Inamrinone Prescribing Information
Package insert / product label
Dosage form: injection
Drug class: Inotropic agents
Medically reviewed by Drugs.com. Last updated on Mar 25, 2024.
On This Page
Inamrinone Description
Inamrinone Injection USP represents a new class of cardiac inotropic agents distinct from digitalis glycosides or catecholamines. Inamrinone lactate is designated chemically as 5-Amino[3,4'-bipyridin]-6(1H)-one 2-hydroxypropanate and has the following structure:
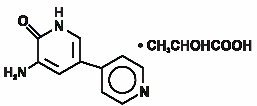
Inamrinone is a pale yellow crystalline compound with a molecular weight of 187.20 and a molecular formula of C10H9N3O. Each mole of lactic acid has a molecular weight of 90.08 and a empirical formula of C3H6O3. The solubilities of inamrinone at pH’s 4.1, 6.0, and 8.0 are 25, 0.9, and 0.7 mg/mL, respectively.
Inamrinone injection is a clear yellow sterile solution available in 20 mL vials for intravenous administration. Each mL contains inamrinone lactate equivalent to 5 mg of inamrinone and 0.25 mg of sodium metabisulfite added as a preservative in Water for Injection. All dosages expressed in the package insert are expressed in terms of the base, inamrinone. The pH is adjusted to between 3.2 to 4.0 with lactic acid or sodium hydroxide. The total concentration of lactic acid can vary between 5 mg and 7.5 mg.
Inamrinone - Clinical Pharmacology
Inamrinone is a positive inotropic agent with vasodilator activity, different in structure and mode of action from either digitalis glycosides or catecholamines.
The mechanism of its inotropic and vasodilator effects has not been fully elucidated.
With respect to its inotropic effect, experimental evidence indicates that it is not a beta-adrenergic agonist. It inhibits myocardial cyclic adenosine monophosphate (c-AMP) phosphodiesterase activity and increases cellular levels of c-AMP. Unlike digitalis, it does not inhibit sodium-potassium adenosine triphosphatase activity.
With respect to its vasodilatory activity, inamrinone reduces afterload and preload by its direct relaxant effect on vascular smooth muscle.
Pharmacokinetics
Following intravenous bolus (1 to 2 minutes) injection of 0.68 mg/kg to 1.2 mg/kg to normal volunteers, inamrinone had a volume of distribution of 1.2 liters/kg, and following a distributive phase half-life of about 4.6 minutes in plasma, had a mean apparent first-order terminal elimination half-life of about 3.6 hours. In patients with congestive heart failure receiving infusions of inamrinone the mean apparent first-order terminal elimination half-life was about 5.8 hours.
Inamrinone has been shown in one study to be 10% to 22% bound to human plasma protein by ultrafiltration in vitro, and in another study 35% to 49% bound by either ultrafiltration or equilibrium dialysis.
The primary route of excretion in man is via the urine as both inamrinone and several metabolites (N-glycolyl, N-acetate, O-glucuronide and N-glucuronide). In normal volunteers, approximately 63% of an oral dose of 14C-Iabelled inamrinone was excreted in the urine over a 96-hour period. In the first 8 hours, 51% of the radioactivity in the urine was inamrinone with 5% as the N-acetate, 8% as the N-glycolate, and less than 5% for each glucuronide. Approximately 18% of the administered dose was excreted in the feces in 72 hours.
In a 24-hour nonradioactive intravenous study, 10% to 40% of the dose was excreted in urine as unchanged inamrinone with the N-acetyl metabolite representing less than 2% of the dose.
In congestive heart failure patients, after a loading bolus dose, steady-state plasma levels of about 2.4 mcg/mL were able to be maintained by an infusion of 5 mcg/kg/min to 10 mcg/kg/min. In some congestive heart failure patients, with associated compromised renal and hepatic perfusion, it is possible that plasma levels of inamrinone may rise during the infusion period; therefore, in these patients, it may be necessary to monitor the hemodynamic response and/or drug level. The principal measures of patient response include cardiac index, pulmonary capillary wedge pressure, central venous pressure, and their relationship to plasma concentrations. Additionally, measurements of blood pressure, urine output, and body weight may prove useful, as may such clinical symptoms as orthopnea, dyspnea, and fatigue.
Pharmacodynamics
In patients with depressed myocardial function, inamrinone produces a prompt increase in cardiac output due to its inotropic and vasodilator actions.
Following a single intravenous bolus dose of inamrinone of 0.75 mg/kg to 3 mg/kg in patients with congestive heart failure, dose-related maximum increases in cardiac output occur (of about 28% at 0.75 mg/kg to about 61% at 3 mg/kg). The peak effect occurs within 10 minutes at all doses. The duration of effect depends upon dose, lasting about 1/2 hour at 0.75 mg/kg and approximately 2 hours at 3 mg/kg.
Over the same range of doses, pulmonary capillary wedge pressure and total peripheral resistance show dose-related decreases (mean maximum decreases of 29% in pulmonary capillary wedge pressure and 29% in systemic vascular resistance). At doses up to 3 mg/kg dose-related decreases in diastolic pressure (up to 13%) have been observed. Mean arterial pressure decreases (9.7%) at a dose of 3 mg/kg. The heart rate is generally unchanged.
The changes in hemodynamic parameters are maintained during continuous intravenous infusion and for several hours thereafter.
Inamrinone is effective in fully digitalized patients without causing signs of cardiac glycoside toxicity. Its inotropic effects are additive to those of digitalis. In cases of atrial flutter/fibrillation, it is possible that inamrinone may increase ventricular response rate because of its slight enhancement of A/V conduction. In these cases, prior treatment with digitalis is recommended.
Improvement in left ventricular function and relief of congestive heart failure in patients with ischemic heart disease have been observed. The improvement has occurred without inducing symptoms or electrocardiographic signs of myocardial ischemia.
At constant heart rate and blood pressure, increases in cardiac output occur without measurable increases in myocardial oxygen consumption or changes in arteriovenous oxygen difference.
Inotropic activity is maintained following repeated intravenous doses of inamrinone. Inamrinone administration produces hemodynamic and symptomatic benefits to patients not satisfactorily controlled by conventional therapy with diuretics and cardiac glycosides.
Indications and Usage for Inamrinone
Inamrinone injection is for the short-term management of congestive heart failure. Because of limited experience and potential for serious adverse effects (see ADVERSE REACTIONS), inamrinone should be used only in patients who can be closely monitored and who have not responded adequately to digitalis, diuretics, and/or vasodilators. Experience with intravenous inamrinone in controlled trials does not extend beyond 48 hours of repeated boluses and/or continuous infusions.
Whether given orally, continuously intravenously, or intermittently intravenously, neither inamrinone nor any other cyclic-AMP-dependent inotrope has been shown in controlled trials to be safe or effective in the long-term treatment of congestive heart failure. In controlled trials of chronic oral therapy with various such agents (including inamrinone), symptoms were not consistently alleviated, and the cyclic-AMP-dependent inotropes were consistently associated with increased risks of hospitalization and death. Patients with NYHA Class IV symptoms appeared to be at particular risk.
Contraindications
Inamrinone is contraindicated in patients who are hypersensitive to it.
It is also contraindicated in those patients known to be hypersensitive to bisulfites.
Warnings
Contains sodium metabisulfite, a sulfite that may cause allergic-type reactions including anaphylactic symptoms and life-threatening or less severe asthmatic episodes in certain susceptible people. The overall prevalence of sulfite sensitivity in the general population is unknown and probably low. Sulfite sensitivity is seen more frequently in asthmatic than in nonasthmatic people.
Precautions
General
Inamrinone should not be used in patients with severe aortic or pulmonic valvular disease in lieu of surgical relief of the obstruction. Like other inotropic agents, it may aggravate outflow tract obstruction in hypertrophic subaortic stenosis.
During intravenous therapy with inamrinone, blood pressure and heart rate shouId be monitored and the rate of infusion slowed or stopped in patients showing excessive decreases in blood pressure.
Patients who have received vigorous diuretic therapy may have insufficient cardiac filling pressure to respond adequately to inamrinone, in which case cautious liberalization of fluid and electrolyte intake may be indicated.
Supraventricular and ventricular arrhythmias have been observed in the very high-risk population treated. While inamrinone per se has not been shown to be arrhythmogenic, the potential for arrhythmia, present in congestive heart failure itself, may be increased by any drug or combination of drugs.
Thrombocytopenia and hepatotoxicity have been noted (see ADVERSE REACTIONS).
USE IN ACUTE MYOCARDIAL INFARCTION
No clinical trials have been carried out in patients in the acute phase of postmyocardial infarction. Therefore, inamrinone is not recommended in these cases.
Laboratory Tests
Fluid and Electrolytes
Fluid and electrolyte changes and renal function should be carefully monitored during inamrinone therapy. Improvement in cardiac output with resultant diuresis may necessitate a reduction in the dose of diuretic. Potassium loss due to excessive diuresis may predispose digitalized patients to arrhythmias. Therefore, hypokalemia should be corrected by potassium supplementation in advance of or during inamrinone use.
Drug Interactions
In a relatively limited experience, no untoward clinical manifestations have been observed in patients in which inamrinone was used concurrently with the following drugs: digitalis glycosides; lidocaine, quinidine; metoprolol, propranolol; hydralazine, prazosin; isosorbide dinitrate, nitroglycerine; chlorthalidone, ethacrynic acid, furosemide, hydrochlorothiazide, spironolactone; captopril; heparin, warfarin; potassium supplements; insulin; diazepam.
One case report of excessive hypotension has been reported when inamrinone was used concurrently with disopyramide.
Until additional experience is available, concurrent administration with disopyramide should be undertaken with caution.
Chemical Interactions
A chemical interaction occurs slowly over a 24-hour period when the intravenous solution of inamrinone is mixed directly with dextrose (glucose)-containing solutions. THEREFORE, INAMRINONE SHOULD NOT BE DILUTED WITH SOLUTIONS THAT CONTAIN DEXTROSE (GLUCOSE) PRIOR TO INJECTION.
A chemical interaction occurs immediately, which is evidenced by the formation of a precipitate when furosemide is injected into an intravenous line of an infusion of inamrinone. Therefore, furosemide should not be administered in intravenous lines containing inamrinone.
Carcinogenesis, Mutagenesis, Impairment of Fertility
There was no suggestion of a carcinogenic potential with inamrinone when administered orally for up to two years to rats and mice at dose levels up to the maximally tolerated dose of 80 mg/kg/day.
The mouse micronucleus test (at 7.5 to 10 times the maximum human dose) and the Chinese hamster ovary chromosome aberration assay were positive indicating both clastogenic potential and suppression of the number of polychromatic erythrocytes. However, the Ames Salmonella assay, mouse lymphoma study, and cultured human lymphocyte metaphase analysis were all negative. The clastogenic effects are in contrast to negative results obtained in the rat male and female fertility studies, and a three-generation study in rats, both with oral dosing.
Slight prolongation of the rat gestation period was seen in these studies at dose levels of 50 mg/kg/day and 100 mg/kg/day. Dystocia occurred in dams receiving 100 mg/kg/day resulting in increased numbers of stillbirths, decreased litter size, and poor pup survival.
Pregnancy
Teratogenic Effects- Pregnancy Category C
In New Zealand white rabbits, inamrinone has been shown to produce fetal skeletal and gross external malformations at oral doses of 16 mg/kg and 50 mg/kg which were toxic for the rabbit. Studies in French Hy/Cr rabbits using oral doses up to 32 mg/kg/day did not confirm this finding. No malformations were seen in rats receiving inamrinone intravenously at the maximum dose used, 15 mg/kg/day (approximately the recommended daily intravenous dose for patients with congestive heart failure). There are no adequate and well-controlled studies in pregnant women. Inamrinone should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Adverse Reactions/Side Effects
Thrombocytopenia: Intravenous injection of inamrinone resulted in platelet count reductions to below 100,000/mm3 or normal limits in 2.4 percent of the patients.
It is more common in patients receiving prolonged therapy. To date, in closely-monitored clinical trials, in patients whose platelet counts were not allowed to remain depressed, no bleeding phenomena have been observed.
Platelet reduction is dose dependent and appears due to a decrease in platelet survival time. Several patients who developed thrombocytopenia while receiving inamrinone had bone marrow examinations which were normal. There is no evidence relating platelet reduction to immune response or to a platelet activating factor.
Gastrointestinal Effects: Gastrointestinal adverse reactions reported with inamrinone during clinical use included nausea (1.7%), vomiting (0.9%), abdominal pain (0.4%), and anorexia (0.4%).
Cardiovascular Effects: Cardiovascular adverse reactions reported with inamrinone include arrhythmia (3%) and hypotension (1.3%).
Hepatic Toxicity: In dogs, at IV doses between 9 mg/kg/day and 32 mg/kg/day, inamrinone showed dose-related hepatotoxicity manifested either as enzyme elevation or hepatic cell necrosis or both. Hepatotoxicity has been observed in man following long-term oral dosing and has been observed, in a limited experience (0.2%), following intravenous administration of inamrinone. There have also been rare reports of enzyme and bilirubin elevation and jaundice.
Hypersensitivity: There have been reports of several apparent hypersensitivity reactions in patients treated with oral inamrinone for about two weeks. Signs and symptoms were variable but included pericarditis, pleuritis and ascites (1 case), myositis with interstitial shadowing on chest x-ray and elevated sedimentation rate (1 case) and vasculitis with nodular pulmonary densities, hypoxemia, and jaundice (1 case). The first patient died, not necessarily of the possible reaction, while the last two resolved with discontinuation of therapy. None of the cases were rechallenged so that attribution to inamrinone is not certain, but possible hypersensitivity reactions should be considered in any patient maintained for a prolonged period on inamrinone.
General: Additional adverse reactions observed in intravenous inamrinone clinical studies include fever (0.9%), chest pain (0.2%), and burning at the site of injection (0.2%).
Management of Adverse Reactions
Platelet Count Reductions: Asymptomatic platelet count reduction (to <150,000/mm3) may be reversed within one week of a decrease in drug dosage. Further, with no change in drug dosage, the count may stabilize at lower than pre-drug levels without any clinical sequelae. Pre-drug platelet counts and frequent platelet counts during therapy are recommended to assist in decisions regarding dosage modifications.
Should a platelet count less than 150,000/mm3 occur, the following actions may be considered:
- •
- Maintain total daily dose unchanged, since in some cases counts have either stabilized or returned to pretreatment levels.
- •
- Decrease total daily dose.
- •
- Discontinue inamrinone if, in the clinical judgment of the physician, risk exceeds the potential benefit.
Gastrointestinal Side Effects: While gastrointestinal side effects were seen infrequently with intravenous therapy, should severe or debilitating ones occur, the physician may wish to reduce dosage or discontinue the drug based on the usual benefit-to-risk considerations.
Hepatic Toxicity: In clinical experience to date with intravenous administration, hepatotoxicity has been observed rarely. If acute marked alterations in liver enzymes occur together with clinical symptoms suggesting an idiosyncratic hypersensitivity reaction, inamrinone therapy should be promptly discontinued.
If less than marked enzyme alterations occur without clinical symptoms, these nonspecific changes should be evaluated on an individual basis. The clinician may wish to continue inamrinone, reduce dosage, or discontinue the drug based on the usual benefit/risk considerations.
Overdosage
A death has been reported with a massive accidental overdose (840 mg over three hours by initial bolus and infusion) of inamrinone, although causal relation is uncertain. Diligence should be exercised during product preparation and administration.
Doses of inamrinone may produce hypotension because of its vasodilator effect. If this occurs, inamrinone administration should be reduced or discontinued. No specific antidote is known, but general measures for circulatory support should be taken.
In rats, the LD50 of inamrinone, as the lactate salt, was 102 mg/kg or 130 mg/kg intravenously in two different studies and 132 mg/kg orally (intragastrically); as a suspension in aqueous gum tragacanth the oral LD50 was 239 mg/kg.
Inamrinone Dosage and Administration
Loading doses of inamrinone injection should be administered as supplied (undiluted). Infusions of inamrinone may be administered in normal or half normal saline solution to a concentration of 1 mg/mL to 3 mg/mL. Diluted solutions should be used within 24 hours.
Inamrinone injection may be administered into running dextrose (glucose) infusions through a Y-Connector or directly into the tubing where preferable.
Chemical Interactions
A chemical interaction occurs slowly over a 24-hour period when the intravenous solution of inamrinone is mixed directly with dextrose (glucose)-containing solutions. THEREFORE, INAMRINONE SHOULD NOT BE DILUTED WITH SOLUTIONS THAT CONTAIN DEXTROSE (GLUCOSE) PRIOR TO INJECTION.
A chemical interaction occurs immediately, which is evidenced by the formation of a precipitate when furosemide is injected into an intravenous line of an infusion of inamrinone. Therefore, furosemide should not be administered in intravenous lines containing inamrinone.
The following procedure is recommended for the administration of inamrinone injection:
1. Initiate therapy with a 0.75 mg/kg loading dose given slowly over 2 to 3 minutes.
|
LOADING DOSE DETERMINATION 0.75 mg/kg (undiluted) |
||||||||||
|
Patient Weight in kg |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
100 |
110 |
120 |
|
mL of undiluted Inamrinone Injection |
4.5 |
6 |
7.5 |
9 |
10.5 |
12 |
13.5 |
15 |
16.5 |
18 |
2. Continue therapy with a maintenance infusion between 5 mcg/kg/min and 10 mcg/kg/min.
3. Based on clinical response, an additional loading dose of 0.75 mg/kg may be given 30 minutes after the initiation of therapy.
4. The rate of infusion usually ranges from 5 mcg/kg/min to 10 mcg/kg/min such that the recommended total daily dose (including loading doses) does not exceed 10 mg/kg. A limited number of patients studied at higher doses support a dosage regimen up to 18 mg/kg/day for shortened durations of therapy.
The following infusion rate chart may be used to assure that the calculations are made correctly.
To utilize the chart, the concentration of inamrinone infusion solution used must be 2.5 mg/mL (2500 mcg/mL). This concentration is prepared by mixing the inamrinone solution with an equal volume of diluent (normal or half normal saline).
|
||||||||||
|
INAMRINONE IV INFUSION RATE (mL/hr) CHART Using 2.5 mg/mL Infusion Concentration* |
||||||||||
|
Patient Weight in kg |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
100 |
110 |
120 |
|
Dosage: 5.0 mcg/kg/min |
4 |
5 |
6 |
7 |
8 |
10 |
11 |
12 |
13 |
14 |
|
7.5 mcg/kg/min |
5 |
7 |
9 |
11 |
13 |
14 |
16 |
18 |
20 |
22 |
|
10.0 mcg/kg/min |
7 |
10 |
12 |
14 |
17 |
19 |
22 |
24 |
26 |
29 |
Example: A 70 kg patient would require a loading dose of 10.5 mL of undiluted inamrinone. If the physician selects a dose of 7.5 mcg/kg/min for the infusion, the flow rate would be 13 mL/hr at the 2.5 mg/mL concentration of inamrinone.
5. The rate of administration and the duration of therapy should be adjusted according to the response of the patient. The physician may wish to reduce or titrate the infusion downward based on clinical responsiveness or untoward effects.
The above dosing regimens can be expected to place most patients’ plasma concentration of inamrinone at approximately 3 mcg/mL. Increases in cardiac index show a linear relationship to plasma concentration of a range of 0.5 mcg/mL to 7 mcg/mL. No observations have been made at greater plasma concentrations.
Patient improvement may be reflected by increases in cardiac output, reduction in pulmonary capillary wedge pressure, and such clinical responses as a lessening of dyspnea and an improvement in other symptoms of heart failure, such as orthopnea and fatigue.
Monitoring central venous pressure (CVP) may be valuable in the assessment of hypotension and fluid balance management. Prior correction or adjustment of fluid/electrolytes is essential to obtain satisfactory response with inamrinone.
Parenteral drug products should be inspected visually and should not be used if particulate matter or discoloration is observed.
How is Inamrinone supplied
Inamrinone Injection USP is supplied in single-dose vials of 20 mL sterile, clear yellow solution individually boxed. NDC 55390-042-10.
Each 1 mL contains inamrinone lactate equivalent to 5 mg of inamrinone.
Protect from light. Packaging is light resistant for protection during storage. Retain in carton until time of use.
Store at controlled room temperature 15° to 30°C (59° to 86°F).
Manufactured by: Manufactured for:
Ben Venue Laboratories, Inc. Bedford Laboratories™
Bedford, OH 44146 Bedford, OH 44146
August 2002 AMR-P01

| INAMRINONE
inamrinone injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Bedford Laboratories (884528407) |
| Registrant - Ben Venue Laboratories (004327953) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Ben Venue Laboratories Inc. | 004327953 | MANUFACTURE(55390-042) | |
More about inamrinone
- Check interactions
- Compare alternatives
- Side effects
- Dosage information
- During pregnancy
- Drug class: inotropic agents