (4.1%), infusion reactions (3.4%), altered mental state (2.7%), and in 1.4% each, uveitis, myocarditis, thyroiditis, febrile neutropenia, sepsis, and tumor flare.
Table 19 summarizes laboratory abnormalities in KEYNOTE-204.
| Laboratory Abnormality* | KEYTRUDA 200 mg every 3 weeks | Brentuximab Vedotin 1.8 mg/kg every 3 weeks |
||
|---|---|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) | All Grades†
(%) | Grades 3-4 (%) |
|
| Chemistry | ||||
| Hyperglycemia | 46 | 4.1 | 36 | 2.0 |
| Increased AST | 39 | 5 | 41 | 3.9 |
| Increased ALT | 34 | 6 | 45 | 5 |
| Hypophosphatemia | 31 | 5 | 18 | 2.7 |
| Increased creatinine | 28 | 3.4 | 14 | 2.6 |
| Hypomagnesemia | 25 | 0 | 12 | 0 |
| Hyponatremia | 24 | 4.1 | 20 | 3.3 |
| Hypocalcemia | 22 | 2.0 | 16 | 0 |
| Increased alkaline phosphatase | 21 | 2.1 | 22 | 2.6 |
| Hyperbilirubinemia | 16 | 2.0 | 9 | 1.3 |
| Hypoalbuminemia | 16 | 0.7 | 19 | 0.7 |
| Hyperkalemia | 15 | 1.4 | 8 | 0 |
| Hematology | ||||
| Lymphopenia | 35 | 9 | 32 | 13 |
| Thrombocytopenia | 34 | 10 | 26 | 5 |
| Neutropenia | 28 | 8 | 43 | 17 |
| Anemia | 24 | 5 | 33 | 8 |
KEYNOTE-087
Among the 210 patients with cHL who received KEYTRUDA in KEYNOTE-087 [see Clinical Studies (14.4)], the median duration of exposure to KEYTRUDA was 8.4 months (range: 1 day to 15.2 months). Serious adverse reactions occurred in 16% of patients who received KEYTRUDA. Serious adverse reactions that occurred in ≥1% of patients included pneumonia, pneumonitis, pyrexia, dyspnea, graft versus host disease (GVHD) and herpes zoster. Two patients died from causes other than disease progression; one from GVHD after subsequent allogeneic HSCT and one from septic shock.
Permanent discontinuation of KEYTRUDA due to an adverse reaction occurred in 5% of patients and dosage interruption due to an adverse reaction occurred in 26%. Fifteen percent of patients had an adverse reaction requiring systemic corticosteroid therapy. Tables 20 and 21 summarize adverse reactions and laboratory abnormalities, respectively, in KEYNOTE-087.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks N=210 |
|
|---|---|---|
| All Grades*
(%) | Grade 3 (%) |
|
|
||
| General | ||
| Fatigue† | 26 | 1.0 |
| Pyrexia | 24 | 1.0 |
| Respiratory, Thoracic and Mediastinal | ||
| Cough‡ | 24 | 0.5 |
| Dyspnea§ | 11 | 1.0 |
| Musculoskeletal and Connective Tissue | ||
| Musculoskeletal pain¶ | 21 | 1.0 |
| Arthralgia | 10 | 0.5 |
| Gastrointestinal | ||
| Diarrhea# | 20 | 1.4 |
| Vomiting | 15 | 0 |
| Nausea | 13 | 0 |
| Skin and Subcutaneous Tissue | ||
| Rash Þ | 20 | 0.5 |
| Pruritus | 11 | 0 |
| Endocrine | ||
| Hypothyroidism | 14 | 0.5 |
| Infections | ||
| Upper respiratory tract infection | 13 | 0 |
| Nervous System | ||
| Headache | 11 | 0.5 |
| Peripheral neuropathyß | 10 | 0 |
Clinically relevant adverse reactions in <10% of patients who received KEYTRUDA included infusion reactions (9%), hyperthyroidism (3%), pneumonitis (3%), uveitis and myositis (1% each), and myelitis and myocarditis (0.5% each).
| Laboratory Abnormality* | KEYTRUDA 200 mg every 3 weeks |
|
|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) |
|
| Chemistry | ||
| Hypertransaminasemia‡ | 34 | 2 |
| Increased alkaline phosphatase | 17 | 0 |
| Increased creatinine | 15 | 0.5 |
| Hematology | ||
| Anemia | 30 | 6 |
| Thrombocytopenia | 27 | 4 |
| Neutropenia | 24 | 7 |
Hyperbilirubinemia occurred in less than 15% of patients on KEYNOTE-087 (10% all Grades, 2.4% Grade 3-4).
PMBCL
Among the 53 patients with PMBCL who received KEYTRUDA in KEYNOTE-170 [see Clinical Studies (14.5)], the median duration of exposure to KEYTRUDA was 3.5 months (range: 1 day to 22.8 months). Serious adverse reactions occurred in 26% of patients. Serious adverse reactions that occurred in >2% of patients included arrhythmia (4%), cardiac tamponade (2%), myocardial infarction (2%), pericardial effusion (2%), and pericarditis (2%). Six (11%) patients died within 30 days of start of treatment. Permanent discontinuation of KEYTRUDA due to an adverse reaction occurred in 8% of patients and dosage interruption due to an adverse reaction occurred in 15%. Twenty-five percent of patients had an adverse reaction requiring systemic corticosteroid therapy. Tables 22 and 23 summarize adverse reactions and laboratory abnormalities, respectively, in KEYNOTE-170.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks N=53 |
|
|---|---|---|
| All Grades*
(%) | Grades 3-4 (%) |
|
|
||
| Musculoskeletal and Connective Tissue | ||
| Musculoskeletal pain† | 30 | 0 |
| Infections | ||
| Upper respiratory tract infection‡ | 28 | 0 |
| General | ||
| Pyrexia | 28 | 0 |
| Fatigue§ | 23 | 2 |
| Respiratory, Thoracic and Mediastinal | ||
| Cough¶ | 26 | 2 |
| Dyspnea | 21 | 11 |
| Gastrointestinal | ||
| Diarrhea# | 13 | 2 |
| Abdominal pain Þ | 13 | 0 |
| Nausea | 11 | 0 |
| Cardiac | ||
| Arrhythmia ß | 11 | 4 |
| Nervous System | ||
| Headache | 11 | 0 |
Clinically relevant adverse reactions in <10% of patients who received KEYTRUDA included hypothyroidism (8%), hyperthyroidism and pericarditis (4% each), and thyroiditis, pericardial effusion, pneumonitis, arthritis and acute kidney injury (2% each).
| Laboratory Abnormality* | KEYTRUDA 200 mg every 3 weeks |
|
|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) |
|
| Hematology | ||
| Anemia | 47 | 0 |
| Leukopenia | 35 | 9 |
| Lymphopenia | 32 | 18 |
| Neutropenia | 30 | 11 |
| Chemistry | ||
| Hyperglycemia | 38 | 4 |
| Hypophosphatemia | 29 | 10 |
| Hypertransaminasemia‡ | 27 | 4 |
| Hypoglycemia | 19 | 0 |
| Increased alkaline phosphatase | 17 | 0 |
| Increased creatinine | 17 | 0 |
| Hypocalcemia | 15 | 4 |
| Hypokalemia | 15 | 4 |
Urothelial Cancer
Patients with urothelial cancer in combination with enfortumab vedotin
The safety of KEYTRUDA in combination with enfortumab vedotin was investigated in KEYNOTE-A39 in patients with locally advanced or metastatic urothelial cancer [see Clinical Studies (14.6)]. A total of 440 patients received KEYTRUDA 200 mg on Day 1 and enfortumab vedotin 1.25 mg/kg on Days 1 and 8 of each 21-day cycle compared to 433 patients who received gemcitabine on Days 1 and 8 and investigator’s choice of cisplatin or carboplatin on Day 1 of each 21-day cycle. Among patients who received KEYTRUDA and enfortumab vedotin, the median duration of exposure to KEYTRUDA was 8.5 months (range: 9 days to 28.5 months).
Fatal adverse reactions occurred in 3.9% of patients treated with KEYTRUDA in combination with enfortumab vedotin including acute respiratory failure (0.7%), pneumonia (0.5%), and pneumonitis/ILD (0.2%).
Serious adverse reactions occurred in 50% of patients receiving KEYTRUDA in combination with enfortumab vedotin. Serious adverse reactions in ≥2% of patients receiving KEYTRUDA in combination with enfortumab vedotin were rash (6%), acute kidney injury (5%), pneumonitis/ILD (4.5%), urinary tract infection (3.6%), diarrhea (3.2%), pneumonia (2.3%), pyrexia (2%), and hyperglycemia (2%).
Permanent discontinuation of KEYTRUDA occurred in 27% of patients. The most common adverse reactions (≥2%) resulting in permanent discontinuation of KEYTRUDA were pneumonitis/ILD (4.8%) and rash (3.4%).
Dose interruptions of KEYTRUDA occurred in 61% of patients. The most common adverse reactions (≥2%) resulting in interruption of KEYTRUDA were rash (17%), peripheral neuropathy (7%), COVID-19 (5%), diarrhea (4.3%), pneumonitis/ILD (3.6%), neutropenia (3.4%), fatigue (3%), alanine aminotransferase increased (2.7%), hyperglycemia (2.5%), pneumonia (2%), and pruritus (2%).
Tables 24 and 25 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in combination with enfortumab vedotin in KEYNOTE-A39.
| Adverse Reaction | KEYTRUDA in combination with Enfortumab Vedotin n=440 | Chemotherapy n=433 |
||
|---|---|---|---|---|
| All Grades*
% | Grades 3-4 % | All Grades*
% | Grades 3-4 % |
|
| Skin and subcutaneous tissue disorders | ||||
| Rash† | 68 | 15 | 15 | 0 |
| Pruritus | 41 | 1.1 | 7 | 0 |
| Alopecia | 35 | 0.5 | 8 | 0.2 |
| General disorders and administration site conditions | ||||
| Fatigue† | 51 | 6 | 57 | 7 |
| Nervous system disorders | ||||
| Peripheral neuropathy† | 67 | 8 | 14 | 0 |
| Dysgeusia | 21 | 0 | 9 | 0 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 33 | 1.8 | 26 | 1.8 |
| Gastrointestinal disorders | ||||
| Diarrhea | 38 | 4.5 | 16 | 1.4 |
| Nausea | 26 | 1.6 | 41 | 2.8 |
| Constipation | 26 | 0 | 34 | 0.7 |
| Investigations | ||||
| Weight loss | 33 | 3.6 | 9 | 0.2 |
| Eye disorders | ||||
| Dry eye† | 24 | 0 | 2.1 | 0 |
| Infections and infestations | ||||
| Urinary tract infection | 21 | 5 | 19 | 8 |
Clinically relevant adverse reactions (<20%) include pyrexia (18%), dry skin (17%), vomiting (12%), pneumonitis/ILD (10%), hypothyroidism (10%), blurred vision (6%), infusion site extravasation (2%), and myositis (0.5%).
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks and Enfortumab Vedotin | Chemotherapy | ||
|---|---|---|---|---|
| All Grades†
% | Grades 3-4 % | All Grades†
% | Grades 3-4 % |
|
| Chemistry | ||||
| Increased aspartate aminotransferase | 75 | 4.6 | 39 | 3.3 |
| Increased creatinine | 71 | 3.2 | 68 | 2.6 |
| Hyperglycemia | 66 | 14 | 54 | 4.7 |
| Increased alanine aminotransferase | 59 | 5 | 49 | 3.3 |
| Hyponatremia | 46 | 13 | 47 | 13 |
| Hypophosphatemia | 44 | 9 | 36 | 9 |
| Hypoalbuminemia | 39 | 1.8 | 35 | 0.5 |
| Hypokalemia | 26 | 5 | 16 | 3.1 |
| Hyperkalemia | 24 | 1.4 | 36 | 4.0 |
| Hypercalcemia | 21 | 1.2 | 14 | 0.2 |
| Hematology | ||||
| Lymphopenia | 58 | 15 | 59 | 17 |
| Anemia | 53 | 7 | 89 | 33 |
| Neutropenia | 30 | 9 | 80 | 50 |
Cisplatin-ineligible patients with urothelial cancer in combination with enfortumab vedotin
The safety of KEYTRUDA in combination with enfortumab vedotin was investigated in KEYNOTE-869 in patients with locally advanced or metastatic urothelial cancer and who are not eligible for cisplatin-based chemotherapy [see Clinical Studies (14.6)]. A total of 121 patients received KEYTRUDA 200 mg on Day 1, and enfortumab vedotin 1.25 mg/kg on days 1 and 8 of each 21-day cycle. The median duration of exposure to KEYTRUDA was 6.9 months (range 1 day to 29.6 months).
Fatal adverse reactions occurred in 5% of patients treated with KEYTRUDA in combination with enfortumab vedotin, including sepsis (1.6%), bullous dermatitis (0.8%), myasthenia gravis (0.8%), and pneumonitis (0.8%).
Serious adverse reactions occurred in 50% of patients receiving KEYTRUDA and enfortumab vedotin. Serious adverse reactions in ≥2% of patients receiving KEYTRUDA in combination with enfortumab vedotin were acute kidney injury (7%), urinary tract infection (7%), urosepsis (5%), hematuria (3.3%), pneumonia (3.3%), pneumonitis (3.3%), sepsis (3.3%), anemia (2.5%), diarrhea (2.5%), hypotension (2.5%), myasthenia gravis (2.5%), myositis (2.5%), and urinary retention (2.5%).
Permanent discontinuation of KEYTRUDA occurred in 32% of patients. The most common adverse reactions (≥2%) resulting in permanent discontinuation of KEYTRUDA were pneumonitis (5%), peripheral neuropathy (5%), rash (3.3%), and myasthenia gravis (2.5%).
Dose interruptions of KEYTRUDA occurred in 69% of patients. The most common adverse reactions (≥2%) resulting in interruption of KEYTRUDA were peripheral neuropathy (22%), rash (17%), neutropenia (7%), fatigue (6%), diarrhea (5%), lipase increased (5%), acute kidney injury (3.3%), ALT increased (2.5%), and COVID-19 (2.5%).
Tables 26 and 27 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in combination with enfortumab vedotin in KEYNOTE-869.
| Adverse Reaction | KEYTRUDA in combination with Enfortumab Vedotin n=121 |
|
|---|---|---|
| All Grades*
% | Grade 3-4 % |
|
|
||
| Skin and subcutaneous tissue disorders | ||
| Rash† | 71 | 21 |
| Alopecia | 52 | 0 |
| Pruritus | 40 | 3.3 |
| Dry skin | 21 | 0.8 |
| Nervous system disorders | ||
| Peripheral neuropathy‡ | 65 | 3.3 |
| Dysgeusia | 35 | 0 |
| Dizziness | 23 | 0 |
| General disorders and administration site conditions | ||
| Fatigue | 60 | 11 |
| Peripheral edema | 26 | 0 |
| Investigations | ||
| Weight loss | 48 | 5 |
| Gastrointestinal disorders | ||
| Diarrhea | 45 | 7 |
| Nausea | 36 | 0.8 |
| Constipation | 27 | 0 |
| Metabolism and nutrition disorders | ||
| Decreased appetite | 38 | 0.8 |
| Infections and infestations | ||
| Urinary tract infection | 30 | 12 |
| Eye disorders | ||
| Dry eye | 25 | 0 |
| Musculoskeletal and connective tissue disorders | ||
| Arthralgia | 23 | 1.7 |
Clinically relevant adverse reactions (<20%) include vomiting (19.8%), fever (18%), hypothyroidism (11%), pneumonitis/ILD (10%), myositis (3.3%), myasthenia gravis (2.5%), and infusion site extravasation (0.8%).
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks and Enfortumab Vedotin |
|
|---|---|---|
| All Grades†
% | Grades 3-4 % |
|
| Chemistry | ||
| Hyperglycemia | 74 | 13 |
| Increased aspartate aminotransferase | 73 | 9 |
| Increased creatinine | 69 | 3.3 |
| Hyponatremia | 60 | 19 |
| Increased alanine aminotransferase | 60 | 7 |
| Increased lipase | 59 | 32 |
| Hypoalbuminemia | 59 | 4.2 |
| Hypophosphatemia | 51 | 15 |
| Hypokalemia | 35 | 8 |
| Increased potassium | 27 | 1.7 |
| Increased calcium | 27 | 4.2 |
| Hematology | ||
| Anemia | 69 | 15 |
| Lymphopenia | 64 | 17 |
| Neutropenia | 32 | 12 |
Platinum-Ineligible Patients with Urothelial Carcinoma
The safety of KEYTRUDA was investigated in KEYNOTE-052, a single-arm trial that enrolled 370 patients with locally advanced or metastatic urothelial carcinoma who had one or more comorbidities. Patients with autoimmune disease or medical conditions that required systemic corticosteroids or other immunosuppressive medications were ineligible [see Clinical Studies (14.6)]. Patients received KEYTRUDA 200 mg every 3 weeks until unacceptable toxicity or either radiographic or clinical disease progression.
The median duration of exposure to KEYTRUDA was 2.8 months (range: 1 day to 15.8 months).
KEYTRUDA was discontinued due to adverse reactions in 11% of patients. Eighteen patients (5%) died from causes other than disease progression. Five patients (1.4%) who were treated with KEYTRUDA experienced sepsis which led to death, and three patients (0.8%) experienced pneumonia which led to death. Adverse reactions leading to interruption of KEYTRUDA occurred in 22% of patients; the most common (≥1%) were liver enzyme increase, diarrhea, urinary tract infection, acute kidney injury, fatigue, joint pain, and pneumonia. Serious adverse reactions occurred in 42% of patients. The most frequent serious adverse reactions (≥2%) were urinary tract infection, hematuria, acute kidney injury, pneumonia, and urosepsis.
Immune-related adverse reactions that required systemic glucocorticoids occurred in 8% of patients, use of hormonal supplementation due to an immune-related adverse reaction occurred in 8% of patients, and 5% of patients required at least one steroid dose ≥40 mg oral prednisone equivalent.
Table 28 summarizes adverse reactions in patients on KEYTRUDA in KEYNOTE-052.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks N=370 |
|
|---|---|---|
| All Grades*
(%) | Grades 3–4 (%) |
|
|
||
| General | ||
| Fatigue† | 38 | 6 |
| Pyrexia | 11 | 0.5 |
| Weight loss | 10 | 0 |
| Musculoskeletal and Connective Tissue | ||
| Musculoskeletal pain‡ | 24 | 4.9 |
| Arthralgia | 10 | 1.1 |
| Metabolism and Nutrition | ||
| Decreased appetite | 22 | 1.6 |
| Hyponatremia | 10 | 4.1 |
| Gastrointestinal | ||
| Constipation | 21 | 1.1 |
| Diarrhea§ | 20 | 2.4 |
| Nausea | 18 | 1.1 |
| Abdominal pain¶ | 18 | 2.7 |
| Elevated LFTs# | 13 | 3.5 |
| Vomiting | 12 | 0 |
| Skin and Subcutaneous Tissue | ||
| RashÞ | 21 | 0.5 |
| Pruritus | 19 | 0.3 |
| Edema peripheralß | 14 | 1.1 |
| Infections | ||
| Urinary tract infection | 19 | 9 |
| Blood and Lymphatic System | ||
| Anemia | 17 | 7 |
| Respiratory, Thoracic, and Mediastinal | ||
| Cough | 14 | 0 |
| Dyspnea | 11 | 0.5 |
| Renal and Urinary | ||
| Increased blood creatinine | 11 | 1.1 |
| Hematuria | 13 | 3.0 |
Previously Treated Urothelial Carcinoma
The safety of KEYTRUDA for the treatment of patients with locally advanced or metastatic urothelial carcinoma with disease progression following platinum-containing chemotherapy was investigated in KEYNOTE-045. KEYNOTE-045 was a multicenter, open-label, randomized (1:1), active-controlled trial in which 266 patients received KEYTRUDA 200 mg every 3 weeks or investigator's choice of chemotherapy (n=255), consisting of paclitaxel (n=84), docetaxel (n=84) or vinflunine (n=87) [see Clinical Studies (14.6)]. Patients with autoimmune disease or a medical condition that required systemic corticosteroids or other immunosuppressive medications were ineligible.
The median duration of exposure was 3.5 months (range: 1 day to 20 months) in patients who received KEYTRUDA and 1.5 months (range: 1 day to 14 months) in patients who received chemotherapy.
KEYTRUDA was discontinued due to adverse reactions in 8% of patients. The most common adverse reaction resulting in permanent discontinuation of KEYTRUDA was pneumonitis (1.9%). Adverse reactions leading to interruption of KEYTRUDA occurred in 20% of patients; the most common (≥1%) were urinary tract infection (1.5%), diarrhea (1.5%), and colitis (1.1%). Serious adverse reactions occurred in 39% of KEYTRUDA-treated patients. The most frequent serious adverse reactions (≥2%) in KEYTRUDA-treated patients were urinary tract infection, pneumonia, anemia, and pneumonitis. Tables 29 and 30 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-045.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks | Chemotherapy* | ||
|---|---|---|---|---|
| n=266 | n=255 | |||
| All Grades†
(%) | Grades 3-4 (%) | All Grades†
(%) | Grades 3-4 (%) |
|
|
||||
| General | ||||
| Fatigue‡ | 38 | 4.5 | 56 | 11 |
| Pyrexia | 14 | 0.8 | 13 | 1.2 |
| Musculoskeletal and Connective Tissue | ||||
| Musculoskeletal pain§ | 32 | 3.0 | 27 | 2.0 |
| Skin and Subcutaneous Tissue | ||||
| Pruritus | 23 | 0 | 6 | 0.4 |
| Rash¶ | 20 | 0.4 | 13 | 0.4 |
| Gastrointestinal | ||||
| Nausea | 21 | 1.1 | 29 | 1.6 |
| Constipation | 19 | 1.1 | 32 | 3.1 |
| Diarrhea# | 18 | 2.3 | 19 | 1.6 |
| Vomiting | 15 | 0.4 | 13 | 0.4 |
| Abdominal pain | 13 | 1.1 | 13 | 2.7 |
| Infections | ||||
| Urinary tract infection | 15 | 4.9 | 14 | 4.3 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 21 | 3.8 | 21 | 1.2 |
| Respiratory, Thoracic and Mediastinal | ||||
| CoughÞ | 15 | 0.4 | 9 | 0 |
| Dyspneaß | 14 | 1.9 | 12 | 1.2 |
| Renal and Urinary | ||||
| Hematuria à | 12 | 2.3 | 8 | 1.6 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks | Chemotherapy | ||
|---|---|---|---|---|
| All Grades†
% | Grades 3-4 % | All Grades†
% | Grades 3-4 % |
|
|
||||
| Chemistry | ||||
| Hyperglycemia | 52 | 8 | 60 | 7 |
| Anemia | 52 | 13 | 68 | 18 |
| Lymphopenia | 45 | 15 | 53 | 25 |
| Hypoalbuminemia | 43 | 1.7 | 50 | 3.8 |
| Hyponatremia | 37 | 9 | 47 | 13 |
| Increased alkaline phosphatase | 37 | 7 | 33 | 4.9 |
| Increased creatinine | 35 | 4.4 | 28 | 2.9 |
| Hypophosphatemia | 29 | 8 | 34 | 14 |
| Increased AST | 28 | 4.1 | 20 | 2.5 |
| Hyperkalemia | 28 | 0.8 | 27 | 6 |
| Hypocalcemia | 26 | 1.6 | 34 | 2.1 |
BCG-unresponsive High-risk NMIBC
The safety of KEYTRUDA was investigated in KEYNOTE-057, a multicenter, open-label, single-arm trial that enrolled 148 patients with high-risk non-muscle invasive bladder cancer (NMIBC), 96 of whom had BCG-unresponsive carcinoma in situ (CIS) with or without papillary tumors. Patients received KEYTRUDA 200 mg every 3 weeks until unacceptable toxicity, persistent or recurrent high-risk NMIBC or progressive disease, or up to 24 months of therapy without disease progression.
The median duration of exposure to KEYTRUDA was 4.3 months (range: 1 day to 25.6 months).
KEYTRUDA was discontinued due to adverse reactions in 11% of patients. The most common adverse (>1%) reaction resulting in permanent discontinuation of KEYTRUDA was pneumonitis (1.4%). Adverse reactions leading to interruption of KEYTRUDA occurred in 22% of patients; the most common (≥2%) were diarrhea (4%) and urinary tract infection (2%). Serious adverse reactions occurred in 28% of KEYTRUDA-treated patients. The most frequent serious adverse reactions (≥2%) in KEYTRUDA-treated patients were pneumonia (3%), cardiac ischemia (2%), colitis (2%), pulmonary embolism (2%), sepsis (2%), and urinary tract infection (2%). Tables 31 and 32 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-057.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks N=148 |
|
|---|---|---|
| All Grades*
(%) | Grades 3–4 (%) |
|
|
||
| General | ||
| Fatigue† | 29 | 0.7 |
| Peripheral edema‡ | 11 | 0 |
| Gastrointestinal | ||
| Diarrhea§ | 24 | 2.0 |
| Nausea | 13 | 0 |
| Constipation | 12 | 0 |
| Skin and Subcutaneous Tissue | ||
| Rash¶ | 24 | 0.7 |
| Pruritus | 19 | 0.7 |
| Musculoskeletal and Connective Tissue | ||
| Musculoskeletal pain# | 19 | 0 |
| Arthralgia | 14 | 1.4 |
| Renal and Urinary | ||
| Hematuria | 19 | 1.4 |
| Respiratory, Thoracic, and Mediastinal | ||
| CoughÞ | 19 | 0 |
| Infections | ||
| Urinary tract infection | 12 | 2.0 |
| Nasopharyngitis | 10 | 0 |
| Endocrine | ||
| Hypothyroidism | 11 | 0 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks |
|
|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) |
|
| Chemistry | ||
| Hyperglycemia | 59 | 8 |
| Increased ALT | 25 | 3.4 |
| Hyponatremia | 24 | 7 |
| Hypophosphatemia | 24 | 6 |
| Hypoalbuminemia | 24 | 2.1 |
| Hyperkalemia | 23 | 1.4 |
| Hypocalcemia | 22 | 0.7 |
| Increased AST | 20 | 3.4 |
| Increased creatinine | 20 | 0.7 |
| Hematology | ||
| Anemia | 35 | 1.4 |
| Lymphopenia | 29 | 1.6 |
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
The safety of KEYTRUDA was investigated in 504 patients with MSI-H or dMMR cancer enrolled in KEYNOTE-158, KEYNOTE-164, and KEYNOTE-051 [see Clinical Studies (14.7)]. The median duration of exposure to KEYTRUDA was 6.2 months (range: 1 day to 53.5 months). Adverse reactions occurring in patients with MSI-H or dMMR cancer were similar to those occurring in patients with other solid tumors who received KEYTRUDA as a single agent.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
Among the 153 patients with MSI-H or dMMR CRC enrolled in KEYNOTE-177 [see Clinical Studies (14.8)] treated with KEYTRUDA, the median duration of exposure to KEYTRUDA was 11.1 months (range: 1 day to 30.6 months). Patients with autoimmune disease or a medical condition that required immunosuppression were ineligible. Adverse reactions occurring in patients with MSI-H or dMMR CRC were similar to those occurring in 2799 patients with melanoma or NSCLC treated with KEYTRUDA as a single agent.
Gastric Cancer
First-line Treatment of Locally Advanced Unresectable or Metastatic HER2-Positive Gastric or Gastroesophageal Junction Adenocarcinoma
The safety of KEYTRUDA was evaluated in 433 patients with HER2-positive gastric or GEJ cancer enrolled in KEYNOTE-811, which included 217 patients treated with KEYTRUDA 200 mg, trastuzumab, and CAPOX (n=189) or FP (n=28) every 3 weeks, compared to 216 patients treated with placebo, trastuzumab, and CAPOX (n=187) or FP (n=29) every 3 weeks [see Clinical Studies (14.9)].
The median duration of exposure to KEYTRUDA was 5.8 months (range: 1 day to 17.7 months).
The study population characteristics were: median age of 63 years (range: 19 to 84), 43% age 65 or older; 81% male; 58% White, 35% Asian, and 0.9% Black; 44% ECOG PS of 0 and 56% ECOG PS of 1.
KEYTRUDA and placebo were discontinued due to adverse reactions in 6% of patients in each arm. The most common adverse reaction resulting in permanent discontinuation of KEYTRUDA was pneumonitis (1.4%). Adverse reactions leading to interruption of KEYTRUDA occurred in 58% of patients; the most common adverse reactions or laboratory abnormalities leading to interruption of KEYTRUDA (≥2%) were neutropenia (18%), thrombocytopenia (12%), diarrhea (6%), anemia (3.7%), hypokalemia (3.7%), fatigue/asthenia (3.2%), decreased appetite (3.2%), increased AST (2.8%), increased blood bilirubin (2.8%), pneumonia (2.8%), increased ALT (2.3%), and vomiting (2.3%).
In the KEYTRUDA arm versus placebo, there was a difference of ≥5% incidence between patients treated with KEYTRUDA versus standard of care for diarrhea (53% vs 44%) and nausea (49% vs 44%). There were no clinically meaningful differences in incidence of Grade 3-4 toxicity between arms.
There was a difference of ≥5% incidence between patients treated with KEYTRUDA versus standard of care for increased ALT (34% vs 29%) and increased creatinine (20% vs 10%). There were no clinically meaningful differences in incidence of Grade 3-4 toxicity between arms.
First-line Treatment of Locally Advanced Unresectable or Metastatic HER2-Negative Gastric or Gastroesophageal Junction Adenocarcinoma
The safety of KEYTRUDA was evaluated in 1572 patients with HER2-negative gastric or GEJ cancer enrolled in KEYNOTE-859, which included 785 patients treated with KEYTRUDA 200 mg and FP (n=106) or CAPOX (n=674) every 3 weeks, compared to 787 patients who received placebo and FP (n=107) or CAPOX (n=679) every 3 weeks [see Clinical Studies (14.9)].
The median duration of exposure to KEYTRUDA was 6.2 months (range: 1 day to 33.7 months).
Serious adverse reactions occurred in 45% of patients receiving KEYTRUDA. Serious adverse reactions in >2% of patients included pneumonia (4.1%), diarrhea (3.9%), hemorrhage (3.9%), and vomiting (2.4%). Fatal adverse reactions occurred in 8% of patients who received KEYTRUDA, including infection (2.3%) and thromboembolism (1.3%).
Permanent discontinuation of KEYTRUDA due to adverse reactions occurred in 15% of patients. Adverse reaction resulting in permanent discontinuation of KEYTRUDA in ≥1% were infections (1.8%) and diarrhea (1.0%).
Dosage interruptions of KEYTRUDA due to an adverse reaction occurred in 65% of patients. Adverse reactions or laboratory abnormalities leading to interruption of KEYTRUDA (≥2%) were neutropenia (21%), thrombocytopenia (13%), diarrhea (5.5%), fatigue (4.8%), infection (4.8%), anemia (4.5%), increased AST (4.3%), increased ALT (3.8%), increased blood bilirubin (3.3%), white blood cell count decreased (2.2%), nausea (2%), palmar-plantar erythrodysesthesia syndrome (2%), and vomiting (2%).
Tables 33 and 34 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-859.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks and FP or CAPOX n=785 | Placebo and FP or CAPOX n=787 |
||
|---|---|---|---|---|
| All Grades*
(%) | Grades 3-4
(%) | All Grades*
(%) | Grades 3-4
(%) |
|
|
||||
| Nervous System | ||||
| Peripheral neuropathy† | 47 | 5 | 48 | 6 |
| Gastrointestinal | ||||
| Nausea | 46 | 3.7 | 46 | 4.4 |
| Diarrhea | 36 | 6 | 32 | 5 |
| Vomiting | 34 | 5 | 27 | 5 |
| Abdominal Pain‡ | 26 | 2.8 | 24 | 2.9 |
| Constipation | 22 | 0.5 | 21 | 0.8 |
| General | ||||
| Fatigue§ | 40 | 8 | 39 | 9 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 29 | 3.3 | 29 | 2.5 |
| Skin and Subcutaneous Tissue | ||||
| Palmar-plantar erythrodysesthesia syndrome | 25 | 3.1 | 22 | 1.8 |
| Investigations | ||||
| Weight loss | 20 | 2.8 | 19 | 2.7 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks and FP or CAPOX | Placebo and FP or CAPOX |
||
|---|---|---|---|---|
| All Grades†
% | Grades 3-4 % | All Grades†
% | Grades 3-4 % |
|
| Hematology | ||||
| Anemia | 65 | 15 | 69 | 13 |
| Thrombocytopenia | 64 | 12 | 62 | 10 |
| Neutropenia | 63 | 25 | 58 | 20 |
| Leukopenia | 59 | 7 | 56 | 6 |
| Lymphopenia | 57 | 20 | 51 | 16 |
| Chemistry | ||||
| Increased AST | 57 | 4.7 | 48 | 3.6 |
| Hypoalbuminemia | 55 | 4.1 | 52 | 2.9 |
| Hyperglycemia | 53 | 6 | 52 | 4.6 |
| Hypocalcemia | 49 | 3.6 | 45 | 3.3 |
| Increased alkaline phosphatase | 48 | 6 | 41 | 5 |
| Hyponatremia | 40 | 13 | 40 | 12 |
| Increased ALT | 40 | 4.2 | 29 | 2.9 |
| Hypokalemia | 35 | 10 | 27 | 9 |
| Bilirubin increased | 32 | 5 | 30 | 5 |
| Hypophosphatemia | 30 | 10 | 27 | 8 |
| Hypomagnesemia | 29 | 0.3 | 22 | 0.7 |
| Increased creatinine | 21 | 3.5 | 18 | 1.7 |
| Hyperkalemia | 20 | 3.7 | 18 | 2.9 |
| Increased INR | 20 | 1.4 | 22 | 0 |
Esophageal Cancer
First-line Treatment of Locally Advanced Unresectable or Metastatic Esophageal Cancer/Gastroesophageal Junction
The safety of KEYTRUDA, in combination with cisplatin and FU chemotherapy was investigated in KEYNOTE-590, a multicenter, double-blind, randomized (1:1), placebo-controlled trial for the first-line treatment in patients with metastatic or locally advanced esophageal or gastroesophageal junction (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma who were not candidates for surgical resection or definitive chemoradiation [see Clinical Studies (14.10)]. A total of 740 patients received either KEYTRUDA 200 mg (n=370) or placebo (n=370) every 3 weeks for up to 35 cycles, both in combination with up to 6 cycles of cisplatin and up to 35 cycles of FU.
The median duration of exposure was 5.7 months (range: 1 day to 26 months) in the KEYTRUDA combination arm and 5.1 months (range: 3 days to 27 months) in the chemotherapy arm.
KEYTRUDA was discontinued for adverse reactions in 15% of patients. The most common adverse reactions resulting in permanent discontinuation of KEYTRUDA (≥1%) were pneumonitis (1.6%), acute kidney injury (1.1%), and pneumonia (1.1%). Adverse reactions leading to interruption of KEYTRUDA occurred in 67% of patients. The most common adverse reactions leading to interruption of KEYTRUDA (≥2%) were neutropenia (19%), fatigue/asthenia (8%), decreased white blood cell count (5%), pneumonia (5%), decreased appetite (4.3%), anemia (3.2%), increased blood creatinine (3.2%), stomatitis (3.2%), malaise (3.0%), thrombocytopenia (3%), pneumonitis (2.7%), diarrhea (2.4%), dysphagia (2.2%), and nausea (2.2%).
Tables 35 and 36 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-590.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks Cisplatin FU n=370 | Placebo Cisplatin FU n=370 |
||
|---|---|---|---|---|
| All Grades*
(%) | Grades 3-4†
(%) | All Grades*
(%) | Grades 3-4†
(%) |
|
| Gastrointestinal | ||||
| Nausea | 67 | 7 | 63 | 7 |
| Constipation | 40 | 0 | 40 | 0 |
| Diarrhea | 36 | 4.1 | 33 | 3 |
| Vomiting | 34 | 7 | 32 | 5 |
| Stomatitis | 27 | 6 | 26 | 3.8 |
| General | ||||
| Fatigue‡ | 57 | 12 | 46 | 9 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 44 | 4.1 | 38 | 5 |
| Investigations | ||||
| Weight loss | 24 | 3.0 | 24 | 5 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks Cisplatin FU | Chemotherapy (Cisplatin and FU) |
||
|---|---|---|---|---|
| All Grades†
% | Grades 3-4 % | All Grades†
% | Grades 3-4 % |
|
| Hematology | ||||
| Anemia | 83 | 21 | 86 | 24 |
| Neutropenia | 74 | 43 | 71 | 41 |
| Leukopenia | 72 | 21 | 73 | 17 |
| Lymphopenia | 55 | 22 | 53 | 18 |
| Thrombocytopenia | 43 | 5 | 46 | 8 |
| Chemistry | ||||
| Hyperglycemia | 56 | 7 | 55 | 6 |
| Hyponatremia | 53 | 19 | 54 | 19 |
| Hypoalbuminemia | 52 | 2.8 | 52 | 2.3 |
| Increased creatinine | 45 | 2.5 | 42 | 2.5 |
| Hypocalcemia | 44 | 3.9 | 38 | 2 |
| Hypophosphatemia | 37 | 9 | 31 | 10 |
| Hypokalemia | 30 | 12 | 34 | 15 |
| Increased alkaline phosphatase | 29 | 1.9 | 29 | 1.7 |
| Hyperkalemia | 28 | 3.6 | 27 | 2.6 |
| Increased AST | 25 | 4.4 | 22 | 2.8 |
| Increased ALT | 23 | 3.6 | 18 | 1.7 |
Previously Treated Recurrent Locally Advanced or Metastatic Esophageal Cancer
Among the 314 patients with esophageal cancer enrolled in KEYNOTE-181 [see Clinical Studies (14.10)] treated with KEYTRUDA, the median duration of exposure to KEYTRUDA was 2.1 months (range: 1 day to 24.4 months). Patients with autoimmune disease or a medical condition that required immunosuppression were ineligible. Adverse reactions occurring in patients with esophageal cancer were similar to those occurring in 2799 patients with melanoma or NSCLC treated with KEYTRUDA as a single agent.
Cervical Cancer
FIGO 2014 Stage III-IVA Cervical Cancer with Chemoradiotherapy
The safety of KEYTRUDA in combination with CRT (cisplatin plus external beam radiation therapy [EBRT] followed by brachytherapy [BT]) was investigated in KEYNOTE-A18, a placebo-controlled, randomized (1:1), multicenter, double-blind trial including 594 patients with FIGO 2014 Stage III-IVA cervical cancer [see Clinical Studies (14.11)]. Two hundred ninety-two patients received KEYTRUDA in combination with chemoradiotherapy and 302 patients received placebo in combination with chemoradiotherapy.
The median duration of exposure to KEYTRUDA was 12.1 months (range: 1 day to 27 months).
Fatal adverse reactions occurred in 1.4% of patients receiving KEYTRUDA in combination with chemoradiotherapy, including 1 case each (0.3%) of large intestinal perforation, urosepsis, sepsis, and vaginal hemorrhage.
Serious adverse reactions occurred in 30% of patients receiving KEYTRUDA in combination with chemoradiotherapy. Serious adverse reactions occurring in ≥1% of patients included urinary tract infection (2.7%), urosepsis (1.4%), and sepsis (1%).
KEYTRUDA was discontinued for adverse reactions in 7% of patients. The most common adverse reaction (≥1%) resulting in permanent discontinuation was diarrhea (1%).
Adverse reactions leading to interruption of KEYTRUDA occurred in 43% of patients; the most common adverse reactions leading to interruption of KEYTRUDA (≥2%) were anemia (8%), COVID-19 (6%), SARS-CoV-2 test positive (3.1%), decreased neutrophil count (2.7%), diarrhea (2.7%), urinary tract infection (2.7%), and increased ALT (2.4%).
Table 37 and Table 38 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-A18.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks and 400 mg every 6 weeks with chemoradiotherapy n=292 | Placebo with chemoradiotherapy n=302 |
||
|---|---|---|---|---|
| All Grades*
(%) | Grades 3-4 (%) | All Grades*
(%) | Grades 3-4 (%) |
|
|
||||
| Gastrointestinal | ||||
| Nausea | 56 | 0 | 61 | 2.3 |
| Diarrhea | 50 | 3.8 | 50 | 4.3 |
| Vomiting | 33 | 1 | 34 | 1.7 |
| Constipation | 18 | 0 | 18 | 0.7 |
| Abdominal pain | 12 | 0.7 | 12 | 1.7 |
| Infections | ||||
| Urinary tract infection† | 32 | 4.1 | 31 | 4.6 |
| General | ||||
| Fatigue‡ | 26 | 1 | 27 | 1.3 |
| Pyrexia | 12 | 0.3 | 13 | 0 |
| Endocrine | ||||
| Hypothyroidism§ | 20 | 0.7 | 5 | 0 |
| Hyperthyroidism | 11 | 0.3 | 2.6 | 0 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 17 | 0.7 | 17 | 0.3 |
| Investigations | ||||
| Weight loss | 17 | 1.4 | 18 | 1 |
| Renal and Urinary | ||||
| Dysuria | 11 | 0.3 | 12 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Rash¶ | 11 | 0.7 | 7 | 0.3 |
| Reproductive System | ||||
| Pelvic pain | 10 | 1 | 13 | 1.3 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks and 400 mg every 6 weeks with chemoradiotherapy | Placebo with chemoradiotherapy |
||
|---|---|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) | All Grades†
(%) | Grades 3-4 (%) |
|
|
||||
| Hematology | ||||
| Lymphopenia | 99 | 96 | 99 | 92 |
| Leukopenia | 96 | 46 | 94 | 49 |
| Anemia | 88 | 31 | 81 | 25 |
| Neutropenia | 75 | 32 | 74 | 33 |
| Thrombocytopenia | 65 | 8 | 61 | 6 |
| Chemistry | ||||
| Hypomagnesemia | 59 | 4.2 | 63 | 3.4 |
| Hyponatremia | 54 | 3.8 | 47 | 4 |
| Increased AST | 45 | 1 | 39 | 1.7 |
| Increased ALT | 44 | 2.1 | 44 | 1 |
| Hypocalcemia | 43 | 4.8 | 40 | 4.3 |
| Hypokalemia | 42 | 14 | 38 | 10 |
| Increased creatinine | 41 | 6 | 43 | 6 |
| Hypoalbuminemia | 37 | 0.7 | 35 | 1.7 |
| Increased alkaline phosphatase | 34 | 0.3 | 33 | 0.3 |
Persistent, Recurrent, or Metastatic Cervical Cancer
The safety of KEYTRUDA in combination with paclitaxel and cisplatin or paclitaxel and carboplatin, with or without bevacizumab, was investigated in KEYNOTE-826, a multicenter, double-blind, randomized (1:1), placebo-controlled trial in patients with persistent, recurrent, or first-line metastatic cervical cancer who had not been treated with chemotherapy except when used concurrently as a radio-sensitizing agent [see Clinical Studies (14.11)]. A total of 616 patients, regardless of tumor PD-L1 expression, received KEYTRUDA 200 mg and chemotherapy with or without bevacizumab (n=307) every 3 weeks or placebo and chemotherapy with or without bevacizumab (n=309) every 3 weeks.
The median duration of exposure to KEYTRUDA was 9.9 months (range: 1 day to 26 months).
Fatal adverse reactions occurred in 4.6% of patients receiving KEYTRUDA in combination with chemotherapy with or without bevacizumab, including 3 cases of hemorrhage, 2 cases of sepsis, 2 cases due to unknown causes, and 1 case each of acute myocardial infarction, autoimmune encephalitis, cardiac arrest, cerebrovascular accident, femur fracture with perioperative pulmonary embolus, intestinal perforation, and pelvic infection.
Serious adverse reactions occurred in 50% of patients receiving KEYTRUDA in combination with chemotherapy with or without bevacizumab. Serious adverse reactions in ≥3% of patients included febrile neutropenia (6.8%), urinary tract infection (5.2%), anemia (4.6%), acute kidney injury (3.3%), and sepsis (3.3%).
KEYTRUDA was discontinued for adverse reactions in 15% of patients. The most common adverse reaction resulting in permanent discontinuation of KEYTRUDA (≥1%) was colitis (1%).
Adverse reactions leading to interruption of KEYTRUDA occurred in 66% of patients; the most common adverse reactions or laboratory abnormalities leading to interruption of KEYTRUDA (≥2%) were thrombocytopenia (15%), neutropenia (14%), anemia (11%), increased ALT (6%), leukopenia (5%), fatigue/asthenia (4.2%), urinary tract infection (3.6%), increased AST (3.3%), pyrexia (3.3%), diarrhea (2.6%), acute kidney injury (2.6%), increased blood creatinine (2.6%), colitis (2.3%), decreased appetite (2%), and cough (2%).
For patients treated with KEYTRUDA, chemotherapy, and bevacizumab (n=196), the most common (≥20%) adverse reactions were peripheral neuropathy (62%), alopecia (58%), anemia (55%), fatigue/asthenia (53%), nausea (41%), neutropenia (41%), diarrhea (39%), hypertension (35%), thrombocytopenia (35%), constipation (31%), arthralgia (31%), vomiting (30%), urinary tract infection (27%), rash (26%), leukopenia (24%), hypothyroidism (22%), and decreased appetite (21%).
Table 39 and Table 40 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-826.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks and chemotherapy* with or without bevacizumab n=307 | Placebo and chemotherapy* with or without bevacizumab n=309 |
||
|---|---|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) | All Grades†
(%) | Grades 3-4 (%) |
|
|
||||
| Nervous System | ||||
| Peripheral neuropathy‡ | 58 | 4.2 | 57 | 6 |
| Skin and Subcutaneous Tissue | ||||
| Alopecia | 56 | 0 | 58 | 0 |
| Rash§ | 22 | 3.6 | 15 | 0.3 |
| General | ||||
| Fatigue¶ | 47 | 7 | 46 | 6 |
| Gastrointestinal | ||||
| Nausea | 40 | 2 | 44 | 1.6 |
| Diarrhea | 36 | 2 | 30 | 2.6 |
| Constipation | 28 | 0.3 | 33 | 1 |
| Vomiting | 26 | 2.6 | 27 | 1.9 |
| Musculoskeletal and Connective Tissue | ||||
| Arthralgia | 27 | 0.7 | 26 | 1.3 |
| Vascular | ||||
| Hypertension | 24 | 9 | 23 | 11 |
| Infections | ||||
| Urinary tract infection | 24 | 9 | 26 | 8 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks and chemotherapy† with or without bevacizumab n=307 | Placebo and chemotherapy† with or without bevacizumab n=309 |
||
|---|---|---|---|---|
| All Grades‡
(%) | Grades 3-4 (%) | All Grades‡
(%) | Grades 3-4 (%) |
|
|
||||
| Hematology | ||||
| Anemia | 80 | 35 | 77 | 33 |
| Leukopenia | 76 | 27 | 69 | 19 |
| Neutropenia | 66 | 39 | 58 | 31 |
| Lymphopenia | 61 | 33 | 56 | 33 |
| Thrombocytopenia | 57 | 19 | 53 | 15 |
| Chemistry | ||||
| Hyperglycemia | 51 | 4.7 | 46 | 2.3 |
| Hypoalbuminemia | 46 | 1.3 | 38 | 5 |
| Hyponatremia | 40 | 14 | 38 | 11 |
| Increased ALT | 40 | 7 | 38 | 6 |
| Increased AST | 40 | 6 | 36 | 3.0 |
| Increased alkaline phosphatase | 38 | 3.4 | 40 | 2.3 |
| Hypocalcemia | 37 | 4.0 | 31 | 5 |
| Increased creatinine | 34 | 5 | 32 | 6 |
| Hypokalemia | 29 | 7 | 26 | 7 |
| Hyperkalemia | 23 | 3.7 | 27 | 4.7 |
| Hypercalcemia | 21 | 1.0 | 20 | 1.3 |
Previously Treated Recurrent or Metastatic Cervical Cancer
Among the 98 patients with cervical cancer enrolled in Cohort E of KEYNOTE-158 [see Clinical Studies (14.11)], the median duration of exposure to KEYTRUDA was 2.9 months (range: 1 day to 22.1 months). Patients with autoimmune disease or a medical condition that required immunosuppression were ineligible.
KEYTRUDA was discontinued due to adverse reactions in 8% of patients. Serious adverse reactions occurred in 39% of patients receiving KEYTRUDA. The most frequent serious adverse reactions reported included anemia (7%), fistula (4.1%), hemorrhage (4.1%), and infections [except UTIs] (4.1%). Tables 41 and 42 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-158.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks N=98 |
|
|---|---|---|
| All Grades*
(%) | Grades 3–4 (%) |
|
|
||
| General | ||
| Fatigue† | 43 | 5 |
| Pain‡ | 22 | 2.0 |
| Pyrexia | 19 | 1.0 |
| Edema peripheral§ | 15 | 2.0 |
| Musculoskeletal and Connective Tissue | ||
| Musculoskeletal pain¶ | 27 | 5 |
| Gastrointestinal | ||
| Diarrhea# | 23 | 2.0 |
| Abdominal painÞ | 22 | 3.1 |
| Nausea | 19 | 0 |
| Vomiting | 19 | 1.0 |
| Constipation | 14 | 0 |
| Metabolism and Nutrition | ||
| Decreased appetite | 21 | 0 |
| Vascular | ||
| Hemorrhageß | 19 | 5 |
| Infections | ||
| UTIà | 18 | 6 |
| Infection (except UTI)è | 16 | 4.1 |
| Skin and Subcutaneous Tissue | ||
| Rashð | 17 | 2.0 |
| Endocrine | ||
| Hypothyroidism | 11 | 0 |
| Nervous System | ||
| Headache | 11 | 2.0 |
| Respiratory, Thoracic and Mediastinal | ||
| Dyspnea | 10 | 1.0 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks |
|
|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) |
|
| Hematology | ||
| Anemia | 54 | 24 |
| Lymphopenia | 47 | 9 |
| Chemistry | ||
| Hypoalbuminemia | 44 | 5 |
| Increased alkaline phosphatase | 42 | 2.6 |
| Hyponatremia | 38 | 13 |
| Hyperglycemia | 38 | 1.3 |
| Increased AST | 34 | 3.9 |
| Increased creatinine | 32 | 5 |
| Hypocalcemia | 27 | 0 |
| Increased ALT | 21 | 3.9 |
| Hypokalemia | 20 | 6 |
Other laboratory abnormalities occurring in ≥10% of patients receiving KEYTRUDA were hypophosphatemia (19% all Grades; 6% Grades 3-4), increased INR (19% all Grades; 0% Grades 3-4), hypercalcemia (14% all Grades; 2.6% Grades 3-4), platelet count decreased (14% all Grades; 1.3% Grades 3-4), activated partial thromboplastin time prolonged (14% all Grades; 0% Grades 3-4), hypoglycemia (13% all Grades; 1.3% Grades 3-4), white blood cell decreased (13% all Grades; 2.6% Grades 3-4), and hyperkalemia (13% all Grades; 1.3% Grades 3-4).
HCC
Previously Treated HCC
The safety of KEYTRUDA was investigated in KEYNOTE-394, a multicenter, double-blind, randomized, placebo-controlled trial that enrolled patients with previously treated HCC. Patients were randomized (2:1) and received KEYTRUDA 200 mg (n=299) or placebo (n=153) intravenously every 3 weeks for up to 35 cycles [see Clinical Studies (14.12)].
The median duration of exposure was 3.3 months (range: 1 day to 27.3 months) in the KEYTRUDA arm and 2.2 months (range: 1 day to 15.5 months) in the placebo arm. KEYTRUDA was discontinued due to adverse reactions in 13% of patients. The most common adverse reaction resulting in permanent discontinuation of KEYTRUDA was ascites (2.3%). Adverse reactions leading to interruption of KEYTRUDA occurred in 26% of patients; the most common adverse reactions or laboratory abnormalities leading to interruption of KEYTRUDA (≥2%) were increased blood bilirubin (9%), increased AST (5%), and increased ALT (2%).
Tables 43 and 44 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-394.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks n=299 | Placebo n=153 |
||
|---|---|---|---|---|
| All Grades*
(%) | Grades 3-5 (%) | All Grades*
(%) | Grades 3-5 (%) |
|
| General | ||||
| Pyrexia | 18 | 0.7 | 14 | 0 |
| Skin and Subcutaneous Tissue | ||||
| Rash† | 18 | 0.7 | 7 | 0 |
| Pruritus | 12 | 0 | 4 | 0 |
| Gastrointestinal | ||||
| Diarrhea | 16 | 1.7 | 9 | 0 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 15 | 0.3 | 9 | 0 |
| Infections | ||||
| Upper respiratory tract infection | 11 | 1.0 | 7 | 0.7 |
| Respiratory, Thoracic, and Mediastinal | ||||
| Cough | 11 | 0 | 9 | 0 |
| Endocrine | ||||
| Hypothyroidism | 10 | 0 | 7 | 0 |
| Laboratory Test* | KEYTRUDA | Placebo | ||
|---|---|---|---|---|
| All Grades†
% | Grades 3-4 % | All Grades†
% | Grades 3-4 % |
|
| Chemistry | ||||
| Increased AST | 54 | 14 | 44 | 12 |
| Increased bilirubin | 47 | 11 | 36 | 7 |
| Increased ALT | 47 | 7 | 32 | 4.6 |
| Increased gamma-glutamyl transferase (GGT) | 40 | 20 | 39 | 15 |
| Hypoalbuminemia | 40 | 0.7 | 20 | 0.7 |
| Increased alkaline phosphatase | 39 | 4.1 | 34 | 4 |
| Hyperglycemia | 36 | 3.3 | 26 | 1.4 |
| Hyponatremia | 36 | 11 | 28 | 5 |
| Hypophosphatemia | 30 | 6 | 17 | 4 |
| Hypocalcemia | 24 | 1.4 | 15 | 0.7 |
| Hematology | ||||
| Lymphopenia | 44 | 11 | 34 | 4.6 |
| Anemia | 36 | 7 | 30 | 3.3 |
| Decreased platelets | 32 | 4.7 | 29 | 2 |
| Leukopenia | 30 | 1.3 | 21 | 0.7 |
| Neutropenia | 25 | 4.4 | 21 | 2 |
BTC
The safety of KEYTRUDA in combination with gemcitabine and cisplatin, was investigated in KEYNOTE-966, a multicenter, double-blind, randomized, placebo-controlled trial in patients with locally advanced unresectable or metastatic BTC who had not received prior systemic therapy in the advanced disease setting [see Clinical Studies (14.13)]. A total of 1063 patients received either KEYTRUDA 200 mg plus gemcitabine and cisplatin chemotherapy (n=529) or placebo plus gemcitabine and cisplatin chemotherapy (n=534) every 3 weeks.
The median duration of exposure to KEYTRUDA was 6 months (range: 1 day to 28 months).
KEYTRUDA was discontinued for adverse reactions in 15% of patients. The most common adverse reaction resulting in permanent discontinuation of KEYTRUDA (≥1%) was pneumonitis (1.3%).
Adverse reactions leading to the interruption of KEYTRUDA occurred in 55% of patients. The most common adverse reactions or laboratory abnormalities leading to interruption of KEYTRUDA (≥2%) were decreased neutrophil count (18%), decreased platelet count (10%), anemia (6%), decreased white blood count (4%), pyrexia (3.8%), fatigue (3.0%), cholangitis (2.8%), increased ALT (2.6%), increased AST (2.5%), and biliary obstruction (2.3%).
In the KEYTRUDA plus chemotherapy versus placebo plus chemotherapy arms, there was a difference of ≥5% incidence in adverse reactions between patients treated with KEYTRUDA versus placebo for pyrexia (26% vs 20%), rash (21% vs 13%), pruritus (15% vs 10%), and hypothyroidism (9% vs. 2.6%). There were no clinically meaningful differences in incidence of Grade 3-4 toxicity between arms.
There was a difference of ≥5% incidence in laboratory abnormalities between patients treated with KEYTRUDA plus chemotherapy versus placebo plus chemotherapy for decreased lymphocytes (69% vs 61%). There were no clinically meaningful differences in incidence of Grade 3-4 toxicity between arms.
MCC
Among the 105 patients with MCC enrolled in KEYNOTE-017 and KEYNOTE-913 [see Clinical Studies (14.14)], the median duration of exposure to KEYTRUDA was 6.3 months (range 1 day to 28 months). Patients with autoimmune disease or a medical condition that required immunosuppression were ineligible. Adverse reactions occurring in patients with MCC were similar to those occurring in 2799 patients with melanoma or NSCLC treated with KEYTRUDA as a single agent. Laboratory abnormalities (Grades 3-4) that occurred at a higher incidence included increased lipase (17%).
RCC
In combination with axitinib in the first-line treatment of advanced RCC (KEYNOTE-426)
The safety of KEYTRUDA in combination with axitinib was investigated in KEYNOTE-426 [see Clinical Studies (14.15)]. Patients with medical conditions that required systemic corticosteroids or other immunosuppressive medications or had a history of severe autoimmune disease other than type 1 diabetes, vitiligo, Sjogren's syndrome, and hypothyroidism stable on hormone replacement were ineligible. Patients received KEYTRUDA 200 mg intravenously every 3 weeks and axitinib 5 mg orally twice daily, or sunitinib 50 mg once daily for 4 weeks and then off treatment for 2 weeks. The median duration of exposure to the combination therapy of KEYTRUDA and axitinib was 10.4 months (range: 1 day to 21.2 months).
The study population characteristics were: median age of 62 years (range: 30 to 89), 40% age 65 or older; 71% male; 80% White; and 80% Karnofsky Performance Status (KPS) of 90-100 and 20% KPS of 70-80.
Fatal adverse reactions occurred in 3.3% of patients receiving KEYTRUDA in combination with axitinib. These included 3 cases of cardiac arrest, 2 cases of pulmonary embolism and 1 case each of cardiac failure, death due to unknown cause, myasthenia gravis, myocarditis, Fournier's gangrene, plasma cell myeloma, pleural effusion, pneumonitis, and respiratory failure.
Serious adverse reactions occurred in 40% of patients receiving KEYTRUDA in combination with axitinib. Serious adverse reactions in ≥1% of patients receiving KEYTRUDA in combination with axitinib included hepatotoxicity (7%), diarrhea (4.2%), acute kidney injury (2.3%), dehydration (1%), and pneumonitis (1%).
Permanent discontinuation due to an adverse reaction of either KEYTRUDA or axitinib occurred in 31% of patients; 13% KEYTRUDA only, 13% axitinib only, and 8% both drugs. The most common adverse reaction (>1%) resulting in permanent discontinuation of KEYTRUDA, axitinib, or the combination was hepatotoxicity (13%), diarrhea/colitis (1.9%), acute kidney injury (1.6%), and cerebrovascular accident (1.2%).
Dose interruptions or reductions due to an adverse reaction, excluding temporary interruptions of KEYTRUDA infusions due to infusion-related reactions, occurred in 76% of patients receiving KEYTRUDA in combination with axitinib. This includes interruption of KEYTRUDA in 50% of patients. Axitinib was interrupted in 64% of patients and dose reduced in 22% of patients. The most common adverse reactions (>10%) resulting in interruption of KEYTRUDA were hepatotoxicity (14%) and diarrhea (11%), and the most common adverse reactions (>10%) resulting in either interruption or reduction of axitinib were hepatotoxicity (21%), diarrhea (19%), and hypertension (18%).
The most common adverse reactions (≥20%) in patients receiving KEYTRUDA and axitinib were diarrhea, fatigue/asthenia, hypertension, hypothyroidism, decreased appetite, hepatotoxicity, palmar-plantar erythrodysesthesia, nausea, stomatitis/mucosal inflammation, dysphonia, rash, cough, and constipation.
Twenty-seven percent (27%) of patients treated with KEYTRUDA in combination with axitinib received an oral prednisone dose equivalent to ≥40 mg daily for an immune-mediated adverse reaction.
Tables 45 and 46 summarize the adverse reactions and laboratory abnormalities, respectively, that occurred in at least 20% of patients treated with KEYTRUDA and axitinib in KEYNOTE-426.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks and Axitinib n=429 | Sunitinib n=425 |
||
|---|---|---|---|---|
| All Grades*
(%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) |
|
|
||||
| Gastrointestinal | ||||
| Diarrhea† | 56 | 11 | 45 | 5 |
| Nausea | 28 | 0.9 | 32 | 0.9 |
| Constipation | 21 | 0 | 15 | 0.2 |
| General | ||||
| Fatigue/Asthenia | 52 | 5 | 51 | 10 |
| Vascular | ||||
| Hypertension‡ | 48 | 24 | 48 | 20 |
| Hepatobiliary | ||||
| Hepatotoxicity§ | 39 | 20 | 25 | 4.9 |
| Endocrine | ||||
| Hypothyroidism | 35 | 0.2 | 32 | 0.2 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 30 | 2.8 | 29 | 0.7 |
| Skin and Subcutaneous Tissue | ||||
| Palmar-plantar erythrodysesthesia syndrome | 28 | 5 | 40 | 3.8 |
| Stomatitis/Mucosal inflammation | 27 | 1.6 | 41 | 4 |
| Rash¶ | 25 | 1.4 | 21 | 0.7 |
| Respiratory, Thoracic and Mediastinal | ||||
| Dysphonia | 25 | 0.2 | 3.3 | 0 |
| Cough | 21 | 0.2 | 14 | 0.5 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks and Axitinib | Sunitinib | ||
|---|---|---|---|---|
| All Grades†
% | Grades 3-4 % | All Grades % | Grades 3-4 % |
|
|
||||
| Chemistry | ||||
| Hyperglycemia | 62 | 9 | 54 | 3.2 |
| Increased ALT | 60 | 20 | 44 | 5 |
| Increased AST | 57 | 13 | 56 | 5 |
| Increased creatinine | 43 | 4.3 | 40 | 2.4 |
| Hyponatremia | 35 | 8 | 29 | 8 |
| Hyperkalemia | 34 | 6 | 22 | 1.7 |
| Hypoalbuminemia | 32 | 0.5 | 34 | 1.7 |
| Hypercalcemia | 27 | 0.7 | 15 | 1.9 |
| Hypophosphatemia | 26 | 6 | 49 | 17 |
| Increased alkaline phosphatase | 26 | 1.7 | 30 | 2.7 |
| Hypocalcemia‡ | 22 | 0.2 | 29 | 0.7 |
| Blood bilirubin increased | 22 | 2.1 | 21 | 1.9 |
| Activated partial thromboplastin time prolonged§ | 22 | 1.2 | 14 | 0 |
| Hematology | ||||
| Lymphopenia | 33 | 11 | 46 | 8 |
| Anemia | 29 | 2.1 | 65 | 8 |
| Thrombocytopenia | 27 | 1.4 | 78 | 14 |
In combination with lenvatinib in the first-line treatment of advanced RCC (KEYNOTE-581)
The safety of KEYTRUDA was evaluated in KEYNOTE-581 [see Clinical Studies (14.15)]. Patients received KEYTRUDA 200 mg intravenously every 3 weeks in combination with lenvatinib 20 mg orally once daily (n=352), or lenvatinib 18 mg orally once daily in combination with everolimus 5 mg orally once daily (n=355), or sunitinib 50 mg orally once daily for 4 weeks then off treatment for 2 weeks (n=340). The median duration of exposure to the combination therapy of KEYTRUDA and lenvatinib was 17 months (range: 0.1 to 39).
Fatal adverse reactions occurred in 4.3% of patients treated with KEYTRUDA in combination with lenvatinib, including cardio-respiratory arrest (0.9%), sepsis (0.9%), and one case (0.3%) each of arrhythmia, autoimmune hepatitis, dyspnea, hypertensive crisis, increased blood creatinine, multiple organ dysfunction syndrome, myasthenic syndrome, myocarditis, nephritis, pneumonitis, ruptured aneurysm, and subarachnoid hemorrhage.
Serious adverse reactions occurred in 51% of patients receiving KEYTRUDA and lenvatinib. Serious adverse reactions in ≥2% of patients were hemorrhagic events (5%), diarrhea (4%), hypertension (3%), myocardial infarction (3%), pneumonitis (3%), vomiting (3%), acute kidney injury (2%), adrenal insufficiency (2%), dyspnea (2%), and pneumonia (2%).
Permanent discontinuation of either of KEYTRUDA, lenvatinib or both due to an adverse reaction occurred in 37% of patients receiving KEYTRUDA in combination with lenvatinib; 29% KEYTRUDA only, 26% lenvatinib only, and 13% both. The most common adverse reactions (≥2%) resulting in permanent discontinuation of KEYTRUDA, lenvatinib, or the combination were pneumonitis (3%), myocardial infarction (3%), hepatotoxicity (3%), acute kidney injury (3%), rash (3%), and diarrhea (2%).
Dose interruptions of KEYTRUDA, lenvatinib, or both due to an adverse reaction occurred in 78% of patients receiving KEYTRUDA in combination with lenvatinib. KEYTRUDA was interrupted in 55% of patients and both drugs were interrupted in 39% of patients. The most common adverse reactions (≥3%) resulting in interruption of KEYTRUDA were diarrhea (10%), hepatotoxicity (8%), fatigue (7%), lipase increased (5%), amylase increased (4%), musculoskeletal pain (3%), hypertension (3%), rash (3%), acute kidney injury (3%), and decreased appetite (3%).
Fifteen percent (15%) of patients treated with KEYTRUDA in combination with lenvatinib received an oral prednisone equivalent to ≥40 mg daily for an immune-mediated adverse reaction.
Tables 47 and 48 summarize the adverse reactions and laboratory abnormalities, respectively, that occurred in ≥20% of patients treated with KEYTRUDA and lenvatinib in KEYNOTE-581.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks with Lenvatinib N=352 | Sunitinib 50 mg N=340 |
||
|---|---|---|---|---|
| All Grades (%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) |
|
|
||||
| General | ||||
| Fatigue* | 63 | 9 | 56 | 8 |
| Gastrointestinal | ||||
| Diarrhea† | 62 | 10 | 50 | 6 |
| Stomatitis‡ | 43 | 2 | 43 | 2 |
| Nausea | 36 | 3 | 33 | 1 |
| Abdominal pain§ | 27 | 2 | 18 | 1 |
| Vomiting | 26 | 3 | 20 | 1 |
| Constipation | 25 | 1 | 19 | 0 |
| Musculoskeletal and Connective Tissue | ||||
| Musculoskeletal disorders¶ | 58 | 4 | 41 | 3 |
| Endocrine | ||||
| Hypothyroidism# | 57 | 1 | 32 | 0 |
| Vascular | ||||
| HypertensionÞ | 56 | 29 | 43 | 20 |
| Hemorrhagic eventsß | 27 | 5 | 26 | 4 |
| Metabolism | ||||
| Decreased appetiteà | 41 | 4 | 31 | 1 |
| Skin and Subcutaneous Tissue | ||||
| Rashè | 37 | 5 | 17 | 1 |
| Palmar-plantar erythrodysesthesia syndromeð | 29 | 4 | 38 | 4 |
| Investigations | ||||
| Weight loss | 30 | 8 | 9 | 0.3 |
| Respiratory, Thoracic and Mediastinal | ||||
| Dysphonia | 30 | 0 | 4 | 0 |
| Renal and Urinary | ||||
| Proteinuriaø | 30 | 8 | 13 | 3 |
| Acute kidney injuryý | 21 | 5 | 16 | 2 |
| Hepatobiliary | ||||
| Hepatotoxicity£ | 25 | 9 | 21 | 5 |
| Nervous System | ||||
| Headache | 23 | 1 | 16 | 1 |
Clinically relevant adverse reactions (<20%) that occurred in patients receiving KEYTRUDA with lenvatinib were myocardial infarction (3%) and angina pectoris (1%).
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks with Lenvatinib | Sunitinib 50 mg | ||
|---|---|---|---|---|
| All Grades
%† | Grade 3-4 %† | All Grades %† | Grade 3-4 %† |
|
|
||||
| Chemistry | ||||
| Hypertriglyceridemia | 80 | 15 | 71 | 15 |
| Hypercholesterolemia | 64 | 5 | 43 | 1 |
| Increased lipase | 61 | 34 | 59 | 28 |
| Increased creatinine | 61 | 5 | 61 | 2 |
| Increased amylase | 59 | 17 | 41 | 9 |
| Increased AST | 58 | 7 | 57 | 3 |
| Hyperglycemia | 55 | 7 | 48 | 3 |
| Increased ALT | 52 | 7 | 49 | 4 |
| Hyperkalemia | 44 | 9 | 28 | 6 |
| Hypoglycemia | 44 | 2 | 27 | 1 |
| Hyponatremia | 41 | 12 | 28 | 9 |
| Decreased albumin | 34 | 0.3 | 22 | 0 |
| Increased alkaline phosphatase | 32 | 4 | 32 | 1 |
| Hypocalcemia | 30 | 2 | 22 | 1 |
| Hypophosphatemia | 29 | 7 | 50 | 8 |
| Hypomagnesemia | 25 | 2 | 15 | 3 |
| Increased creatine phosphokinase | 24 | 6 | 36 | 5 |
| Hypermagnesemia | 23 | 2 | 22 | 3 |
| Hypercalcemia | 21 | 1 | 11 | 1 |
| Hematology | ||||
| Lymphopenia | 54 | 9 | 66 | 15 |
| Thrombocytopenia | 39 | 2 | 73 | 13 |
| Anemia | 38 | 3 | 66 | 8 |
| Leukopenia | 34 | 1 | 77 | 8 |
| Neutropenia | 31 | 4 | 72 | 16 |
Grade 3 and 4 increased ALT or AST was seen in 9% of patients. Grade ≥2 increased ALT or AST was reported in 64 (18%) patients, of whom 20 (31%) received ≥40 mg daily oral prednisone equivalent. Recurrence of Grade ≥2 increased ALT or AST was observed on rechallenge in 10 patients receiving both KEYTRUDA and lenvatinib (n=38) and was not observed on rechallenge with KEYTRUDA alone (n=3).
Adjuvant treatment of RCC
The safety of KEYTRUDA as a single agent was investigated in KEYNOTE-564, a randomized (1:1) double-blind placebo-controlled trial in which 984 patients who had undergone nephrectomy for RCC received 200 mg of KEYTRUDA by intravenous infusion every 3 weeks (n=488) or placebo (n=496) for up to one year [see Clinical Studies (14.15)]. The median duration of exposure to KEYTRUDA was 11.1 months (range: 1 day to 14.3 months). Patients with active autoimmune disease or a medical condition that required immunosuppression were ineligible.
Serious adverse reactions occurred in 20% of these patients receiving KEYTRUDA. Serious adverse reactions (≥1%) were acute kidney injury, adrenal insufficiency, pneumonia, colitis, and diabetic ketoacidosis (1% each). Fatal adverse reactions occurred in 0.2% of those treated with KEYTRUDA, including one case of pneumonia.
Discontinuation of KEYTRUDA due to an adverse reaction occurred in 21% of patients; the most common (≥1%) were increased ALT (1.6%), colitis (1%), and adrenal insufficiency (1%).
Dose interruptions of KEYTRUDA due to an adverse reaction occurred in 26% of patients; the most common (≥1%) were increased AST (2.3%), arthralgia (1.6%), hypothyroidism (1.6%), diarrhea (1.4%), increased ALT (1.4%), fatigue (1.4%), rash, decreased appetite, and vomiting (1% each). Tables 49 and 50 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in KEYNOTE-564.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks n=488 | Placebo n=496 |
||
|---|---|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) |
|
|
||||
| Musculoskeletal and Connective Tissue | ||||
| Musculoskeletal pain‡ | 41 | 1.2 | 36 | 0.6 |
| General | ||||
| Fatigue§ | 40 | 1.2 | 31 | 0.2 |
| Skin and Subcutaneous Tissue | ||||
| Rash¶ | 30 | 1.4 | 15 | 0.4 |
| Pruritus | 23 | 0.2 | 13 | 0 |
| Gastrointestinal | ||||
| Diarrhea# | 27 | 2.7 | 23 | 0.2 |
| Nausea | 16 | 0.4 | 10 | 0 |
| Abdominal painÞ | 11 | 0.4 | 13 | 0.2 |
| Endocrine | ||||
| Hypothyroidism | 21 | 0.2 | 3.6 | 0 |
| Hyperthyroidism | 12 | 0.2 | 0.2 | 0 |
| Respiratory, Thoracic and Mediastinal | ||||
| Cough ß | 17 | 0 | 12 | 0 |
| Nervous System | ||||
| Headacheà | 15 | 0.2 | 13 | 0 |
| Hepatobiliary | ||||
| Hepatotoxicityè | 14 | 3.7 | 7 | 0.6 |
| Renal and Urinary | ||||
| Acute kidney injuryð | 13 | 1.2 | 10 | 0.2 |
| Laboratory Test† | KEYTRUDA 200 mg every 3 weeks | Placebo | ||
|---|---|---|---|---|
| All Grades‡
% | Grades 3-4 % | All Grades % | Grades 3-4 % |
|
|
||||
| Chemistry | ||||
| Increased glucose | 48 | 8 | 45 | 4.5 |
| Increased creatinine | 40 | 1.1 | 28 | 0.2 |
| Increased INR | 27 | 0.9 | 20 | 0.8 |
| Hyponatremia | 21 | 3.3 | 13 | 1.9 |
| Increased ALT | 20 | 3.8 | 11 | 0.2 |
| Hematology | ||||
| Anemia | 28 | 0.5 | 20 | 0.4 |
Endometrial Carcinoma
Primary Advanced or Recurrent Endometrial Carcinoma
The safety of KEYTRUDA in combination with chemotherapy (paclitaxel and carboplatin) was investigated in KEYNOTE-868, a randomized (1:1), multicenter, double-blind, placebo-controlled trial that enrolled patients with advanced or recurrent endometrial carcinoma [see Clinical Studies (14.16)]. A total of 759 patients received KEYTRUDA 200 mg every 3 weeks and chemotherapy for 6 cycles followed by KEYTRUDA 400 mg every 6 weeks for up to 14 cycles (n=382) or placebo and chemotherapy for 6 cycles followed by placebo for up to 14 cycles (n=377). The median duration of exposure to KEYTRUDA was 5.6 months (range: 1 day to 24.0 months).
Serious adverse reactions occurred in 35% of patients receiving KEYTRUDA in combination with chemotherapy, compared to 19% of patients receiving placebo in combination with chemotherapy.
Fatal adverse reactions occurred in 1.6% of patients receiving KEYTRUDA in combination with chemotherapy, including COVID-19 (0.5%), and cardiac arrest (0.3%).
KEYTRUDA was discontinued for an adverse reaction in 14% of patients. Chemotherapy dose reduction was required in 29% of patients receiving KEYTRUDA in combination with chemotherapy, compared to 23% of patients receiving placebo in combination with chemotherapy. There were no clinically meaningful differences in chemotherapy discontinuations or interruptions between arms.
Adverse reactions occurring in patients treated with KEYTRUDA and chemotherapy were generally similar to those observed with KEYTRUDA alone or chemotherapy alone with the exception of rash (33% all Grades; 2.9% Grades 3-4).
In Combination with Lenvatinib for the Treatment of Advanced Endometrial Carcinoma That Is pMMR or Not MSI-H.
The safety of KEYTRUDA in combination with lenvatinib was investigated in KEYNOTE-775, a multicenter, open-label, randomized (1:1), active-controlled trial in patients with advanced endometrial carcinoma previously treated with at least one prior platinum-based chemotherapy regimen in any setting, including in the neoadjuvant and adjuvant settings [see Clinical Studies (14.16)]. Patients with endometrial carcinoma that is pMMR or not MSI-H received KEYTRUDA 200 mg every 3 weeks in combination with lenvatinib 20mg orally once daily (n=342) or received doxorubicin or paclitaxel (n=325).
For patients with pMMR or not MSI-H tumor status, the median duration of study treatment was 7.2 months (range 1 day to 26.8 months) and the median duration of exposure to KEYTRUDA was 6.8 months (range: 1 day to 25.8 months).
Fatal adverse reactions among these patients occurred in 4.7% of those treated with KEYTRUDA and lenvatinib, including 2 cases of pneumonia, and 1 case of the following: acute kidney injury, acute myocardial infarction, colitis, decreased appetite, intestinal perforation, lower gastrointestinal hemorrhage, malignant gastrointestinal obstruction, multiple organ dysfunction syndrome, myelodysplastic syndrome, pulmonary embolism, and right ventricular dysfunction.
Serious adverse reactions occurred in 50% of these patients receiving KEYTRUDA and lenvatinib. Serious adverse reactions (≥3%) were hypertension (4.4%) and urinary tract infections (3.2%).
Discontinuation of KEYTRUDA due to an adverse reaction occurred in 15% of these patients. The most common adverse reaction leading to discontinuation of KEYTRUDA (≥1%) was increased ALT (1.2%).
Dose interruptions of KEYTRUDA due to an adverse reaction occurred in 48% of these patients. The most common adverse reactions leading to interruption of KEYTRUDA (≥3%) were diarrhea (8%), increased ALT (4.4%), increased AST (3.8%), and hypertension (3.5%).
Tables 51 and 52 summarize adverse reactions and laboratory abnormalities, respectively, in patients on KEYTRUDA in combination with lenvatinib in KEYNOTE-775.
| Endometrial Carcinoma (pMMR or not MSI-H) | ||||
|---|---|---|---|---|
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks and Lenvatinib n=342 | Doxorubicin or Paclitaxel n=325 |
||
| All Grades*
(%) | Grades 3-4 (%) | All Grades*
(%) | Grades 3-4 (%) |
|
|
||||
| Endocrine | ||||
| Hypothyroidism† | 67 | 0.9 | 0.9 | 0 |
| Vascular | ||||
| Hypertension‡ | 67 | 39 | 6 | 2.5 |
| Hemorrhagic events§ | 25 | 2.6 | 15 | 0.9 |
| General | ||||
| Fatigue¶ | 58 | 11 | 54 | 6 |
| Gastrointestinal | ||||
| Diarrhea# | 55 | 8 | 20 | 2.8 |
| Nausea | 49 | 2.9 | 47 | 1.5 |
| Vomiting | 37 | 2.3 | 21 | 2.2 |
| StomatitisÞ | 35 | 2.6 | 26 | 1.2 |
| Abdominal painß | 34 | 2.6 | 21 | 1.2 |
| Constipation | 27 | 0 | 25 | 0.6 |
| Musculoskeletal and Connective Tissue | ||||
| Musculoskeletal disordersà | 53 | 5 | 27 | 0.6 |
| Metabolism | ||||
| Decreased appetiteè | 44 | 7 | 21 | 0 |
| Investigations | ||||
| Weight loss | 34 | 10 | 6 | 0.3 |
| Renal and Urinary | ||||
| Proteinuriað | 29 | 6 | 3.4 | 0.3 |
| Infections | ||||
| Urinary tract infectionø | 31 | 5 | 13 | 1.2 |
| Nervous System | ||||
| Headache | 26 | 0.6 | 9 | 0.3 |
| Respiratory, Thoracic and Mediastinal | ||||
| Dysphonia | 22 | 0 | 0.6 | 0 |
| Skin and Subcutaneous Tissue | ||||
| Palmar-plantar erythrodysesthesiaý | 23 | 2.9 | 0.9 | 0 |
| Rash£ | 20 | 2.3 | 4.9 | 0 |
| Endometrial Carcinoma (pMMR or not MSI-H) | ||||
|---|---|---|---|---|
| Laboratory Test† | KEYTRUDA 200 mg every 3 weeks and Lenvatinib | Doxorubicin or Paclitaxel |
||
| All Grades‡
% | Grades 3-4 % | All Grades‡
% | Grades 3-4 % |
|
|
||||
| Chemistry | ||||
| Hypertriglyceridemia | 70 | 6 | 45 | 1.7 |
| Hypoalbuminemia | 60 | 2.7 | 42 | 1.6 |
| Increased aspartate aminotransferase | 58 | 9 | 23 | 1.6 |
| Hyperglycemia | 58 | 8 | 45 | 4.4 |
| Hypomagnesemia | 53 | 6 | 32 | 3.8 |
| Increased alanine aminotransferase | 55 | 9 | 21 | 1.2 |
| Hypercholesteremia | 53 | 3.2 | 23 | 0.7 |
| Hyponatremia | 46 | 15 | 28 | 7 |
| Increased alkaline phosphatase | 43 | 4.7 | 18 | 0.9 |
| Hypocalcemia | 40 | 4.7 | 21 | 1.9 |
| Increased lipase | 36 | 14 | 13 | 3.9 |
| Increased creatinine | 35 | 4.7 | 18 | 1.9 |
| Hypokalemia | 34 | 10 | 24 | 5 |
| Hypophosphatemia | 26 | 8 | 17 | 3.2 |
| Increased amylase | 25 | 7 | 8 | 1 |
| Hyperkalemia | 23 | 2.4 | 12 | 1.2 |
| Increased creatine kinase | 19 | 3.7 | 7 | 0 |
| Increased bilirubin | 18 | 3.6 | 6 | 1.6 |
| Hematology | ||||
| Lymphopenia | 50 | 16 | 65 | 20 |
| Thrombocytopenia | 50 | 8 | 30 | 4.7 |
| Anemia | 49 | 8 | 84 | 14 |
| Leukopenia | 43 | 3.5 | 83 | 43 |
| Neutropenia | 31 | 6 | 76 | 58 |
As a Single Agent for the Treatment of Advanced MSI-H or dMMR Endometrial Carcinoma
Among the 90 patients with MSI-H or dMMR endometrial carcinoma enrolled in KEYNOTE-158 [see Clinical Studies (14.16)] treated with KEYTRUDA as a single agent, the median duration of exposure to KEYTRUDA was 8.3 months (range: 1 day to 26.9 months). Adverse reactions occurring in patients with endometrial carcinoma were similar to those occurring in 2799 patients with melanoma or NSCLC treated with KEYTRUDA as a single agent.
TMB-H Cancer
The safety of KEYTRUDA was investigated in 105 patients with TMB-H cancer enrolled in KEYNOTE-158 [see Clinical Studies (14.17)]. The median duration of exposure to KEYTRUDA was 4.9 months (range: 0.03 to 35.2 months). Adverse reactions occurring in patients with TMB-H cancer were similar to those occurring in patients with other solid tumors who received KEYTRUDA as a single agent.
cSCC
Among the 159 patients with advanced cSCC (recurrent or metastatic or locally advanced disease) enrolled in KEYNOTE-629 [see Clinical Studies (14.18)], the median duration of exposure to KEYTRUDA was 6.9 months (range 1 day to 28.9 months). Patients with autoimmune disease or a medical condition that required systemic corticosteroids or other immunosuppressive medications were ineligible. Adverse reactions occurring in patients with recurrent or metastatic cSCC or locally advanced cSCC were similar to those occurring in 2799 patients with melanoma or NSCLC treated with KEYTRUDA as a single agent. Laboratory abnormalities (Grades 3-4) that occurred at a higher incidence included lymphopenia (10%) and decreased sodium (10%).
TNBC
Neoadjuvant and Adjuvant Treatment of High-Risk Early-Stage TNBC
The safety of KEYTRUDA in combination with neoadjuvant chemotherapy (carboplatin and paclitaxel followed by doxorubicin or epirubicin and cyclophosphamide) followed by surgery and continued adjuvant treatment with KEYTRUDA as a single agent was investigated in KEYNOTE-522, a randomized (2:1), multicenter, double-blind, placebo-controlled trial in patients with newly diagnosed, previously untreated, high-risk early-stage TNBC.
A total of 778 patients on the KEYTRUDA arm received at least 1 dose of KEYTRUDA in combination with neoadjuvant chemotherapy followed by KEYTRUDA as adjuvant treatment after surgery, compared to 389 patients who received at least 1 dose of placebo in combination with neoadjuvant chemotherapy followed by placebo as adjuvant treatment after surgery [see Clinical Studies (14.19)].
The median duration of exposure to KEYTRUDA 200 mg every 3 weeks was 13.3 months (range: 1 day to 21.9 months).
Fatal adverse reactions occurred in 0.9% of patients receiving KEYTRUDA, including 1 each of adrenal crisis, autoimmune encephalitis, hepatitis, pneumonia, pneumonitis, pulmonary embolism, and sepsis in association with multiple organ dysfunction syndrome and myocardial infarction.
Serious adverse reactions occurred in 44% of patients receiving KEYTRUDA. Serious adverse reactions in ≥2% of patients who received KEYTRUDA included febrile neutropenia (15%), pyrexia (3.7%), anemia (2.6%), and neutropenia (2.2%).
KEYTRUDA was discontinued for adverse reactions in 20% of patients. The most common adverse reactions (≥1%) resulting in permanent discontinuation of KEYTRUDA were increased ALT (2.7%), increased AST (1.5%), and rash (1%). Adverse reactions leading to the interruption of KEYTRUDA occurred in 57% of patients. The most common adverse reactions leading to interruption of KEYTRUDA (≥2%) were neutropenia (26%), thrombocytopenia (6%), increased ALT (6%), increased AST (3.7%), anemia (3.5%), rash (3.2%), febrile neutropenia (2.8%), leukopenia (2.8%), upper respiratory tract infection (2.6%), pyrexia (2.2%), and fatigue (2.1%).
Tables 53 and 54 summarize the adverse reactions and laboratory abnormalities, respectively, in patients treated with KEYTRUDA in KEYNOTE-522.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks with chemotherapy*/KEYTRUDA n=778 | Placebo with chemotherapy*/Placebo n=389 |
||
|---|---|---|---|---|
| All Grades†
(%) | Grades 3-4 (%) | All Grades†
(%) | Grades 3-4 (%) |
|
|
||||
| General | ||||
| Fatigue‡ | 70 | 8 | 66 | 3.9 |
| Pyrexia | 28 | 1.3 | 19 | 0.3 |
| Gastrointestinal | ||||
| Nausea | 67 | 3.7 | 66 | 1.8 |
| Constipation | 42 | 0 | 39 | 0.3 |
| Diarrhea | 41 | 3.2 | 34 | 1.8 |
| Stomatitis§ | 34 | 2.7 | 29 | 1 |
| Vomiting | 31 | 2.7 | 28 | 1.5 |
| Abdominal pain¶ | 24 | 0.5 | 23 | 0.8 |
| Skin and Subcutaneous Tissue | ||||
| Alopecia | 61 | 0 | 58 | 0 |
| Rash# | 52 | 5 | 41 | 0.5 |
| Nervous System | ||||
| Peripheral neuropathyÞ | 41 | 3.3 | 42 | 2.3 |
| Headache | 30 | 0.5 | 29 | 1 |
| Musculoskeletal and Connective Tissue | ||||
| Arthralgia | 29 | 0.5 | 31 | 0.3 |
| Myalgia | 20 | 0.5 | 19 | 0 |
| Respiratory, Thoracic and Mediastinal | ||||
| Coughß | 26 | 0.1 | 24 | 0 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 23 | 0.9 | 17 | 0.3 |
| Psychiatric | ||||
| Insomnia | 21 | 0.5 | 19 | 0 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks with chemotherapy†/KEYTRUDA | Placebo with chemotherapy†/Placebo |
||
|---|---|---|---|---|
| All Grades‡
% | Grades 3-4 % | All Grades‡
% | Grades 3-4 % |
|
|
||||
| Hematology | ||||
| Anemia | 97 | 22 | 96 | 19 |
| Leukopenia | 93 | 41 | 91 | 32 |
| Neutropenia | 88 | 62 | 89 | 62 |
| Lymphopenia | 80 | 28 | 74 | 22 |
| Thrombocytopenia | 58 | 11 | 57 | 9 |
| Chemistry | ||||
| Increased ALT | 71 | 9 | 69 | 4.6 |
| Increased AST | 66 | 6 | 58 | 1.8 |
| Hyperglycemia | 65 | 5 | 62 | 2.8 |
| Increased alkaline phosphatase | 41 | 1 | 37 | 0.8 |
| Hyponatremia | 38 | 9 | 28 | 6 |
| Hypoalbuminemia | 36 | 1.2 | 30 | 1.5 |
| Hypocalcemia | 32 | 3.2 | 29 | 4.4 |
| Hypokalemia | 32 | 6 | 24 | 2.8 |
| Hypophosphatemia | 23 | 6 | 18 | 4.5 |
| Hypercalcemia | 21 | 3 | 24 | 3.4 |
Locally Recurrent Unresectable or Metastatic TNBC
The safety of KEYTRUDA in combination with paclitaxel, paclitaxel protein-bound, or gemcitabine and carboplatin was investigated in KEYNOTE-355, a multicenter, double-blind, randomized (2:1), placebo-controlled trial in patients with locally recurrent unresectable or metastatic TNBC who had not been previously treated with chemotherapy in the metastatic setting [see Clinical Studies (14.19)]. A total of 596 patients (including 34 patients from a safety run-in) received KEYTRUDA 200 mg every 3 weeks in combination with paclitaxel, paclitaxel protein-bound, or gemcitabine and carboplatin.
The median duration of exposure to KEYTRUDA was 5.7 months (range: 1 day to 33.0 months).
Fatal adverse reactions occurred in 2.5% of patients receiving KEYTRUDA in combination with chemotherapy, including cardio-respiratory arrest (0.7%) and septic shock (0.3%).
Serious adverse reactions occurred in 30% of patients receiving KEYTRUDA in combination with paclitaxel, paclitaxel protein-bound, or gemcitabine and carboplatin. Serious adverse reactions in ≥2% of patients were pneumonia (2.9%), anemia (2.2%), and thrombocytopenia (2%).
KEYTRUDA was discontinued for adverse reactions in 11% of patients. The most common adverse reactions resulting in permanent discontinuation of KEYTRUDA (≥1%) were increased ALT (2.2%), increased AST (1.5%), and pneumonitis (1.2%). Adverse reactions leading to the interruption of KEYTRUDA occurred in 50% of patients. The most common adverse reactions leading to interruption of KEYTRUDA (≥2%) were neutropenia (22%), thrombocytopenia (14%), anemia (7%), increased ALT (6%), leukopenia (5%), increased AST (5%), decreased white blood cell count (3.9%), and diarrhea (2%).
Tables 55 and 56 summarize the adverse reactions and laboratory abnormalities in patients on KEYTRUDA in KEYNOTE-355.
| Adverse Reaction | KEYTRUDA 200 mg every 3 weeks with chemotherapy n=596 | Placebo every 3 weeks with chemotherapy n=281 |
||
|---|---|---|---|---|
| All Grades*
(%) | Grades 3-4 (%) | All Grades*
(%) | Grades 3-4 (%) |
|
|
||||
| General | ||||
| Fatigue† | 48 | 5 | 49 | 4.3 |
| Gastrointestinal | ||||
| Nausea | 44 | 1.7 | 47 | 1.8 |
| Diarrhea | 28 | 1.8 | 23 | 1.8 |
| Constipation | 28 | 0.5 | 27 | 0.4 |
| Vomiting | 26 | 2.7 | 22 | 3.2 |
| Skin and Subcutaneous Tissue | ||||
| Alopecia | 34 | 0.8 | 35 | 1.1 |
| Rash‡ | 26 | 2 | 16 | 0 |
| Respiratory, Thoracic and Mediastinal | ||||
| Cough§ | 23 | 0 | 20 | 0.4 |
| Metabolism and Nutrition | ||||
| Decreased appetite | 21 | 0.8 | 14 | 0.4 |
| Nervous System | ||||
| Headache¶ | 20 | 0.7 | 23 | 0.7 |
| Laboratory Test* | KEYTRUDA 200 mg every 3 weeks with chemotherapy | Placebo every 3 weeks with chemotherapy |
||
|---|---|---|---|---|
| All Grades†
% | Grades 3-4 % | All Grades†
% | Grades 3-4 % |
|
| Hematology | ||||
| Anemia | 90 | 20 | 85 | 19 |
| Leukopenia | 85 | 39 | 86 | 39 |
| Neutropenia | 76 | 49 | 77 | 52 |
| Lymphopenia | 70 | 26 | 70 | 19 |
| Thrombocytopenia | 54 | 19 | 53 | 21 |
| Chemistry | ||||
| Increased ALT | 60 | 11 | 58 | 8 |
| Increased AST | 57 | 9 | 55 | 6 |
| Hyperglycemia | 52 | 4.4 | 51 | 2.2 |
| Hypoalbuminemia | 37 | 2.2 | 32 | 2.2 |
| Increased alkaline phosphatase | 35 | 3.9 | 39 | 2.2 |
| Hypocalcemia | 29 | 3.3 | 27 | 1.8 |
| Hyponatremia | 28 | 5 | 26 | 6 |
| Hypophosphatemia | 21 | 7 | 18 | 4.8 |
| Hypokalemia | 20 | 4.4 | 18 | 4.0 |
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of KEYTRUDA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal: Exocrine pancreatic insufficiency
Hepatobiliary: sclerosing cholangitis
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, KEYTRUDA can cause fetal harm when administered to a pregnant woman. There are no available human data informing the risk of embryo-fetal toxicity. In animal models, the PD-1/PD-L1 signaling pathway is important in the maintenance of pregnancy through induction of maternal immune tolerance to fetal tissue (see Data). Human IgG4 (immunoglobulins) are known to cross the placenta; therefore, pembrolizumab has the potential to be transmitted from the mother to the developing fetus. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Animal reproduction studies have not been conducted with KEYTRUDA to evaluate its effect on reproduction and fetal development. A literature-based assessment of the effects of the PD-1 pathway on reproduction demonstrated that a central function of the PD-1/PD-L1 pathway is to preserve pregnancy by maintaining maternal immune tolerance to the fetus. Blockade of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to the fetus and to result in an increase in fetal loss; therefore, potential risks of administering KEYTRUDA during pregnancy include increased rates of abortion or stillbirth. As reported in the literature, there were no malformations related to the blockade of PD-1 signaling in the offspring of these animals; however, immune-mediated disorders occurred in PD-1 knockout mice. Based on its mechanism of action, fetal exposure to pembrolizumab may increase the risk of developing immune-mediated disorders or of altering the normal immune response.
8.2 Lactation
Risk Summary
There are no data on the presence of pembrolizumab in either animal or human milk or its effects on the breastfed child or on milk production. Maternal IgG is known to be present in human milk. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed child to KEYTRUDA are unknown. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with KEYTRUDA and for 4 months after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating KEYTRUDA [see Use in Specific Populations (8.1)].
Contraception
KEYTRUDA can cause fetal harm when administered to a pregnant woman [see Warnings and Precautions (5.5), Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with KEYTRUDA and for 4 months after the last dose.
8.4 Pediatric Use
The safety and effectiveness of KEYTRUDA as a single agent have been established in pediatric patients with melanoma, cHL, PMBCL, MCC, MSI-H or dMMR cancer, and TMB-H cancer. Use of KEYTRUDA in pediatric patients for these indications is supported by evidence from adequate and well-controlled studies in adults with additional pharmacokinetic and safety data in pediatric patients [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.1, 14.4, 14.5, 14.7, 14.14, 14.17)].
In KEYNOTE-051, 173 pediatric patients (65 pediatric patients aged 6 months to younger than 12 years and 108 pediatric patients aged 12 to 17 years) with advanced melanoma, lymphoma, or PD-L1 positive solid tumors received KEYTRUDA 2 mg/kg every 3 weeks. The median duration of exposure was 2.1 months (range: 1 day to 25 months). Adverse reactions that occurred at a ≥10% higher rate in pediatric patients when compared to adults included pyrexia (33%), vomiting (29%), headache (25%), abdominal pain (23%), decreased lymphocyte count (13%), and decreased white blood cell count (11%). Laboratory abnormalities that occurred at a ≥10% higher rate in pediatric patients when compared to adults were leukopenia (31%), neutropenia (28%), thrombocytopenia (22%), and Grade 3 anemia (17%).
The safety and effectiveness of KEYTRUDA in pediatric patients have not been established in the other approved indications [see Indications and Usage (1)].
8.5 Geriatric Use
Of 3781 patients with melanoma, NSCLC, HNSCC, or urothelial carcinoma who were treated with KEYTRUDA in clinical studies, 48% were 65 years and over and 17% were 75 years and over. No overall differences in safety or effectiveness were observed between elderly patients and younger patients.
Of 389 adult patients with cHL who were treated with KEYTRUDA in clinical studies, 46 (12%) were 65 years and over. Patients aged 65 years and over had a higher incidence of serious adverse reactions (50%) than patients aged younger than 65 years (24%). Clinical studies of KEYTRUDA in cHL did not include sufficient numbers of patients aged 65 years and over to determine whether effectiveness differs from that in younger patients.
Of 506 adult patients with Stage IB (T2a ≥4 cm), II, or IIIA NSCLC following complete resection and platinum-based chemotherapy who were treated with KEYTRUDA in KEYNOTE-091, 242 (48%) were 65 years and over. No overall differences in safety or effectiveness were observed between elderly patients and younger patients.
Of 596 adult patients with TNBC who were treated with KEYTRUDA in combination with paclitaxel, paclitaxel protein-bound, or gemcitabine and carboplatin in KEYNOTE-355, 137 (23%) were 65 years and over. No overall differences in safety or effectiveness were observed between elderly patients and younger patients.
Of 406 adult patients with endometrial carcinoma who were treated with KEYTRUDA in combination with lenvatinib in KEYNOTE-775, 201 (50%) were 65 years and over. No overall differences in safety or effectiveness were observed between elderly patients and younger patients.
Of the 564 patients with locally advanced or metastatic urothelial cancer treated with KEYTRUDA in combination with enfortumab vedotin, 44% (n=247) were 65-74 years and 26% (n=144) were 75 years or older. No overall differences in safety or effectiveness were observed between patients 65 years of age or older and younger patients. Patients 75 years of age or older treated with KEYTRUDA in combination with enfortumab vedotin experienced a higher incidence of fatal adverse reactions than younger patients. The incidence of fatal adverse reactions was 4% in patients younger than 75 and 7% in patients 75 years or older.
Of the 432 patients randomized to KEYTRUDA in combination with axitinib in the KEYNOTE-426 trial, 40% were 65 years or older. No overall difference in safety or efficacy was reported between patients who were ≥65 years of age and younger.
Of 292 adult patients with FIGO 2014 Stage III-IVA cervical cancer who were treated with KEYTRUDA in combination with CRT in KEYNOTE-A18, 42 (14%) were 65 years and over. No overall differences in safety or efficacy were observed between elderly and younger patients.
11. Keytruda Description
Pembrolizumab is a programmed death receptor-1 (PD 1)-blocking antibody. Pembrolizumab is a humanized monoclonal IgG4 kappa antibody with an approximate molecular weight of 149 kDa. Pembrolizumab is produced in recombinant Chinese hamster ovary (CHO) cells.
KEYTRUDA (pembrolizumab) injection is a sterile, preservative-free, clear to slightly opalescent, colorless to slightly yellow solution for intravenous use. Each vial contains 100 mg of pembrolizumab in 4 mL of solution. Each 1 mL of solution contains 25 mg of pembrolizumab and is formulated in: L-histidine (1.55 mg), polysorbate 80 (0.2 mg), sucrose (70 mg), and Water for Injection, USP.
12. Keytruda - Clinical Pharmacology
12.1 Mechanism of Action
Binding of the PD-1 ligands, PD-L1 and PD-L2, to the PD-1 receptor found on T cells, inhibits T cell proliferation and cytokine production. Upregulation of PD-1 ligands occurs in some tumors and signaling through this pathway can contribute to inhibition of active T-cell immune surveillance of tumors. Pembrolizumab is a monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, releasing PD-1 pathway-mediated inhibition of the immune response, including the anti-tumor immune response. In syngeneic mouse tumor models, blocking PD-1 activity resulted in decreased tumor growth.
In syngeneic mouse tumor models, combination treatment of a PD-1 blocking antibody and kinase inhibitor lenvatinib decreased tumor-associated macrophages, increased activated cytotoxic T cells, and reduced tumor growth compared to either treatment alone.
12.2 Pharmacodynamics
There are no clinically significant exposure-response relationships for efficacy or safety at pembrolizumab dosages of 200 mg or 2 mg/kg every 3 weeks regardless of cancer type. There are no clinically significant exposure-response relationships for efficacy or safety at pembrolizumab dosages of 200 mg or 2 mg/kg every 3 weeks and 400 mg every 6 weeks in patients with solid tumors based on observed data in adult patients with melanoma. The exposure-response relationships for efficacy or safety at pembrolizumab dosages of 400 mg every 6 weeks in patients with classical Hodgkin lymphoma or mediastinal large B-cell lymphoma have not been fully characterized.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of pembrolizumab was characterized using a population PK analysis with concentration data collected from 2993 patients with various cancers who received pembrolizumab doses of 1 to 10 mg/kg every 2 weeks, 2 to 10 mg/kg every 3 weeks, or 200 mg every 3 weeks.
Steady-state concentrations of pembrolizumab were reached by 16 weeks of repeated dosing with an every 3-week regimen and the systemic accumulation was 2.1-fold. The peak concentration (Cmax), trough concentration (Cmin), and area under the plasma concentration versus time curve at steady state (AUCss) of pembrolizumab increased dose proportionally in the dose range of 2 to 10 mg/kg every 3 weeks.
Distribution
The geometric mean value (CV%) for volume of distribution at steady state is 6.0 L (20%).
Elimination
Pembrolizumab clearance (CV%) is approximately 23% lower [geometric mean, 195 mL/day (40%)] at steady state than that after the first dose [252 mL/day (37%)]; this decrease in clearance with time is not considered clinically important. The terminal half-life (t1/2) is 22 days (32%).
Specific Populations
The following factors had no clinically important effect on the CL of pembrolizumab: age (range: 15 to 94 years), sex, race (89% White), renal impairment (eGFR ≥15 mL/min/1.73 m2), mild to moderate hepatic impairment (total bilirubin ≤3 times ULN and any AST), or tumor burden. The impact of severe hepatic impairment (total bilirubin >3 times ULN and any AST) on the pharmacokinetics of pembrolizumab is unknown.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described in this section with the incidence of ADA in other studies, including those of KEYTRUDA or of other pembrolizumab products.
Trough levels of pembrolizumab interfere with the electrochemiluminescent (ECL) assay results; therefore, a subset analysis was performed in the KEYTRUDA-treated patients with a pembrolizumab concentration below the drug tolerance level of the ADA assay.
In clinical studies in patients treated with KEYTRUDA at a dosage of 2 mg/kg every 3 weeks, 200 mg every 3 weeks, or 10 mg/kg every 2 or 3 weeks, 27 (2.1%) of 1,289 evaluable patients tested positive for treatment-emergent anti-pembrolizumab antibodies of whom 6 (0.5%) patients had neutralizing antibodies against pembrolizumab. There were no identified clinically significant effects of ADA on pembrolizumab pharmacokinetics or on the risk of infusion reactions. Because of the low occurrence of ADA, the effect of these ADA on the effectiveness of KEYTRUDA is unknown.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been performed to test the potential of pembrolizumab for carcinogenicity or genotoxicity.
Fertility studies have not been conducted with pembrolizumab. In 1-month and 6-month repeat-dose toxicology studies in monkeys, there were no notable effects in the male and female reproductive organs; however, most animals in these studies were not sexually mature.
13.2 Animal Toxicology and/or Pharmacology
In animal models, inhibition of PD-1/PD-L1 signaling increased the severity of some infections and enhanced inflammatory responses. Mycobacterium tuberculosis-infected PD-1 knockout mice exhibit markedly decreased survival compared with wild-type controls, which correlated with increased bacterial proliferation and inflammatory responses in these animals. PD-1 blockade using a primate anti-PD-1 antibody was also shown to exacerbate M. tuberculosis infection in rhesus macaques. PD-1 and PD-L1 knockout mice and mice receiving PD-L1-blocking antibody have also shown decreased survival following infection with lymphocytic choriomeningitis virus. Administration of pembrolizumab in chimpanzees with naturally occurring chronic hepatitis B infection resulted in two out of four animals with significantly increased levels of serum ALT, AST, and GGT, which persisted for at least 1 month after discontinuation of pembrolizumab.
14. Clinical Studies
14.1 Melanoma
Ipilimumab-Naive Melanoma
The efficacy of KEYTRUDA was investigated in KEYNOTE-006 (NCT01866319), a randomized (1:1:1), open-label, multicenter, active-controlled trial in 834 patients. Patients were randomized to receive KEYTRUDA at a dose of 10 mg/kg intravenously every 2 weeks or 10 mg/kg intravenously every 3 weeks until disease progression or unacceptable toxicity or to ipilimumab 3 mg/kg intravenously every 3 weeks for 4 doses unless discontinued earlier for disease progression or unacceptable toxicity. Patients with disease progression could receive additional doses of treatment unless disease progression was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging. Randomization was stratified by line of therapy (0 vs. 1), ECOG PS (0 vs. 1), and PD-L1 expression (≥1% of tumor cells [positive] vs. <1% of tumor cells [negative]) according to an investigational use only (IUO) assay. Key eligibility criteria were unresectable or metastatic melanoma; no prior ipilimumab; and no more than one prior systemic treatment for metastatic melanoma. Patients with BRAF V600E mutation-positive melanoma were not required to have received prior BRAF inhibitor therapy. Patients with autoimmune disease; a medical condition that required immunosuppression; previous severe hypersensitivity to other monoclonal antibodies; and HIV, hepatitis B or hepatitis C infection, were ineligible. Assessment of tumor status was performed at 12 weeks, then every 6 weeks through Week 48, followed by every 12 weeks thereafter. The major efficacy outcome measures were overall survival (OS) and progression-free survival (PFS; as assessed by blinded independent central review [BICR] using Response Evaluation Criteria in Solid Tumors [RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ]). Additional efficacy outcome measures were objective response rate (ORR) and duration of response (DoR).
The study population characteristics were: median age of 62 years (range: 18 to 89); 60% male; 98% White; 66% had no prior systemic therapy for metastatic disease; 69% ECOG PS of 0; 80% had PD-L1 positive melanoma, 18% had PD-L1 negative melanoma, and 2% had unknown PD-L1 status using the IUO assay; 65% had M1c stage disease; 68% with normal LDH; 36% with reported BRAF mutation-positive melanoma; and 9% with a history of brain metastases. Among patients with BRAF mutation-positive melanoma, 139 (46%) were previously treated with a BRAF inhibitor.
The study demonstrated statistically significant improvements in OS and PFS for patients randomized to KEYTRUDA as compared to ipilimumab. Among the 91 patients randomized to KEYTRUDA 10 mg/kg every 3 weeks with an objective response, response durations ranged from 1.4+ to 8.1+ months. Among the 94 patients randomized to KEYTRUDA 10 mg/kg every 2 weeks with an objective response, response durations ranged from 1.4+ to 8.2 months. Efficacy results are summarized in Table 57 and Figure 1.
| Endpoint | KEYTRUDA 10 mg/kg every 3 weeks n=277 | KEYTRUDA 10 mg/kg every 2 weeks n=279 | Ipilimumab 3 mg/kg every 3 weeks n=278 |
|---|---|---|---|
|
|||
| OS | |||
| Deaths (%) | 92 (33%) | 85 (30%) | 112 (40%) |
| Hazard ratio* (95% CI) | 0.69 (0.52, 0.90) | 0.63 (0.47, 0.83) | --- |
| p-Value (stratified log-rank) | 0.004 | <0.001 | --- |
| PFS by BICR | |||
| Events (%) | 157 (57%) | 157 (56%) | 188 (68%) |
| Median in months (95% CI) | 4.1 (2.9, 6.9) | 5.5 (3.4, 6.9) | 2.8 (2.8, 2.9) |
| Hazard ratio* (95% CI) | 0.58 (0.47, 0.72) | 0.58 (0.46, 0.72) | --- |
| p-Value (stratified log-rank) | <0.001 | <0.001 | --- |
| Best objective response by BICR | |||
| ORR (95% CI) | 33% (27, 39) | 34% (28, 40) | 12% (8, 16) |
| Complete response rate | 6% | 5% | 1% |
| Partial response rate | 27% | 29% | 10% |
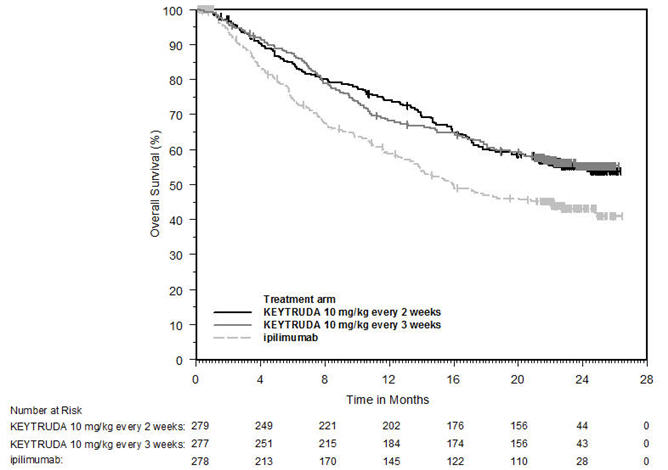
Ipilimumab-Refractory Melanoma
The efficacy of KEYTRUDA was investigated in KEYNOTE-002 (NCT01704287), a multicenter, randomized (1:1:1), active-controlled trial in 540 patients randomized to receive one of two doses of KEYTRUDA in a blinded fashion or investigator's choice chemotherapy. The treatment arms consisted of KEYTRUDA 2 mg/kg or 10 mg/kg intravenously every 3 weeks or investigator's choice of any of the following chemotherapy regimens: dacarbazine 1000 mg/m2 intravenously every 3 weeks (26%), temozolomide 200 mg/m2 orally once daily for 5 days every 28 days (25%), carboplatin AUC 6 mg/mL/min intravenously plus paclitaxel 225 mg/m2 intravenously every 3 weeks for four cycles then carboplatin AUC of 5 mg/mL/min plus paclitaxel 175 mg/m2 every 3 weeks (25%), paclitaxel 175 mg/m2 intravenously every 3 weeks (16%), or carboplatin AUC 5 or 6 mg/mL/min intravenously every 3 weeks (8%). Randomization was stratified by ECOG PS (0 vs. 1), LDH levels (normal vs. elevated [≥110% ULN]) and BRAF V600 mutation status (wild-type [WT] or V600E). The trial included patients with unresectable or metastatic melanoma with progression of disease; refractory to two or more doses of ipilimumab (3 mg/kg or higher) and, if BRAF V600 mutation-positive, a BRAF or MEK inhibitor; and disease progression within 24 weeks following the last dose of ipilimumab. The trial excluded patients with uveal melanoma and active brain metastasis. Patients received KEYTRUDA until unacceptable toxicity; disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at 4 to 6 weeks with repeat imaging; withdrawal of consent; or physician's decision to stop therapy for the patient. Assessment of tumor status was performed at 12 weeks after randomization, then every 6 weeks through week 48, followed by every 12 weeks thereafter. Patients on chemotherapy who experienced progression of disease were offered KEYTRUDA. The major efficacy outcomes were PFS as assessed by BICR per RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, and OS. Additional efficacy outcome measures were confirmed ORR as assessed by BICR per RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, and DoR.
The study population characteristics were: median age of 62 years (range: 15 to 89), 43% age 65 or older; 61% male; 98% White; and 55% ECOG PS of 0 and 45% ECOG PS of 1. Twenty-three percent of patients were BRAF V600 mutation positive, 40% had elevated LDH at baseline, 82% had M1c disease, and 73% had two or more prior therapies for advanced or metastatic disease.
The study demonstrated a statistically significant improvement in PFS for patients randomized to KEYTRUDA as compared to control arm. There was no statistically significant difference between KEYTRUDA 2 mg/kg and chemotherapy or between KEYTRUDA 10 mg/kg and chemotherapy in the OS analysis in which 55% of the patients who had been randomized to receive chemotherapy had crossed over to receive KEYTRUDA. Among the 38 patients randomized to KEYTRUDA 2 mg/kg with an objective response, response durations ranged from 1.3+ to 11.5+ months. Among the 46 patients randomized to KEYTRUDA 10 mg/kg with an objective response, response durations ranged from 1.1+ to 11.1+ months. Efficacy results are summarized in Table 58 and Figure 2.
| Endpoint | KEYTRUDA 2 mg/kg every 3 weeks | KEYTRUDA 10 mg/kg every 3 weeks | Chemotherapy |
|---|---|---|---|
| n=180 | n=181 | n=179 | |
| PFS | |||
| Number of Events, n (%) | 129 (72%) | 126 (70%) | 155 (87%) |
| Progression, n (%) | 105 (58%) | 107 (59%) | 134 (75%) |
| Death, n (%) | 24 (13%) | 19 (10%) | 21 (12%) |
| Median in months (95% CI) | 2.9 (2.8, 3.8) | 2.9 (2.8, 4.7) | 2.7 (2.5, 2.8) |
| p-Value (stratified log-rank) | <0.001 | <0.001 | --- |
| Hazard ratio* (95% CI) | 0.57 (0.45, 0.73) | 0.50 (0.39, 0.64) | --- |
| OS† | |||
| Deaths (%) | 123 (68%) | 117 (65%) | 128 (72%) |
| Hazard ratio* (95% CI) | 0.86 (0.67, 1.10) | 0.74 (0.57, 0.96) | --- |
| p-Value (stratified log-rank) | 0.117 | 0.011‡ | --- |
| Median in months (95% CI) | 13.4 (11.0, 16.4) | 14.7 (11.3, 19.5) | 11.0 (8.9, 13.8) |
| Objective Response Rate | |||
| ORR (95% CI) | 21% (15, 28) | 25% (19, 32) | 4% (2, 9) |
| Complete response rate | 2% | 3% | 0% |
| Partial response rate | 19% | 23% | 4% |
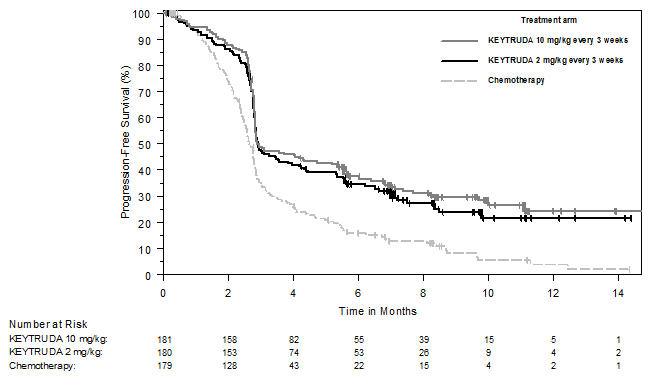 |
Adjuvant Treatment of Resected Stage IIB or IIC Melanoma
The efficacy of KEYTRUDA was investigated in KEYNOTE-716 (NCT03553836), a multicenter, randomized (1:1), double-blind, placebo-controlled trial in patients with completely resected Stage IIB or IIC melanoma. Patients were randomized to KEYTRUDA 200 mg or the pediatric (≥12 years old) dose of KEYTRUDA 2 mg/kg intravenously (up to a maximum of 200 mg) every three weeks or placebo for up to one year until disease recurrence or unacceptable toxicity. Randomization was stratified by AJCC 8th edition T Stage (>2.0-4.0 mm with ulceration vs. >4.0 mm without ulceration vs. >4.0 mm with ulceration). Patients must not have been previously treated for melanoma beyond complete surgical resection for their melanoma prior to study entry. The major efficacy outcome measure was investigator-assessed recurrence-free survival (RFS) (defined as the time between the date of randomization and the date of first recurrence [local, in-transit or regional lymph nodes or distant recurrence] or death, whichever occurred first). New primary melanomas were excluded from the definition of RFS. Distant metastasis-free survival (DMFS), defined as a spread of tumor to distant organs or distant lymph nodes, was an additional efficacy outcome measure. Patients underwent imaging every six months for one year from randomization, every 6 months from years 2 to 4, and then once in year 5 from randomization or until recurrence, whichever came first.
The study population characteristics were: median age of 61 years (range: 16 to 87), 39% age 65 or older; 60% male; 98% White; and 93% ECOG PS of 0 and 7% ECOG PS of 1. Sixty-four percent had Stage IIB and 35% had Stage IIC.
The trial demonstrated a statistically significant improvement in RFS and DMFS for patients randomized to the KEYTRUDA arm compared with placebo. Efficacy results are summarized in Table 59 and Figure 3.
| Endpoint | KEYTRUDA 200 mg every 3 weeks n=487 | Placebo n=489 |
|---|---|---|
| NR = not reached | ||
|
||
| RFS | ||
| Number (%) of patients with event | 54 (11%) | 82 (17%) |
| Median in months (95% CI) | NR (22.6, NR) | NR (NR, NR) |
| Hazard ratio*,† (95% CI) | 0.65 (0.46, 0.92) | |
| p-Value† | 0.0132‡ | |
| DMFS | ||
| Number (%) of patients with event | 63 (13%) | 95 (19%) |
| Median in months (95% CI) | NR (NR, NR) | NR (NR, NR) |
| Hazard ratio*,† (95% CI) | 0.64 (0.47, 0.88) | |
| p-Value† | 0.0058§ | |
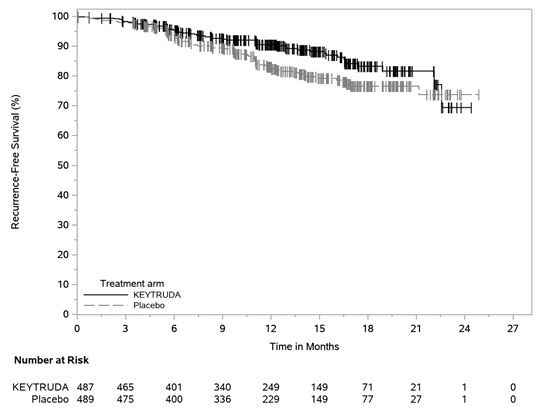 |
Adjuvant Treatment of Stage III Resected Melanoma
The efficacy of KEYTRUDA was investigated in KEYNOTE-054 (NCT02362594), a multicenter, randomized (1:1), double-blind, placebo-controlled trial in patients with completely resected Stage IIIA (>1 mm lymph node metastasis), IIIB, or IIIC melanoma. Patients were randomized to KEYTRUDA 200 mg intravenously every three weeks or placebo for up to one year until disease recurrence or unacceptable toxicity. Randomization was stratified by American Joint Committee on Cancer 7th edition (AJCC) stage (IIIA vs. IIIB vs. IIIC 1-3 positive lymph nodes vs. IIIC ≥4 positive lymph nodes) and geographic region (North America, European countries, Australia, and other countries as designated). Patients must have undergone lymph node dissection and, if indicated, radiotherapy within 13 weeks prior to starting treatment. The major efficacy outcome measure was investigator-assessed recurrence-free survival (RFS) in the whole population and in the population with PD-L1 positive tumors where RFS was defined as the time between the date of randomization and the date of first recurrence (local, regional, or distant metastasis) or death, whichever occurs first. New primary melanomas were excluded from the definition of RFS. DMFS in the whole population and in the population with PD-L1 positive tumors were additional efficacy outcome measures. DMFS was defined as a spread of tumor to distant organs or distant lymph nodes. Patients underwent imaging every 12 weeks after the first dose of KEYTRUDA for the first two years, then every 6 months from year 3 to 5, and then annually.
The study population characteristics were: median age of 54 years (range: 19 to 88), 25% age 65 or older; 62% male; and 94% ECOG PS of 0 and 6% ECOG PS of 1. Sixteen percent had Stage IIIA, 46% had Stage IIIB, 18% had Stage IIIC (1-3 positive lymph nodes), and 20% had Stage IIIC (≥4 positive lymph nodes); 50% were BRAF V600 mutation positive and 44% were BRAF wild-type; and 84% had PD-L1 positive melanoma with TPS ≥1% according to an IUO assay.
The trial demonstrated a statistically significant improvement in RFS and DMFS for patients randomized to the KEYTRUDA arm compared with placebo. Efficacy results are summarized in Table 60 and Figure 4.
| Endpoint | KEYTRUDA 200 mg every 3 weeks n=514 | Placebo n=505 |
|---|---|---|
| NR = not reached | ||
| RFS | ||
| Number (%) of patients with event | 135 (26%) | 216 (43%) |
| Median in months (95% CI) | NR | 20.4 (16.2, NR) |
| Hazard ratio*,† (95% CI) | 0.57 (0.46, 0.70) | |
| p-Value† (log-rank) | <0.001‡ | |
| DMFS | ||
| Number (%) of patients with event | 173 (34%) | 245 (49%) |
| Median in months (95% CI) | NR (49.6, NR) | 40.0 (27.7, NR) |
| Hazard ratio*,† (95% CI) | 0.60 (0.49, 0.73) | |
| p-Value† (log-rank) | <0.0001§ | |
For patients with PD-L1 positive tumors, the RFS HR was 0.54 (95% CI: 0.42, 0.69); p<0.0001. For patients with PD-L1 positive tumors, the DMFS HR was 0.61 (95% CI: 0.49, 0.76); p<0.0001. The RFS and DMFS benefit for KEYTRUDA compared to placebo was observed regardless of tumor PD-L1 expression.
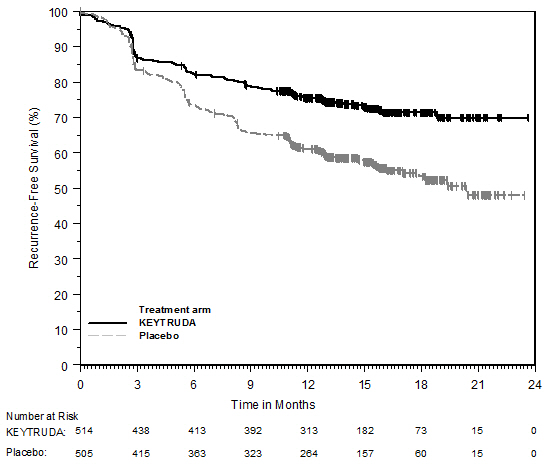 |
14.2 Non-Small Cell Lung Cancer
First-line treatment of metastatic nonsquamous NSCLC with pemetrexed and platinum chemotherapy
The efficacy of KEYTRUDA in combination with pemetrexed and platinum chemotherapy was investigated in KEYNOTE-189 (NCT02578680), a randomized, multicenter, double-blind, active-controlled trial conducted in 616 patients with metastatic nonsquamous NSCLC, regardless of PD-L1 tumor expression status, who had not previously received systemic therapy for metastatic disease and in whom there were no EGFR or ALK genomic tumor aberrations. Patients with autoimmune disease that required systemic therapy within 2 years of treatment; a medical condition that required immunosuppression; or who had received more than 30 Gy of thoracic radiation within the prior 26 weeks were ineligible. Randomization was stratified by smoking status (never vs. former/current), choice of platinum (cisplatin vs. carboplatin), and tumor PD-L1 status (TPS <1% [negative] vs. TPS ≥1%). Patients were randomized (2:1) to one of the following treatment arms:
- KEYTRUDA 200 mg, pemetrexed 500 mg/m2, and investigator's choice of cisplatin 75 mg/m2 or carboplatin AUC 5 mg/mL/min intravenously on Day 1 of each 21-day cycle for 4 cycles followed by KEYTRUDA 200 mg and pemetrexed 500 mg/m2 intravenously every 3 weeks. KEYTRUDA was administered prior to chemotherapy on Day 1.
- Placebo, pemetrexed 500 mg/m2, and investigator's choice of cisplatin 75 mg/m2 or carboplatin AUC 5 mg/mL/min intravenously on Day 1 of each 21-day cycle for 4 cycles followed by placebo and pemetrexed 500 mg/m2 intravenously every 3 weeks.
Treatment with KEYTRUDA continued until RECIST v1.1 (modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ)-defined progression of disease as determined by the investigator, unacceptable toxicity, or a maximum of 24 months. Administration of KEYTRUDA was permitted beyond RECIST-defined disease progression if the patient was clinically stable and considered to be deriving clinical benefit by the investigator. Patients randomized to placebo and chemotherapy were offered KEYTRUDA as a single agent at the time of disease progression. Assessment of tumor status was performed at Week 6, Week 12, and then every 9 weeks thereafter. The main efficacy outcome measures were OS and PFS as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ. Additional efficacy outcome measures were ORR and DoR, as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
The study population characteristics were: median age of 64 years (range: 34 to 84), 49% age 65 or older; 59% male; 94% White and 3% Asian; 56% ECOG PS of 1; and 18% with history of brain metastases. Thirty-one percent had tumor PD-L1 expression TPS <1% [negative]. Seventy-two percent received carboplatin and 12% were never smokers. A total of 85 patients in the placebo and chemotherapy arm received an anti-PD-1/PD-L1 monoclonal antibody at the time of disease progression.
The trial demonstrated a statistically significant improvement in OS and PFS for patients randomized to KEYTRUDA in combination with pemetrexed and platinum chemotherapy compared with placebo, pemetrexed, and platinum chemotherapy. Table 61 and Figure 5 summarize the efficacy results for KEYNOTE-189.
| Endpoint | KEYTRUDA 200 mg every 3 weeks Pemetrexed Platinum Chemotherapy n=410 | Placebo Pemetrexed Platinum Chemotherapy n=206 |
|---|---|---|
| NR = not reached | ||
| OS | ||
| Number (%) of patients with event | 127 (31%) | 108 (52%) |
| Median in months (95% CI) | NR (NR, NR) | 11.3 (8.7, 15.1) |
| Hazard ratio* (95% CI) | 0.49 (0.38, 0.64) | |
| p-Value† | <0.0001 | |
| PFS | ||
| Number of patients with event (%) | 245 (60%) | 166 (81%) |
| Median in months (95% CI) | 8.8 (7.6, 9.2) | 4.9 (4.7, 5.5) |
| Hazard ratio* (95% CI) | 0.52 (0.43, 0.64) | |
| p-Value† | <0.0001 | |
| Objective Response Rate | ||
| ORR‡ (95% CI) | 48% (43, 53) | 19% (14, 25) |
| Complete response | 0.5% | 0.5% |
| Partial response | 47% | 18% |
| p-Value§ | <0.0001 | |
| Duration of Response | ||
| Median in months (range) | 11.2 (1.1+, 18.0+) | 7.8 (2.1+, 16.4+) |
At the protocol-specified final OS analysis, the median in the KEYTRUDA in combination with pemetrexed and platinum chemotherapy arm was 22.0 months (95% CI: 19.5, 24.5) compared to 10.6 months (95% CI: 8.7, 13.6) in the placebo with pemetrexed and platinum chemotherapy arm, with an HR of 0.56 (95% CI: 0.46, 0.69).
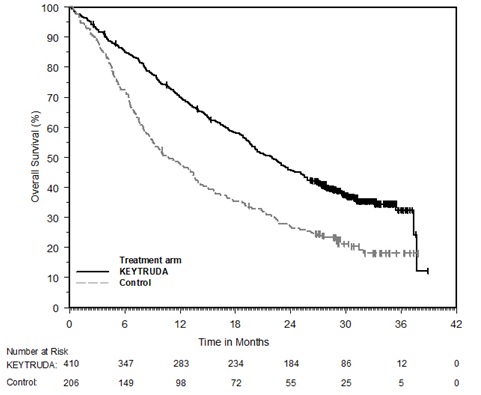
First-line treatment of metastatic squamous NSCLC with carboplatin and either paclitaxel or paclitaxel protein-bound chemotherapy
The efficacy of KEYTRUDA in combination with carboplatin and investigator's choice of either paclitaxel or paclitaxel protein-bound was investigated in KEYNOTE-407 (NCT02775435), a randomized, multi-center, double-blind, placebo-controlled trial conducted in 559 patients with metastatic squamous NSCLC, regardless of PD-L1 tumor expression status, who had not previously received systemic therapy for metastatic disease. Patients with autoimmune disease that required systemic therapy within 2 years of treatment; a medical condition that required immunosuppression; or who had received more than 30 Gy of thoracic radiation within the prior 26 weeks were ineligible. Randomization was stratified by tumor PD-L1 status (TPS <1% [negative] vs. TPS ≥1%), choice of paclitaxel or paclitaxel protein-bound, and geographic region (East Asia vs. non-East Asia). Patients were randomized (1:1) to one of the following treatment arms; all study medications were administered via intravenous infusion:
- KEYTRUDA 200 mg and carboplatin AUC 6 mg/mL/min on Day 1 of each 21-day cycle for 4 cycles, and paclitaxel 200 mg/m2 on Day 1 of each 21-day cycle for 4 cycles or paclitaxel protein-bound 100 mg/m2 on Days 1, 8 and 15 of each 21-day cycle for 4 cycles, followed by KEYTRUDA 200 mg every 3 weeks. KEYTRUDA was administered prior to chemotherapy on Day 1.
- Placebo and carboplatin AUC 6 mg/mL/min on Day 1 of each 21-day cycle for 4 cycles and paclitaxel 200 mg/m2 on Day 1 of each 21-day cycle for 4 cycles or paclitaxel protein-bound 100 mg/m2 on Days 1, 8 and 15 of each 21-day cycle for 4 cycles, followed by placebo every 3 weeks.
Treatment with KEYTRUDA and chemotherapy or placebo and chemotherapy continued until RECIST v1.1 (modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ)-defined progression of disease as determined by BICR, unacceptable toxicity, or a maximum of 24 months. Administration of KEYTRUDA was permitted beyond RECIST-defined disease progression if the patient was clinically stable and deriving clinical benefit as determined by the investigator. Patients randomized to the placebo and chemotherapy arm were offered KEYTRUDA as a single agent at the time of disease progression. Assessment of tumor status was performed every 6 weeks through Week 18, every 9 weeks through Week 45 and every 12 weeks thereafter. The main efficacy outcome measures were PFS and ORR as assessed by BICR using RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, and OS. An additional efficacy outcome measure was DoR as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
The study population characteristics were: median age of 65 years (range: 29 to 88), 55% age 65 or older; 81% male; 77% White; 71% ECOG PS of 1; and 8% with a history of brain metastases. Thirty-five percent had tumor PD-L1 expression TPS <1%; 19% were from the East Asian region; and 60% received paclitaxel.
The trial demonstrated a statistically significant improvement in OS, PFS and ORR in patients randomized to KEYTRUDA in combination with carboplatin and either paclitaxel or paclitaxel protein-bound chemotherapy compared with patients randomized to placebo with carboplatin and either paclitaxel or paclitaxel protein-bound chemotherapy. Table 62 and Figure 6 summarize the efficacy results for KEYNOTE-407.
| Endpoint | KEYTRUDA 200 mg every 3 weeks Carboplatin Paclitaxel/Paclitaxel protein-bound n=278 | Placebo Carboplatin Paclitaxel/Paclitaxel protein-bound n=281 |
|---|---|---|
| NE = not estimable | ||
| OS | ||
| Number of events (%) | 85 (31%) | 120 (43%) |
| Median in months (95% CI) | 15.9 (13.2, NE) | 11.3 (9.5, 14.8) |
| Hazard ratio* (95% CI) | 0.64 (0.49, 0.85) | |
| p-Value† | 0.0017 | |
| PFS | ||
| Number of events (%) | 152 (55%) | 197 (70%) |
| Median in months (95% CI) | 6.4 (6.2, 8.3) | 4.8 (4.2, 5.7) |
| Hazard ratio* (95% CI) | 0.56 (0.45, 0.70) | |
| p-Value† | <0.0001 | |
| n=101 | n=103 | |
| Objective Response Rate‡ | ||
| ORR (95% CI) | 58% (48, 68) | 35% (26, 45) |
| Difference (95% CI) | 23.6% (9.9, 36.4) | |
| p-Value§ | 0.0008 | |
| Duration of Response‡ | ||
| Median duration of response in months (range) | 7.2 (2.4, 12.4+) | 4.9 (2.0, 12.4+) |
At the protocol-specified final OS analysis, the median in the KEYTRUDA in combination with carboplatin and either paclitaxel or paclitaxel protein-bound chemotherapy arm was 17.1 months (95% CI: 14.4, 19.9) compared to 11.6 months (95% CI: 10.1, 13.7) in the placebo with carboplatin and either paclitaxel or paclitaxel protein-bound chemotherapy arm, with an HR of 0.71 (95% CI: 0.58, 0.88).
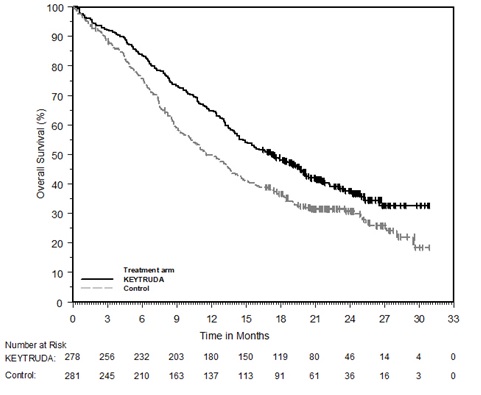
First-line treatment of metastatic NSCLC as a single agent
KEYNOTE-042
The efficacy of KEYTRUDA was investigated in KEYNOTE-042 (NCT02220894), a randomized, multicenter, open-label, active-controlled trial conducted in 1274 patients with Stage III NSCLC who were not candidates for surgical resection or definitive chemoradiation, or patients with metastatic NSCLC. Only patients whose tumors expressed PD-L1 (TPS ≥1%) by an immunohistochemistry assay using the PD-L1 IHC 22C3 pharmDx kit and who had not received prior systemic treatment for metastatic NSCLC were eligible. Patients with EGFR or ALK genomic tumor aberrations; autoimmune disease that required systemic therapy within 2 years of treatment; a medical condition that required immunosuppression; or who had received more than 30 Gy of radiation in the thoracic region within the prior 26 weeks of initiation of study were ineligible. Randomization was stratified by ECOG PS (0 vs. 1), histology (squamous vs. nonsquamous), geographic region (East Asia vs. non-East Asia), and PD-L1 expression (TPS ≥50% vs. TPS 1 to 49%). Patients were randomized (1:1) to receive KEYTRUDA 200 mg intravenously every 3 weeks or investigator's choice of either of the following platinum-containing chemotherapy regimens:
- Pemetrexed 500 mg/m2 every 3 weeks and carboplatin AUC 5 to 6 mg/mL/min every 3 weeks on Day 1 for a maximum of 6 cycles followed by optional pemetrexed 500 mg/m2 every 3 weeks for patients with nonsquamous histologies;
- Paclitaxel 200 mg/m2 every 3 weeks and carboplatin AUC 5 to 6 mg/mL/min every 3 weeks on Day 1 for a maximum of 6 cycles followed by optional pemetrexed 500 mg/m2 every 3 weeks for patients with nonsquamous histologies.
Treatment with KEYTRUDA continued until RECIST v1.1 (modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ)-defined progression of disease, unacceptable toxicity, or a maximum of 24 months. Administration of KEYTRUDA was permitted beyond RECIST-defined disease progression if the patient was clinically stable and deriving clinical benefit as determined by the investigator. Treatment with KEYTRUDA could be reinitiated at the time of subsequent disease progression and administered for up to 12 months. Assessment of tumor status was performed every 9 weeks. The main efficacy outcome measure was OS in the subgroup of patients with TPS ≥50% NSCLC, the subgroup of patients with TPS ≥20% NSCLC, and the overall population with TPS ≥1% NSCLC. Additional efficacy outcome measures were PFS and ORR in the subgroup of patients with TPS ≥50% NSCLC, the subgroup of patients with TPS ≥20% NSCLC, and the overall population with TPS ≥1% NSCLC as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
The study population characteristics were: median age of 63 years (range: 25 to 90), 45% age 65 or older; 71% male; and 64% White, 30% Asian, and 2% Black. Nineteen percent were Hispanic or Latino. Sixty-nine percent had ECOG PS of 1; 39% with squamous and 61% with nonsquamous histology; 87% had M1 disease and 13% had Stage IIIA (2%) or Stage IIIB (11%) and who were not candidates for surgical resection or definitive chemoradiation per investigator assessment; and 5% with treated brain metastases at baseline. Forty-seven percent of patients had TPS ≥50% NSCLC and 53% had TPS 1 to 49% NSCLC.
The trial demonstrated a statistically significant improvement in OS for patients (PD-L1 TPS ≥50%, TPS ≥20%, TPS ≥1%) randomized to KEYTRUDA as compared with chemotherapy. Table 63 and Figure 7 summarize the efficacy results in the subgroup of patients with TPS ≥50% and in all randomized patients with TPS ≥1%.
| TPS ≥1% | TPS ≥50% | |||
|---|---|---|---|---|
| Endpoint | KEYTRUDA 200 mg every 3 weeks | Chemotherapy | KEYTRUDA 200 mg every 3 weeks | Chemotherapy |
| n=637 | n=637 | n=299 | n=300 | |
|
||||
| OS | ||||
| Number of events (%) | 371 (58%) | 438 (69%) | 157 (53%) | 199 (66%) |
| Median in months (95% CI) | 16.7 (13.9, 19.7) | 12.1 (11.3, 13.3) | 20.0 (15.4, 24.9) | 12.2 (10.4, 14.2) |
| Hazard ratio* (95% CI) | 0.81 (0.71, 0.93) | 0.69 (0.56, 0.85) | ||
| p-Value† | 0.0036 | 0.0006 | ||
| PFS | ||||
| Number of events (%) | 507 (80%) | 506 (79%) | 221 (74%) | 233 (78%) |
| Median in months (95% CI) | 5.4 (4.3, 6.2) | 6.5 (6.3, 7.0) | 6.9 (5.9, 9.0) | 6.4 (6.1, 6.9) |
| Hazard ratio*, ‡ (95% CI) | 1.07 (0.94, 1.21) | 0.82 (0.68, 0.99) |
||
| p-Value† | -‡ | NS§ | ||
| Objective Response Rate | ||||
| ORR‡ (95% CI) | 27% (24, 31) | 27% (23, 30) | 39% (33.9, 45.3) | 32% (26.8, 37.6) |
| Complete response rate | 0.5% | 0.5% | 0.7% | 0.3% |
| Partial response rate | 27% | 26% | 39% | 32% |
| Duration of Response | ||||
| % with duration ≥12 months¶ | 47% | 16% | 42% | 17% |
| % with duration ≥18 months¶ | 26% | 6% | 25% | 5% |
The results of all efficacy outcome measures in the subgroup of patients with PD-L1 TPS ≥20% NSCLC were intermediate between the results of those with PD-L1 TPS ≥1% and those with PD-L1 TPS ≥50%. In a pre-specified exploratory subgroup analysis for patients with TPS 1-49% NSCLC, the median OS was 13.4 months (95% CI: 10.7, 18.2) for the pembrolizumab group and 12.1 months (95% CI: 11.0, 14.0) in the chemotherapy group, with an HR of 0.92 (95% CI: 0.77, 1.11).
| Figure 7: Kaplan-Meier Curve for Overall Survival in all Randomized Patients in KEYNOTE-042 (TPS ≥1%) |
|
|
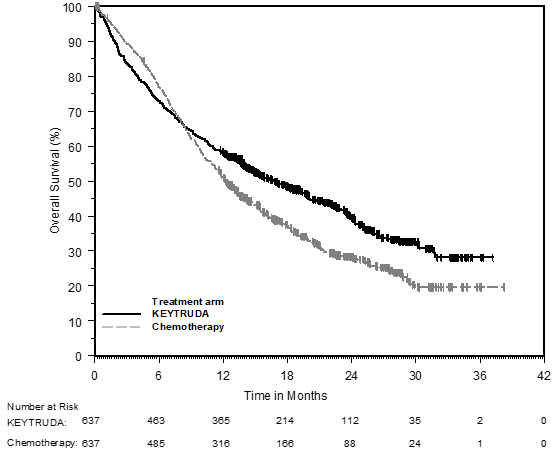
KEYNOTE-024
The efficacy of KEYTRUDA was also investigated in KEYNOTE-024 (NCT02142738), a randomized, multicenter, open-label, active-controlled trial in 305 previously untreated patients with metastatic NSCLC. The study design was similar to that of KEYNOTE-042, except that only patients whose tumors had high PD-L1 expression (TPS of 50% or greater) by an immunohistochemistry assay using the PD-L1 IHC 22C3 pharmDx kit were eligible. Patients were randomized (1:1) to receive KEYTRUDA 200 mg intravenously every 3 weeks or investigator's choice of any of the following platinum-containing chemotherapy regimens:
- Pemetrexed 500 mg/m2 every 3 weeks and carboplatin AUC 5 to 6 mg/mL/min every 3 weeks on Day 1 for 4 to 6 cycles followed by optional pemetrexed 500 mg/m2 every 3 weeks for patients with nonsquamous histologies;
- Pemetrexed 500 mg/m2 every 3 weeks and cisplatin 75 mg/m2 every 3 weeks on Day 1 for 4 to 6 cycles followed by optional pemetrexed 500 mg/m2 every 3 weeks for patients with nonsquamous histologies;
- Gemcitabine 1250 mg/m2 on days 1 and 8 and cisplatin 75 mg/m2 every 3 weeks on Day 1 for 4 to 6 cycles;
- Gemcitabine 1250 mg/m2 on Days 1 and 8 and carboplatin AUC 5 to 6 mg/mL/min every 3 weeks on Day 1 for 4 to 6 cycles;
- Paclitaxel 200 mg/m2 every 3 weeks and carboplatin AUC 5 to 6 mg/mL/min every 3 weeks on Day 1 for 4 to 6 cycles followed by optional pemetrexed maintenance (for nonsquamous histologies).
Patients randomized to chemotherapy were offered KEYTRUDA at the time of disease progression.
The main efficacy outcome measure was PFS as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ. Additional efficacy outcome measures were OS and ORR as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
The study population characteristics were: median age of 65 years (range: 33 to 90), 54% age 65 or older; 61% male; 82% White and 15% Asian; 65% with ECOG PS of 1; 18% with squamous and 82% with nonsquamous histology and 9% with history of brain metastases. A total of 66 patients in the chemotherapy arm received KEYTRUDA at the time of disease progression.
The trial demonstrated a statistically significant improvement in both PFS and OS for patients randomized to KEYTRUDA as compared with chemotherapy. Table 64 and Figure 8 summarize the efficacy results for KEYNOTE-024.
| Endpoint | KEYTRUDA 200 mg every 3 weeks | Chemotherapy |
|---|---|---|
| n=154 | n=151 | |
| NR = not reached | ||
| PFS | ||
| Number (%) of patients with event | 73 (47%) | 116 (77%) |
| Median in months (95% CI) | 10.3 (6.7, NR) | 6.0 (4.2, 6.2) |
| Hazard ratio* (95% CI) | 0.50 (0.37, 0.68) | |
| p-Value (stratified log-rank) | <0.001 | |
| OS | ||
| Number (%) of patients with event | 44 (29%) | 64 (42%) |
| Median in months (95% CI)† | 30.0 (18.3, NR) | 14.2 (9.8, 19.0) |
| Hazard ratio* (95% CI) | 0.60 (0.41, 0.89) | |
| p-Value (stratified log-rank) | 0.005‡ | |
| Objective Response Rate | ||
| ORR (95% CI) | 45% (37, 53) | 28% (21, 36) |
| Complete response rate | 4% | 1% |
| Partial response rate | 41% | 27% |
| p-Value (Miettinen-Nurminen) | 0.001 | |
| Median duration of response in months (range) | NR (1.9+, 14.5+) | 6.3 (2.1+, 12.6+) |
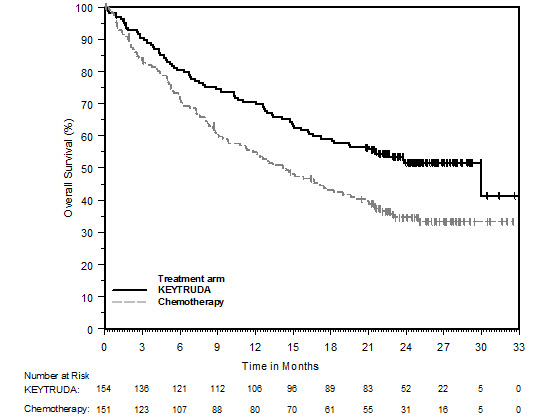
Previously treated NSCLC
The efficacy of KEYTRUDA was investigated in KEYNOTE-010 (NCT01905657), a randomized, multicenter, open-label, active-controlled trial conducted in 1033 patients with metastatic NSCLC that had progressed following platinum-containing chemotherapy, and if appropriate, targeted therapy for EGFR or ALK genomic tumor aberrations. Eligible patients had PD-L1 expression TPS of 1% or greater by an immunohistochemistry assay using the PD-L1 IHC 22C3 pharmDx kit. Patients with autoimmune disease; a medical condition that required immunosuppression; or who had received more than 30 Gy of thoracic radiation within the prior 26 weeks were ineligible. Randomization was stratified by tumor PD-L1 expression (PD-L1 expression TPS ≥50% vs. PD-L1 expression TPS=1-49%), ECOG PS (0 vs. 1), and geographic region (East Asia vs. non-East Asia). Patients were randomized (1:1:1) to receive KEYTRUDA 2 mg/kg intravenously every 3 weeks, KEYTRUDA 10 mg/kg intravenously every 3 weeks or docetaxel intravenously 75 mg/m2 every 3 weeks until unacceptable toxicity or disease progression. Patients randomized to KEYTRUDA were permitted to continue until disease progression that was symptomatic, rapidly progressive, required urgent intervention, occurred with a decline in performance status, or confirmation of progression at 4 to 6 weeks with repeat imaging or for up to 24 months without disease progression. Assessment of tumor status was performed every 9 weeks. The main efficacy outcome measures were OS and PFS as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, in the subgroup of patients with TPS ≥50% and the overall population with TPS ≥1%. Additional efficacy outcome measures were ORR and DoR in the subgroup of patients with TPS ≥50% and the overall population with TPS ≥1%.
The study population characteristics were: median age of 63 years (range: 20 to 88), 42% age 65 or older; 61% male; 72% White and 21% Asian; 66% ECOG PS of 1; 43% with high PD-L1 tumor expression; 21% with squamous, 70% with nonsquamous, and 8% with mixed, other or unknown histology; 91% metastatic (M1) disease; 15% with history of brain metastases; and 8% and 1% with EGFR and ALK genomic aberrations, respectively. All patients had received prior therapy with a platinum-doublet regimen, 29% received two or more prior therapies for their metastatic disease.
Tables 65 and 66 and Figure 9 summarize efficacy results in the subgroup with TPS ≥50% population and in all patients, respectively.
| Endpoint | KEYTRUDA 2 mg/kg every 3 weeks n=139 | KEYTRUDA 10 mg/kg every 3 weeks n=151 | Docetaxel 75 mg/m2 every 3 weeks n=152 |
|---|---|---|---|
| NR = not reached | |||
| OS | |||
| Deaths (%) | 58 (42%) | 60 (40%) | 86 (57%) |
| Median in months (95% CI) | 14.9 (10.4, NR) | 17.3 (11.8, NR) | 8.2 (6.4, 10.7) |
| Hazard ratio* (95% CI) | 0.54 (0.38, 0.77) | 0.50 (0.36, 0.70) | --- |
| p-Value (stratified log-rank) | <0.001 | <0.001 | --- |
| PFS | |||
| Events (%) | 89 (64%) | 97 (64%) | 118 (78%) |
| Median in months (95% CI) | 5.2 (4.0, 6.5) | 5.2 (4.1, 8.1) | 4.1 (3.6, 4.3) |
| Hazard ratio* (95% CI) | 0.58 (0.43, 0.77) | 0.59 (0.45, 0.78) | --- |
| p-Value (stratified log-rank) | <0.001 | <0.001 | --- |
| Objective Response Rate | |||
| ORR† (95% CI) | 30% (23, 39) | 29% (22, 37) | 8% (4, 13) |
| p-Value (Miettinen-Nurminen) | <0.001 | <0.001 | --- |
| Median duration of response in months (range) | NR (0.7+, 16.8+) | NR (2.1+, 17.8+) | 8.1 (2.1+, 8.8+) |
| Endpoint | KEYTRUDA 2 mg/kg every 3 weeks n=344 | KEYTRUDA 10 mg/kg every 3 weeks n=346 | Docetaxel 75 mg/m2 every 3 weeks n=343 |
|---|---|---|---|
| NR = not reached | |||
| OS | |||
| Deaths (%) | 172 (50%) | 156 (45%) | 193 (56%) |
| Median in months (95% CI) | 10.4 (9.4, 11.9) | 12.7 (10.0, 17.3) | 8.5 (7.5, 9.8) |
| Hazard ratio* (95% CI) | 0.71 (0.58, 0.88) | 0.61 (0.49, 0.75) | --- |
| p-Value (stratified log-rank) | <0.001 | <0.001 | --- |
| PFS | |||
| Events (%) | 266 (77%) | 255 (74%) | 257 (75%) |
| Median in months (95% CI) | 3.9 (3.1, 4.1) | 4.0 (2.6, 4.3) | 4.0 (3.1, 4.2) |
| Hazard ratio* (95% CI) | 0.88 (0.73, 1.04) | 0.79 (0.66, 0.94) | --- |
| p-Value (stratified log-rank) | 0.068 | 0.005 | --- |
| Objective Response Rate | |||
| ORR† (95% CI) | 18% (14, 23) | 19% (15, 23) | 9% (7, 13) |
| p-Value (Miettinen-Nurminen) | <0.001 | <0.001 | --- |
| Median duration of response in months (range) | NR (0.7+, 20.1+) | NR (2.1+, 17.8+) | 6.2 (1.4+, 8.8+) |
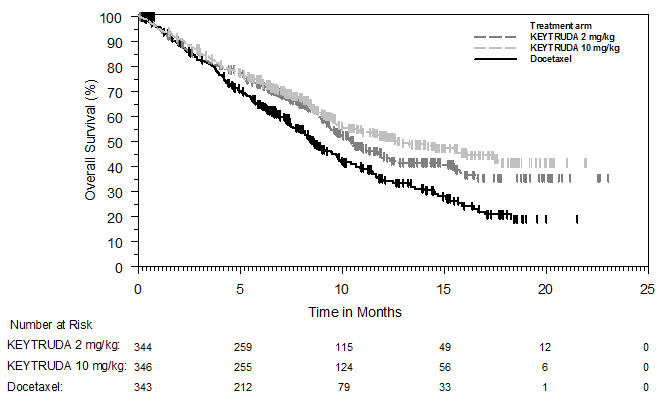 |
Neoadjuvant and adjuvant treatment of resectable NSCLC
The efficacy of KEYTRUDA in combination with neoadjuvant chemotherapy followed by surgery and continued adjuvant treatment with KEYTRUDA as a single agent was investigated in KEYNOTE-671 (NCT03425643), a multicenter, randomized, double-blind, placebo-controlled trial conducted in 797 patients with previously untreated and resectable Stage II, IIIA, or IIIB (N2) NSCLC by AJCC 8th edition. Patients were enrolled regardless of tumor PD-L1 expression. Patients with active autoimmune disease that required systemic therapy within 2 years of treatment, a medical condition that required immunosuppression, or a history of interstitial lung disease or pneumonitis that required steroids were ineligible. Randomization was stratified by stage (II vs. III), tumor PD-L1 expression (TPS ≥50% or <50%), histology (squamous vs. nonsquamous), and geographic region (East Asia vs. non-East Asia).
Patients were randomized (1:1) to one of the following treatment arms:
- Treatment Arm A: neoadjuvant KEYTRUDA 200 mg on Day 1 in combination with cisplatin 75 mg/m2 and either pemetrexed 500 mg/m2 on Day 1 or gemcitabine 1000 mg/m2 on Days 1 and 8 of each 21-day cycle for up to 4 cycles. Within 4-12 weeks following surgery, KEYTRUDA 200 mg was administered every 3 weeks for up to 13 cycles.
- Treatment Arm B: neoadjuvant placebo on Day 1 in combination with cisplatin 75 mg/m2 and either pemetrexed 500 mg/m2 on Day 1 or gemcitabine 1000 mg/m2 on Days 1 and 8 of each 21-day cycle for up to 4 cycles. Within 4-12 weeks following surgery, placebo was administered every 3 weeks for up to 13 cycles.
All study medications were administered via intravenous infusion. Treatment with KEYTRUDA or placebo continued until completion of the treatment (17 cycles), disease progression that precluded definitive surgery, disease recurrence in the adjuvant phase, disease progression for those who did not undergo surgery or had incomplete resection and entered the adjuvant phase, or unacceptable toxicity. Assessment of tumor status was performed at baseline, Week 7, and Week 13 in the neoadjuvant phase and within 4 weeks prior to the start of the adjuvant phase. Following the start of the adjuvant phase, assessment of tumor status was performed every 16 weeks through the end of Year 3, and then every 6 months thereafter.
The trial was not designed to isolate the effect of KEYTRUDA in each phase (neoadjuvant or adjuvant) of treatment.
The major efficacy outcome measures were OS and investigator-assessed event-free survival (EFS). Additional efficacy outcome measures were pathological complete response (pCR) rate and major pathological response (mPR) rate as assessed by blinded independent pathology review.
The study population characteristics were: median age of 64 years (range: 26 to 83); 45% age 65 or older and 7% age 75 or older; 71% male; 61% White, 31% Asian, 2% Black, 4% race not reported; 9% Hispanic or Latino; 63% ECOG PS of 0 and 37% ECOG PS of 1. Thirty percent had Stage II and 70% had Stage III disease; 33% had TPS ≥50% and 67% had TPS <50%; 43% had tumors with squamous histology and 57% had tumors with non-squamous histology; 31% were from the East Asian region.
Eighty-one percent of patients in the KEYTRUDA in combination with platinum-containing chemotherapy arm received definitive surgery compared to 76% of patients in the placebo in combination with platinum-containing chemotherapy arm.
The trial demonstrated statistically significant improvements in OS and EFS for patients randomized to KEYTRUDA in combination with platinum-containing chemotherapy followed by KEYTRUDA as a single agent compared with patients randomized to placebo in combination with platinum-containing chemotherapy followed by placebo alone.
Table 67 and Figure 10 summarize the efficacy results for KEYNOTE-671.
| Endpoint | KEYTRUDA 200 mg every 3 weeks with chemotherapy/KEYTRUDA n=397 | Placebo with chemotherapy/Placebo n=400 |
|---|---|---|
| NR = not reached | ||
|
||
| OS | ||
| Number of patients with event (%) | 110 (28%) | 144 (36%) |
| Median in months* (95% CI) | NR (NR, NR) | 52.4 (45.7, NR) |
| Hazard ratio† (95% CI) | 0.72 (0.56, 0.93) | |
| p-Value‡,§ | 0.0103 | |
| EFS | ||
| Number of patients with event (%) | 139 (35%) | 205 (51%) |
| Median in months* (95% CI) | NR (34.1, NR) | 17.0 (14.3, 22.0) |
| Hazard ratio† (95% CI) | 0.58 (0.46, 0.72) | |
| p-Value‡,¶ | <0.0001 | |
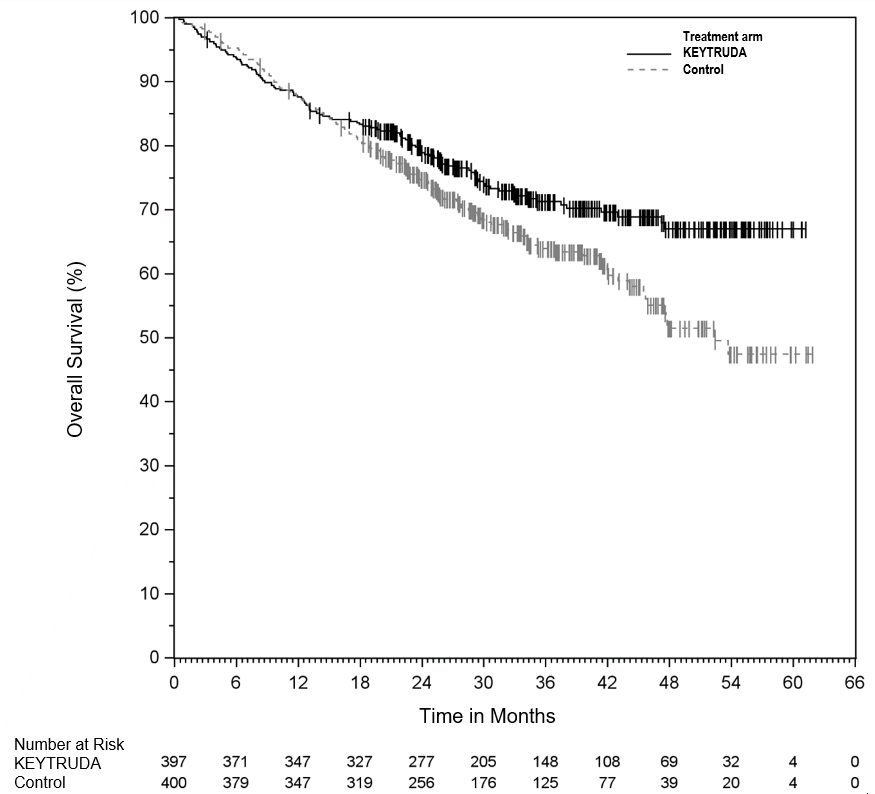 |
The trial demonstrated a statistically significant difference in pCR rate (18.1% vs. 4.0%; p<0.0001) and mPR rate (30.2% vs. 11.0%; p<0.0001).
Adjuvant treatment of resected NSCLC
The efficacy of KEYTRUDA was investigated in KEYNOTE-091 (NCT02504372), a multicenter, randomized, triple-blind, placebo-controlled trial conducted in 1177 patients with completely resected Stage IB (T2a ≥4 cm), II, or IIIA NSCLC by AJCC 7th edition. Patients had not received neoadjuvant radiotherapy or chemotherapy. Adjuvant chemotherapy up to 4 cycles was optional. Patients were ineligible if they had active autoimmune disease, were on chronic immunosuppressive agents, or had a history of interstitial lung disease or pneumonitis. Randomization was stratified by stage (IB vs. II vs. IIIA), receipt of adjuvant chemotherapy (yes vs. no), PD-L1 status (TPS <1% [negative] vs. TPS 1-49% vs. TPS ≥50%), and geographic region (Western Europe vs. Eastern Europe vs. Asia vs. Rest of World). Patients were randomized (1:1) to receive KEYTRUDA 200 mg or placebo intravenously every 3 weeks.
Treatment continued until RECIST v1.1-defined disease recurrence as determined by the investigator, unacceptable toxicity or up to one year. Tumor assessments were conducted every 12 weeks for the first year, then every 6 months for years 2 to 3, and then annually through year 5. After year 5, imaging was performed as per local standard of care. The major efficacy outcome measure was investigator-assessed disease-free survival (DFS). An additional efficacy outcome measure was OS.
Of 1177 patients randomized, 1010 (86%) received adjuvant platinum-based chemotherapy following resection. Among these 1010 patients, the median age was 64 years (range: 35 to 84), 49% age 65 or older; 68% male; 77% White, 18% Asian; 86% current or former smokers; and 39% with ECOG PS of 1. Eleven percent had Stage IB, 57% had Stage II, and 31% had Stage IIIA disease. Thirty-nine percent had PD-L1 TPS <1% [negative], 33% had TPS 1-49%, and 28% had TPS ≥50%. Fifty-two percent were from Western Europe, 20% from Eastern Europe, 17% from Asia, and 11% from Rest of World.
The trial met its primary endpoint, demonstrating a statistically significant improvement in DFS in the overall population for patients randomized to the KEYTRUDA arm compared to patients randomized to the placebo arm. In an exploratory subgroup analysis of the 167 patients (14%) who did not receive adjuvant chemotherapy, the DFS HR was 1.25 (95% CI: 0.76, 2.05). OS results were not mature with only 42% of pre-specified OS events in the overall population.
Table 68 and Figure 11 summarize the efficacy results for KEYNOTE-091 in patients who received adjuvant chemotherapy.
| Endpoint | KEYTRUDA 200 mg every 3 weeks n=506 | Placebo n=504 |
|---|---|---|
| NR = not reached | ||
|
||
| DFS | ||
| Number (%) of patients with event | 177 (35%) | 231 (46%) |
| Median in months (95% CI) | 58.7 (39.2, NR) | 34.9 (28.6, NR) |
| Hazard ratio* (95% CI) | 0.73 (0.60, 0.89) | |
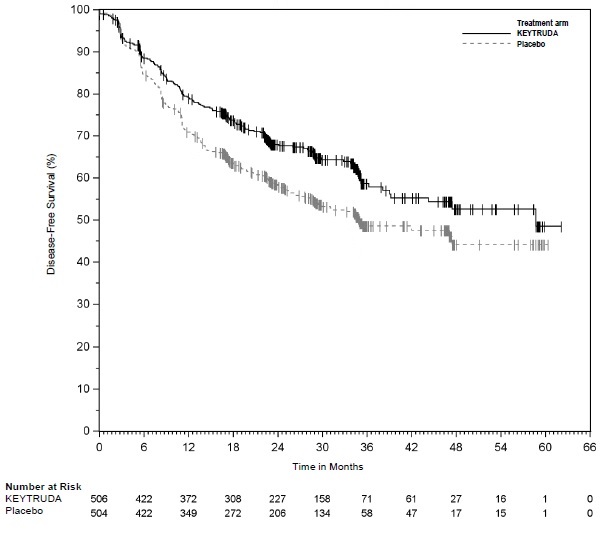 |
14.3 Head and Neck Squamous Cell Cancer
First-line treatment of metastatic or unresectable, recurrent HNSCC
The efficacy of KEYTRUDA was investigated in KEYNOTE-048 (NCT02358031), a randomized, multicenter, open-label, active-controlled trial conducted in 882 patients with metastatic HNSCC who had not previously received systemic therapy for metastatic disease or with recurrent disease who were considered incurable by local therapies. Patients with active autoimmune disease that required systemic therapy within two years of treatment or a medical condition that required immunosuppression were ineligible. Randomization was stratified by tumor PD-L1 expression (TPS ≥50% or <50%) according to the PD-L1 IHC 22C3 pharmDx kit, HPV status according to p16 IHC (positive or negative), and ECOG PS (0 vs. 1). Patients were randomized 1:1:1 to one of the following treatment arms:
- KEYTRUDA 200 mg intravenously every 3 weeks
- KEYTRUDA 200 mg intravenously every 3 weeks, carboplatin AUC 5 mg/mL/min intravenously every 3 weeks or cisplatin 100 mg/m2 intravenously every 3 weeks, and FU 1000 mg/m2/day as a continuous intravenous infusion over 96 hours every 3 weeks (maximum of 6 cycles of platinum and FU)
- Cetuximab 400 mg/m2 intravenously as the initial dose then 250 mg/m2 intravenously once weekly, carboplatin AUC 5 mg/mL/min intravenously every 3 weeks or cisplatin 100 mg/m2 intravenously every 3 weeks, and FU 1000 mg/m2/day as a continuous intravenous infusion over 96 hours every 3 weeks (maximum of 6 cycles of platinum and FU)
Treatment with KEYTRUDA continued until RECIST v1.1-defined progression of disease as determined by the investigator, unacceptable toxicity, or a maximum of 24 months. Administration of KEYTRUDA was permitted beyond RECIST-defined disease progression if the patient was clinically stable and considered to be deriving clinical benefit by the investigator. Assessment of tumor status was performed at Week 9 and then every 6 weeks for the first year, followed by every 9 weeks through 24 months. A retrospective re-classification of patients' tumor PD-L1 status according to CPS using the PD-L1 IHC 22C3 pharmDx kit was conducted using the tumor specimens used for randomization.
The main efficacy outcome measures were OS and PFS as assessed by BICR according to RECIST v1.1 (modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ) sequentially tested in the subgroup of patients with CPS ≥20, the subgroup of patients with CPS ≥1, and the overall population.
The study population characteristics were: median age of 61 years (range: 20 to 94), 36% age 65 or older; 83% male; 73% White, 20% Asian and 2.4% Black; 61% had ECOG PS of 1; and 79% were former/current smokers. Twenty-two percent of patients' tumors were HPV-positive, 23% had PD-L1 TPS ≥50%, and 95% had Stage IV disease (Stage IVA 19%, Stage IVB 6%, and Stage IVC 70%). Eighty-five percent of patients' tumors had PD-L1 expression of CPS ≥1 and 43% had CPS ≥20.
The trial demonstrated a statistically significant improvement in OS for patients randomized to KEYTRUDA in combination with chemotherapy compared to those randomized to cetuximab in combination with chemotherapy at a pre-specified interim analysis in the overall population. Table 69 and Figure 12 summarize efficacy results for KEYTRUDA in combination with chemotherapy.
| Endpoint | KEYTRUDA 200 mg every 3 weeks Platinum FU | Cetuximab Platinum FU |
|---|---|---|
| n=281 | n=278 | |
| OS | ||
| Number (%) of patients with event | 197 (70%) | 223 (80%) |
| Median in months (95% CI) | 13.0 (10.9, 14.7) | 10.7 (9.3, 11.7) |
| Hazard ratio† (95% CI) | 0.77 (0.63, 0.93) | |
| p-Value‡ | 0.0067 | |
| PFS | ||
| Number of patients with event (%) | 244 (87%) | 253 (91%) |
| Median in months (95% CI) | 4.9 (4.7, 6.0) | 5.1 (4.9, 6.0) |
| Hazard ratio† (95% CI) | 0.92 (0.77, 1.10) | |
| p-Value‡ | 0.3394 | |
| Objective Response Rate | ||
| ORR§ (95% CI) | 36% (30.0, 41.5) | 36% (30.7, 42.3) |
| Complete response rate | 6% | 3% |
| Partial response rate | 30% | 33% |
| Duration of Response | ||
| Median in months (range) | 6.7 (1.6+, 30.4+) | 4.3 (1.2+, 27.9+) |
At the pre-specified final OS analysis for the ITT population, the hazard ratio was 0.72 (95% CI: 0.60, 0.87). In addition, KEYNOTE-048 demonstrated a statistically significant improvement in OS for the subgroups of patients with PD-L1 CPS ≥1 (HR=0.65, 95% CI: 0.53, 0.80) and CPS ≥20 (HR=0.60, 95% CI: 0.45, 0.82).
|
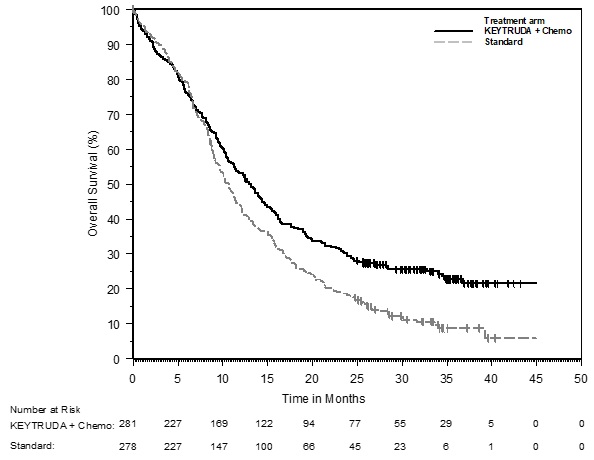 |
The trial also demonstrated a statistically significant improvement in OS for the subgroup of patients with PD-L1 CPS ≥1 randomized to KEYTRUDA as a single agent compared to those randomized to cetuximab in combination with chemotherapy at a pre-specified interim analysis. At the time of the interim and final analyses, there was no significant difference in OS between the KEYTRUDA single agent arm and the control arm for the overall population.
Table 70 summarizes efficacy results for KEYTRUDA as a single agent in the subgroups of patients with CPS ≥1 HNSCC and CPS ≥20 HNSCC. Figure 13 summarizes the OS results in the subgroup of patients with CPS ≥1 HNSCC.
| Endpoint | CPS ≥1 | CPS ≥20 | ||
|---|---|---|---|---|
| KEYTRUDA 200 mg every 3 weeks | Cetuximab Platinum FU | KEYTRUDA 200 mg every 3 weeks | Cetuximab Platinum FU |
|
| n=257 | n=255 | n=133 | n=122 | |
| OS | ||||
| Number of events (%) | 177 (69%) | 206 (81%) | 82 (62%) | 95 (78%) |
| Median in months (95% CI) | 12.3 (10.8, 14.9) | 10.3 (9.0, 11.5) | 14.9 (11.6, 21.5) | 10.7 (8.8, 12.8) |
| Hazard ratio† (95% CI) | 0.78 (0.64, 0.96) | 0.61 (0.45, 0.83) | ||
| p-Value‡ | 0.0171 | 0.0015 | ||
| PFS | ||||
| Number of events (%) | 225 (88%) | 231 (91%) | 113 (85%) | 111 (91%) |
| Median in months (95% CI) | 3.2 (2.2, 3.4) | 5.0 (4.8, 5.8) | 3.4 (3.2, 3.8) | 5.0 (4.8, 6.2) |
| Hazard ratio† (95% CI) | 1.15 (0.95, 1.38) | 0.97 (0.74, 1.27) | ||
| Objective Response Rate | ||||
| ORR§ (95% CI) | 19% (14.5, 24.4) | 35% (29.1, 41.1) | 23% (16.4, 31.4) | 36% (27.6, 45.3) |
| Complete response rate | 5% | 3% | 8% | 3% |
| Partial response rate | 14% | 32% | 16% | 33% |
| Duration of Response | ||||
| Median in months (range) | 20.9 (1.5+, 34.8+) | 4.5 (1.2+, 28.6+) | 20.9 (2.7, 34.8+) | 4.2 (1.2+, 22.3+) |
At the pre-specified final OS analysis comparing KEYTRUDA as a single agent to cetuximab in combination with chemotherapy, the hazard ratio for the subgroup of patients with CPS ≥1 was 0.74 (95% CI: 0.61, 0.90) and the hazard ratio for the subgroup of patients with CPS ≥20 was 0.58 (95% CI: 0.44, 0.78).
In an exploratory subgroup analysis for patients with CPS 1-19 HNSCC at the time of the pre-specified final OS analysis, the median OS was 10.8 months (95% CI: 9.0, 12.6) for KEYTRUDA as a single agent and 10.1 months (95% CI: 8.7, 12.1) for cetuximab in combination with chemotherapy, with an HR of 0.86 (95% CI: 0.66, 1.12).
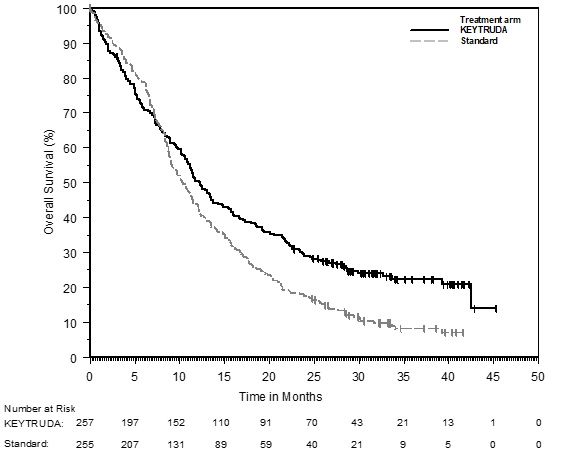
Previously treated recurrent or metastatic HNSCC
The efficacy of KEYTRUDA was investigated in KEYNOTE-012 (NCT01848834), a multicenter, non-randomized, open-label, multi-cohort study that enrolled 174 patients with recurrent or metastatic HNSCC who had disease progression on or after platinum-containing chemotherapy administered for recurrent or metastatic HNSCC or following platinum-containing chemotherapy administered as part of induction, concurrent, or adjuvant therapy. Patients with active autoimmune disease, a medical condition that required immunosuppression, evidence of interstitial lung disease, or ECOG PS ≥2 were ineligible.
Patients received KEYTRUDA 10 mg/kg every 2 weeks (n=53) or 200 mg every 3 weeks (n=121) until unacceptable toxicity or disease progression that was symptomatic, was rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at least 4 weeks later with repeat imaging. Patients without disease progression were treated for up to 24 months. Treatment with pembrolizumab could be reinitiated for subsequent disease progression and administered for up to 1 additional year. Assessment of tumor status was performed every 8 weeks. The major efficacy outcome measures were ORR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, as assessed by BICR, and DoR.
The study population characteristics were median age of 60 years, 32% age 65 or older; 82% male; 75% White, 16% Asian, and 6% Black; 87% had M1 disease; 33% had HPV positive tumors; 63% had prior cetuximab; 29% had an ECOG PS of 0 and 71% had an ECOG PS of 1; and the median number of prior lines of therapy administered for the treatment of HNSCC was 2.
The ORR was 16% (95% CI: 11, 22) with a complete response rate of 5%. The median follow-up time was 8.9 months. Among the 28 responding patients, the median DoR had not been reached (range: 2.4+ to 27.7+ months), with 23 patients having responses of 6 months or longer. The ORR and DoR were similar irrespective of dosage regimen (10 mg/kg every 2 weeks or 200 mg every 3 weeks) or HPV status.
14.4 Classical Hodgkin Lymphoma
KEYNOTE-204
The efficacy of KEYTRUDA was investigated in KEYNOTE-204 (NCT02684292), a randomized, open-label, active controlled trial conducted in 304 patients with relapsed or refractory cHL. The trial enrolled adults with relapsed or refractory disease after at least one multi-agent chemotherapy regimen. Patients were randomized (1:1) to receive:
- KEYTRUDA 200 mg intravenously every 3 weeks or
- Brentuximab vedotin (BV) 1.8 mg/kg intravenously every 3 weeks
Treatment was continued until unacceptable toxicity, disease progression, or a maximum of 35 cycles (up to approximately 2 years). Disease assessment was performed every 12 weeks. Randomization was stratified by prior autologous HSCT (yes vs. no) and disease status after frontline therapy (primary refractory vs. relapse <12 months after completion vs. relapse ≥12 months after completion). The main efficacy measure was PFS as assessed by BICR using 2007 revised International Working Group criteria.
The study population characteristics were: median age of 35 years (range: 18 to 84); 57% male; 77% White, 9% Asian, 3.9% Black. The median number of prior therapies was 2 (range: 1 to 10) in the KEYTRUDA arm and 3 (range: 1 to 11) in the BV arm, with 18% in both arms having 1 prior line. Forty-two percent of patients were refractory to the last prior therapy, 29% had primary refractory disease, 37% had prior autologous HSCT, 5% had received prior BV, and 39% had prior radiation therapy.
Efficacy is summarized in Table 71 and Figure 14.
| Endpoint | KEYTRUDA 200 mg every 3 weeks n=151 | Brentuximab Vedotin 1.8 mg/kg every 3 weeks n=153 |
|---|---|---|
| + Denotes a censored value. | ||
| PFS | ||
| Number of patients with event (%) | 81 (54%) | 88 (58%) |
| Median in months (95% CI)* | 13.2 (10.9, 19.4) | 8.3 (5.7, 8.8) |
| Hazard ratio† (95% CI) | 0.65 (0.48, 0.88) | |
| p-Value‡ | 0.0027 | |
| Objective Response Rate | ||
| ORR§ (95% CI) | 66% (57, 73) | 54% (46, 62) |
| Complete response | 25% | 24% |
| Partial response | 41% | 30% |
| Duration of Response | ||
| Median in months (range)* | 20.7 (0.0+, 33.2+) | 13.8 (0.0+, 33.9+) |
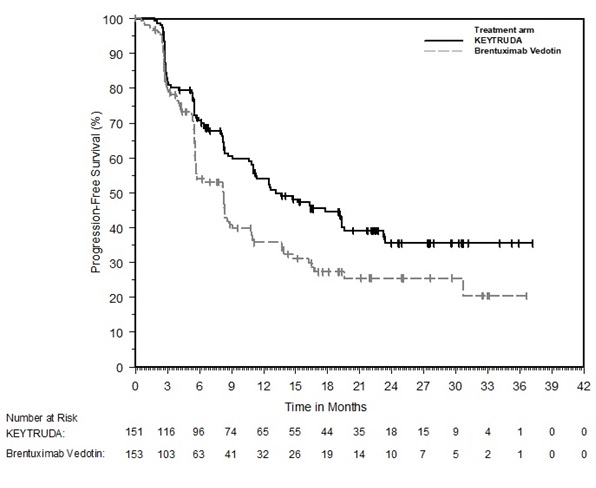 |
KEYNOTE-087
The efficacy of KEYTRUDA was investigated in KEYNOTE-087 (NCT02453594), a multicenter, non-randomized, open-label trial in 210 patients with relapsed or refractory cHL. Patients with active, non-infectious pneumonitis, an allogeneic HSCT within the past 5 years (or >5 years but with symptoms of GVHD), active autoimmune disease, a medical condition that required immunosuppression, or an active infection requiring systemic therapy were ineligible for the trial. Patients received KEYTRUDA 200 mg intravenously every 3 weeks until unacceptable toxicity or documented disease progression, or for up to 24 months in patients who did not progress. Disease assessment was performed every 12 weeks. The major efficacy outcome measures (ORR, Complete Response Rate, and DoR) were assessed by BICR according to the 2007 revised International Working Group (IWG) criteria.
The study population characteristics were: median age of 35 years (range: 18 to 76), 9% age 65 or older; 54% male; 88% White; and 49% ECOG PS of 0 and 51% ECOG PS of 1. The median number of prior lines of therapy administered for the treatment of cHL was 4 (range: 1 to 12). Fifty-eight percent were refractory to the last prior therapy, including 35% with primary refractory disease and 14% whose disease was chemo-refractory to all prior regimens. Sixty-one percent of patients had undergone prior autologous HSCT, 83% had received prior brentuximab vedotin and 36% of patients had prior radiation therapy.
Efficacy results for KEYNOTE-087 are summarized in Table 72.
14.5 Primary Mediastinal Large B-Cell Lymphoma
The efficacy of KEYTRUDA was investigated in KEYNOTE-170 (NCT02576990), a multicenter, open-label, single-arm trial in 53 patients with relapsed or refractory PMBCL. Patients were not eligible if they had active non-infectious pneumonitis, allogeneic HSCT within the past 5 years (or >5 years but with symptoms of GVHD), active autoimmune disease, a medical condition that required immunosuppression, or an active infection requiring systemic therapy. Patients were treated with KEYTRUDA 200 mg intravenously every 3 weeks until unacceptable toxicity or documented disease progression, or for up to 24 months for patients who did not progress. Disease assessments were performed every 12 weeks and assessed by BICR according to the 2007 revised IWG criteria. The efficacy outcome measures were ORR and DoR.
The study population characteristics were: median age of 33 years (range: 20 to 61 years); 43% male; 92% White; and 43% ECOG PS of 0 and 57% ECOG PS of 1. The median number of prior lines of therapy administered for the treatment of PMBCL was 3 (range 2 to 8). Thirty-six percent had primary refractory disease, 49% had relapsed disease refractory to the last prior therapy, and 15% had untreated relapse. Twenty-six percent of patients had undergone prior autologous HSCT, and 32% of patients had prior radiation therapy. All patients had received rituximab as part of a prior line of therapy.
For the 24 responders, the median time to first objective response (complete or partial response) was 2.8 months (range 2.1 to 8.5 months). Efficacy results for KEYNOTE-170 are summarized in Table 73.
14.6 Urothelial Cancer
In Combination with Enfortumab Vedotin for the Treatment of Patients with Urothelial Cancer
The efficacy of KEYTRUDA in combination with enfortumab vedotin was evaluated in KEYNOTE-A39 (NCT04223856), an open-label, randomized, multicenter trial that enrolled 886 patients with locally advanced or metastatic urothelial cancer who received no prior systemic therapy for locally advanced or metastatic disease. Patients with active CNS metastases, ongoing sensory or motor neuropathy Grade ≥2, or uncontrolled diabetes defined as hemoglobin A1C (HbA1c) ≥8% or HbA1c ≥7% with associated diabetes symptoms were excluded.
Patients were randomized 1:1 to receive either:
- KEYTRUDA 200 mg over 30 minutes on Day 1 and enfortumab vedotin 1.25 mg/kg on Days 1 and 8 of each 21-day cycle. KEYTRUDA was given approximately 30 minutes after enfortumab vedotin. Treatment was continued until disease progression or unacceptable toxicity. In the absence of disease progression or unacceptable toxicity, KEYTRUDA was continued for up to 2 years.
- Gemcitabine 1000 mg/m2 on Days 1 and 8 of a 21-day cycle with cisplatin 70 mg/m2 or carboplatin (AUC = 4.5 or 5) on Day 1 of a 21-day cycle. Treatment was continued until disease progression or unacceptable toxicity for up to 6 cycles.
Randomization was stratified by cisplatin eligibility, PD-L1 expression, and presence of liver metastases.
The median age was 69 years (range: 22 to 91); 77% were male; 67% were White, 22% were Asian, 1% were Black or African American, and 10% were unknown or other; 12% were Hispanic or Latino. Patients had a baseline ECOG performance status of 0 (49%), 1 (47%), or 2 (3%). Forty-seven percent of patients had a documented baseline HbA1c of <5.7%. At baseline, 95% of patients had metastatic urothelial cancer, including 72% with visceral and 22% with liver metastases, and 5% had locally advanced urothelial cancer. Eighty-five percent of patients had urothelial carcinoma (UC) histology including 6% with UC mixed squamous differentiation and 2% with UC mixed other histologic variants. Forty-six percent of patients were considered cisplatin-ineligible and 54% were considered cisplatin-eligible at time of randomization.
The major efficacy outcome measures were OS and PFS as assessed by BICR according to RECIST v1.1. Additional efficacy outcome measures included ORR as assessed by BICR.
The trial demonstrated statistically significant improvements in OS, PFS, and ORR for patients randomized to KEYTRUDA in combination with enfortumab vedotin as compared to platinum-based chemotherapy. Efficacy results were consistent across all stratified patient subgroups.
Table 74 and Figures 15 and 16 summarize the efficacy results for KEYNOTE-A39.
| Endpoint | KEYTRUDA 200 mg every 3 weeks in combination with Enfortumab Vedotin n=442 | Cisplatin or carboplatin with gemcitabine n=444 |
|---|---|---|
| NR = not reached | ||
|
||
| OS | ||
| Number (%) of patients with event | 133 (30%) | 226 (51%) |
| Median in months (95% CI) | 31.5 (25.4, NR) | 16.1 (13.9, 18.3) |
| Hazard ratio* (95% CI) | 0.47 (0.38, 0.58) | |
| p-Value† | <0.0001 | |
| PFS | ||
| Number (%) of patients with event | 223 (50%) | 307 (69%) |
| Median in months (95% CI) | 12.5 (10.4, 16.6) | 6.3 (6.2, 6.5) |
| Hazard ratio* (95% CI) | 0.45 (0.38, 0.54) | |
| p-Value† | <0.0001 | |
| Confirmed Objective Response Rate‡ | ||
| ORR§ % (95% CI) | 68% (63, 72) | 44% (40, 49) |
| p-Value¶ | <0.0001 | |
| Complete response | 29% | 12% |
| Partial response | 39% | 32% |
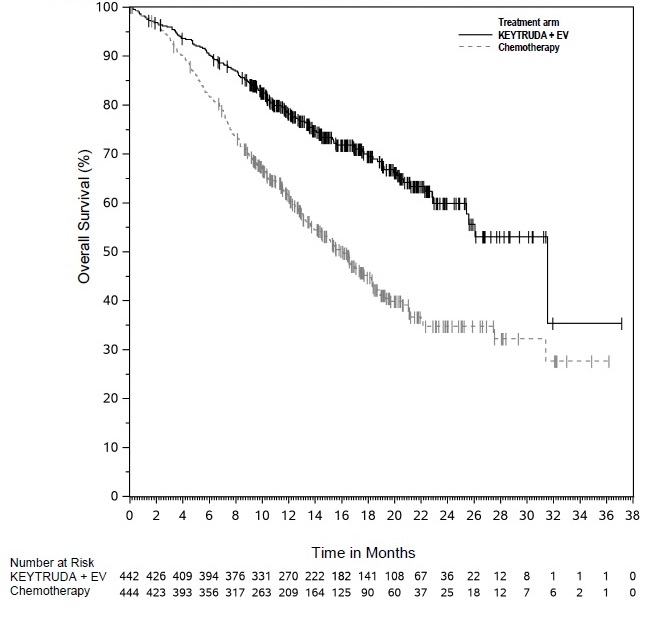 |
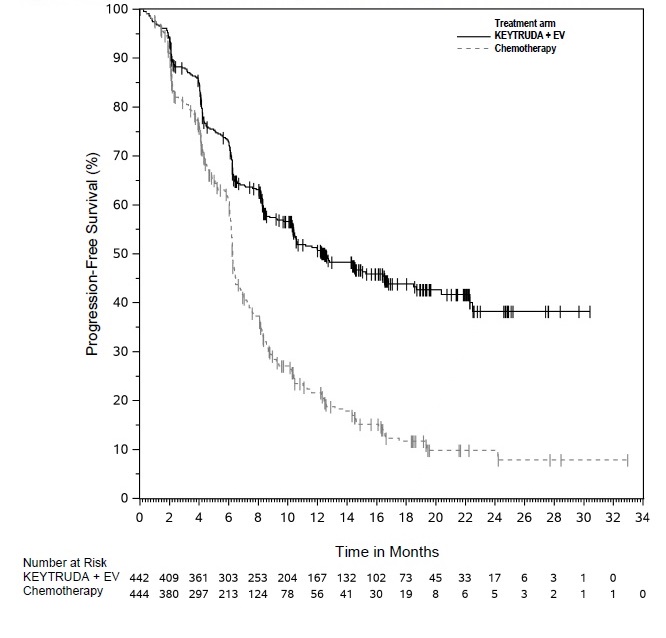 |
In Combination with Enfortumab Vedotin for the Treatment of Cisplatin-Ineligible Patients with Urothelial Cancer
The efficacy of KEYTRUDA in combination with enfortumab vedotin was evaluated in KEYNOTE-869 (NCT03288545), an open-label, multi-cohort (dose escalation cohort, Cohort A, Cohort K) study in patients with locally advanced or metastatic urothelial cancer who were ineligible for cisplatin-containing chemotherapy and received no prior systemic therapy for locally advanced or metastatic disease. Patients with active CNS metastases, ongoing sensory or motor neuropathy Grade ≥2, or uncontrolled diabetes defined as hemoglobin A1C (HbA1c) ≥8% or HbA1c ≥7% with associated diabetes symptoms were excluded from participating in the study.
Patients in the dose escalation cohort (n=5), Cohort A (n=40), and Cohort K (n=76) received enfortumab vedotin 1.25 mg/kg as an IV infusion over 30 minutes on Days 1 and 8 of a 21-day cycle followed by KEYTRUDA 200 mg as an IV infusion on Day 1 of a 21-day cycle approximately 30 minutes after enfortumab vedotin. Patients were treated until disease progression or unacceptable toxicity.
A total of 121 patients received KEYTRUDA in combination with enfortumab vedotin. The median age was 71 years (range: 51 to 91); 74% were male; 85% were White, 5% were Black, 4% were Asian and 6% were other, unknown or not reported. Ten percent of patients were Hispanic or Latino. Forty-five percent of patients had an ECOG performance status of 1 and 15% had an ECOG performance status of 2. Forty-seven percent of patients had a documented baseline HbA1c of <5.7%. Reasons for cisplatin-ineligibility included: 60% with baseline creatinine clearance of 30-59 mL/min, 10% with ECOG PS of 2, 13% with Grade 2 or greater hearing loss, and 16% with more than one cisplatin-ineligibility criteria.
At baseline, 97.5% of patients had metastatic urothelial cancer and 2.5% of patients had locally advanced urothelial cancer. Thirty-seven percent of patients had upper tract disease. Eighty-four percent of patients had visceral metastasis at baseline, including 22% with liver metastases. Thirty-nine percent of patients had TCC histology; 13% had TCC with squamous differentiation, and 48% had TCC with other histologic variants.
The major efficacy outcome measures were ORR and DoR as assessed by BICR according to RECIST v1.1.
The median follow-up time for the dose escalation cohort + Cohort A was 44.7 months (range 0.7 to 52.4) and for Cohort K was 14.8 months (range: 0.6 to 26.2).
Efficacy results are presented in Table 75 below.
| Endpoint | KEYTRUDA in combination with Enfortumab Vedotin n=121 |
|---|---|
| Confirmed ORR (95% CI) | 68% (58.7, 76.0) |
| Complete response rate | 12% |
| Partial response rate | 55% |
The median duration of response for the dose escalation cohort + Cohort A was 22.1 months (range: 1.0+ to 46.3+) and for Cohort K was not reached (range: 1.2 to 24.1+).
Platinum-Ineligible Patients with Urothelial Carcinoma
The efficacy of KEYTRUDA was investigated in KEYNOTE-052 (NCT02335424), a multicenter, open-label, single-arm trial in 370 patients with locally advanced or metastatic urothelial carcinoma who had one or more comorbidities, including patients who were not eligible for any platinum-containing chemotherapy. The trial excluded patients with autoimmune disease or a medical condition that required immunosuppression. Patients received KEYTRUDA 200 mg every 3 weeks until unacceptable toxicity or disease progression. Patients with initial radiographic disease progression could receive additional doses of treatment during confirmation of progression unless disease progression was symptomatic, was rapidly progressive, required urgent intervention, or occurred with a decline in performance status. Patients without disease progression could be treated for up to 24 months. Tumor response assessments were performed at 9 weeks after the first dose, then every 6 weeks for the first year, and then every 12 weeks thereafter. The major efficacy outcome measures were ORR and DoR as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
The study population characteristics were: median age of 74 years; 77% male; and 89% White. Eighty-seven percent had M1 disease, and 13% had M0 disease. Eighty-one percent had a primary tumor in the lower tract, and 19% of patients had a primary tumor in the upper tract. Eighty-five percent of patients had visceral metastases, including 21% with liver metastases. Fifty percent of patients had baseline creatinine clearance of <60 mL/min, 32% had ECOG PS of 2, 9% had ECOG PS of 2 and baseline creatinine clearance of <60 mL/min, and 9% had one or more of Class III heart failure, Grade 2 or greater peripheral neuropathy, and Grade 2 or greater hearing loss. Ninety percent of patients were treatment naïve, and 10% received prior adjuvant or neoadjuvant platinum-based chemotherapy.
The median follow-up time for 370 patients treated with KEYTRUDA was 11.4 months (range 0.1 to 63.8 months). Efficacy results are summarized in Table 76.
| Endpoint | KEYTRUDA 200 mg every 3 weeks |
|---|---|
| All Subjects n=370 |
|
| + Denotes ongoing response | |
| Objective Response Rate | |
| ORR (95% CI) | 29% (24, 34) |
| Complete response rate | 10% |
| Partial response rate | 20% |
| Duration of Response | |
| Median in months (range) | 33.4 (1.4+, 60.7+) |
Platinum-Eligible Patients with Previously Untreated Urothelial Carcinoma
The efficacy of KEYTRUDA for the first-line treatment of platinum-eligible patients with locally advanced or metastatic urothelial carcinoma was investigated in KEYNOTE-361 (NCT02853305), a multicenter, randomized, open-label, active-controlled study in 1010 previously untreated patients. The safety and efficacy of KEYTRUDA in combination with platinum-based chemotherapy for previously untreated patients with locally advanced or metastatic urothelial carcinoma has not been established.
The study compared KEYTRUDA with or without platinum-based chemotherapy (i.e., cisplatin or carboplatin with gemcitabine) to platinum-based chemotherapy alone. Among the patients receiving KEYTRUDA plus platinum-based chemotherapy, 44% received cisplatin and 56% received carboplatin.
The study did not meet its major efficacy outcome measures of improved PFS or OS in the KEYTRUDA plus chemotherapy arm compared to the chemotherapy-alone arm. Additional efficacy endpoints, including improvement of OS in the KEYTRUDA monotherapy arm, could not be formally tested.
Previously Treated Urothelial Carcinoma
The efficacy of KEYTRUDA was investigated in KEYNOTE-045 (NCT02256436), a multicenter, randomized (1:1), active-controlled trial in 542 patients with locally advanced or metastatic urothelial carcinoma with disease progression on or after platinum-containing chemotherapy. The trial excluded patients with autoimmune disease or a medical condition that required immunosuppression.
Patients were randomized to receive either KEYTRUDA 200 mg every 3 weeks (n=270) or investigator's choice of any of the following chemotherapy regimens all given intravenously every 3 weeks (n=272): paclitaxel 175 mg/m2 (n=90), docetaxel 75 mg/m2 (n=92), or vinflunine 320 mg/m2 (n=90). Treatment continued until unacceptable toxicity or disease progression. Patients with initial radiographic disease progression could receive additional doses of treatment during confirmation of progression unless disease progression was symptomatic, was rapidly progressive, required urgent intervention, or occurred with a decline in performance status. Patients without disease progression could be treated for up to 24 months. Assessment of tumor status was performed at 9 weeks after randomization, then every 6 weeks through the first year, followed by every 12 weeks thereafter. The major efficacy outcomes were OS and PFS as assessed by BICR per RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ. Additional efficacy outcome measures were ORR as assessed by BICR per RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, and DoR.
The study population characteristics were: median age of 66 years (range: 26 to 88), 58% age 65 or older; 74% male; 72% White and 23% Asian; 42% ECOG PS of 0 and 56% ECOG PS of 1; and 96% M1 disease and 4% M0 disease. Eighty-seven percent of patients had visceral metastases, including 34% with liver metastases. Eighty-six percent had a primary tumor in the lower tract and 14% had a primary tumor in the upper tract. Fifteen percent of patients had disease progression following prior platinum-containing neoadjuvant or adjuvant chemotherapy. Twenty-one percent had received 2 or more prior systemic regimens in the metastatic setting. Seventy-six percent of patients received prior cisplatin, 23% had prior carboplatin, and 1% were treated with other platinum-based regimens.
The study demonstrated statistically significant improvements in OS and ORR for patients randomized to KEYTRUDA as compared to chemotherapy. There was no statistically significant difference between KEYTRUDA and chemotherapy with respect to PFS. The median follow-up time for this trial was 9.0 months (range: 0.2 to 20.8 months). Table 77 and Figure 17 summarize the efficacy results for KEYNOTE-045.
| KEYTRUDA 200 mg every 3 weeks | Chemotherapy | |
|---|---|---|
| n=270 | n=272 | |
| + Denotes ongoing response NR = not reached |
||
|
||
| OS | ||
| Deaths (%) | 155 (57%) | 179 (66%) |
| Median in months (95% CI) | 10.3 (8.0, 11.8) | 7.4 (6.1, 8.3) |
| Hazard ratio* (95% CI) | 0.73 (0.59, 0.91) | |
| p-Value (stratified log-rank) | 0.004 | |
| PFS by BICR | ||
| Events (%) | 218 (81%) | 219 (81%) |
| Median in months (95% CI) | 2.1 (2.0, 2.2) | 3.3 (2.3, 3.5) |
| Hazard ratio* (95% CI) | 0.98 (0.81, 1.19) | |
| p-Value (stratified log-rank) | 0.833 | |
| Objective Response Rate | ||
| ORR (95% CI) | 21% (16, 27) | 11% (8, 16) |
| Complete response rate | 7% | 3% |
| Partial response rate | 14% | 8% |
| p-Value (Miettinen-Nurminen) | 0.002 | |
| Median duration of response in months (range) | NR (1.6+, 15.6+) | 4.3 (1.4+, 15.4+) |
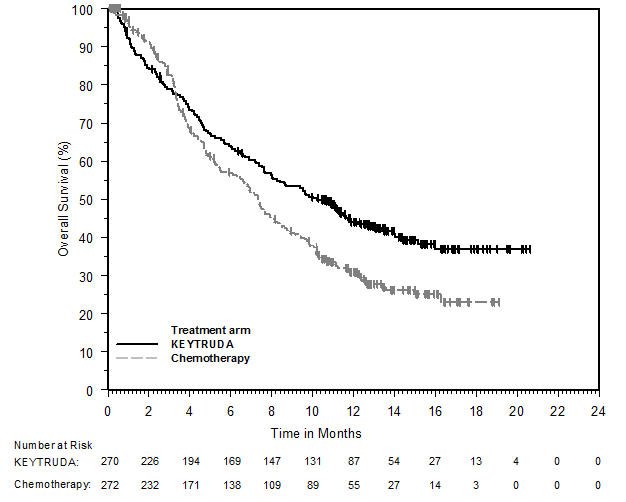 |
BCG-unresponsive High-Risk Non-Muscle Invasive Bladder Cancer
The efficacy of KEYTRUDA was investigated in KEYNOTE-057 (NCT02625961), a multicenter, open-label, single-arm trial in 96 patients with Bacillus Calmette-Guerin (BCG)-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS) with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy. BCG-unresponsive high-risk NMIBC was defined as persistent disease despite adequate BCG therapy, disease recurrence after an initial tumor-free state following adequate BCG therapy, or T1 disease following a single induction course of BCG. Adequate BCG therapy was defined as administration of at least five of six doses of an initial induction course plus either of: at least two of three doses of maintenance therapy or at least two of six doses of a second induction course. Prior to treatment, all patients had undergone transurethral resection of bladder tumor (TURBT) to remove all resectable disease (Ta and T1 components). Residual CIS (Tis components) not amenable to complete resection was allowed. The trial excluded patients with muscle invasive (i.e., T2, T3, T4) locally advanced non-resectable or metastatic urothelial carcinoma, concurrent extra-vesical (i.e., urethra, ureter or renal pelvis) non-muscle invasive transitional cell carcinoma of the urothelium, or autoimmune disease or a medical condition that required immunosuppression.
Patients received KEYTRUDA 200 mg every 3 weeks until unacceptable toxicity, persistent or recurrent high-risk NMIBC, or progressive disease. Assessment of tumor status was performed every 12 weeks for two years and then every 24 weeks for three years, and patients without disease progression could be treated for up to 24 months. The major efficacy outcome measures were complete response (as defined by negative results for cystoscopy [with TURBT/biopsies as applicable], urine cytology, and computed tomography urography [CTU] imaging) and duration of response.
The study population characteristics were: median age of 73 years (range: 44 to 92); 44% age ≥75; 84% male; 67% White; and 73% and 27% with an ECOG performance status of 0 or 1, respectively. Tumor pattern at study entry was CIS with T1 (13%), CIS with high grade TA (25%), and CIS (63%). Baseline high-risk NMIBC disease status was 27% persistent and 73% recurrent. The median number of prior instillations of BCG was 12.
The median follow-up time was 28.0 months (range: 4.6 to 40.5 months). Efficacy results are summarized in Table 78.
14.7 Microsatellite Instability-High or Mismatch Repair Deficient Cancer
The efficacy of KEYTRUDA was investigated in 504 patients with MSI-H or dMMR cancers enrolled in three multicenter, non-randomized, open-label, multi-cohort trials: KEYNOTE-164 (NCT02460198), KEYNOTE-158 (NCT02628067), and KEYNOTE-051 (NCT02332668). All trials excluded patients with autoimmune disease or a medical condition that required immunosuppression. Regardless of histology, MSI or MMR tumor status was determined using polymerase chain reaction (PCR; local or central) or immunohistochemistry (IHC; local or central), respectively.
- KEYNOTE-164 enrolled 124 patients with advanced MSI-H or dMMR colorectal cancer (CRC) that progressed following treatment with fluoropyrimidine and either oxaliplatin or irinotecan +/- anti-VEGF/EGFR mAb-based therapy.
- KEYNOTE-158 enrolled 373 patients with advanced MSI-H or dMMR non-colorectal cancers (non-CRC) who had disease progression following prior therapy. Patients were either prospectively enrolled with MSI-H/dMMR tumors (Cohort K) or retrospectively identified in one of 10 solid tumor cohorts (Cohorts A-J).
- KEYNOTE-051 enrolled 7 pediatric patients with MSI-H or dMMR cancers.
Adult patients received KEYTRUDA 200 mg every 3 weeks (pediatric patients received 2 mg/kg every 3 weeks) until unacceptable toxicity, disease progression, or a maximum of 24 months. In KEYNOTE-164 and KEYNOTE-158, assessment of tumor status was performed every 9 weeks through the first year, then every 12 weeks thereafter. In KEYNOTE-051, assessment of tumor status was performed every 8 weeks for 24 weeks, and then every 12 weeks thereafter. The major efficacy outcome measures were ORR and DoR as assessed by BICR according to RECIST v1.1 (modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ in KEYNOTE-158) and as assessed by the investigator according to RECIST v1.1 in KEYNOTE-051.
In KEYNOTE-164 and KEYNOTE-158, the study population characteristics were median age of 60 years, 36% age 65 or older; 44% male; 78% White, 14% Asian, 4% American Indian or Alaska Native, and 3% Black; and 45% ECOG PS of 0 and 55% ECOG PS of 1. Ninety-two percent of patients had metastatic disease and 4% had locally advanced, unresectable disease. Thirty-seven percent of patients received one prior line of therapy and 61% received two or more prior lines of therapy.
In KEYNOTE-051, the study population characteristics were median age of 11 years (range: 3 to 16); 71% female; 86% White and 14% Asian; and 57% had a Lansky/Karnofsky Score of 100. Seventy-one percent of patients had Stage IV and 14% had Stage III disease. Fifty-seven percent of patients received one prior line of therapy and 29% received two prior lines of therapy.
Discordant results were observed between local MSI-H or dMMR tests and central testing among patients enrolled in Cohort K of KEYNOTE-158. Among 104 tumor samples that were MSI-H or dMMR by local testing and also tested using the FoundationOne®CDx (F1CDx) test, 59 (56.7%) were MSI-H and 45 (43.3%) were not MSI-H. Among 169 tumor samples that were MSI-H or dMMR by local testing and also tested using the VENTANA MMR RxDx Panel, 105 (62.1%) were dMMR and 64 (37.9%) were pMMR.
Efficacy results are summarized in Tables 79 and 80.
| Endpoint | KEYTRUDA n=504* |
|---|---|
| + Denotes ongoing response | |
| Objective Response Rate | |
| ORR (95% CI)† | 33.3% (29.2, 37.6) |
| Complete response rate | 10.3% |
| Partial response rate | 23.0% |
| Duration of Response | n=168 |
| Median in months (range) | 63.2 (1.9+, 63.9+) |
| % with duration ≥12 months | 77% |
| % with duration ≥36 months | 39% |
| Objective Response Rate | Duration of Response range |
|||
|---|---|---|---|---|
| N | n (%) | 95% CI | (months) | |
| + Denotes ongoing response | ||||
|
||||
| CRC | 124 | 42 (34%) | (26%, 43%) | (4.4, 58.5+) |
| Non-CRC* | 380 | 126 (33%) | (28%, 38%) | (1.9+, 63.9+) |
| Endometrial cancer | 94 | 47 (50%) | (40%, 61%) | (2.9, 63.2) |
| Gastric or GE junction cancer | 51 | 20 (39%) | (26%, 54%) | (1.9+, 63.0+) |
| Small intestinal cancer | 27 | 16 (59%) | (39%, 78%) | (3.7+, 57.3+) |
| Brain cancer | 27† | 1 (4%)‡ | (0%, 19%) | 18.9 |
| Ovarian cancer | 25 | 8 (32%) | (15%, 54%) | (4.2, 56.6+) |
| Biliary cancer | 22 | 9 (41%) | (21%, 64%) | (6.2, 49.0+) |
| Pancreatic cancer | 22 | 4 (18%) | (5%, 40%) | (8.1, 24.3+) |
| Sarcoma | 14 | 3 (21%) | (5%, 51%) | (35.4+, 57.2+) |
| Breast cancer | 13 | 1 (8%) | (0%,36%) | 24.3+ |
| Other§ | 13 | 4 (31%) | (9%, 61%) | (6.2+, 32.3+) |
| Cervical cancer | 11 | 1 (9%) | (0%, 41%) | 63.9+ |
| Neuroendocrine cancer | 11 | 1 (9%) | (0%, 41%) | 13.3 |
| Prostate cancer | 8 | 1 (13%) | (0%, 53%) | 24.5+ |
| Adrenocortical cancer | 7 | 1 (14%) | (0%, 58%) | 4.2 |
| Mesothelioma | 7 | 0 (0%) | (0%, 41%) | |
| Thyroid cancer | 7 | 1 (14%) | (0%, 58%) | 8.2 |
| Small cell lung cancer | 6 | 2 (33%) | (4%, 78%) | (20.0, 47.5) |
| Bladder cancer | 6 | 3 (50%) | (12%, 88%) | (35.6+, 57.5+) |
| Salivary cancer | 5 | 2 (40%) | (5%, 85%) | (42.6+, 57.8+) |
| Renal cell cancer | 4 | 1 (25%) | (0%, 81%) | 22.0 |
Exploratory analysis by TMB
In an exploratory analysis performed in 138 patients (Cohort K of KEYNOTE-158) who were tested retrospectively for tumor mutation burden (TMB) using an FDA-approved test, 45 (33%) had tumors with TMB score of <10 mut/Mb; ORR in these 45 patients was 6.7% (95% CI: 1.4, 18.3). Among the 45 patients with TMB score of <10 mut/Mb, 39 of the patients were not MSI-H/dMMR when tested using an FDA-approved test.
14.8 Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
The efficacy of KEYTRUDA was investigated in KEYNOTE-177 (NCT02563002), a multicenter, randomized, open-label, active-controlled trial that enrolled 307 patients with previously untreated unresectable or metastatic MSI-H or dMMR CRC. MSI or MMR tumor status was determined locally using polymerase chain reaction (PCR) or immunohistochemistry (IHC), respectively. Patients with autoimmune disease or a medical condition that required immunosuppression were ineligible.
Patients were randomized (1:1) to receive KEYTRUDA 200 mg intravenously every 3 weeks or investigator’s choice of the following chemotherapy regimens given intravenously every 2 weeks:
- mFOLFOX6 (oxaliplatin, leucovorin, and FU) or mFOLFOX6 in combination with either bevacizumab or cetuximab: Oxaliplatin 85 mg/m2, leucovorin 400 mg/m2 (or levoleucovorin 200 mg/m2), and FU 400 mg/m2 bolus on Day 1, then FU 2400 mg/m2 over 46-48 hours. Bevacizumab 5 mg/kg on Day 1 or cetuximab 400 mg/m2 on first infusion, then 250 mg/m2 weekly.
- FOLFIRI (irinotecan, leucovorin, and FU) or FOLFIRI in combination with either bevacizumab or cetuximab: Irinotecan 180 mg/m2, leucovorin 400 mg/m2 (or levoleucovorin 200 mg/m2), and FU 400 mg/m2 bolus on Day 1, then FU 2400 mg/m2 over 46-48 hours. Bevacizumab 5 mg/kg on Day 1 or cetuximab 400 mg/m2 on first infusion, then 250 mg/m2 weekly.
Treatment with KEYTRUDA or chemotherapy continued until RECIST v1.1-defined progression of disease as determined by the investigator or unacceptable toxicity. Patients treated with KEYTRUDA without disease progression could be treated for up to 24 months. Assessment of tumor status was performed every 9 weeks. Patients randomized to chemotherapy were offered KEYTRUDA at the time of disease progression. The main efficacy outcome measures were PFS (as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ) and OS. Additional efficacy outcome measures were ORR and DoR.
A total of 307 patients were enrolled and randomized to KEYTRUDA (n=153) or chemotherapy (n=154). The baseline characteristics of these 307 patients were: median age of 63 years (range: 24 to 93), 47% age 65 or older; 50% male; 75% White and 16% Asian; 52% had an ECOG PS of 0 and 48% had an ECOG PS of 1; and 27% received prior adjuvant or neoadjuvant chemotherapy. Among 154 patients randomized to receive chemotherapy,143 received chemotherapy per the protocol. Of the 143 patients, 56% received mFOLFOX6, 44% received FOLFIRI, 70% received bevacizumab plus mFOLFOX6 or FOLFIRI, and 11% received cetuximab plus mFOLFOX6 or FOLFIRI.
The trial demonstrated a statistically significant improvement in PFS for patients randomized to KEYTRUDA compared with chemotherapy. There was no statistically significant difference between KEYTRUDA and chemotherapy in the final OS analysis. Sixty percent of the patients who had been randomized to receive chemotherapy had crossed over to receive subsequent anti-PD-1/PD-L1 therapies including KEYTRUDA. The median follow-up time at the final analysis was 38.1 months (range: 0.2 to 58.7 months). Table 81 and Figure 18 summarize the key efficacy measures for KEYNOTE-177.
| Endpoint | KEYTRUDA 200 mg every 3 weeks n=153 | Chemotherapy n=154 |
|---|---|---|
| + Denotes ongoing response NR = not reached |
||
|
||
| PFS | ||
| Number (%) of patients with event | 82 (54%) | 113 (73%) |
| Median in months (95% CI) | 16.5 (5.4, 32.4) | 8.2 (6.1, 10.2) |
| Hazard ratio* (95% CI) | 0.60 (0.45, 0.80) | |
| p-Value† | 0.0004 | |
| OS‡ | ||
| Number (%) of patients with event | 62 (41%) | 78 (51%) |
| Median in months (95% CI) | NR (49.2, NR) | 36.7 (27.6, NR) |
| Hazard ratio* (95% CI) | 0.74 (0.53, 1.03) | |
| p-Value§ | 0.0718 | |
| Objective Response Rate¶ | ||
| ORR (95% CI) | 44% (35.8, 52.0) | 33% (25.8, 41.1) |
| Complete response rate | 11% | 4% |
| Partial response rate | 33% | 29% |
| Duration of Response¶,# | ||
| Median in months (range) | NR (2.3+, 41.4+) | 10.6 (2.8, 37.5+) |
| % with duration ≥12 monthsÞ | 75% | 37% |
| % with duration ≥24 monthsÞ | 43% | 18% |
|
|
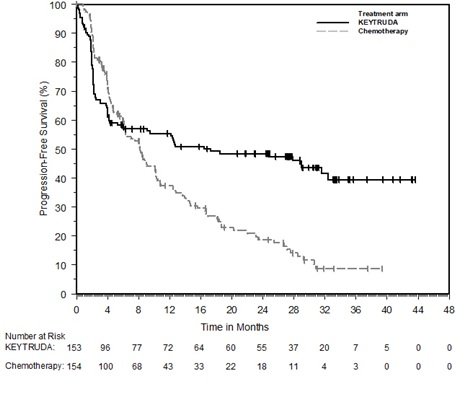
14.9 Gastric Cancer
First-line Treatment of Locally Advanced Unresectable or Metastatic HER2-Positive Gastric or Gastroesophageal Junction Adenocarcinoma
The efficacy of KEYTRUDA in combination with trastuzumab plus fluoropyrimidine and platinum chemotherapy was investigated in KEYNOTE-811 (NCT03615326), a multicenter, randomized, double-blind, placebo-controlled trial that enrolled 698 patients with HER2-positive advanced gastric or gastroesophageal junction (GEJ) adenocarcinoma who had not previously received systemic therapy for metastatic disease. PD-L1 status was determined using the PD-L1 IHC 22C3 pharmDx™ kit. Patients with an autoimmune disease that required systemic therapy within 2 years of treatment or a medical condition that required immunosuppression were ineligible. Randomization was stratified by PD-L1 expression (CPS ≥1 or CPS <1), chemotherapy regimen (5-FU plus cisplatin [FP] or capecitabine plus oxaliplatin [CAPOX]), and geographic region (Europe/Israel/North America/Australia, Asia, or Rest of the World). Patients were randomized (1:1) to one of the following treatment arms:
- KEYTRUDA 200 mg, trastuzumab 8 mg/kg on first infusion and 6 mg/kg in subsequent cycles, followed by investigator’s choice of combination chemotherapy of cisplatin 80 mg/m2 for up to 6 cycles and 5-FU 800 mg/m2/day for 5 days (FP) or oxaliplatin 130 mg/m2 up to 6-8 cycles and capecitabine 1000 mg/m2 bid for 14 days (CAPOX). KEYTRUDA was administered prior to trastuzumab and chemotherapy on Day 1 of each cycle.
- Placebo, trastuzumab 8 mg/kg on first infusion and 6 mg/kg in subsequent cycles, followed by investigator’s choice of combination chemotherapy of cisplatin 80 mg/m2 for up to 6 cycles and 5-FU 800 mg/m2/day for 5 days (FP) or oxaliplatin 130 mg/m2 up to 6-8 cycles and capecitabine 1000 mg/m2 bid for 14 days (CAPOX).
All study medications, except oral capecitabine, were administered as an intravenous infusion every 3-week cycle. Treatment with KEYTRUDA continued until RECIST v1.1-defined progression of disease as determined by BICR, unacceptable toxicity, or a maximum of 24 months. In an interim efficacy analysis, the major outcome measures assessed were ORR and DoR by BICR using RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
At the time of the interim analysis, ORR and DoR were assessed in the first 264 patients randomized. Among the 264 patients, the population characteristics were: median age of 62 years (range: 19 to 84), 41% age 65 or older; 82% male; 63% White, 31% Asian, and 0.8% Black; 47% ECOG PS of 0 and 53% ECOG PS of 1. Ninety-seven percent of patients had metastatic disease (Stage IV) and 3% had locally advanced unresectable disease. Ninety-one percent (n=240) had tumors that were not MSI-H, 1% (n=2) had tumors that were MSI-H, and in 8% (n=22) the status was not known. Eighty-seven percent of patients received CAPOX.
A statistically significant improvement in ORR was demonstrated in patients randomized to KEYTRUDA in combination with trastuzumab and chemotherapy compared with placebo in combination with trastuzumab and chemotherapy. Efficacy results are summarized in Table 82.
| Endpoint | KEYTRUDA 200 mg every 3 weeks Trastuzumab Fluoropyrimidine and Platinum Chemotherapy n=133 | Placebo Trastuzumab Fluoropyrimidine and Platinum Chemotherapy n=131 |
|---|---|---|
| + Denotes ongoing response | ||
| Objective Response Rate | ||
| ORR* (95% CI) | 74% (66, 82) | 52% (43, 61) |
| Complete response rate | 11% | 3.1% |
| Partial response rate | 63% | 49% |
| p-Value† | <0.0001 | |
| Duration of Response | n=99 | n=68 |
| Median in months (range) | 10.6 (1.1+, 16.5+) | 9.5 (1.4+, 15.4+) |
| % with duration ≥6 months‡ | 65% | 53% |
In a pre-specified subgroup analysis of ORR based on PD-L1 status, the ORR in patients with PD-L1-positive disease (CPS ≥1) was 76% (95% CI: 67, 83) in the pembrolizumab arm (n=117) versus 51% (95% CI: 41, 60) in the control arm (n=112). In patients with tumors that were PD-L1 CPS<1, the ORR was 63% (95% CI: 35, 85) in the pembrolizumab arm (n=16) versus 58% (95% CI: 34, 80) in the control arm (n=19).
In a subsequent interim analysis of pre-specified subgroups based on PD-L1 status in the full study population (n=698), the HR for PFS and OS in patients with PD-L1 CPS<1 (N=104) was 1.03 (95% CI: 0.65, 1.64) and 1.41 (95% CI: 0.90, 2.20), respectively.
First-line Treatment of Locally Unresectable or Metastatic HER2-Negative Gastric or Gastroesophageal Junction Adenocarcinoma
The efficacy of KEYTRUDA in combination with fluoropyrimidine- and platinum-containing chemotherapy was investigated in KEYNOTE-859 (NCT03675737), a multicenter, randomized, double-blind, placebo-controlled trial that enrolled 1579 patients with HER2-negative advanced gastric or GEJ adenocarcinoma who had not previously received systemic therapy for metastatic disease. Patients with an autoimmune disease that required systemic therapy within 2 years of treatment or a medical condition that required immunosuppression were ineligible. Randomization was stratified by PD-L1 expression (CPS ≥1 or CPS <1), chemotherapy regimen (FP or CAPOX), and geographic region (Europe/Israel/North America/Australia, Asia, or Rest of the World). Patients were randomized (1:1) to one of the following treatment arms; treatment was administered prior to chemotherapy on Day 1 of each cycle:
- KEYTRUDA 200 mg, investigator’s choice of combination chemotherapy of cisplatin 80 mg/m2 and 5-FU 800 mg/m2/day for 5 days (FP) or oxaliplatin 130 mg/m2 and capecitabine 1000 mg/m2 bid for 14 days (CAPOX).
- Placebo, investigator’s choice of combination chemotherapy of cisplatin 80 mg/m2 and 5-FU 800 mg/m2/day for 5 days (FP) or oxaliplatin 130 mg/m2 and capecitabine 1000 mg/m2 bid for 14 days (CAPOX).
All study medications, except oral capecitabine, were administered as an intravenous infusion every 3-week cycle. Platinum agents could be administered for 6 or more cycles following local guidelines. Treatment with KEYTRUDA continued until RECIST v1.1-defined progression of disease as determined by BICR, unacceptable toxicity, or a maximum of 24 months. The major efficacy outcome measure was OS. Additional secondary efficacy outcome measures included PFS, ORR, and DoR as assessed by BICR using RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
The population characteristics were: median age of 62 years (range: 21 to 86), 39% age 65 or older; 68% male and 32% female; 55% White, 34% Asian, 4.6% Multiple, 4.2% American Indian or Alaskan Native, 1.3% Black, and 0.2% Native Hawaiian or other Pacific Islander; 76% Not Hispanic or Latino and 21% Hispanic or Latino; 37% ECOG PS of 0 and 63% ECOG PS of 1. Ninety-seven percent of patients had metastatic disease (Stage IV) and 3% had locally advanced unresectable disease. Seventy-eight percent had tumors that expressed PD-L1 with a CPS ≥1 and 5% (n=74) had tumors that were MSI-H. Eighty-six percent of patients received CAPOX.
A statistically significant improvement in OS, PFS, and ORR was demonstrated in patients randomized to KEYTRUDA in combination with chemotherapy compared with placebo in combination with chemotherapy at the time of a pre-specified interim analysis of OS. Efficacy results are summarized in Table 83 and Figures 19 and 20.
| Endpoint | KEYTRUDA 200 mg every 3 weeks and FP or CAPOX n=790 | Placebo and FP or CAPOX n=789 | KEYTRUDA 200 mg every 3 weeks and FP or CAPOX n=618 | Placebo and FP or CAPOX n=617 | KEYTRUDA 200 mg every 3 weeks and FP or CAPOX n=279 | Placebo and FP or CAPOX n=272 |
|---|---|---|---|---|---|---|
| All Patients | CPS≥1 | CPS≥10 | ||||
| + Denotes ongoing response | ||||||
|
||||||
| OS | ||||||
| Number (%) of patients with event | 603 (76) | 666 (84) | 464 (75) | 526 (85) | 188 (67) | 226 (83) |
| Median in months (95% CI) | 12.9 (11.9, 14.0) | 11.5 (10.6, 12.1) | 13.0 (11.6, 14.2) | 11.4 (10.5, 12.0) | 15.7 (13.8, 19.3) | 11.8 (10.3, 12.7) |
| Hazard ratio† (95% CI) | 0.78 (0.70, 0.87) | 0.74 (0.65, 0.84) | 0.65 (0.53, 0.79) | |||
| p-Value (stratified log-rank)‡ | <0.0001 | <0.0001 | <0.0001 | |||
| PFS | ||||||
| Number (%) of patients with event | 572 (72) | 608 (77) | 443 (72%) | 483 (78%) | 190 (68) | 210 (77) |
| Median in months (95% CI) | 6.9 (6.3, 7.2) | 5.6 (5.5, 5.7) | 6.9 (6.0, 7.2) | 5.6 (5.4, 5.7) | 8.1 (6.8, 8.5) | 5.6 (5.4, 6.7) |
| Hazard ratio† (95% CI) | 0.76 (0.67, 0.85) | 0.72 (0.63, 0.82) | 0.62 (0.51, 0.76) | |||
| p-Value (stratified log-rank)‡ | <0.0001 | <0.0001 | <0.0001 | |||
| Objective Response Rate | ||||||
| ORR§ (95% CI) | 51% (48, 55) | 42% (38, 45) | 52% (48, 56) | 43% (39, 47) | 61% (55, 66) | 43% (37, 49) |
| Complete response rate | 9% | 6% | 10% | 6% | 13% | 5% |
| Partial response rate | 42% | 36% | 42% | 37% | 48% | 38% |
| p-Value¶ | <0.0001 | 0.0004 | <0.0001 | |||
| Duration of Response | n=405 | n=331 | n=322 | n=263 | n=169 | n=117 |
| Median in months# (95% CI) | 8.0 (7.0, 9.7) | 5.7 (5.5, 6.9) | 8.3 (7.0, 10.9) | 5.6 (5.4, 6.9) | 10.9 (8.0, 13.8) | 5.8 (5.3, 7.0) |
| Range in months | 1.2+, 41.5+ | 1.3+, 34.7+ | 1.2+, 41.5+ | 1.3+, 34.2+ | 1.2+, 41.5+ | 1.4+, 31.2+ |
|
|
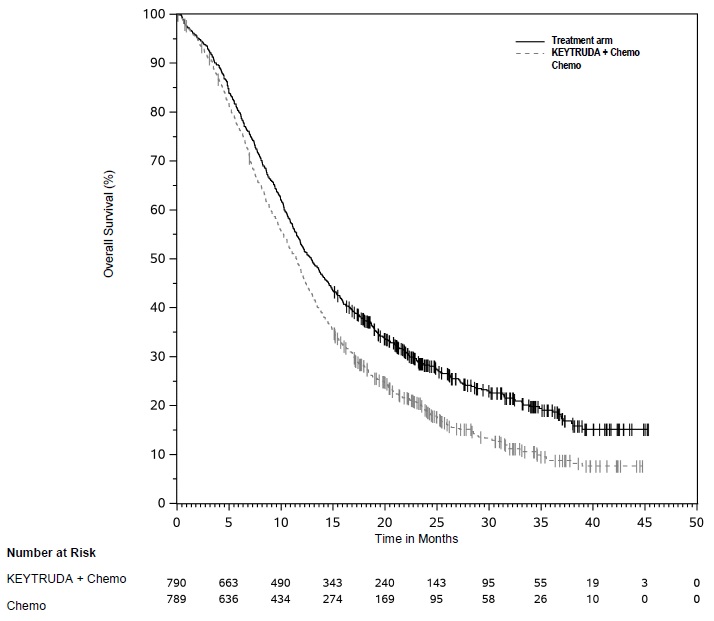
|
|
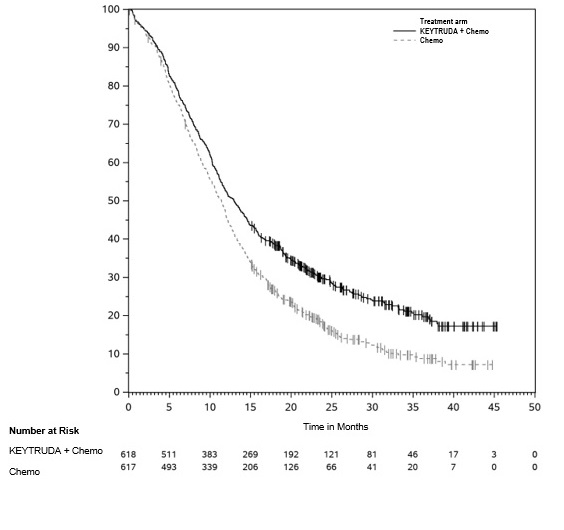
In an exploratory subgroup analysis in patients with PD-L1 CPS<1 (n=344) at the time of the pre-specified interim analysis of OS, the median OS was 12.7 months (95% CI: 11.4, 15.0) for the KEYTRUDA arm and 12.2 months (95% CI: 9.5, 14.0) for the placebo arm, with a HR of 0.92 (95% CI: 0.73, 1.17).
14.10 Esophageal Cancer
First-line Treatment of Locally Advanced Unresectable or Metastatic Esophageal/Gastroesophageal Junction Cancer
KEYNOTE-590
The efficacy of KEYTRUDA was investigated in KEYNOTE-590 (NCT03189719), a multicenter, randomized, placebo-controlled trial that enrolled 749 patients with metastatic or locally advanced esophageal or gastroesophageal junction (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma who were not candidates for surgical resection or definitive chemoradiation. PD-L1 status was centrally determined in tumor specimens in all patients using the PD-L1 IHC 22C3 pharmDx kit. Patients with active autoimmune disease, a medical condition that required immunosuppression, or who received prior systemic therapy in the locally advanced or metastatic setting were ineligible. Randomization was stratified by tumor histology (squamous cell carcinoma vs. adenocarcinoma), geographic region (Asia vs. ex-Asia), and ECOG performance status (0 vs. 1).
Patients were randomized (1:1) to one of the following treatment arms; all study medications were administered via intravenous infusion:
- KEYTRUDA 200 mg on Day 1 of each three-week cycle in combination with cisplatin 80 mg/m2 IV on Day 1 of each three-week cycle for up to six cycles and FU 800 mg/m2 IV per day on Day 1 to Day 5 of each three-week cycle, or per local standard for FU administration, for up to 24 months.
- Placebo on Day 1 of each three-week cycle in combination with cisplatin 80 mg/m2 IV on Day 1 of each three-week cycle for up to six cycles and FU 800 mg/m2 IV per day on Day 1 to Day 5 of each three-week cycle, or per local standard for FU administration, for up to 24 months.
Treatment with KEYTRUDA or chemotherapy continued until unacceptable toxicity or disease progression. Patients could be treated with KEYTRUDA for up to 24 months in the absence of disease progression. The major efficacy outcome measures were OS and PFS as assessed by the investigator according to RECIST v1.1 (modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ). The study pre-specified analyses of OS and PFS based on squamous cell histology, CPS ≥10, and in all patients. Additional efficacy outcome measures were ORR and DoR, according to modified RECIST v1.1, as assessed by the investigator.
The study population characteristics were: median age of 63 years (range: 27 to 94), 43% age 65 or older; 83% male; 37% White, 53% Asian, and 1% Black; 40% had an ECOG PS of 0 and 60% had an ECOG PS of 1. Ninety-one percent had M1 disease and 9% had M0 disease. Seventy-three percent had a tumor histology of squamous cell carcinoma, and 27% had adenocarcinoma.
The trial demonstrated a statistically significant improvement in OS and PFS for patients randomized to KEYTRUDA in combination with chemotherapy, compared to chemotherapy.
Table 84 and Figure 21 summarize the efficacy results for KEYNOTE-590 in all patients.
| Endpoint | KEYTRUDA 200 mg every 3 weeks Cisplatin FU n=373 | Placebo Cisplatin FU n=376 |
|---|---|---|
| OS | ||
| Number (%) of events | 262 (70) | 309 (82) |
| Median in months (95% CI) | 12.4 (10.5, 14.0) | 9.8 (8.8, 10.8) |
| Hazard ratio* (95% CI) | 0.73 (0.62, 0.86) | |
| p-Value† | <0.0001 | |
| PFS | ||
| Number of events (%) | 297 (80) | 333 (89) |
| Median in months (95% CI) | 6.3 (6.2, 6.9) | 5.8 (5.0, 6.0) |
| Hazard ratio* (95% CI) | 0.65 (0.55, 0.76) | |
| p-Value† | <0.0001 | |
| Objective Response Rate | ||
| ORR, %‡
(95% CI) | 45 (40, 50) | 29 (25, 34) |
| Number (%) of complete responses | 24 (6) | 9 (2.4) |
| Number (%) of partial responses | 144 (39) | 101 (27) |
| p-Value§ | <0.0001 | |
| Duration of Response | ||
| Median in months (range) | 8.3 (1.2+, 31.0+) | 6.0 (1.5+, 25.0+) |
|
|
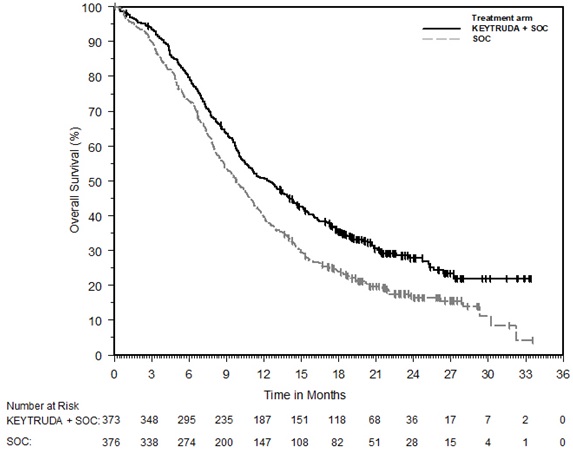
In a pre-specified formal test of OS in patients with PD-L1 CPS ≥ 10 (n=383), the median was 13.5 months (95% CI: 11.1, 15.6) for the KEYTRUDA arm and 9.4 months (95% CI: 8.0, 10.7) for the placebo arm, with a HR of 0.62 (95% CI: 0.49, 0.78; p-Value < 0.0001). In an exploratory analysis, in patients with PD-L1 CPS < 10 (n=347), the median OS was 10.5 months (95% CI: 9.7, 13.5) for the KEYTRUDA arm and 10.6 months (95% CI: 8.8, 12.0) for the placebo arm, with a HR of 0.86 (95% CI: 0.68, 1.10).
Previously Treated Recurrent Locally Advanced or Metastatic Esophageal Cancer
KEYNOTE-181
The efficacy of KEYTRUDA was investigated in KEYNOTE-181 (NCT02564263), a multicenter, randomized, open-label, active-controlled trial that enrolled 628 patients with recurrent locally advanced or metastatic esophageal cancer who progressed on or after one prior line of systemic treatment for advanced disease. Patients with HER2/neu positive esophageal cancer were required to have received treatment with approved HER2/neu targeted therapy. All patients were required to have tumor specimens for PD-L1 testing at a central laboratory; PD-L1 status was determined using the PD-L1 IHC 22C3 pharmDx kit. Patients with a history of non-infectious pneumonitis that required steroids or current pneumonitis, active autoimmune disease, or a medical condition that required immunosuppression were ineligible.
Patients were randomized (1:1) to receive either KEYTRUDA 200 mg every 3 weeks or investigator's choice of any of the following chemotherapy regimens, all given intravenously: paclitaxel 80-100 mg/m2 on Days 1, 8, and 15 of every 4-week cycle, docetaxel 75 mg/m2 every 3 weeks, or irinotecan 180 mg/m2 every 2 weeks. Randomization was stratified by tumor histology (esophageal squamous cell carcinoma [ESCC] vs. esophageal adenocarcinoma [EAC]/Siewert type I EAC of the gastroesophageal junction [GEJ]), and geographic region (Asia vs. ex-Asia). Treatment with KEYTRUDA or chemotherapy continued until unacceptable toxicity or disease progression. Patients randomized to KEYTRUDA were permitted to continue beyond the first RECIST v1.1 (modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ)-defined disease progression if clinically stable until the first radiographic evidence of disease progression was confirmed at least 4 weeks later with repeat imaging. Patients treated with KEYTRUDA without disease progression could be treated for up to 24 months. Assessment of tumor status was performed every 9 weeks. The major efficacy outcome measure was OS evaluated in the following co-primary populations: patients with ESCC, patients with tumors expressing PD-L1 CPS ≥10, and all randomized patients. Additional efficacy outcome measures were PFS, ORR, and DoR, according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, as assessed by BICR.
A total of 628 patients were enrolled and randomized to KEYTRUDA (n=314) or investigator's treatment of choice (n=314). Of these 628 patients, 167 (27%) had ESCC that expressed PD-L1 with a CPS ≥10. Of these 167 patients, 85 patients were randomized to KEYTRUDA and 82 patients to investigator's treatment of choice [paclitaxel (n=50), docetaxel (n=19), or irinotecan (n=13)]. The baseline characteristics of these 167 patients were: median age of 65 years (range: 33 to 80), 51% age 65 or older; 84% male; 32% White and 68% Asian; 38% had an ECOG PS of 0 and 62% had an ECOG PS of 1. Ninety percent had M1 disease and 10% had M0 disease. Prior to enrollment, 99% of patients had received platinum-based treatment and 84% had also received treatment with a fluoropyrimidine. Thirty-three percent of patients received prior treatment with a taxane.
The observed OS hazard ratio was 0.77 (95% CI: 0.63, 0.96) in patients with ESCC, 0.70 (95% CI: 0.52, 0.94) in patients with tumors expressing PD-L1 CPS ≥10, and 0.89 (95% CI: 0.75, 1.05) in all randomized patients. On further examination in patients whose ESCC tumors expressed PD-L1 (CPS ≥10), an improvement in OS was observed among patients randomized to KEYTRUDA as compared with chemotherapy. Table 85 and Figure 22 summarize the key efficacy measures for KEYNOTE-181 for patients with ESCC CPS ≥10.
| Endpoint | KEYTRUDA 200 mg every 3 weeks n=85 | Chemotherapy n=82 |
|---|---|---|
|
||
| OS | ||
| Number (%) of patients with event | 68 (80%) | 72 (88%) |
| Median in months (95% CI) | 10.3 (7.0, 13.5) | 6.7 (4.8, 8.6) |
| Hazard ratio* (95% CI) | 0.64 (0.46, 0.90) | |
| PFS | ||
| Number (%) of patients with event | 76 (89%) | 76 (93%) |
| Median in months (95% CI) | 3.2 (2.1, 4.4) | 2.3 (2.1, 3.4) |
| Hazard ratio* (95% CI) | 0.66 (0.48, 0.92) | |
| Objective Response Rate | ||
| ORR (95% CI) | 22 (14, 33) | 7 (3, 15) |
| Number (%) of complete responses | 4 (5) | 1 (1) |
| Number (%) of partial responses | 15 (18) | 5 (6) |
| Median duration of response in months (range) | 9.3 (2.1+, 18.8+) | 7.7 (4.3, 16.8+) |
|
|
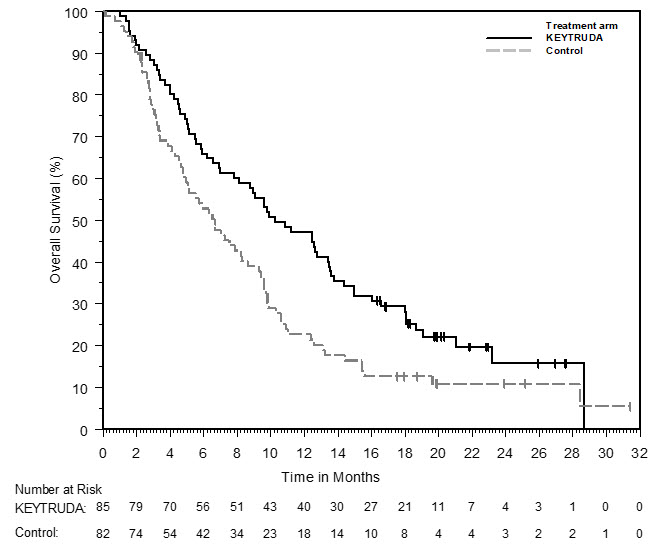
KEYNOTE-180
The efficacy of KEYTRUDA was investigated in KEYNOTE-180 (NCT02559687), a multicenter, non-randomized, open-label trial that enrolled 121 patients with locally advanced or metastatic esophageal cancer who progressed on or after at least 2 prior systemic treatments for advanced disease. With the exception of the number of prior lines of treatment, the eligibility criteria were similar to and the dosage regimen identical to KEYNOTE-181.
The major efficacy outcome measures were ORR and DoR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, as assessed by BICR.
Among the 121 patients enrolled, 29% (n=35) had ESCC that expressed PD-L1 CPS ≥10. The baseline characteristics of these 35 patients were: median age of 65 years (range: 47 to 81), 51% age 65 or older; 71% male; 26% White and 69% Asian; 40% had an ECOG PS of 0 and 60% had an ECOG PS of 1. One hundred percent had M1 disease.
The ORR in the 35 patients with ESCC expressing PD-L1 was 20% (95% CI: 8, 37). Among the 7 responding patients, the DoR ranged from 4.2 to 25.1+ months, with 5 patients (71%) having responses of 6 months or longer and 3 patients (57%) having responses of 12 months or longer.
14.11 Cervical Cancer
FIGO 2014 Stage III-IVA Cervical Cancer with Chemoradiotherapy
The efficacy of KEYTRUDA in combination with CRT (cisplatin and external beam radiation therapy [EBRT] followed by brachytherapy [BT]) was investigated in KEYNOTE-A18 (NCT04221945), a multicenter, randomized, double-blind, placebo-controlled trial that enrolled 1060 patients with cervical cancer who had not previously received any definitive surgery, radiation, or systemic therapy for cervical cancer. There were 596 patients with FIGO 2014 Stage III-IVA (tumor involvement of the lower vagina with or without extension onto pelvic sidewall or hydronephrosis/non-functioning kidney or has spread to adjacent pelvic organs) with either node-positive or node-negative disease, and 462 patients with FIGO 2014 Stage IB2-IIB (tumor lesions >4 cm or clinically visible lesions that have spread beyond the uterus but have not extended onto the pelvic wall or to the lower third of vagina) with node-positive disease; two patients had FIGO 2014 Stage IVB disease. Randomization was stratified by planned type of EBRT (Intensity-modulated radiation therapy [IMRT] or volumetric modulated arc therapy [VMAT] vs. non-IMRT and non-VMAT), stage at screening of cervical cancer (FIGO 2014 Stage IB2-IIB vs. FIGO 2014 Stage III-IVA), and planned total radiotherapy dose (EBRT + brachytherapy dose of <70 Gy vs. ≥70 Gy as per equivalent dose [EQD2]).
Patients were randomized (1:1) to one of two treatment arms:
- KEYTRUDA 200 mg IV every 3 weeks (5 cycles) concurrent with cisplatin 40 mg/m2 IV weekly (5 cycles, an optional sixth infusion could be administered per local practice), and radiotherapy (EBRT followed by BT), followed by KEYTRUDA 400 mg IV every 6 weeks (15 cycles)
- Placebo IV every 3 weeks (5 cycles) concurrent with cisplatin 40 mg/m2 IV weekly (5 cycles, an optional sixth infusion could be administered per local practice), and radiotherapy (EBRT followed by BT), followed by placebo IV every 6 weeks (15 cycles)
Treatment continued until RECIST v1.1-defined progression of disease as determined by investigator or unacceptable toxicity.
Assessment of tumor status was performed every 12 weeks from completion of CRT for the first two years, followed by every 24 weeks in year 3, and then annually. The major efficacy outcome measures were PFS as assessed by investigator according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, or histopathologic confirmation, and OS.
Among the 596 patients with FIGO 2014 Stage III-IVA disease, the baseline characteristics were: median age of 52 years (range: 22 to 87), 17% age 65 or older; 36% White, 34% Asian, 1% Black; 38% Hispanic or Latino; 68% ECOG PS 0 and 32% ECOG PS 1; 93% with CPS ≥1; 70% had positive pelvic and/or positive para-aortic lymph node(s) and 30% had neither positive pelvic nor para-aortic lymph node(s); 83% had squamous cell carcinoma and 17% had non-squamous histology. Regarding radiation, 85% of patients received IMRT or VMAT EBRT, and the median EQD2 dose was 87 Gy (range: 7 to 114).
The trial demonstrated a statistically significant improvement in PFS in the overall population. In an exploratory subgroup analysis for the 462 patients (44%) with FIGO 2014 Stage IB2-IIB disease, the PFS HR estimate was 0.91 (95% CI: 0.63, 1.31), indicating that the PFS improvement in the overall population was primarily attributed to the results seen in the subgroup of patients with FIGO 2014 Stage III-IVA disease. OS data were not mature at the time of PFS analysis, with 10% deaths in the overall population.
The efficacy results in the exploratory subgroup analysis of 596 patients with FIGO 2014 Stage III-IVA disease are summarized in Table 86 and Figure 23.
| Endpoint | KEYTRUDA 200 mg every 3 weeks and 400 mg every 6 weeks with CRT n=293 | Placebo with CRT n=303 |
|---|---|---|
| CRT = Chemoradiotherapy NR = not reached |
||
|
||
| PFS by Investigator | ||
| Number of patients with event (%) | 61 (21%) | 94 (31%) |
| Median in months (95% CI) | NR (NR, NR) | NR (18.8, NR) |
| 12-month PFS rate (95% CI) | 81% (75, 85) | 70% (64, 76) |
| Hazard ratio* (95% CI) | 0.59 (0.43, 0.82) | |
|
|
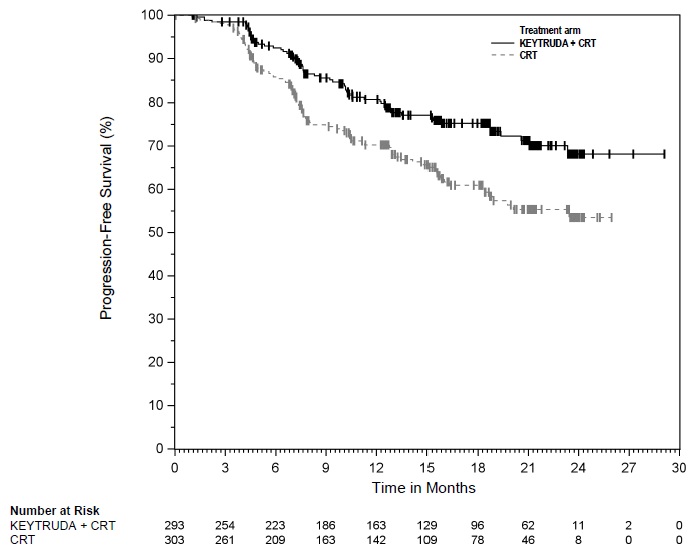
Persistent, Recurrent, or Metastatic Cervical Cancer
The efficacy of KEYTRUDA in combination with paclitaxel and cisplatin or paclitaxel and carboplatin, with or without bevacizumab, was investigated in KEYNOTE-826 (NCT03635567), a multicenter, randomized, double-blind, placebo-controlled trial that enrolled 617 patients with persistent, recurrent, or first-line metastatic cervical cancer who had not been treated with chemotherapy except when used concurrently as a radio-sensitizing agent. Patients were enrolled regardless of tumor PD-L1 expression status. Patients with autoimmune disease that required systemic therapy within 2 years of treatment or a medical condition that required immunosuppression were ineligible. Randomization was stratified by metastatic status at initial diagnosis, investigator decision to use bevacizumab, and PD-L1 status (CPS <1 vs. CPS 1 to <10 vs. CPS ≥10). Patients were randomized (1:1) to one of the two treatment groups:
- Treatment Group 1: KEYTRUDA 200 mg plus chemotherapy with or without bevacizumab
- Treatment Group 2: Placebo plus chemotherapy with or without bevacizumab
The investigator selected one of the following four treatment regimens prior to randomization:
- Paclitaxel 175 mg/m2 + cisplatin 50 mg/m2
- Paclitaxel 175 mg/m2 + cisplatin 50 mg/m2 + bevacizumab 15 mg/kg
- Paclitaxel 175 mg/m2 + carboplatin AUC 5 mg/mL/min
- Paclitaxel 175 mg/m2 + carboplatin AUC 5 mg/mL/min + bevacizumab 15 mg/kg
All study medications were administered as an intravenous infusion. All study treatments were administered on Day 1 of each 3-week treatment cycle. Cisplatin could be administered on Day 2 of each 3-week treatment cycle. Treatment with KEYTRUDA continued until RECIST v1.1-defined progression of disease, unacceptable toxicity, or a maximum of 24 months. Administration of KEYTRUDA was permitted beyond RECIST-defined disease progression if the patient was clinically stable and considered to be deriving clinical benefit by the investigator. Assessment of tumor status was performed every 9 weeks for the first year, followed by every 12 weeks thereafter. The main efficacy outcome measures were OS and PFS as assessed by investigator according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ. Additional efficacy outcome measures were ORR and DoR, according to RECIST v1.1, as assessed by investigator.
Of the 617 enrolled patients, 548 patients (89%) had tumors expressing PD-L1 with a CPS ≥1. Among these 548 enrolled patients with tumors expressing PD-L1, 273 patients were randomized to KEYTRUDA in combination with chemotherapy with or without bevacizumab, and 275 patients were randomized to placebo in combination with chemotherapy with or without bevacizumab. Sixty-three percent of the 548 patients received bevacizumab as part of study treatment. The baseline characteristics of the 548 patients were: median age of 51 years (range: 22 to 82), 16% age 65 or older; 59% White, 18% Asian, 6% American Indian or Alaska Native, and 1% Black; 37% Hispanic or Latino; 56% ECOG performance status 0 and 43% ECOG performance status 1. Seventy-five percent had squamous cell carcinoma, 21% adenocarcinoma, and 5% adenosquamous histology, and 32% of patients had metastatic disease at diagnosis. At study entry, 21% of patients had metastatic disease only and 79% had persistent or recurrent disease with or without distant metastases, of whom 39% had received prior chemoradiation only and 17% had received prior chemoradiation plus surgery.
A statistically significant improvement in OS and PFS was demonstrated in patients randomized to receive KEYTRUDA compared with patients randomized to receive placebo. An updated OS analysis was conducted at the time of final analysis when 354 deaths in the CPS ≥1 population were observed. Table 87 and Figure 24 summarize the key efficacy measures for KEYNOTE-826 for patients with tumors expressing PD-L1 (CPS ≥1).
| Endpoint | KEYTRUDA 200 mg every 3 weeks and chemotherapy* with or without bevacizumab n=273 | Placebo and chemotherapy* with or without bevacizumab n=275 |
|---|---|---|
| + Denotes ongoing response NR = not reached |
||
|
||
| OS | ||
| Number of patients with event (%) | 118 (43.2) | 154 (56.0) |
| Median in months (95% CI) | NR (19.8, NR) | 16.3 (14.5, 19.4) |
| Hazard ratio† (95% CI) | 0.64 (0.50, 0.81) | |
| p-Value‡ | 0.0001 | |
| Updated OS | ||
| Number of patients with event (%) | 153 (56.0%) | 201 (73.1%) |
| Median in months (95% CI) | 28.6 (22.1, 38.0) | 16.5 (14.5, 20.0) |
| Hazard ratio† (95% CI) | 0.60 (0.49, 0.74) | |
| PFS | ||
| Number of patients with event (%) | 157 (57.5) | 198 (72.0) |
| Median in months (95% CI) | 10.4 (9.7, 12.3) | 8.2 (6.3, 8.5) |
| Hazard ratio† (95% CI) | 0.62 (0.50, 0.77) | |
| p-Value§ | < 0.0001 | |
| Objective Response Rate | ||
| ORR¶ (95% CI) | 68% (62, 74) | 50% (44, 56) |
| Complete response rate | 23% | 13% |
| Partial response rate | 45% | 37% |
| Duration of Response | ||
| Median in months (range) | 18.0 (1.3+, 24.2+) | 10.4 (1.5+, 22.0+) |
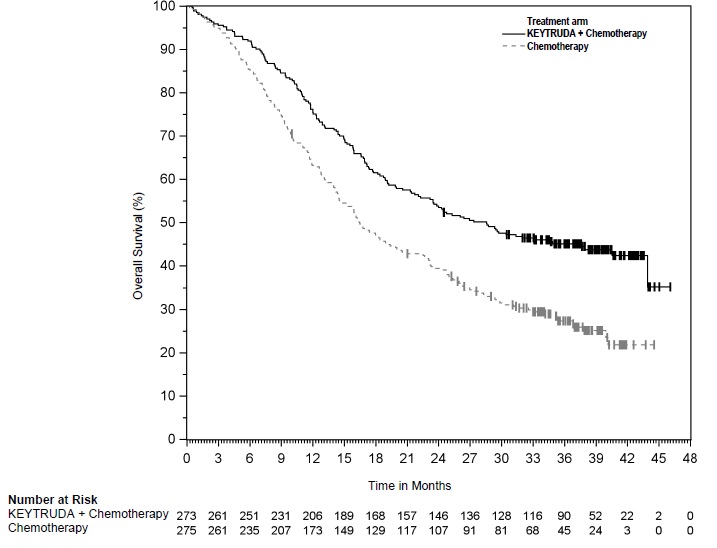
Previously Treated Recurrent or Metastatic Cervical Cancer
The efficacy of KEYTRUDA was investigated in 98 patients with recurrent or metastatic cervical cancer enrolled in a single cohort (Cohort E) in KEYNOTE-158 (NCT02628067), a multicenter, non-randomized, open-label, multi-cohort trial. The trial excluded patients with autoimmune disease or a medical condition that required immunosuppression. Patients received KEYTRUDA 200 mg intravenously every 3 weeks until unacceptable toxicity or documented disease progression. Patients with initial radiographic disease progression could receive additional doses of treatment during confirmation of progression unless disease progression was symptomatic, was rapidly progressive, required urgent intervention, or occurred with a decline in performance status. Patients without disease progression could be treated for up to 24 months. Assessment of tumor status was performed every 9 weeks for the first 12 months, and every 12 weeks thereafter. The major efficacy outcome measures were ORR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, as assessed by BICR, and DoR.
Among the 98 patients in Cohort E, 77 (79%) had tumors that expressed PD-L1 with a CPS ≥ 1 and received at least one line of chemotherapy in the metastatic setting. PD-L1 status was determined using the IHC 22C3 pharmDx kit. The baseline characteristics of these 77 patients were: median age of 45 years (range: 27 to 75); 81% White, 14% Asian, and 3% Black; 32% ECOG PS of 0 and 68% ECOG PS of 1; 92% had squamous cell carcinoma, 6% adenocarcinoma, and 1% adenosquamous histology; 95% had M1 disease and 5% had recurrent disease; and 35% had one and 65% had two or more prior lines of therapy in the recurrent or metastatic setting.
No responses were observed in patients whose tumors did not have PD-L1 expression (CPS <1). Efficacy results are summarized in Table 88 for patients with PD-L1 expression (CPS ≥1).
| Endpoint | KEYTRUDA 200 mg every 3 weeks n=77* |
|---|---|
| + Denotes ongoing response NR = not reached |
|
| Objective Response Rate | |
| ORR (95% CI) | 14.3% (7.4, 24.1) |
| Complete response rate | 2.6% |
| Partial response rate | 11.7% |
| Duration of Response | |
| Median in months (range) | NR (4.1, 18.6+)† |
| % with duration ≥6 months | 91% |
14.12 Hepatocellular Carcinoma
Previously Treated HCC
The efficacy of KEYTRUDA was investigated in KEYNOTE-394 (NCT03062358), a multicenter, randomized, placebo-controlled, double-blind trial conducted in Asia in patients with Barcelona Clinic Liver Cancer (BCLC) Stage B or C HCC, who were previously treated with sorafenib or oxaliplatin-based chemotherapy and who were not amenable to or were refractory to local-regional therapy. Patients were also required to have Child-Pugh A liver function.
Patients with hepatitis B had treated controlled disease (HBV viral load <2000 IU/mL or <104 copies/mL). Patients with an autoimmune disease that required systemic therapy within 2 years of treatment or a medical condition that required immunosuppression were ineligible. Patients with hepatic encephalopathy, main branch portal venous invasion, clinically apparent ascites, or esophageal or gastric variceal bleeding within the last 6 months were also ineligible.
Randomization was stratified by prior treatment: sorafenib vs. oxaliplatin-based chemotherapy, macrovascular invasion, and etiology (active HBV vs. others (active HCV, non-infected)). Patients were randomized (2:1) to receive pembrolizumab 200 mg intravenously every 3 weeks or placebo.
Treatment with KEYTRUDA continued until RECIST v1.1-defined progression of disease as determined by BICR, unacceptable toxicity, or a maximum of 24 months. Assessment of tumor status was performed every 6 weeks. The main efficacy outcome measure was OS. Additional efficacy outcome measures were PFS, ORR, and DoR, as assessed by BICR using RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
The study enrolled 453 patients, and 360 (79%) had active hepatitis B. The population characteristics in patients with active hepatitis B were: median age of 52 years (range: 23 to 82), 16% age 65 or older; 86% male; 100% Asian; 42% ECOG PS of 0 and 58% ECOG PS of 1; 90% received prior sorafenib and 10% received prior oxaliplatin-based chemotherapy. Patient characteristics also included extrahepatic disease (77%), macrovascular invasion (10%), BCLC stage C (93%) and B (7%), and baseline AFP ≥200 ng/mL (57%).
KEYNOTE-394 demonstrated improved OS in patients with HCC secondary to hepatitis B randomized to KEYTRUDA compared with placebo. Efficacy results are summarized in Table 89 and Figure 25.
| Endpoint | KEYTRUDA 200 mg every 3 weeks n=236 | Placebo n=124 |
|---|---|---|
| + Denotes ongoing response | ||
| OS* | ||
| Number (%) of patients with events | 172 (73) | 105 (85) |
| Median in months (95% CI) | 13.9 (12.5, 17.9) | 13.0 (10.1, 15.6) |
| Hazard ratio† (95% CI) | 0.78 (0.61, 0.99) | |
| PFS‡ | ||
| Number (%) of patients with events | 189 (80) | 108 (87) |
| Median in months (95% CI) | 2 (1.4, 2.7) | 2.3 (1.4, 2.8) |
| Hazard ratio† (95% CI) | 0.78 (0.61, 1.00) | |
| Objective Response Rate‡ | ||
| ORR§ (95% CI) | 11% (7, 16) | 1.6% (0.2, 5.7) |
| Number (%) of complete responses | 2 (0.9%) | 1 (0.8%) |
| Number (%) of partial responses | 24 (10%) | 1 (0.8%) |
| Duration of Response* | n=28 | n=2 |
| Median in months¶ (range) | 23.9 (2.6+, 44.4+) | 5.6 (3.0+, 5.6) |
| Figure 25: Kaplan-Meier Curve for Overall Survival in KEYNOTE-394 |
|
|
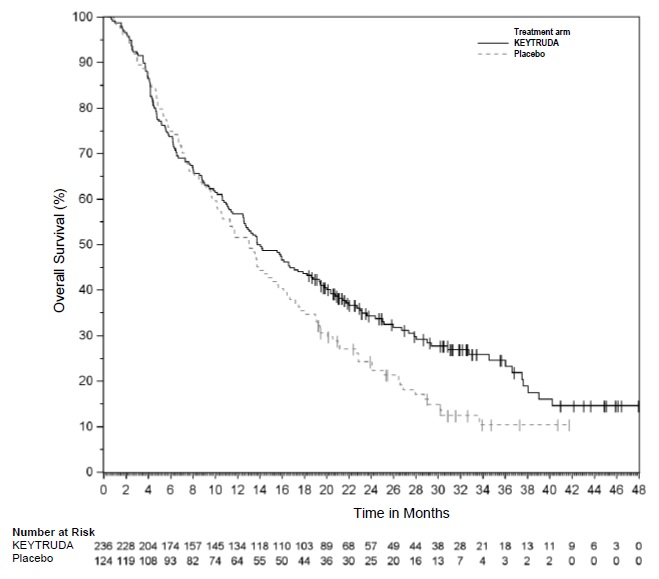
14.13 Biliary Tract Cancer
The efficacy of KEYTRUDA in combination with gemcitabine and cisplatin chemotherapy was investigated in KEYNOTE-966 (NCT04003636), a multicenter, randomized, double-blind, placebo-controlled trial that enrolled 1069 patients with locally advanced unresectable or metastatic BTC, who had not received prior systemic therapy in the advanced disease setting. Patients with autoimmune disease that required systemic therapy within 2 years of treatment or a medical condition that required immunosuppression were ineligible. Randomization was stratified by region (Asia vs. non-Asia), locally advanced versus metastatic, and site of origin (gallbladder, intrahepatic or extrahepatic cholangiocarcinoma).
Patients were randomized (1:1) to KEYTRUDA 200 mg on Day 1 plus gemcitabine 1000 mg/m2 and cisplatin 25 mg/m2 on Day 1 and Day 8 every 3 weeks, or placebo on Day 1 plus gemcitabine 1000 mg/m2 and cisplatin 25 mg/m2 on Day 1 and Day 8 every 3 weeks. Study medications were administered via intravenous infusion. Treatment continued until unacceptable toxicity or disease progression. For pembrolizumab, treatment continued for a maximum of 35 cycles, or approximately 24 months. For gemcitabine, treatment could be continued beyond 8 cycles while for cisplatin, treatment could be administered for a maximum of 8 cycles.
Administration of KEYTRUDA with chemotherapy was permitted beyond RECIST-defined disease progression if the patient was clinically stable and considered by the investigator to be deriving clinical benefit. Assessment of tumor status was performed at baseline and then every 6 weeks through 54 weeks, followed by every 12 weeks thereafter.
Study population characteristics were median age of 64 years (range: 23 to 85), 47% age 65 or older; 52% male; 49% White, 46% Asian, 1.3% Black or African American; 10% Hispanic or Latino; 46% ECOG PS of 0 and 54% ECOG PS of 1; 31% of patients had a history of hepatitis B infection, and 3% had a history of hepatitis C infection.
The major efficacy outcome measure was OS. Additional efficacy outcome measures were PFS, ORR and DoR as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
Table 90 and Figure 26 summarize the efficacy results for KEYNOTE-966.
| Endpoint | KEYTRUDA 200 mg every 3 weeks with gemcitabine/cisplatin n=533 | Placebo with gemcitabine/cisplatin n=536 |
|---|---|---|
| NS = not significant | ||
|
||
| OS* | ||
| Number of patients with event (%) | 414 (78%) | 443 (83%) |
| Median in months (95% CI) | 12.7 (11.5, 13.6) | 10.9 (9.9, 11.6) |
| Hazard ratio† (95% CI) | 0.83 (0.72, 0.95) | |
| p-Value‡ | 0.0034 | |
| PFS§ | ||
| Number (%) of patients with event | 361 (68%) | 391 (73%) |
| Median in months (95% CI) | 6.5 (5.7, 6.9) | 5.6 (5.1, 6.6) |
| Hazard ratio† (95% CI) | 0.86 (0.75, 1.00) | |
| p-Value‡ | NS | |
| Objective Response Rate§ | ||
| ORR¶ (95% CI) | 29% (25, 33) | 29% (25, 33) |
| Number (%) of complete responses | 11 (2.1%) | 7 (1.3%) |
| Number (%) of partial responses | 142 (27%) | 146 (27%) |
| p-Value # | NS | |
| Duration of Response* | n=156 | n=152 |
| Median in months Þ (95% CI) | 8.3 (6.9, 10.2) | 6.8 (5.7, 7.1) |
| Figure 26: Kaplan-Meier Curve for Overall Survival in KEYNOTE-966 |
|
|
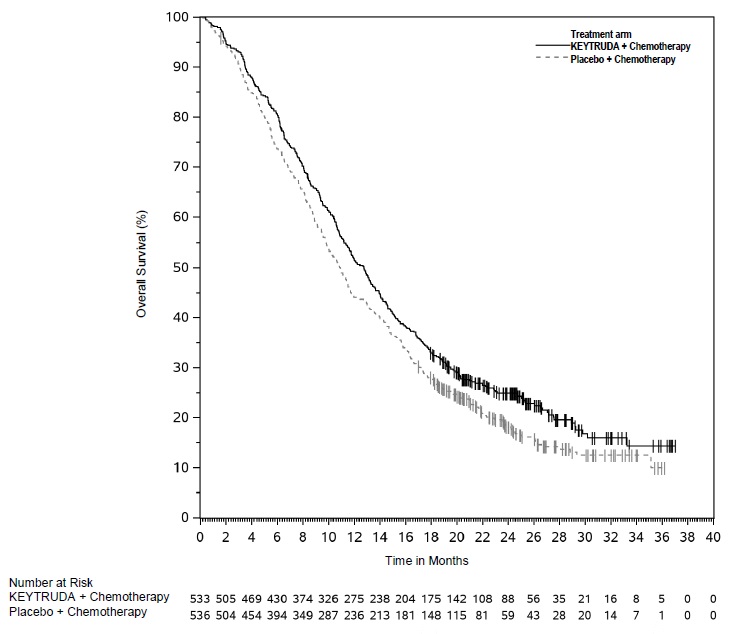
14.14 Merkel Cell Carcinoma
The efficacy of KEYTRUDA was investigated in KEYNOTE-017 (NCT02267603) and KEYNOTE-913 (NCT03783078), two multicenter, non-randomized, open-label trials that enrolled 105 patients with recurrent locally advanced or metastatic MCC who had not received prior systemic therapy for their advanced disease. Patients with active autoimmune disease or a medical condition that required immunosuppression were ineligible.
Patients received KEYTRUDA 2 mg/kg (KEYNOTE-017) or 200 mg (KEYNOTE-913) every 3 weeks until unacceptable toxicity or disease progression that was symptomatic, rapidly progressive, required urgent intervention, occurred with a decline in performance status, or was confirmed at least 4 weeks later with repeat imaging. Patients without disease progression were treated for up to 24 months.
The major efficacy outcome measures were ORR and DoR as assessed by BICR per RECIST v1.1.
Among the 105 patients enrolled, the median age was 73 years (range: 38 to 91), 79% were age 65 or older; 62% were male; 80% were White, race in 19% was unknown or missing, and 1% were Asian; 53% had ECOG PS of 0, and 47% had ECOG PS of 1. Thirteen percent had stage IIIB disease and 84% had stage IV. Seventy-six percent of patients had prior surgery and 51% had prior radiation therapy.
Efficacy results are summarized in Table 91.
| Endpoint | KEYNOTE-017 KEYTRUDA 2 mg/kg every 3 weeks n=50 | KEYNOTE-913 KEYTRUDA 200 mg or 2 mg/kg every 3 weeks n=55 |
|---|---|---|
| + Denotes ongoing response | ||
| NR = not reached | ||
| Objective Response Rate | ||
| ORR (95% CI) | 56% (41, 70) | 49% (35, 63) |
| Complete responses, n (%) | 12 (24%) | 9 (16%) |
| Partial responses, n (%) | 16 (32%) | 18 (33%) |
| Duration of Response | n=28 | n=27 |
| Median DoR in months (range) | NR (5.9, 34.5+) | NR (4.8, 25.4+) |
| Patients with duration ≥6 months, n (%) | 27 (96%) | 25 (93%) |
| Patients with duration ≥12 months, n (%) | 15 (54%) | 19 (70%) |
14.15 Renal Cell Carcinoma
First-line treatment with axitinib
KEYNOTE-426
The efficacy of KEYTRUDA in combination with axitinib was investigated in KEYNOTE-426 (NCT02853331), a randomized, multicenter, open-label trial conducted in 861 patients who had not received systemic therapy for advanced RCC. Patients were enrolled regardless of PD-L1 tumor expression status. Patients with active autoimmune disease requiring systemic immunosuppression within the last 2 years were ineligible. Randomization was stratified by International Metastatic RCC Database Consortium (IMDC) risk categories (favorable versus intermediate versus poor) and geographic region (North America versus Western Europe versus "Rest of the World").
Patients were randomized (1:1) to one of the following treatment arms:
- KEYTRUDA 200 mg intravenously every 3 weeks up to 24 months in combination with axitinib 5 mg orally, twice daily. Patients who tolerated axitinib 5 mg twice daily for 2 consecutive cycles (6 weeks) could increase to 7 mg and then subsequently to 10 mg twice daily. Axitinib could be interrupted or reduced to 3 mg twice daily and subsequently to 2 mg twice daily to manage toxicity.
- Sunitinib 50 mg orally, once daily for 4 weeks and then off treatment for 2 weeks.
Treatment with KEYTRUDA and axitinib continued until RECIST v1.1-defined progression of disease or unacceptable toxicity. Administration of KEYTRUDA and axitinib was permitted beyond RECIST-defined disease progression if the patient was clinically stable and considered to be deriving clinical benefit by the investigator. Assessment of tumor status was performed at baseline, after randomization at Week 12, then every 6 weeks thereafter until Week 54, and then every 12 weeks thereafter.
The study population characteristics were: median age of 62 years (range: 26 to 90), 38% age 65 or older; 73% male; 79% White and 16% Asian; 20% and 80% of patients had a baseline KPS of 70 to 80 and 90 to 100, respectively; and patient distribution by IMDC risk categories was 31% favorable, 56% intermediate, and 13% poor.
The main efficacy outcome measures were OS and PFS as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ. Additional efficacy outcome measures included ORR, as assessed by BICR. A statistically significant improvement in OS was demonstrated at the first pre-specified interim analysis in patients randomized to KEYTRUDA in combination with axitinib compared with sunitinib. The trial also demonstrated statistically significant improvements in PFS and ORR. An updated OS analysis was conducted when 418 deaths were observed based on the planned number of deaths for the pre-specified final analysis. Table 92 and Figure 27 summarize the efficacy results for KEYNOTE-426.
| Endpoint | KEYTRUDA 200 mg every 3 weeks and Axitinib | Sunitinib |
|---|---|---|
| n=432 | n=429 | |
| NR = not reached | ||
|
||
| OS | ||
| Number of patients with event (%) | 59 (14%) | 97 (23%) |
| Median in months (95% CI) | NR (NR, NR) | NR (NR, NR) |
| Hazard ratio* (95% CI) | 0.53 (0.38, 0.74) | |
| p-Value† | <0.0001‡ | |
| Updated OS | ||
| Number of patients with event (%) | 193 (45%) | 225 (52%) |
| Median in months (95% CI) | 45.7 (43.6, NR) | 40.1 (34.3, 44.2) |
| Hazard ratio* (95% CI) | 0.73 (0.60, 0.88) | |
| PFS | ||
| Number of patients with event (%) | 183 (42%) | 213 (50%) |
| Median in months (95% CI) | 15.1 (12.6, 17.7) | 11.0 (8.7, 12.5) |
| Hazard ratio* (95% CI) | 0.69 (0.56, 0.84) | |
| p-Value† | 0.0001§ | |
| Objective Response Rate | ||
| ORR¶ (95% CI) | 59% (54, 64) | 36% (31, 40) |
| Complete response rate | 6% | 2% |
| Partial response rate | 53% | 34% |
| p-Value# | <0.0001 | |
| Figure 27: Kaplan-Meier Curve for Updated Overall Survival in KEYNOTE-426 |
|
|
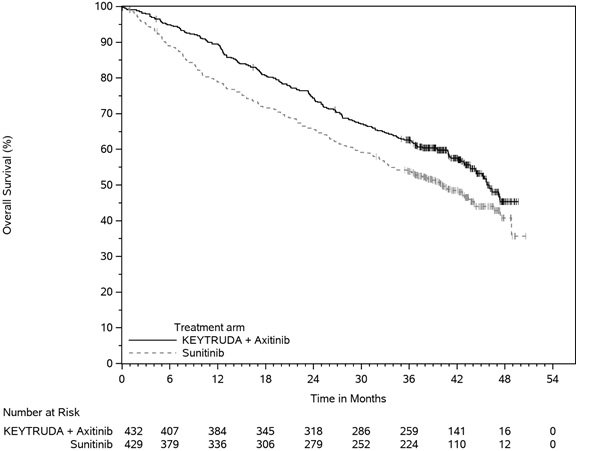
In an exploratory analysis, the updated analysis of OS in patients with IMDC favorable, intermediate, intermediate/poor, and poor risk demonstrated a HR of 1.17 (95% CI: 0.76, 1.80), 0.67 (95% CI: 0.52, 0.86), 0.64 (95% CI: 0.52, 0.80), and 0.51 (95% CI: 0.32, 0.81), respectively.
First-line treatment with lenvatinib
KEYNOTE-581
The efficacy of KEYTRUDA in combination with lenvatinib was investigated in KEYNOTE-581 (NCT02811861), a multicenter, open-label, randomized trial conducted in 1069 patients with advanced RCC in the first-line setting. Patients were enrolled regardless of PD-L1 tumor expression status. Patients with active autoimmune disease or a medical condition that required immunosuppression were ineligible. Randomization was stratified by geographic region (North America versus Western Europe versus "Rest of the World") and Memorial Sloan Kettering Cancer Center (MSKCC) prognostic groups (favorable versus intermediate versus poor risk).
Patients were randomized (1:1:1) to one of the following treatment arms:
- KEYTRUDA 200 mg intravenously every 3 weeks up to 24 months in combination with lenvatinib 20 mg orally once daily.
- Lenvatinib 18 mg orally once daily in combination with everolimus 5 mg orally once daily.
- Sunitinib 50 mg orally once daily for 4 weeks then off treatment for 2 weeks.
Treatment continued until unacceptable toxicity or disease progression. Administration of KEYTRUDA with lenvatinib was permitted beyond RECIST-defined disease progression if the patient was clinically stable and considered by the investigator to be deriving clinical benefit. KEYTRUDA was continued for a maximum of 24 months; however, treatment with lenvatinib could be continued beyond 24 months. Assessment of tumor status was performed at baseline and then every 8 weeks.
The study population characteristics were: median age of 62 years (range: 29 to 88 years), 42% age 65 or older; 75% male; 74% White, 21% Asian, 1% Black, and 2% other races; 18% and 82% of patients had a baseline KPS of 70 to 80 and 90 to 100, respectively; patient distribution by MSKCC risk categories was 27% favorable, 64% intermediate, and 9% poor. Common sites of metastases in patients were lung (68%), lymph node (45%), and bone (25%).
The major efficacy outcome measures were PFS, as assessed by independent radiologic review (IRC) according to RECIST v1.1, and OS. Additional efficacy outcome measures included confirmed ORR as assessed by IRC. KEYTRUDA in combination with lenvatinib demonstrated statistically significant improvements in PFS, OS, and ORR compared with sunitinib. An updated OS analysis was conducted when 304 deaths were observed based on the planned number of deaths for the pre-specified final analysis. Table 93 and Figures 28 and 29 summarize the efficacy results for KEYNOTE-581.
| Endpoint | KEYTRUDA 200 mg every 3 weeks and Lenvatinib | Sunitinib |
|---|---|---|
| n=355 | n=357 | |
| Tumor assessments were based on RECIST 1.1; only confirmed responses are included for ORR. Data cutoff date = 28 Aug 2020, Updated OS cutoff date = 31 July 2022 CI = confidence interval; NR= Not reached |
||
| Progression-Free Survival (PFS) | ||
| Number of events, n (%) | 160 (45%) | 205 (57%) |
| Progressive disease | 145 (41%) | 196 (55%) |
| Death | 15 (4%) | 9 (3%) |
| Median PFS in months (95% CI) | 23.9 (20.8, 27.7) | 9.2 (6.0, 11.0) |
| Hazard ratio* (95% CI) | 0.39 (0.32, 0.49) | |
| p-Value† | <0.0001 | |
| Overall Survival (OS) | ||
| Number of deaths, n (%) | 80 (23%) | 101 (28%) |
| Median OS in months (95% CI) | NR (33.6, NR) | NR (NR, NR) |
| Hazard ratio* (95% CI) | 0.66 (0.49, 0.88) | |
| p-Value† | 0.0049 | |
| Updated OS | ||
| Number of deaths, n (%) | 149 (42%) | 159 (45%) |
| Median OS in months (95% CI) | 53.7 (48.7, NR) | 54.3 (40.9, NR) |
| Hazard ratio* (95% CI) | 0.79 (0.63, 0.99) | |
| Objective Response Rate (Confirmed) | ||
| ORR, n (%) | 252 (71%) | 129 (36%) |
| (95% CI) | (66, 76) | (31, 41) |
| Complete response rate | 16% | 4% |
| Partial response rate | 55% | 32% |
| p-Value‡ | <0.0001 | |
| Figure 28: Kaplan-Meier Curve for PFS in KEYNOTE-581 |
|
|
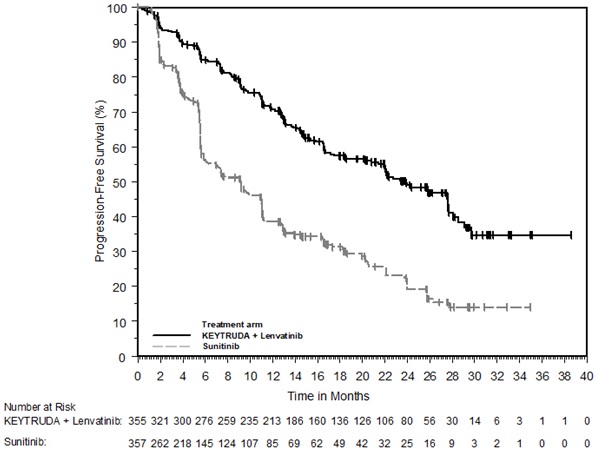
| Figure 29: Kaplan-Meier Curve for Updated Overall Survival in KEYNOTE-581 |
|
|
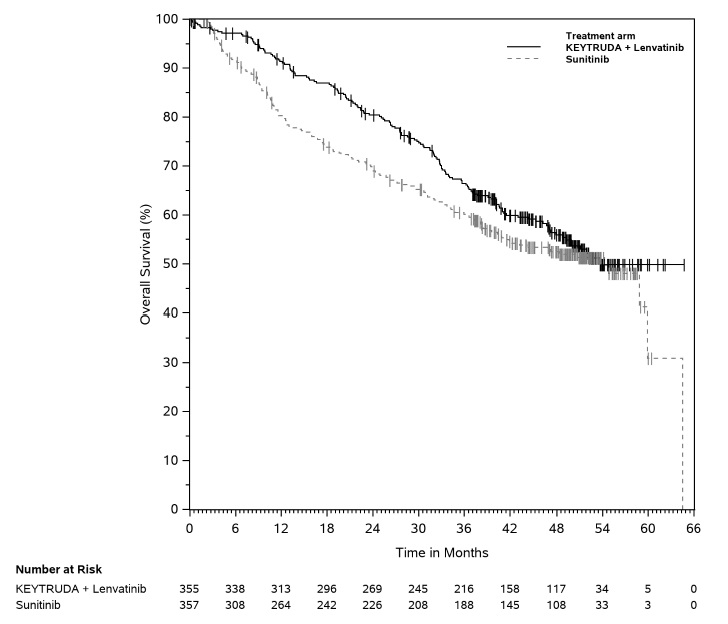
KEYNOTE-B61
The efficacy of KEYTRUDA in combination with lenvatinib was investigated in KEYNOTE-B61 (NCT04704219), a multicenter, single-arm trial that enrolled 160 patients with advanced or metastatic non-clear cell RCC in the first-line setting. Patients with active autoimmune disease or a medical condition that required immunosuppression were ineligible.
Patients received KEYTRUDA 400 mg every 6 weeks in combination with lenvatinib 20 mg orally once daily. KEYTRUDA was continued for a maximum of 24 months; however, lenvatinib could be continued beyond 24 months. Treatment continued until unacceptable toxicity or disease progression. Administration of KEYTRUDA with lenvatinib was permitted beyond RECIST-defined disease progression if the patient was considered by the investigator to be deriving clinical benefit.
Among the 158 treated patients, the baseline characteristics were: median age of 60 years (range: 24 to 87 years); 71% male; 86% White, 8% Asian, and 3% Black; <1% Hispanic or Latino; 22% and 78% of patients had a baseline KPS of 70 to 80 and 90 to 100, respectively; histologic subtypes were 59% papillary, 18% chromophobe, 4% translocation, <1% medullary, 13% unclassified, and 6% other; patient distribution by IMDC risk categories was 35% favorable, 54% intermediate, and 10% poor. Common sites of metastases in patients were lymph node (65%), lung (35%), bone (30%), and liver (21%).
The major efficacy outcome measure was ORR as assessed by BICR using RECIST 1.1. Additional efficacy outcome measures included DOR as assessed by BICR using RECIST 1.1. Efficacy results are summarized in Table 94.
| Endpoint | KEYTRUDA 400 mg every 6 weeks and Lenvatinib n=158 |
|---|---|
| CI = confidence interval + Denotes ongoing response |
|
|
|
| Objective Response Rate (Confirmed) | |
| ORR (95% CI) | 51% (43, 59) |
| Complete response | 8% |
| Partial response | 42% |
| Duration of Response* | |
| Median in months (range) | 19.5 (1.5+, 23.5+) |
Adjuvant Treatment of RCC (KEYNOTE‑564)
The efficacy of KEYTRUDA was investigated as adjuvant therapy for RCC in KEYNOTE-564 (NCT03142334), a multicenter, randomized (1:1), double-blind, placebo-controlled trial in 994 patients with intermediate-high or high risk of recurrence of RCC, or M1 no evidence of disease (NED). The intermediate-high risk category included: pT2 with Grade 4 or sarcomatoid features; pT3, any Grade without nodal involvement (N0) or distant metastases (M0). The high risk category included: pT4, any Grade N0 and M0; any pT, any Grade with nodal involvement and M0. The M1 NED category included patients with metastatic disease who had undergone complete resection of primary and metastatic lesions. Patients must have undergone a partial nephroprotective or radical complete nephrectomy (and complete resection of solid, isolated, soft tissue metastatic lesion(s) in M1 NED participants) with negative surgical margins ≥4 weeks prior to the time of screening. Patients were excluded from the trial if they had received prior systemic therapy for advanced RCC. Patients with active autoimmune disease or a medical condition that required immunosuppression were also ineligible. Patients were randomized to KEYTRUDA 200 mg administered intravenously every 3 weeks or placebo for up to 1 year until disease recurrence or unacceptable toxicity. Randomization was stratified by metastasis status (M0, M1 NED); M0 group was further stratified by ECOG PS (0,1) and geographic region (US, non-US).
The study population characteristics were: median age of 60 years (range: 25 to 84), 33% age 65 or older; 71% male; 75% White, 14% Asian, 9% Unknown, 1% Black or African American, 1% American Indian or Alaska Native, 1% Multiracial; 13% Hispanic or Latino, 78% Not Hispanic or Latino, 8% Unknown; and 85% ECOG PS of 0 and 15% ECOG PS of 1. Ninety-four percent of patients enrolled had N0 disease; 11% had sarcomatoid features; 86% were intermediate-high risk; 8% were high risk; and 6% were M1 NED. Ninety-two percent of patients had a radical nephrectomy, and 8% had a partial nephrectomy.
The major efficacy outcome measure was investigator-assessed disease-free survival (DFS), defined as time to recurrence, metastasis, or death. An additional outcome measure was OS. Statistically significant improvements in DFS and OS were demonstrated at pre-specified interim analyses in patients randomized to the KEYTRUDA arm compared with placebo. Efficacy results are summarized in Table 95 and Figures 30 and 31.
| Endpoint | KEYTRUDA 200 mg every 3 weeks n=496 | Placebo n=498 |
|---|---|---|
| NR = not reached | ||
| DFS | ||
| Number (%) of patients with event | 109 (22%) | 151 (30%) |
| Median in months (95% CI) | NR | NR |
| Hazard ratio* (95% CI) | 0.68 (0.53, 0.87) | |
| p-Value† | 0.0010‡ | |
| 24-month DFS rate (95% CI) | 77% (73, 81) | 68% (64, 72) |
| OS | ||
| Number (%) of patients with event | 55 (11%) | 86 (17%) |
| Median in months (95% CI) | NR (NR, NR) | NR (NR, NR) |
| Hazard ratio* (95% CI) | 0.62 (0.44, 0.87) | |
| p-Value† | 0.0024§ | |
| 48-month OS rate (95% CI) | 91% (88, 93) | 86% (83, 89) |
| Figure 30: Kaplan-Meier Curve for Disease-Free Survival in KEYNOTE-564 |
|
|
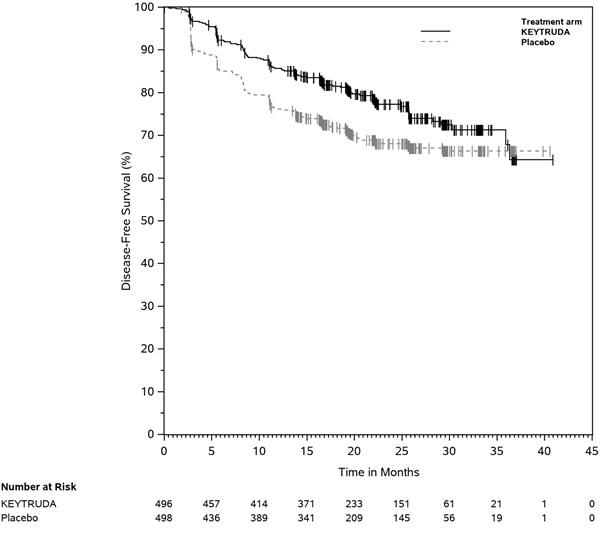
| Figure 31: Kaplan-Meier Curve for Overall Survival in KEYNOTE-564 |
|
|

14.16 Endometrial Carcinoma
In Combination with Paclitaxel and Carboplatin for the Treatment of Primary Advanced or Recurrent Endometrial Carcinoma
The efficacy of KEYTRUDA in combination with paclitaxel and carboplatin was investigated in KEYNOTE-868/NRG-GY018 (NCT03914612), a multicenter, randomized, double-blind, placebo-controlled trial in 810 patients with advanced or recurrent endometrial carcinoma. The study design included two separate cohorts based on MMR status; 222 (27%) patients were in dMMR cohort, 588 (73%) patients were in pMMR cohort. The trial enrolled measurable Stage III, measurable Stage IVA, Stage IVB or recurrent endometrial cancer (with or without measurable disease). Patients who had not received prior systemic therapy or had received prior chemotherapy in the adjuvant setting were eligible. Patients who had received prior adjuvant chemotherapy were only eligible if their chemotherapy-free interval was at least 12 months. Patients with endometrial sarcoma, including carcinosarcoma, or patients with active autoimmune disease or a medical condition that required immunosuppression were ineligible. Randomization was stratified according to MMR status, ECOG PS (0 or 1 vs. 2), and prior adjuvant chemotherapy.
Patients were randomized (1:1) to one of the following treatment arms:
- KEYTRUDA 200 mg every 3 weeks, paclitaxel 175 mg/m2 and carboplatin AUC 5 mg/mL/min for 6 cycles, followed by KEYTRUDA 400 mg every 6 weeks for up to 14 cycles.
- Placebo every 3 weeks, paclitaxel 175 mg/m2 and carboplatin AUC 5 mg/mL/min for 6 cycles, followed by placebo every 6 weeks for up to 14 cycles.
All study medications were administered as an intravenous infusion on Day 1 of each treatment cycle. Treatment continued until disease progression, unacceptable toxicity, or a maximum of 20 cycles (up to approximately 24 months). Patients with measurable disease who had RECIST-defined stable disease or partial response at the completion of cycle 6 were permitted to continue receiving paclitaxel and carboplatin with KEYTRUDA or placebo for up to 10 cycles as determined by the investigator. Assessment of tumor status was performed every 9 weeks for the first 9 months and then every 12 weeks thereafter. The major efficacy outcome measure was PFS as assessed by the investigator according to RECIST 1.1. An additional efficacy outcome measure was OS.
The dMMR population characteristics were: median age of 66 years (range: 37 to 86), 55% age 65 or older; 79% White, 9% Black, and 3% Asian; 5% Hispanic or Latino; 64% ECOG PS of 0, 33% ECOG PS of 1, and 3% ECOG PS of 2; 61% had recurrent disease and 39% had primary or persistent disease; 5% received prior adjuvant chemotherapy and 43% received prior radiotherapy. The histologic subtypes were endometrioid carcinoma (81%), adenocarcinoma NOS (11%), serous carcinoma (2%), and other (6%).
The pMMR population characteristics were: median age of 66 years (range: 29 to 94), 54% age 65 or older; 72% White, 16% Black, and 5% Asian; 6% Hispanic or Latino; 67% ECOG PS of 0, 30% ECOG PS of 1, and 3% ECOG PS of 2; 56% had recurrent disease and 44% had primary or persistent disease; 26% received prior adjuvant chemotherapy and 41% received prior radiotherapy. The histologic subtypes were endometrioid carcinoma (52%), serous carcinoma (26%), adenocarcinoma NOS (10%), clear cell carcinoma (7%), and other (5%).
The trial demonstrated statistically significant improvements in PFS for patients randomized to KEYTRUDA in combination with paclitaxel and carboplatin compared to placebo in combination with paclitaxel and carboplatin in both the dMMR and pMMR populations. Table 96 and Figures 32 and 33 summarize the efficacy results for KEYNOTE-868 by MMR status. At the time of the PFS analysis, OS data were not mature with 12% deaths in the dMMR population and 17% deaths in the pMMR population.
| Endpoint | dMMR Population | pMMR Population | ||
|---|---|---|---|---|
| KEYTRUDA with paclitaxel and carboplatin n=110 | Placebo with paclitaxel and carboplatin n=112 | KEYTRUDA with paclitaxel and carboplatin n=294 | Placebo with paclitaxel and carboplatin n=294 |
|
| NR = not reached | ||||
| PFS* | ||||
| Number (%) of patients with event | 26 (24%) | 57 (51%) | 91 (31%) | 124 (42%) |
| Median in months (95% CI) | NR (30.7, NR) | 6.5 (6.4, 8.7) | 11.1 (8.7, 13.5) | 8.5 (7.2, 8.8) |
| Hazard ratio† (95% CI) | 0.30 (0.19, 0.48) | 0.60 (0.46, 0.78) | ||
| p-Value‡ | <0.0001 | <0.0001 | ||
| Figure 32: Kaplan-Meier Curve for Progression-Free Survival in KEYNOTE-868 (dMMR Population) |
|
|
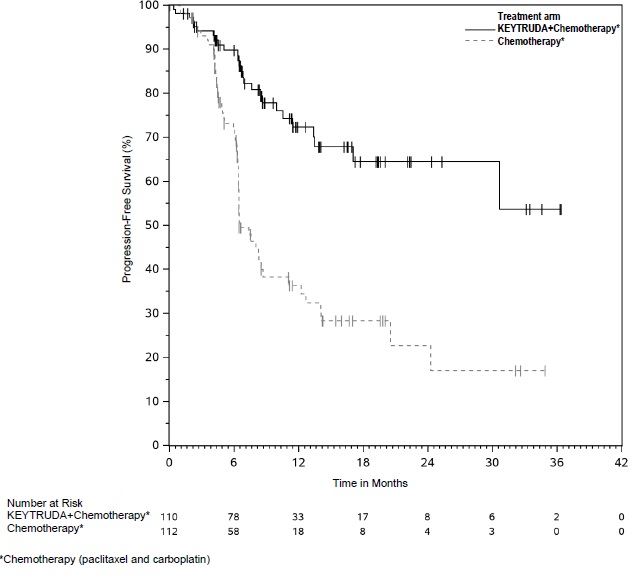
| Figure 33: Kaplan-Meier Curve for Progression-Free Survival in KEYNOTE-868 (pMMR Population) |
|
|
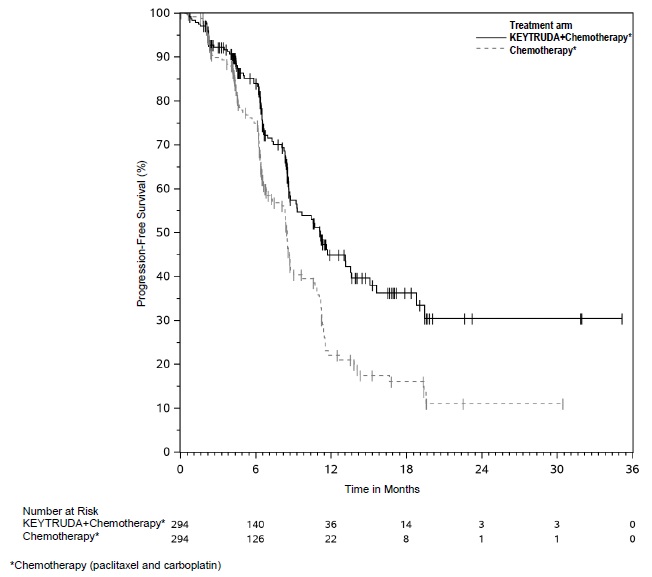
In Combination with Lenvatinib for the Treatment of Advanced Endometrial Carcinoma That Is pMMR or Not MSI-H
The efficacy of KEYTRUDA in combination with lenvatinib was investigated in KEYNOTE-775 (NCT03517449), a multicenter, open-label, randomized, active-controlled trial that enrolled 827 patients with advanced endometrial carcinoma who had been previously treated with at least one prior platinum-based chemotherapy regimen in any setting, including in the neoadjuvant and adjuvant settings. Patients with endometrial sarcoma, including carcinosarcoma, or patients who had active autoimmune disease or a medical condition that required immunosuppression were ineligible. Patients with endometrial carcinoma that were pMMR (using the VENTANA MMR RxDx Panel test) or not MSI-H were stratified by ECOG performance status, geographic region, and history of pelvic radiation. Patients were randomized (1:1) to one of the following treatment arms:
- KEYTRUDA 200 mg intravenously every 3 weeks in combination with lenvatinib 20 mg orally once daily.
- Investigator’s choice, consisting of either doxorubicin 60 mg/m2 every 3 weeks or paclitaxel 80 mg/m2 given weekly, 3 weeks on/1 week off.
Treatment with KEYTRUDA and lenvatinib continued until RECIST v1.1-defined progression of disease as verified by BICR, unacceptable toxicity, or for KEYTRUDA, a maximum of 24 months. Treatment was permitted beyond RECIST v1.1-defined disease progression if the treating investigator considered the patient to be deriving clinical benefit, and the treatment was tolerated. Assessment of tumor status was performed every 8 weeks. The major efficacy outcome measures were OS and PFS as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ. Additional efficacy outcome measures included ORR and DoR, as assessed by BICR.
Among the 697 pMMR patients, 346 patients were randomized to KEYTRUDA in combination with lenvatinib, and 351 patients were randomized to investigator’s choice of doxorubicin (n=254) or paclitaxel (n=97). The pMMR population characteristics were: median age of 65 years (range: 30 to 86), 52% age 65 or older; 62% White, 22% Asian, and 3% Black; 60% ECOG PS of 0 and 40% ECOG PS of 1. The histologic subtypes were endometrioid carcinoma (55%), serous (30%), clear cell carcinoma (7%), mixed (4%), and other (3%). All 697 of these patients received prior systemic therapy for endometrial carcinoma: 67% had one, 30% had two, and 3% had three or more prior systemic therapies. Thirty-seven percent of patients received only prior neoadjuvant or adjuvant therapy.
Efficacy results for the pMMR or not MSI-H patients are summarized in Table 97 and Figures 34 and 35.
| Endometrial Carcinoma (pMMR or not MSI-H) | ||
|---|---|---|
| Endpoint | KEYTRUDA 200 mg every 3 weeks and Lenvatinib n=346 | Doxorubicin or Paclitaxel n=351 |
|
||
| OS | ||
| Number (%) of patients with event | 165 (48%) | 203 (58%) |
| Median in months (95% CI) | 17.4 (14.2, 19.9) | 12.0 (10.8, 13.3) |
| Hazard ratio* (95% CI) | 0.68 (0.56, 0.84) | |
| p-Value† | 0.0001 | |
| PFS | ||
| Number (%) of patients with event | 247 (71%) | 238 (68%) |
| Median in months (95% CI) | 6.6 (5.6, 7.4) | 3.8 (3.6, 5.0) |
| Hazard ratio* (95% CI) | 0.60 (0.50, 0.72) | |
| p-Value† | <0.0001 | |
| Objective Response Rate | ||
| ORR‡ (95% CI) | 30% (26, 36) | 15% (12, 19) |
| Complete response rate | 5% | 3% |
| Partial response rate | 25% | 13% |
| p-Value§ | <0.0001 | |
| Duration of Response | n=105 | n=53 |
| Median in months (range) | 9.2 (1.6+, 23.7+) | 5.7 (0.0+, 24.2+) |
| Figure 34: Kaplan-Meier Curve for Overall Survival in KEYNOTE-775 (pMMR or Not MSI-H) |
|
|
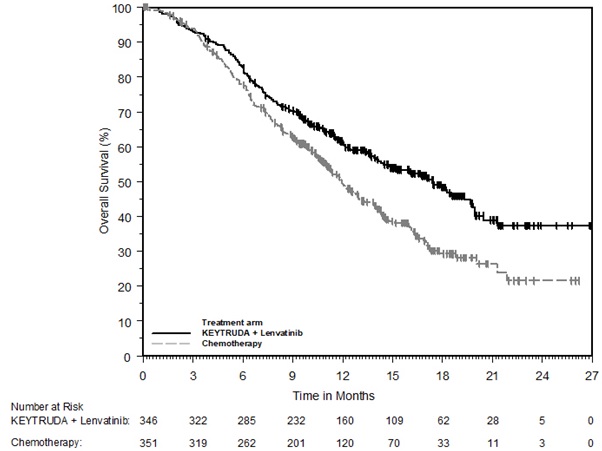
| Figure 35: Kaplan-Meier Curve for Progression-Free Survival in KEYNOTE-775 (pMMR or Not MSI-H) |
|
|
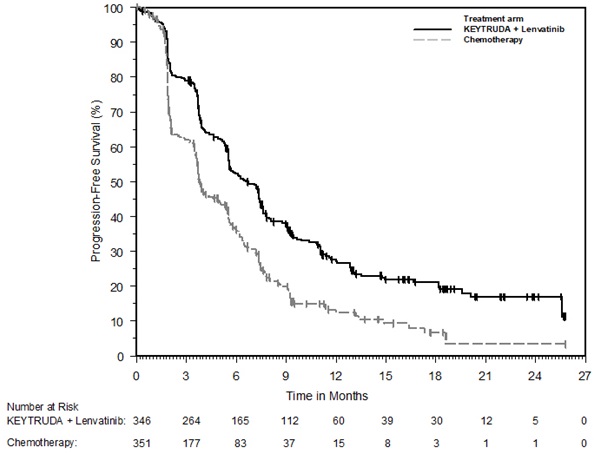
As a Single Agent for the Treatment of Advanced MSI-H or dMMR Endometrial Carcinoma
The efficacy of KEYTRUDA was investigated in KEYNOTE-158 (NCT02628067), a multicenter, non-randomized, open-label, multi-cohort trial. The trial enrolled 90 patients with unresectable or metastatic MSI-H or dMMR endometrial carcinoma in Cohorts D and K who received at least one dose of KEYTRUDA. MSI or MMR tumor status was determined using polymerase chain reaction (PCR) or immunohistochemistry (IHC), respectively. Patients with autoimmune disease or a medical condition that required immunosuppression were ineligible. Patients received KEYTRUDA 200 mg intravenously every 3 weeks until unacceptable toxicity or documented disease progression. Patients treated with KEYTRUDA without disease progression could be treated for up to 24 months. Assessment of tumor status was performed every 9 weeks for the first 12 months, and every 12 weeks thereafter. The major efficacy outcome measures were ORR and DoR as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
Among the 90 patients evaluated, the baseline characteristics were: median age of 64 years (range: 42 to 86); 83% White, 8% Asian, and 3% Black; 12% Hispanic or Latino; 39% ECOG PS of 0 and 61% ECOG PS of 1; 96% had M1 disease and 4% had M0 disease at study entry; and 51% had one and 48% had two or more prior lines of therapy. Nine patients received only adjuvant therapy and one patient received only neoadjuvant and adjuvant therapy before participating in the study.
Efficacy results are summarized in Table 98.
| Endpoint | KEYTRUDA n=90* |
|---|---|
| + Denotes ongoing response NR = not reached |
|
|
|
| Objective Response Rate | |
| ORR (95% CI) | 46% (35, 56) |
| Complete response rate | 12% |
| Partial response rate | 33% |
| Duration of Response | n=41 |
| Median in months (range) | NR (2.9, 55.7+) |
| % with duration ≥12 months | 68% |
| % with duration ≥24 months | 44% |
14.17 Tumor Mutational Burden-High Cancer
The efficacy of KEYTRUDA was investigated in a prospectively-planned retrospective analysis of 10 cohorts (A through J) of patients with various previously treated unresectable or metastatic solid tumors with high tumor mutation burden (TMB-H) who were enrolled in a multicenter, non-randomized, open-label trial, KEYNOTE-158 (NCT02628067). The trial excluded patients who previously received an anti-PD-1 or other immune-modulating monoclonal antibody, or who had an autoimmune disease, or a medical condition that required immunosuppression. Patients received KEYTRUDA 200 mg intravenously every 3 weeks until unacceptable toxicity or documented disease progression. Assessment of tumor status was performed every 9 weeks for the first 12 months and every 12 weeks thereafter.
The statistical analysis plan pre-specified ≥10 and ≥13 mutations per megabase using the FoundationOne CDx assay as cutpoints to assess TMB. Testing of TMB was blinded with respect to clinical outcomes. The major efficacy outcome measures were ORR and DoR in patients who received at least one dose of KEYTRUDA as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
In KEYNOTE-158, 1050 patients were included in the efficacy analysis population. TMB was analyzed in the subset of 790 patients with sufficient tissue for testing based on protocol-specified testing requirements. Of the 790 patients, 102 (13%) had tumors identified as TMB-H, defined as TMB ≥10 mutations per megabase. Among the 102 patients with TMB-H advanced solid tumors, the study population characteristics were: median age of 61 years (range: 27 to 80), 34% age 65 or older; 34% male; 81% White; and 41% ECOG PS of 0 and 58% ECOG PS of 1. Fifty-six percent of patients had at least two prior lines of therapy.
Efficacy results are summarized in Tables 99 and 100.
|
Endpoint | KEYTRUDA 200 mg every 3 weeks |
|
|---|---|---|
| TMB ≥10 mut/Mb n=102* | TMB ≥13 mut/Mb n=70 |
|
| + Denotes ongoing response NR = not reached |
||
| Objective Response Rate | ||
| ORR (95% CI) | 29% (21, 39) | 37% (26, 50) |
| Complete response rate | 4% | 3% |
| Partial response rate | 25% | 34% |
| Duration of Response | n=30 | n=26 |
| Median in months (range)† | NR (2.2+, 34.8+) | NR (2.2+, 34.8+) |
| % with duration ≥12 months | 57% | 58% |
| % with duration ≥24 months | 50% | 50% |
| Objective Response Rate | Duration of Response range | |||
|---|---|---|---|---|
| N | n (%) | 95% CI | (months) | |
| CR = complete response PR = partial response SD = stable disease PD = progressive disease |
||||
|
||||
| Overall* | 102 | 30 (29%) | (21%, 39%) | (2.2+, 34.8+) |
| Small cell lung cancer | 34 | 10 (29%) | (15%, 47%) | (4.1, 32.5+) |
| Cervical cancer | 16 | 5 (31%) | (11%, 59%) | (3.7+, 34.8+) |
| Endometrial cancer | 15 | 7 (47%) | (21%, 73%) | (8.4+, 33.9+) |
| Anal cancer | 14 | 1 (7%) | (0.2%, 34%) | 18.8+ |
| Vulvar cancer | 12 | 2 (17%) | (2%, 48%) | (8.8, 11.0) |
| Neuroendocrine cancer | 5 | 2 (40%) | (5%, 85%) | (2.2+, 32.6+) |
| Salivary cancer | 3 | PR, SD, PD | 31.3+ | |
| Thyroid cancer | 2 | CR, CR | (8.2, 33.2+) | |
| Mesothelioma cancer | 1 | PD | ||
In an exploratory analysis in 32 patients enrolled in KEYNOTE-158 whose cancer had TMB ≥10 mut/Mb and <13 mut/Mb, the ORR was 13% (95% CI: 4%, 29%), including two complete responses and two partial responses.
14.18 Cutaneous Squamous Cell Carcinoma
The efficacy of KEYTRUDA was investigated in patients with recurrent or metastatic cSCC or locally advanced cSCC enrolled in KEYNOTE-629 (NCT03284424), a multicenter, multi-cohort, non-randomized, open-label trial. The trial excluded patients with autoimmune disease or a medical condition that required immunosuppression.
Patients received KEYTRUDA 200 mg intravenously every 3 weeks until documented disease progression, unacceptable toxicity, or a maximum of 24 months. Patients with initial radiographic disease progression could receive additional doses of KEYTRUDA during confirmation of progression unless disease progression was symptomatic, rapidly progressive, required urgent intervention, or occurred with a decline in performance status.
Assessment of tumor status was performed every 6 weeks during the first year, and every 9 weeks during the second year. The major efficacy outcome measures were ORR and DoR as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
Among the 105 patients with recurrent or metastatic cSCC treated, the study population characteristics were: median age of 72 years (range: 29 to 95), 71% age 65 or older; 76% male; 71% White, 25% race unknown; 34% ECOG PS of 0 and 66% ECOG PS of 1. Forty-five percent of patients had locally recurrent only cSCC, 24% had metastatic only cSCC, and 31% had both locally recurrent and metastatic cSCC. Eighty-seven percent received one or more prior lines of therapy; 74% received prior radiation therapy.
Among the 54 patients with locally advanced cSCC treated, the study population characteristics were: median age of 76 years (range: 35 to 95), 80% age 65 or older; 72% male; 83% White, 13% race unknown; 41% ECOG PS of 0 and 59% ECOG PS of 1. Twenty-two percent received one or more prior lines of therapy; 63% received prior radiation therapy.
Efficacy results are summarized in Table 101.
| Endpoint | KEYTRUDA Recurrent or Metastatic cSCC n=105 | KEYTRUDA Locally Advanced cSCC n=54 |
|---|---|---|
| + Denotes ongoing response | ||
|
||
| Objective Response Rate | ||
| ORR (95% CI) | 35% (26, 45) | 50% (36, 64) |
| Complete response rate | 11% | 17% |
| Partial response rate | 25% | 33% |
| Duration of Response* | n=37 | n=27 |
| Median in months (range) | NR (2.7, 30.4+) | NR (1.0+, 17.2+) |
| % with duration ≥6 months | 76% | 81% |
| % with duration ≥12 months | 68% | 37% |
14.19 Triple-Negative Breast Cancer
Neoadjuvant and Adjuvant Treatment of High-Risk Early-Stage TNBC
The efficacy of KEYTRUDA in combination with neoadjuvant chemotherapy followed by surgery and continued adjuvant treatment with KEYTRUDA as a single agent was investigated in KEYNOTE-522 (NCT03036488), a randomized (2:1), multicenter, double-blind, placebo-controlled trial conducted in 1174 patients with newly diagnosed previously untreated high-risk early-stage TNBC (tumor size >1 cm but ≤2 cm in diameter with nodal involvement or tumor size >2 cm in diameter regardless of nodal involvement). Patients were enrolled regardless of tumor PD-L1 expression. Patients with active autoimmune disease that required systemic therapy within two years of treatment or a medical condition that required immunosuppression were ineligible. Randomization was stratified by nodal status (positive vs. negative), tumor size (T1/T2 vs. T3/T4), and choice of carboplatin (dosed every 3 weeks vs. weekly).
Patients were randomized (2:1) to one of the following two treatment arms; all study medications were administered intravenously:
-
Arm 1:
- Four cycles of preoperative KEYTRUDA 200 mg every 3 weeks on Day 1 of cycles 1-4 of treatment regimen in combination with:
- Carboplatin
- AUC 5 mg/mL/min every 3 weeks on Day 1 of cycles 1-4 of treatment regimen
-or- - AUC 1.5 mg/mL/min every week on Days 1, 8, and 15 of cycles 1-4 of treatment regimen
-and-
- AUC 5 mg/mL/min every 3 weeks on Day 1 of cycles 1-4 of treatment regimen
- Paclitaxel 80 mg/m2 every week on Days 1, 8, and 15 of cycles 1-4 of treatment regimen
- Carboplatin
- Followed by four additional cycles of preoperative KEYTRUDA 200 mg every 3 weeks on Day 1 of cycles 5-8 of treatment regimen in combination with:
- Doxorubicin 60 mg/m2 -or- epirubicin 90 mg/m2 every 3 weeks on Day 1 of cycles 5-8 of treatment regimen -and-
- Cyclophosphamide 600 mg/m2 every 3 weeks on Day 1 of cycles 5-8 of treatment regimen
- Following surgery, nine cycles of KEYTRUDA 200 mg every 3 weeks were administered.
- Four cycles of preoperative KEYTRUDA 200 mg every 3 weeks on Day 1 of cycles 1-4 of treatment regimen in combination with:
-
Arm 2:
- Four cycles of preoperative placebo every 3 weeks on Day 1 of cycles 1-4 of treatment regimen in combination with:
- Carboplatin
- AUC 5 mg/mL/min every 3 weeks on Day 1 of cycles 1-4 of treatment regimen
-or- - AUC 1.5 mg/mL/min every week on Days 1, 8, and 15 of cycles 1-4 of treatment regimen
-and-
- AUC 5 mg/mL/min every 3 weeks on Day 1 of cycles 1-4 of treatment regimen
- Paclitaxel 80 mg/m2 every week on Days 1, 8, and 15 of cycles 1-4 of treatment regimen
- Carboplatin
- Followed by four cycles of preoperative placebo every 3 weeks on Day 1 of cycles 5-8 of treatment regimen in combination with:
- Doxorubicin 60 mg/m2 -or- epirubicin 90 mg/m2 every 3 weeks on Day 1 of cycles 5-8 of treatment regimen -and-
- Cyclophosphamide 600 mg/m2 every 3 weeks on Day 1 of cycles 5-8 of treatment regimen
- Following surgery, nine cycles of placebo every 3 weeks were administered.
- Four cycles of preoperative placebo every 3 weeks on Day 1 of cycles 1-4 of treatment regimen in combination with:
The main efficacy outcomes were pCR rate and EFS. pCR was defined as absence of invasive cancer in the breast and lymph nodes (ypT0/Tis ypN0) and was assessed by the blinded local pathologist at the time of definitive surgery. EFS was defined as the time from randomization to the first occurrence of any of the following events: progression of disease that precludes definitive surgery, local or distant recurrence, second primary malignancy, or death due to any cause. An additional efficacy outcome was overall survival (OS).
The study population characteristics were: median age of 49 years (range: 22 to 80), 11% age 65 or older; 99.9% female; 64% White, 20% Asian, 4.5% Black, and 1.8% American Indian or Alaska Native; 87% ECOG PS of 0 and 13% ECOG PS of 1; 56% were pre-menopausal status and 44% were post-menopausal status; 7% were primary Tumor 1 (T1), 68% T2, 19% T3, and 7% T4; 49% were nodal involvement 0 (N0), 40% N1, 11% N2, and 0.2% N3; 75% of patients were overall Stage II and 25% were Stage III.
Table 102 and Figure 36 summarize the efficacy results for KEYNOTE-522. At the protocol pre-specified IA4 interim analysis of OS, OS data were not mature with 45% of the required events for the final analysis.
| Endpoint | KEYTRUDA 200 mg every 3 weeks with chemotherapy/KEYTRUDA n=784 | Placebo with chemotherapy/Placebo n=390 |
|---|---|---|
|
||
| pCR (ypT0/Tis ypN0)* | ||
| Number of patients with pCR | 494 | 217 |
| pCR Rate (%), (95% CI) | 63.0 (59.5, 66.4) | 55.6 (50.6, 60.6) |
| Treatment difference (%) estimate (95% CI)†,‡ | 7.5 (1.6, 13.4) | |
| EFS | ||
| Number of patients with event (%) | 123 (16%) | 93 (24%) |
| Hazard ratio (95% CI)§ | 0.63 (0.48, 0.82) | |
| p-Value¶,# | 0.00031 | |
| Figure 36: Kaplan-Meier Curve for Event-Free Survival in KEYNOTE-522 |
|
|
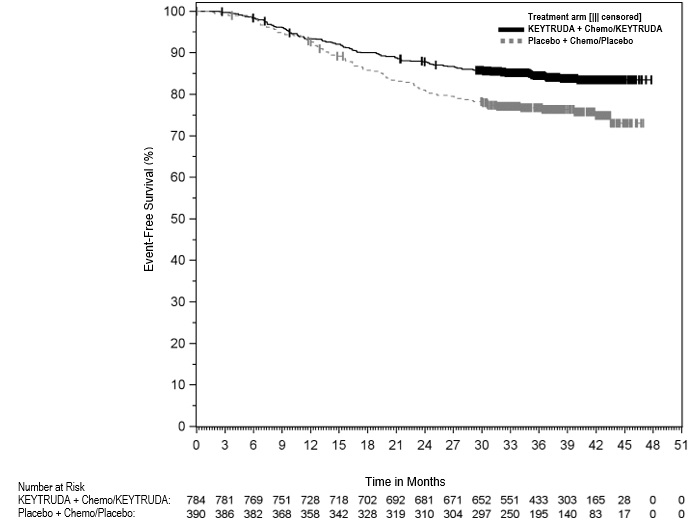
Locally Recurrent Unresectable or Metastatic TNBC
The efficacy of KEYTRUDA in combination with paclitaxel, paclitaxel protein-bound, or gemcitabine and carboplatin was investigated in KEYNOTE-355 (NCT02819518), a multicenter, double-blind, randomized, placebo-controlled trial conducted in 847 patients with locally recurrent unresectable or metastatic TNBC, regardless of tumor PD-L1 expression, who had not been previously treated with chemotherapy in the metastatic setting. Patients with active autoimmune disease that required systemic therapy within 2 years of treatment or a medical condition that required immunosuppression were ineligible. Randomization was stratified by chemotherapy treatment (paclitaxel or paclitaxel protein-bound vs. gemcitabine and carboplatin), tumor PD-L1 expression (CPS ≥1 vs. CPS <1) according to the PD-L1 IHC 22C3 pharmDx kit, and prior treatment with the same class of chemotherapy in the neoadjuvant setting (yes vs. no).
Patients were randomized (2:1) to one of the following treatment arms; all study medications were administered via intravenous infusion:
- KEYTRUDA 200 mg on Day 1 every 3 weeks in combination with paclitaxel protein-bound 100 mg/m2 on Days 1, 8 and 15 every 28 days, paclitaxel 90 mg/m2 on Days 1, 8, and 15 every 28 days, or gemcitabine 1000 mg/m2 and carboplatin AUC 2 mg/mL/min on Days 1 and 8 every 21 days.
- Placebo on Day 1 every 3 weeks in combination with paclitaxel protein-bound 100 mg/m2 on Days 1, 8 and 15 every 28 days, paclitaxel 90 mg/m2 on Days 1, 8, and 15 every 28 days, or gemcitabine 1000 mg/m2 and carboplatin AUC 2 mg/mL/min on Days 1 and 8 every 21 days.
Assessment of tumor status was performed at Weeks 8, 16, and 24, then every 9 weeks for the first year, and every 12 weeks thereafter. The main efficacy outcome measures were OS and PFS as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ, tested in the subgroup of patients with CPS ≥10. Additional efficacy outcome measures were ORR and DoR as assessed by BICR.
The study population characteristics for patients were: median age of 53 years (range: 22 to 85), 21% age 65 or older; 100% female; 68% White, 21% Asian, and 4% Black; 60% ECOG PS of 0 and 40% ECOG PS of 1; and 68% were post-menopausal status. Seventy-five percent of patients had tumor PD-L1 expression CPS ≥1 and 38% had tumor PD-L1 expression CPS ≥10.
Table 103 and Figures 37 and 38 summarize the efficacy results for KEYNOTE-355.
| Endpoint | KEYTRUDA 200 mg every 3 weeks with chemotherapy n=220 | Placebo every 3 weeks with chemotherapy n=103 |
|---|---|---|
|
||
| OS* | ||
| Number of patients with event (%) | 155 (70%) | 84 (82%) |
| Median in months (95% CI) | 23 (19.0, 26.3) | 16.1 (12.6, 18.8) |
| Hazard ratio† (95% CI) | 0.73 (0.55, 0.95) | |
| p-Value‡ | 0.0093 | |
| PFS§ | ||
| Number of patients with event (%) | 136 (62%) | 79 (77%) |
| Median in months (95% CI) | 9.7 (7.6, 11.3) | 5.6 (5.3, 7.5) |
| Hazard ratio† (95% CI) | 0.65 (0.49, 0.86) | |
| p-Value¶ | 0.0012 | |
| Objective Response Rate (Confirmed)* | ||
| ORR (95% CI) | 53% (46, 59) | 41% (31, 51) |
| Complete response rate | 17% | 14% |
| Partial response rate | 35% | 27% |
| Duration of Response* | n=116 | n=42 |
| Median in months (95% CI) | 12.8 (9.9, 25.9) | 7.3 (5.5, 15.4) |
| Figure 37: Kaplan-Meier Curve for Overall Survival in KEYNOTE-355 (CPS ≥10) |
|
|
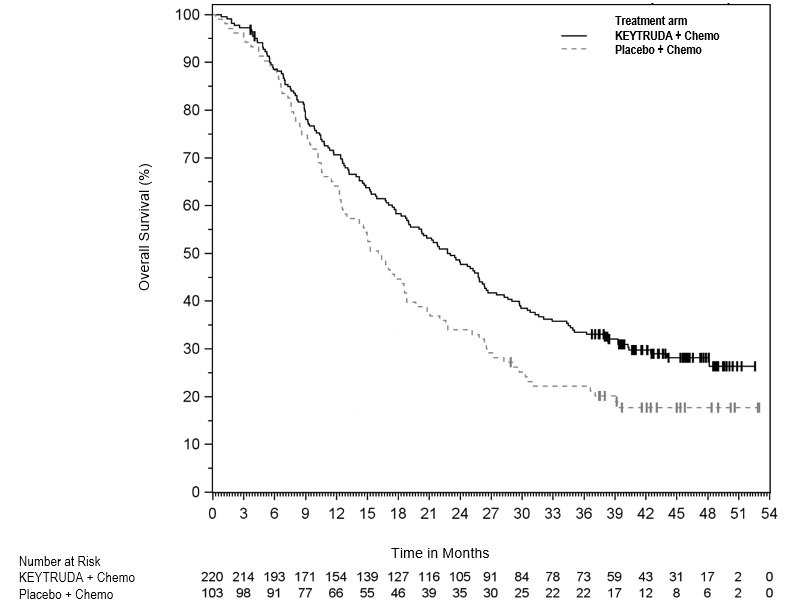
| Figure 38: Kaplan-Meier Curve for Progression-Free Survival in KEYNOTE-355 (CPS ≥10) |
|
|
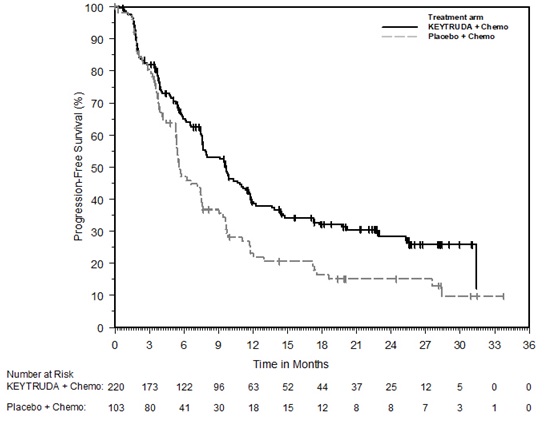
14.20 Adult Classical Hodgkin Lymphoma and Adult Primary Mediastinal Large B-Cell Lymphoma: Additional Dosing Regimen of 400 mg Every 6 Weeks
The efficacy and safety of KEYTRUDA using a dosage of 400 mg every 6 weeks for the classical Hodgkin lymphoma and primary mediastinal large B-cell lymphoma indications for adults was primarily based on the dose/exposure efficacy and safety relationships and observed pharmacokinetic data in patients with melanoma [see Clinical Pharmacology (12.2)].
16. How is Keytruda supplied
KEYTRUDA injection (clear to slightly opalescent, colorless to slightly yellow solution):
Carton containing one 100 mg/4 mL (25 mg/mL), single-dose vial (NDC 0006-3026-02)
Carton containing two 100 mg/4 mL (25 mg/mL), single-dose vials (NDC 0006-3026-04)
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Immune-Mediated Adverse Reactions
- Inform patients of the risk of immune-mediated adverse reactions that may be severe or fatal, may occur after discontinuation of treatment, and may require corticosteroid treatment and interruption or discontinuation of KEYTRUDA. These reactions may include:
- Pneumonitis: Advise patients to contact their healthcare provider immediately for new or worsening cough, chest pain, or shortness of breath [see Warnings and Precautions (5.1)].
- Colitis: Advise patients to contact their healthcare provider immediately for diarrhea or severe abdominal pain [see Warnings and Precautions (5.1)].
- Hepatitis: Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, or easy bruising or bleeding [see Warnings and Precautions (5.1)].
- Endocrinopathies: Advise patients to contact their healthcare provider immediately for signs or symptoms of adrenal insufficiency, hypophysitis, hypothyroidism, hyperthyroidism, or Type 1 diabetes mellitus [see Warnings and Precautions (5.1)].
- Nephritis: Advise patients to contact their healthcare provider immediately for signs or symptoms of nephritis [see Warnings and Precautions (5.1)].
- Severe skin reactions: Advise patients to contact their healthcare provider immediately for any signs or symptoms of severe skin reactions, SJS or TEN [see Warnings and Precautions (5.1)].
- Other immune-mediated adverse reactions:
- Advise patients that immune-mediated adverse reactions can occur and may involve any organ system, and to contact their healthcare provider immediately for any new or worsening signs or symptoms [see Warnings and Precautions (5.1)].
- Advise patients of the risk of solid organ transplant rejection and other transplant (including corneal graft) rejection. Advise patients to contact their healthcare provider immediately for signs or symptoms of organ transplant rejection and other transplant (including corneal graft) rejection [see Warnings and Precautions (5.1)].
Infusion-Related Reactions
- Advise patients to contact their healthcare provider immediately for signs or symptoms of infusion-related reactions [see Warnings and Precautions (5.2)].
Complications of Allogeneic HSCT
- Advise patients of the risk of post-allogeneic hematopoietic stem cell transplantation complications [see Warnings and Precautions (5.3)].
Embryo-Fetal Toxicity
- Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.5), Use in Specific Populations (8.1, 8.3)].
- Advise females of reproductive potential to use effective contraception during treatment with KEYTRUDA and for 4 months after the last dose [see Warnings and Precautions (5.5), Use in Specific Populations (8.1, 8.3)].
Lactation
- Advise women not to breastfeed during treatment with KEYTRUDA and for 4 months after the last dose [see Use in Specific Populations (8.2)].
Laboratory Tests
- Advise patients of the importance of keeping scheduled appointments for blood work or other laboratory tests [see Warnings and Precautions (5.1)].
Manufactured by: Merck Sharp & Dohme LLC
Rahway, NJ 07065, USA
U.S. License No. 0002
For patent information: www.msd.com/research/patent
The trademarks depicted herein are owned by their respective companies.
uspi-mk3475-iv-2408r079
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: August 2024 | ||
| MEDICATION GUIDE KEYTRUDA® (key-true-duh) (pembrolizumab) injection |
|||
| What is the most important information I should know about KEYTRUDA? | |||
| KEYTRUDA is a medicine that may treat certain cancers by working with your immune system. KEYTRUDA can cause your immune system to attack normal organs and tissues in any area of your body and can affect the way they work. These problems can sometimes become severe or life-threatening and can lead to death. You can have more than one of these problems at the same time. These problems may happen anytime during treatment or even after your treatment has ended. | |||
| Call or see your healthcare provider right away if you develop any new or worsening signs or symptoms, including: | |||
| Lung problems | |||
|
|
|
|
| Intestinal problems | |||
|
|||
| Liver problems | |||
|
| ||
| Hormone gland problems | |||
|
|
||
| Kidney problems | |||
|
|
||
| Skin problems | |||
|
|
||
| Problems can also happen in other organs and tissues. These are not all of the signs and symptoms of immune system problems that can happen with KEYTRUDA. Call or see your healthcare provider right away for any new or worsening signs or symptoms, which may include: | |||
|
|||
| Infusion reactions that can sometimes be severe or life-threatening. Signs and symptoms of infusion reactions may include: | |||
|
|
||
| Rejection of a transplanted organ or tissue. Your healthcare provider should tell you what signs and symptoms you should report and monitor you depending on the type of organ or tissue transplant that you have had. | |||
| Complications, including graft-versus-host-disease (GVHD), in people who have received a bone marrow (stem cell) transplant that uses donor stem cells (allogeneic). These complications can be serious and can lead to death. These complications may happen if you underwent transplantation either before or after being treated with KEYTRUDA. Your healthcare provider will monitor you for these complications. | |||
|
Getting medical treatment right away may help keep these problems from becoming more serious. Your healthcare provider will check you for these problems during treatment with KEYTRUDA. Your healthcare provider may treat you with corticosteroid or hormone replacement medicines. Your healthcare provider may also need to delay or completely stop treatment with KEYTRUDA if you have severe side effects. |
|||
| What is KEYTRUDA? | |||
| KEYTRUDA is a prescription medicine used to treat: | |||
|
|||
Before receiving KEYTRUDA, tell your healthcare provider about all of your medical conditions, including if you:
|
|||
| Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. | |||
| How will I receive KEYTRUDA? | |||
|
|||
| What are the possible side effects of KEYTRUDA? | |||
| KEYTRUDA can cause serious side effects. See “What is the most important information I should know about KEYTRUDA?” | |||
| Common side effects of KEYTRUDA when used alone include: feeling tired, pain, including pain in muscles, rash, diarrhea, fever, cough, decreased appetite, itching, shortness of breath, constipation, bones or joints and stomach-area (abdominal) pain, nausea, and low levels of thyroid hormone. | |||
| Side effects of KEYTRUDA when used alone that are more common in children than in adults include: fever, vomiting, headache, stomach area (abdominal) pain, and low levels of white blood cells. | |||
| Common side effects of KEYTRUDA when given with certain chemotherapy or chemotherapy with radiation therapy medicines include: feeling tired or weak, nausea, constipation, diarrhea, decreased appetite, rash, vomiting, cough, trouble breathing, fever, hair loss, inflammation of the nerves that may cause pain, weakness, and paralysis in the arms and legs, swelling of the lining of the mouth, nose, eyes, throat, intestines, or vagina, mouth sores, headache, weight loss, stomach-area (abdominal) pain, joint and muscle pain, trouble sleeping, bleeding, blisters, or rash on the palms of your hands and soles of your feet, urinary tract infection, and low levels of thyroid hormone. | |||
| Common side effects of KEYTRUDA when given with chemotherapy and bevacizumab include: tingling or numbness of the arms or legs, hair loss, low red blood cell count, feeling tired or weak, nausea, low white blood cell count, diarrhea, high blood pressure, decreased platelet count, constipation, joint aches, vomiting, urinary tract infection, rash, low levels of thyroid hormone, and decreased appetite. | |||
| Common side effects of KEYTRUDA when given with axitinib include: diarrhea, feeling tired or weak, high blood pressure, liver problems, low levels of thyroid hormone, decreased appetite, blisters or rash on the palms of your hands and soles of your feet, nausea, mouth sores or swelling of the lining of the mouth, nose, eyes, throat, intestines, or vagina, hoarseness, rash, cough, and constipation. | |||
| Common side effects of KEYTRUDA when given with lenvatinib include: low levels of thyroid hormone, high blood pressure, feeling tired, diarrhea, joint and muscle pain, nausea, decreased appetite, vomiting, mouth sores, weight loss, stomach-area (abdominal) pain, urinary tract infection, protein in your urine, constipation, headache, bleeding, blisters or rash on the palms of your hands and soles of your feet, hoarseness, rash, liver problems, and kidney problems. | |||
| Common side effects of KEYTRUDA when given with enfortumab vedotin include: rash, tingling or numbness of the arms or legs, feeling tired, itching, diarrhea, hair loss, weight loss, decreased appetite, dry eye, nausea, constipation, changes in sense of taste, and urinary tract infection. | |||
| These are not all the possible side effects of KEYTRUDA. | |||
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |||
| General information about the safe and effective use of KEYTRUDA | |||
| Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about KEYTRUDA that is written for health professionals. | |||
| What are the ingredients in KEYTRUDA? | |||
| Active ingredient: pembrolizumab | |||
| Inactive ingredients: KEYTRUDA injection: L-histidine, polysorbate 80, sucrose, and Water for Injection. | |||
| Manufactured by: Merck Sharp & Dohme LLC Rahway, NJ 07065, USA | U.S. License No. 0002 For patent information: www.msd.com/research/patent Copyright © 2014-2024 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates. All rights reserved. usmg-mk3475-iv-2408r065 For more information, go to www.keytruda.com. | ||
PRINCIPAL DISPLAY PANEL - 50 mg Vial Carton
NDC 0006-3029-02
Keytruda®
(pembrolizumab)
for Injection
50 mg / vial
For Intravenous Infusion Only
Dispense the enclosed Medication Guide to each patient.
Sterile lyophilized powder must be reconstituted with Sterile Water for
Injection, USP. Reconstituted solution requires further dilution prior
to administration.
Rx only
Single-dose vial. Discard unused portion.

PRINCIPAL DISPLAY PANEL - 100 mg/4 mL Vial Carton
NDC 0006-3026-02
Keytruda®
(pembrolizumab)
Injection
100 mg/4 mL
(25 mg/mL)
For Intravenous Infusion Only
Dispense the enclosed Medication Guide to each patient.
Requires dilution prior to administration.
Rx only
Single-dose vial. Discard unused portion.

| KEYTRUDA
pembrolizumab injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| KEYTRUDA
pembrolizumab injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Merck Sharp & Dohme LLC (118446553) |
Frequently asked questions
- What is the success rate for treatment?
- How long does it take for Keytruda to work?
- What are monoclonal antibodies?
- What is the difference between Opdivo and Keytruda?
- Pembrolizumab vs. nivolumab: How do they compare?
- Can you take prednisone with Keytruda?
- Is it covered by Medicare / Medicaid?
- How are Inlyta and Keytruda used in kidney cancer?
- How is it administered?
More about Keytruda (pembrolizumab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (230)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- Support group
- FDA approval history
- Drug class: anti-PD-1 and PD-L1 monoclonal antibodies (immune checkpoint inhibitors)
- Breastfeeding
- En español
Patient resources
Professional resources
Related treatment guides
Copyright © 2014-2024 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates.
All rights reserved.