Flebogamma DIF: Package Insert / Prescribing Info
Package insert / product label
Generic name: immune globulin (human)
Dosage form: injection, solution
Drug class: Immune globulins
J Code (medical billing code): J1599 (500 mg, injection)
Medically reviewed by Drugs.com. Last updated on Dec 5, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
FLEBOGAMMA 5% DIF safely and effectively.
See full prescribing information for FLEBOGAMMA 5% DIF.
FLEBOGAMMA 5% DIF (immune globulin intravenous [human]), solution
for intravenous administration
Initial U.S. Approval: 2006
WARNING: THROMBOSIS, RENAL DYSFUNCTION, and ACUTE RENAL FAILURE
See full prescribing information for complete boxed warning.
- Thrombosis may occur with immune globulin products, including FLEBOGAMMA 5% DIF. Risk factors may include: advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling vascular catheters, hyperviscosity, and cardiovascular risk factors.
- For patients at risk of thrombosis administer FLEBOGAMMA 5% DIF at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk of hyperviscosity.
- Renal dysfunction, acute renal failure, osmotic nephrosis and death may occur with the administration of human immune globulin intravenous (IGIV) products, particularly those products that contain sucrose. FLEBOGAMMA 5% DIF does not contain sucrose.
- For patients at risk of renal dysfunction or failure, administer FLEBOGAMMA 5% DIF at the minimum dose and infusion rate practicable. (5.2)
Recent Major Changes
| Dosage and Administration (2 ) | 9/2019 |
Indications and Usage for Flebogamma DIF
Flebogamma 5% DIF is an immune globulin intravenous (human), indicated for treatment of primary (inherited) immunodeficiency (PI) in adults and pediatric patients 2 years of age and older. (1)
Flebogamma DIF Dosage and Administration
For Intravenous Use Only
| Indication | Dose | Initial Infusion Rate | Maintenance Dose Rate (if tolerated) |
| PI | 300-600 mg per kg every 3-4 weeks | 0.01 mL per kg per minute (0.5 mg per kg per min) | Increase to 0.10 mL per kg per minute (5 mg per kg per min) |
- For patients at risk of renal dysfunction or thrombosis, administer Flebogamma 5% DIF at the minimum dose and infusion rate practicable. (5.2, 5.4)
- Ensure that patients with pre-existing renal insufficiency are not volume-depleted and discontinue Flebogamma 5% DIF if renal function deteriorates. (5.2)
Dosage Forms and Strengths
Solution for intravenous injection containing 5% IgG (50 mg per mL). (3)
Contraindications
Warnings and Precautions
- IgA-deficient patients with antibodies to IgA are at greater risk of developing severe hypersensitivity and anaphylactic reactions. (5.1)
- Monitor renal function, including blood urea nitrogen, serum creatinine, and urine output in patients at risk of developing acute renal failure. (5.2)
- Hyperproteinemia, increased serum viscosity, and hyponatremia may occur in patients receiving Flebogamma 5% DIF therapy. (5.3)
- Thrombosis may occur. Monitor patients with known risk factors for thrombosis and consider baseline assessment of blood viscosity for those at risk of hyperviscosity. (5.4)
- Aseptic meningitis syndrome (AMS) may occur in patients receiving Flebogamma 5% DIF therapy, especially with high doses or rapid infusion. (5.5)
- Hemolysis, either intravascular or due to enhanced red blood cell sequestration, can develop subsequent to Flebogamma 5% DIF treatments. Risk factors include high doses and non-O blood group. Monitor patients for hemolysis and hemolytic anemia. (5.6)
- Monitor patients for pulmonary adverse reactions (transfusion-related acute lung injury, TRALI). (5.7)
- Patients receiving Flebogamma 5% DIF for the first time or being restarted on the product after a treatment hiatus of more than 8 weeks may be at a higher risk for development of fever, chills, nausea, and vomiting. (5.8)
- Flebogamma 5% DIF is made from human plasma and may contain infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. (5.9)
- Passive transfer of antibodies may confound serologic testing. (5.11)
- Flebogamma 5% DIF contains sorbitol. The presence of sorbitol presents a risk to those with hereditary fructose intolerance (HFI). (5.12)
Adverse Reactions/Side Effects
The most common adverse reactions (reported in at least 5% of clinical trial adult subjects) were headache, pyrexia/fever, pain, infusion site reactions, diarrhea, rigors or chills, urticaria, and infusion site inflammation. (6)
The most common adverse reactions (reported in at least 5% of clinical trial pediatric subjects) were headache, pyrexia, hypotension, tachycardia, diastolic hypotension, nausea, abdominal pain, diarrhea, pain, and vomiting. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Grifols Biologicals at 1-888-GRIFOLS (1-888-474-3657) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Passive transfer of antibodies may transiently interfere with the immune response to live virus vaccines, such as measles, mumps, and rubella. (7)
Use In Specific Populations
- Pregnancy: No human or animal data. Use only if clearly needed. (8.1)
- Geriatric: In patients over age 65 or in any patient at risk of developing renal insufficiency, do not exceed the recommended dose, and infuse Flebogamma 5% DIF at the minimum dose and infusion rate practicable and at less than 0.06 mL per kg per minute (3 mg per kg per min). (8.5)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 9/2019
Full Prescribing Information
WARNING: THROMBOSIS, RENAL DYSFUNCTION, and ACUTE RENAL FAILURE
- Thrombosis may occur with immune globulin products, including FLEBOGAMMA 5% DIF. Risk factors may include: advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling central vascular catheters, hyperviscosity, and cardiovascular risk factors. Thrombosis may occur in the absence of known risk factors. (see Warnings and Precautions [5.4] and Patient Counseling Information [17])
- For patients at risk of thrombosis, administer FLEBOGAMMA 5% DIF at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity. (see Dosage and Administration [2.3] and Warnings and Precautions [5.4])
- Renal dysfunction, acute renal failure, osmotic nephrosis, and death1have been related to intravenous immune globulin (IGIV) products. Patients predisposed to acute renal failure include patients with any degree of pre-existing renal insufficiency, diabetes mellitus, age greater than 65, volume depletion, sepsis, paraproteinemia, or patients receiving known nephrotoxic drugs.
- Administer FLEBOGAMMA 5% DIF at the minimum dose and rate of infusion practicable in patients at risk for renal dysfunction or failure.
- Reports of renal dysfunction and acute renal failure occur more commonly in patients receiving IGIV products containing sucrose as a stabilizer. They account for a disproportionate share of the total number of reported cases of renal dysfunction and acute renal failure. FLEBOGAMMA 5% DIF does not contain sucrose. (see Dosage and Administration [2.3] and Warnings and Precautions [5.2])
1. Indications and Usage for Flebogamma DIF
Flebogamma 5% DIF is an immune globulin intravenous (human) solution indicated in adults and pediatric patients 2 years of age and older for the treatment of primary immunodeficiency (PI), including the humoral immune defects in common variable immunodeficiency, x-linked agammaglobulinemia, severe combined immunodeficiency, and Wiskott-Aldrich syndrome.
2. Flebogamma DIF Dosage and Administration
For Intravenous Use Only
2.1 Dosage
Treatment of Primary Immunodeficiency (PI)
| Dose | Initial Infusion Rate | Maintenance Dose Rate (if tolerated) |
| 300-600 mg per kg body weight (6.0-12.0 mL per kg) administered every 3-4 weeks | 0.01 mL per kg per minute (0.5 mg per kg per min) | Increase to 0.10 mL per kg per minute (5 mg per kg per min) |
If a patient has been exposed to measles, it may be prudent to administer an extra dose of IGIV as soon as possible and within 6 days of exposure. A dose of 400 mg/kg should provide a serum level > 240 mIU/mL of measles antibodies for at least two weeks.
If a patient is at risk of future measles exposure and receives a dose of less than 530 mg/kg every 3-4 weeks, the dose should be increased to at least 530 mg/kg. This should provide a serum level of 240 mIU/mL of measles antibodies for at least 22 days after infusion.
As there are significant differences in the half-life of IgG among patients with PI, the frequency and amount of immunoglobulin therapy may vary from patient to patient. Adjust the dose according to the clinical response.
Adjust the dosage over time to achieve the desired trough IgG levels and clinical responses. No randomized controlled trial data are available to determine an optimum target trough serum IgG level.
2.2 Preparation and Handling
- Inspect Flebogamma 5% DIF visually for particulate matter and color prior to administration. Do not use the vial if particles are detected. Do not use if turbid.
- Several vials of Flebogamma 5% DIF may be pooled into an empty sterile solution container by using aseptic technique, if large doses are to be administered.
- Do not dilute with intravenous fluids. Do not inject other medications into intravenous tubing being used for Flebogamma 5% DIF.
- Infuse Flebogamma 5% DIF through a separate intravenous line. Do not add any medications or intravenous fluids to the Flebogamma 5% DIF infusion container. Do not mix IGIV products of different formulations or from different manufacturers.
- Discard unused contents and administration devices after use.
- Use promptly any vial that has been entered.
- Discard partially used vials. Do not save for future use because the solution contains no preservative.
- Do not use solution that has been frozen.
2.3 Administration
The recommended initial infusion rate of Flebogamma 5% DIF is 0.01 mL per kg body weight per minute (0.5 mg per kg per min). If the infusion is well-tolerated during the first 30 minutes, the rate may be gradually increased to a maximum of 0.10 mL per kg per minute (5 mg per kg per min).
Monitor patient vital signs throughout the infusion. Slow or stop infusion if adverse reactions occur. If symptoms subside promptly, the infusion may be resumed at a lower rate that is comfortable for the patient.
3. Dosage Forms and Strengths
Flebogamma 5% DIF is a liquid preparation containing 5% IgG (50 mg per mL).
4. Contraindications
- Flebogamma 5% DIF is contraindicated in patients who have had a history of anaphylactic or severe systemic hypersensitivity reactions to the administration of human immune globulin.
- Flebogamma 5% DIF is contraindicated in IgA-deficient patients with antibodies to IgA and a history of hypersensitivity. (see Warnings and Precautions [5.1])
5. Warnings and Precautions
5.1 Hypersensitivity
Severe hypersensitivity reactions and anaphylactic reactions with a fall in blood pressure may occur, even in patients who had tolerated previous treatment with IGIV. (see Contraindications [4]) If hypersensitivity reaction develops, discontinue Flebogamma 5% DIF infusion immediately and institute appropriate treatment.
Flebogamma 5% DIF contains trace amounts of IgA (less than 50 μg/mL). (see Description [11]) Patients with antibodies to IgA have a greater risk of developing potentially severe hypersensitivity and anaphylactic reactions. Flebogamma 5% DIF is contraindicated in patients with antibodies against IgA and a history of hypersensitivity reaction. (see Contraindications [4])
5.2 Renal Dysfunction/Failure
Acute renal dysfunction/failure, acute tubular necrosis, proximal tubular nephropathy, osmotic nephrosis, and death have been reported in patients receiving IGIV, particularly those products containing sucrose2. Flebogamma 5% DIF does not contain sucrose.
Ensure that patients are not volume-depleted before administering Flebogamma 5% DIF. For patients judged to be at risk for developing renal dysfunction, including patients with any degree of pre-existing renal insufficiency, diabetes mellitus, age greater than 65, volume depletion, sepsis, paraproteinemia, or patients receiving known nephrotoxic drugs, administer Flebogamma 5% DIF at the minimum dose and rate of infusion practicable3. (see Boxed Warning, Dosing and Administration [2.3])
Periodic monitoring of renal function and urine output is particularly important in patients judged to be at increased risk of developing acute renal failure1. Assess renal function, including measurement of blood urea nitrogen (BUN) and serum creatinine, before the initial infusion of Flebogamma 5% DIF and at appropriate intervals thereafter. If renal function deteriorates, consider discontinuation of the product.
5.3 Hyperproteinemia, Increased Serum Viscosity, and Hyponatremia
Hyperproteinemia, increased serum viscosity, and hyponatremia may occur in patients receiving Flebogamma 5% DIF therapy. It is clinically critical to distinguish true hyponatremia from a pseudohyponatremia that is temporally or causally related to hyperproteinemia with concomitant decreased calculated serum osmolarity or elevated osmolar gap, because treatment aimed at decreasing serum free water in patients with pseudohyponatremia may lead to volume depletion, a further increase in serum viscosity, and a higher risk of thrombosis.
5.4 Thrombosis
Thrombosis may occur following treatment with immune globulin products, including Flebogamma 5% DIF. Risk factors may include advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling central vascular catheters, hyperviscosity, and cardiovascular risk factors. Thrombosis may occur in the absence of known risk factors.
Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies. (see Warnings and Precautions [5.10]) For patients at risk of thrombosis, administer Flebogamma 5% DIF at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity. (see Boxed Warning, Dosage and Administration [2.3], and Patient Counseling Information [17])
5.5 Aseptic Meningitis Syndrome (AMS)
AMS has been reported to occur following IGIV treatment. Discontinuation of IGIV treatment has resulted in remission of AMS within several days without sequelae4-7. The symptoms of AMS usually begin within several hours to 2 days following IGIV treatment.
AMS is characterized by the following signs and symptoms: severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea, and vomiting. (see Patient Counseling Information [17]) Cerebrospinal fluid (CSF) studies frequently reveal pleocytosis up to several thousand cells per cubic millimeter, predominantly from the granulocytic series, and elevated protein levels up to several hundred mg/dL, but negative culture results. Conduct a thorough neurological examination to patients exhibiting such signs and symptoms, including CSF studies, to rule out other causes of meningitis.
AMS may occur more frequently following high-dose (e.g. over 1.0 g per kg body weight) or rapid infusion of IGIV.
5.6 Hemolysis
Flebogamma 5% DIF may contain blood group antibodies that may act as hemolysins and induce in vivo coating of red blood cells (RBCs) with immunoglobulin, causing a positive direct antiglobulin test (DAT) (Coombs' test) result and hemolysis8-11. Delayed hemolytic anemia can develop subsequent to IGIV therapy due to enhanced RBC sequestration and acute hemolysis, consistent with intravascular hemolysis, has been reported12. Cases of severe hemolysis-related renal dysfunction/failure or disseminated intravascular coagulation have occurred following infusion of IGIV.
The following risk factors may be associated with the development of hemolysis following IGIV administration: high doses (e.g., at least 2 g per kg), given either as a single administration or divided over several days, and non-O blood group13. Other individual patient factors, such as an underlying inflammatory state (as may be reflected by, for example, elevated C-reactive protein or erythrocyte sedimentation rate), have been hypothesized to increase the risk of hemolysis following administration of IGIV14, but their role is uncertain. Hemolysis has been reported following administration of IGIV for a variety of indications, including ITP and PI11. Monitor patients for clinical signs and symptoms of hemolysis, particularly patients with risk factors noted above. Consider appropriate laboratory testing in higher risk patients, including measurement of hemoglobin or hematocrit prior to infusion and within 36 to 96 hours post infusion. If clinical signs and symptoms of hemolysis or a significant drop in hemoglobin or hematocrit have been observed, perform appropriate confirmatory laboratory testing. If transfusion is indicated for patients who develop hemolysis with clinically compromising anemia after receiving IGIV, perform adequate cross-matching to avoid exacerbating on-going hemolysis. (see Patient Counseling Information [17])
5.7 Transfusion-Related Acute Lung Injury (TRALI)
Non-cardiogenic pulmonary edema has been reported in patients following IGIV treatment15. TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever. Symptoms typically appear within 1 to 6 hours after transfusion.
Monitor patients for pulmonary adverse reactions. (see Patient Counseling Information [17]) If TRALI is suspected, perform appropriate tests for the presence of antineutrophil antibodies and anti-HLA antibodies in both the product and patient serum. TRALI may be managed by using oxygen therapy with adequate ventilatory support.
5.8 Infusion Reactions
Individuals receiving Flebogamma 5% DIF for the first time, or being restarted on the product after a treatment hiatus of more than 8 weeks, may be at a higher risk for the development of fever, chills, nausea, and vomiting. Careful monitoring of recipients and adherence to recommendations regarding dosage and administration may reduce the risk of these types of events. (see Dosage and Administration [2.3])
5.9 Transmissible Infectious Agents
Because Flebogamma 5% DIF is made from human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. This also applies to unknown or emerging viruses and other pathogens. No cases of transmission of viral diseases or CJD have been associated with the use of Flebogamma 5% DIF. All infections suspected by a physician possibly to have been transmitted by this product should be reported by the physician or other healthcare provider to Grifols Biologicals at 1-888-474-3657.
Before prescribing or administering Flebogamma 5% DIF, the physician should discuss the risks and benefits of its use with the patient. (see Patient Counseling Information [17])
5.10 Monitoring: Laboratory Tests
- Periodic monitoring of renal function and urine output is particularly important in patients judged to be at increased risk of developing acute renal failure. Assess renal function, including measurement of BUN and serum creatinine, before the initial infusion of Flebogamma 5% DIF and at appropriate intervals thereafter.
- Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies because of the potentially increased risk of thrombosis.
- If signs and/or symptoms of hemolysis are present after an infusion of Flebogamma 5% DIF, perform appropriate laboratory testing for confirmation.
- If TRALI is suspected, perform appropriate tests for the presence of antineutrophil antibodies and anti-HLA antibodies in both the product and patient serum.
5.11 Interference with Laboratory Tests
After infusion of IgG, the transitory rise of the various passively transferred antibodies in the patient's blood may yield positive serological testing results, with the potential for misleading interpretation. Passive transmission of antibodies to erythrocyte antigens (e.g. A, B, and D) may cause a positive direct or indirect antiglobulin (Coombs') test.
5.12 Hereditary Fructose Intolerance
Flebogamma 5% DIF contains sorbitol. The presence of sorbitol presents a risk to those with hereditary fructose intolerance (HFI). The incidence of HFI is estimated at 1 in 20,000 births and is usually diagnosed at the time of weaning when fructose or sucrose is introduced into the diet. Clinical symptoms include recurrent vomiting, abdominal pain and hypoglycemia. Flebogamma 5% DIF must not be administered to subjects with HFI.
6. Adverse Reactions/Side Effects
The most common adverse reactions (reported in at least 5% of clinical trial adult subjects) were headache, pyrexia/fever, pain, infusion site reactions, diarrhea, rigors or chills, urticaria, and infusion site inflammation.
The most common adverse reactions (reported in at least 5% of clinical trial pediatric subjects) were headache, pyrexia, hypotension, tachycardia, diastolic hypotension, nausea, abdominal pain, diarrhea, pain, and vomiting.
To report SUSPECTED ADVERSE REACTIONS, contact Grifols Biologicals at 1-888-GRIFOLS (1-888-474-3657) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adverse reactions were reported in a study of 46 individuals with PI receiving infusions every 3 to 4 weeks of 300-600 mg per kg body weight. Thirty-one subjects (67.4%) had at least one adverse reaction at some time during the study that was considered product-related. None of the 46 subjects who participated in this study discontinued the study prematurely due to an adverse drug reaction.
Adverse reactions that occurred with an incidence of at least 5% on a per-subject basis are summarized in Table 1.
|
a. include combined reported terms of pyrexia and body temperature increase. |
||
|
b. include combined reported terms of pain such as pain (not otherwise specified), injection site pain, back pain, abdominal pain (not otherwise specified), and abdominal pain upper. |
||
|
c. include combined reported terms of infusion site inflammation and others such as injection site oedema and injection site swelling. |
||
| Adverse Reaction | Subjects (%) N=46 | Number of Events |
| Headache | 10 (21.7) | 24 |
| Pyrexiaa | 9 (19.6) | 12 |
| Painb | 7 (15.2) | 11 |
| Injection site reaction | 6 (13.0) | 10 |
| Diarrhea | 4 (8.7) | 5 |
| Rigors | 4 (8.7) | 7 |
| Urticaria | 3 (6.5) | 3 |
| Infusion site inflammationc | 3 (6.6) | 3 |
Other common adverse drug reactions reported in fewer than 5% of the subjects included hypertension, sinusitis, nausea/vomiting, positive Coombs test, arthralgia, myalgia, dizziness, bronchitis, and hypotension.
The total number of adverse reactions reported whose onset were within 72 hours after the end of an infusion of Flebogamma 5% DIF was 94. There were a total of 709 infusions, resulting in a ratio of 0.13 temporally associated adverse reactions per infusion (the upper bound of the 1-sided 95% confidence interval = 0.18). Of the 709 total infusions, 70 (9.7%, 1-sided 95% upper bound CI = 12.4%) were associated with at least one adverse reaction that began within 72 hours after the completion of an infusion. In this analysis, each infusion is only counted once, regardless of the number of adverse reactions that occurred during the infusion, when during the 72-hour period after the infusion the adverse reaction started, or the intensity of those adverse reactions.
Factoring adverse reaction intensity into the analysis of the 709 infusions shows that there were 58 infusions with at least one mild adverse reaction (8.2% [upper bound 95% CI=10.5%]), 25 infusions with at least one moderate adverse reaction (3.5% [upper bound 95% CI=5.2%]), and 1 infusion with a severe adverse reaction (0.1% [upper bound 95% CI=0.8%]). In this analysis, if a subject reported multiple events with different intensities during the same infusion (e.g., mild headache and moderate pyrexia), that infusion would be counted in all relevant categories. Therefore, the number of infusions counted is 84.
Three subjects (6.5%) experienced a treatment-emergent rise in AST (> 3x the upper-limit of normal), and 1 subject (2.2%) experienced a treatment-emergent rise in ALT (> 3x the upper-limit of normal). None of these abnormal lab values were long-lasting (i.e. they occurred at 1 or 2 infusions), and none of these subjects had a concomitant treatment-emergent rise in total bilirubin.
A clinical study with Flebogamma 5% DIF for the treatment of PI was conducted in 24 pediatric subjects aged 2-16 years to determine whether they respond differently from adult subjects. Pediatric subjects received intravenous infusions of 262-625 mg per kg body weight every 3-4 weeks. Twenty subjects (83.3%) had at least one adverse reaction at some time during the study that was considered product-related. There were no deaths or serious adverse reactions.
In 317 infusions, 20 pediatric subjects reported 159 treatment-related adverse drug reactions (ADR). Treatment-related adverse reactions that occurred with an incidence of at least 5% on a per-subject basis included headache (42%), pyrexia (29%), hypotension (25%), tachycardia (25%), diastolic hypotension (21%), nausea (8%), abdominal pain (8%), diarrhea (8%), pain (8%), and vomiting (8%). Of these, 99 ADRs were mild, 54 were moderate, and 6 were severe in intensity. The most common severe ADR was headache. Tachycardia was defined as mild, moderate, or severe by ratio of heart rate over baseline of 1.2-1.4, 1.41-1.6, or >1.6, respectively. Two episodes of tachycardia were moderate and 14 were mild, 1 mild case had the infusion interrupted and the rest had no action taken, and all resolved. Hypotension was defined as mild, moderate, or severe by the decrease in pressure below baseline of 10-15%, 16-25%, and >25%, respectively. Ten episodes of hypotension/diastolic hypotension were moderate and 29 were mild, one moderate case had the infusion interrupted and the rest had no action taken, and all resolved. None of the ADRs related to fluctuation in vital signs were severe, all resolved without sequelae, and none were considered clinically significant.
One subject (6.5%) experienced a treatment-emergent rise in ALT (over 2.5x the upper-limit of normal). This was considered a mild treatment-related AE and resolved without sequelae. All other lab values were within normal limits. One subject experienced one positive Coombs' test result after baseline (experienced 14 days after the final infusion). No subjects experienced clinically significant abnormal lab values for LDH, bilirubin, serum creatinine. In addition, no subjects experienced positive test results for HBsAg, HCV, or HIV.
6.2 Post-marketing Experience
Because adverse reactions are reported voluntarily post-approval from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to product exposure.
The following adverse reactions have been identified during the post-approval use of IGIV products16, 17, including Flebogamma 5% DIF.
| Infusion Reactions | Hypersensitivity (e.g. anaphylaxis), headache, diarrhea, tachycardia, fever, fatigue, dizziness, malaise, chills, flushing, urticaria or other skin reactions, wheezing or other chest discomfort, nausea, vomiting, rigors, back pain, myalgia, arthralgia, and changes in blood pressure |
| Renal | Acute renal dysfunction/failure, osmotic nephropathy |
| Respiratory | Apnea, Acute Respiratory Distress Syndrome (ARDS), Transfusion-Related Acute Lung Injury (TRALI), cyanosis, hypoxemia, pulmonary edema, dyspnea, bronchospasm |
| Cardiovascular | Cardiac arrest, thromboembolism, vascular collapse, hypotension |
| Neurological | Coma, loss of consciousness, seizures, tremor, aseptic meningitis syndrome |
| Integumentary | Stevens-Johnson Syndrome, epidermolysis, erythema multiformae, dermatitis (e.g. bullous dermatitis) |
| Hematologic | Pancytopenia, leukopenia, hemolysis, positive direct antiglobulin (Coombs) test |
| Musculoskeletal | Back pain |
| Gastrointestinal | Hepatic dysfunction, abdominal pain |
| General/Body as a Whole | Pyrexia, rigors |
Related/similar drugs
Hizentra
Hizentra (immune globulin) is used as a replacement therapy in patients with primary humoral ...
Privigen
Privigen is used to treat primary immune deficiency conditions and idiopathic thrombocytopenic ...
Nplate
Nplate is used to prevent bleeding episodes in people with chronic immune thrombocytopenic purpura ...
Gammagard Liquid
Gammagard (immune globulin) is used to treat primary immune deficiency and multifocal motor ...
Octagam
Octagam (immune globulin intravenous) is used to treat primary immunodeficiency diseases, immune ...
Hyqvia
Hyqvia is used for chronic inflammatory demyelinating polyradiculoneuropathy, primary ...
Xembify
Xembify (immune globulin subcutaneous) is used as a replacement therapy in patients with primary ...
Gamunex-C
Gamunex-C (immune globulin) is used to treat primary immunodeficiency, chronic inflammatory ...
7. Drug Interactions
Passive transfer of antibodies may transiently impair the immune response to live attenuated virus vaccines, such as measles, mumps, and rubella. Inform the immunizing physician of recent therapy with Flebogamma 5% DIF so that appropriate measures can be taken (see Patient Counseling Information [17])
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no studies of Flebogamma 5% DIF use in pregnant women. Animal reproduction studies have not been performed with Flebogamma 5% DIF. It is also not known whether Flebogamma 5% DIF can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Immunoglobulins cross the placenta from maternal circulation increasingly after 30 weeks of gestation. Flebogamma 5% DIF should be given to a pregnant woman only if clearly needed.
8.2 Lactation
Risk Summary
There is no information regarding the presence of Flebogamma 5% DIF in human milk, its effects on the breastfed infant, or its effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Flebogamma 5% DIF and any potential adverse effects on the breastfed infant from Flebogamma 5% DIF or from the underlying maternal condition.
8.4 Pediatric Use
Flebogamma 5% DIF was studied in a multicenter clinical trial for the treatment of PI in 24 subjects aged 2-16 years (seven were 2-5 years of age, seven were 6-11 years, and ten were 12-16 years), and found to be efficacious for the prevention of acute serious bacterial infections. No pediatric-specific dose requirements were necessary to achieve the desired serum IgG levels. Twenty subjects (83.3%) had at least one adverse reaction at some time during the study that was considered product-related. There were no deaths or serious adverse reactions. Treatment-related adverse reactions that occurred with an incidence of at least 5% on a per-subject basis included headache (42%), pyrexia (29%), hypotension (25%), tachycardia (25%), diastolic hypotension (21%), nausea (8%), abdominal pain (8%), diarrhea (8%), pain (8%), and vomiting (8%).
Safety and efficacy of Flebogamma 5% DIF in pediatric patients below the age of 2 years have not been established.
8.5 Geriatric Use
Limited information is available for the geriatric use of Flebogamma 5% DIF. Clinical studies of Flebogamma 5% DIF did not include sufficient numbers of subjects over the age of 65 to determine whether they respond differently from younger subjects. Use caution when administering Flebogamma 5% DIF to patients age 65 and over who are judged to be at increased risk for developing thrombosis or renal insufficiency. Do not exceed recommended dose, and administer Flebogamma 5% DIF at the minimum dose and infusion rate practicable, and at less than 0.06 mL per kg per minute (3 mg per kg per min). (see Boxed Warning, Warning and Precautions [5.2, 5.4], and Dosage and Administration [2.3])
10. Overdosage
Overdose may lead to fluid overload and hyperviscosity. Patients at particular risk of complications of fluid overload and hyperviscosity include elderly patients and patients with cardiac or renal impairment.
11. Flebogamma DIF Description
Flebogamma 5% DIF is a ready to use, sterile, clear or slightly opalescent and colorless to pale yellow, liquid preparation of purified immunoglobulin (IgG) obtained from human plasma pools. The purification process includes cold ethanol fractionation, polyethylene glycol precipitation, ion exchange chromatography, low pH treatment, pasteurization, solvent detergent treatment, and Planova nanofiltration using 20 nanometer (nm) filters.
Flebogamma 5% DIF is a purified (at least 97% IgG), unmodified, human IgG. The distribution of the four IgG subclasses is approximately 66.6% IgG1, 28.5% IgG2, 2.7% IgG3, and 2.2% IgG4. Flebogamma 5% DIF contains trace amounts of IgA (typically less than 50 μg/mL) and trace amounts of sodium and IgM.
Flebogamma 5% DIF contains 5 g human normal immunoglobulin and 5 g D-sorbitol (as stabilizer) in 100 mL of water for injection, and ≤ 3 mg/mL polyethylene glycol. There is no preservative in the formulation. The pH of the solution ranges from 5 to 6 and the osmolality from 240 to 370 mOsm/kg, which is within the normal physiological range.
Screening against potentially infectious agents begins with the donor selection process and continues throughout plasma collections and plasma preparation. Each individual plasma donation used in the manufacture of Flebogamma 5% DIF is collected only at FDA-approved blood establishments and is tested by FDA-licensed serological test for hepatitis B virus (HBV) surface antigen (HBsAg), and for antibodies to human immunodeficiency virus (HIV-1/HIV-2) and hepatitis C virus (HCV) in accordance with U.S. regulatory requirements. As an additional safety measure, mini-pools of plasma are tested for the presence of HBV, HIV-1 and HCV by FDA-licensed nucleic acid testing (NAT) and found to be negative. In addition, plasma is tested by in-process NAT for hepatitis A virus (HAV) and parvovirus B19 (B19) on mini-pools and the viral load limit for B19 in the manufacturing pool is set not to exceed 104 IU/mL. NAT for the presence of HCV and HIV in the manufacturing plasma pool is also performed and found to be negative.
To further improve the margin of safety, three dedicated, independent virus inactivation/removal steps have been integrated into the manufacturing and formulation processes, namely pasteurization at 60 ºC, 10 hours, solvent-detergent treatment for 6 hours, and nanofiltration down to 20 nm Planova filters.
In vitro virus spiking studies have been used to validate the capability of the manufacturing process to inactivate and remove viruses. To establish the minimum applicable virus clearance capacity of the manufacturing process, these virus clearance studies were performed on seven steps of the production process (pasteurization, solvent-detergent treatment, nanofiltration, Fraction I precipitation, Fraction II+III precipitation, 4% PEG precipitation, and pH treatment for 4 hours at 37 ºC).
The viral reduction data (in log10) from these experiments are summarized in Table 2.
|
* When the RF is < 1 log10, it is not taken into account for the calculation of the overall reduction capacity. |
||||||||
|
≥ is no residual infectivity detected; nd = not done; na = non-applicable, since the virus is theoretically resistant to this treatment. |
||||||||
|
Abbreviations: HIV = Human immunodeficiency virus; PRV = Pseudorabies virus; IBR = Infectious bovine rhinotracheitis virus; BVDV = Bovine viral diarrhea virus; SINDBIS = Sindbis virus; WNV = West Nile virus; EMC = Encephalomyocarditis virus; PPV = Porcine parvovirus. |
||||||||
| Target virus | HIV-1, HIV-2
(env. RNA) | HBV Herpesvirus
(env. DNA) | HCV
(env. RNA) | WNV
(env. RNA) | HAV
(non-env. RNA) | Virus B19
(non-env. DNA) |
||
| Model virus | HIV-1 | PRV | IBR | BVDV | SINDBIS | WNV | EMC | PPV |
| Fraction I precipitation | < 1.00* | nd | nd | nd | nd | 2.78 | nd | < 1.00* |
| Ethanol incubation (Fraction II+III) | 1.48 | nd | nd | nd | nd | < 1.00* | nd | nd |
| PEG precipitation | ≥ 6.10 | ≥ 5.92 | nd | ≥ 5.78 | nd | nd | ≥ 6.41 | 6.35 |
| Acid pH treatment | 2.47 | ≥ 5.32 | nd | < 1.00* | nd | nd | 1.36 | na |
| Pasteurization | ≥ 5.64 | ≥ 4.96 | ≥ 6.33 | ≥ 4.69 | ≥ 6.49 | ≥ 5.42 | ≥ 5.56 | 4.08 |
| Solvent Detergent | ≥ 4.61 | ≥ 6.95 | nd | ≥ 6.14 | nd | ≥ 5.59 | na | na |
| Nanofiltration
20 nanometer | ≥ 4.81 | ≥ 4.63 | nd | ≥ 4.67 | nd | ≥ 3.63 | ≥ 5.92 | 4.61 |
| Overall Reduction Capacity | ≥ 25.11 | ≥ 27.78 | ≥ 6.33 | ≥ 21.28 | ≥ 6.49 | ≥ 17.42 | ≥ 19.25 | 15.04 |
Additionally, the manufacturing process was investigated for its capacity to decrease infectivity of an experimental agent of transmissible spongiform encephalopathy (TSE), considered as a model for the vCJD and CJD agents. Several individual production steps in the Flebogamma 5% DIF manufacturing process have been shown to decrease TSE infectivity of an experimental model agent. TSE reduction steps include 4% polyethylene glycol precipitation [at least 6.19 log10] and Planova nanofiltration using a 20 nanometer filter [at least 5.45 log10]. These studies provide reasonable assurance that low levels of CJD/vCJD agent infectivity, if present in the starting material, would be removed.
12. Flebogamma DIF - Clinical Pharmacology
12.1 Mechanism of Action
Flebogamma 5% DIF, immune globulin intravenous (human), is a replacement therapy for PI. It supplies a broad spectrum of opsonizing and neutralizing IgG antibodies against a wide variety of bacterial and viral agents. Flebogamma 5% DIF also contains a spectrum of antibodies capable of reacting with cells such as erythrocytes. The role of these antibodies and the mechanisms of action of IgG in Flebogamma 5% DIF have not been fully elucidated.
12.3 Pharmacokinetics
In the clinical study assessing safety and efficacy in PI, Flebogamma 5% DIF was administered as an IV infusion (300-600 mg per kg) to subjects every 3 (n = 8) or 4 (n = 12) weeks for 12 months. The pharmacokinetics of total IgG was determined after the 7th infusion for the 3-week dosing interval and after the 5th infusion for the 4-week dosing interval (Table 3).
|
a. This half-life is an apparent value derived from a period of measurement of 28 days. |
||||
|
b. For subjects on the 3-week schedule, the average of the trough levels from Infusion 7 to the end of the study was calculated; for those on a 4-week schedule, the average of the trough levels from Infusion 5 to the end of the study was calculated. The means of the subject means are presented in this table. |
||||
| Variable | 3-Week Dosing Interval (n = 8) | 4-Week Dosing Interval (n = 12) | ||
| Mean [Range] | SD | Mean [Range] | SD | |
| Cmax (mg/dL) | 1,929 [1,300-2,420] | 441 | 2,069 [1,590-2,800] | 338 |
| AUC0 - last(day·mg/dL) | 31,159 [20,458-40,104] | 6,572 | 32,894 [27,650-41,814] | 3,886 |
| Clearance (mL/day) | 139 [81-243] | 57 | 109 [59-161] | 33 |
| Half-life (days)a | 30 [19-41] | 9 | 32 [25-39] | 5 |
| Trough IgG level (mg/dL)b | 951.38 [773.17-1143.15] | 132.42 | 899.89 [776.70-1,137.14] | 92.03 |
There were 3 adolescent (up to 16 years of age) subjects who underwent pharmacokinetic testing, all of whom were on the 3-week infusion schedule. There were no clinically relevant differences among the adults and adolescents that were tested.
13. Nonclinical Toxicology
13.1 Carcinogenicity, Mutagenesis, Impairment of Fertility
No animal studies were conducted to evaluate the carcinogenic or mutagenic effect of Flebogamma 5% DIF or its effects on fertility.
13.2 Animal Toxicology and/or Pharmacology
Acute toxicity studies were performed in mice and rats at doses up to 2.5 g per kg body weight with infusion rates 6-37 times higher than the maximum rates recommended for humans. The most common clinical observations in mice studies were piloerection, ptosis, ataxia, and increase in respiration all lasting 90 minutes or less. No relevant adverse effects could be confirmed affecting respiratory, circulatory, renal, autonomic and central nervous systems, somatomotor activity, and behavior of the treated mice and rats.
Five of the 25 rats treated with the highest dose, approximately 8 times the maximum infusion rate recommended for humans, showed a transient “reddish urine” sign which was not confirmed as a relevant toxicity-causing phenomenon after renal macroscopic and microscopic analysis. This phenomenon was ascribed to hemolysis when serum was analyzed, suggesting a possible relation to cross reactivity of rodent red cells with human antibodies. No “reddish urine” was detected in any mouse, a much smaller animal where the rate of infusion was comparatively much higher than in rats. The macroscopic inspection of all treated mice did not show any renal alteration.
14. Clinical Studies
A multicenter, open-label, historically controlled study was conducted in the United States to assess the efficacy, safety, and pharmacokinetics of Flebogamma 5% DIF in adult and pediatric subjects with PI. A total of 46 subjects aged 15-75 years (63% male, 37% female) were enrolled and treated with Flebogamma 5% DIF at a dose of 300-600 mg per kg per infusion every 3-4 weeks for 12 months.
Since the subjects in the clinical study were assigned to two different treatment intervals (3-week vs. 4-week infusion schedules), the dosage had to be adjusted to ensure that the subjects received approximately the same dosage on an annualized basis. Therefore, subjects in the 3-week schedule received 75% of the monthly (4-week) dosage per infusion. This resulted in a mean annualized dosage of 451 mg per kg per month for subjects in the 3-week schedule (n=13, range 288-588 mg per kg per month) and 448 mg per kg per month for subjects in the 4-week schedule (n=33, range 298-591 mg per kg per month).
During the study period, the annual rate of acute serious bacterial infection, defined as bacterial pneumonia, bacteremia or sepsis, osteomyelitis/septic arthritis, visceral abscess, and bacterial meningitis per subject per year, was 0.021 (with an upper 1-sided 98% confidence interval of 0.112). One subject had one episode of bacterial pneumonia and there were no other episodes of serious bacterial infections reported (Table 4).
|
a. Estimate = Total episodes/Total subject years. |
||||
|
b. The confidence interval is obtained by using a generalized linear model procedure for Poisson distribution. |
||||
| Infections | Subjects (N=46)
N (%) | Episodes | Estimatesa | 98% CIb |
| Bacterial pneumonia | 1 (2.2) | 1 | ||
| Bacteremia or sepsis | 0 (0.0) | 0 | ||
| Osteomyelitis/septic arthritis | 0 (0.0) | 0 | ||
| Bacterial meningitis | 0 (0.0) | 0 | ||
| Total subjects | 1 (2.2) | 1 | 0.021 | (0.001-0.112) |
The number of days of work/school missed, hospitalizations and days of each hospitalization, the number of visits to physicians or emergency rooms, other infections documented by positive radiographic findings and fever, and days on therapeutic and prophylactic oral/parenteral antibiotic use were also evaluated. These variables were annualized by using the subject-years exposure data of those subjects experiencing the events, but not the entire study cohort. With regard to the number of other validated infections, the mean rate was less than 2 days per subject per year (this calculation used all subjects, including those who had no infections). (Table 5)
|
a. Days of work/school missed per subject year are derived as total days of work/school missed divided by total days in study multiplied by 365. If data are missing for a period (e.g. between Infusion 2 and Infusion 3), then number of days in this period is not counted in the denominator. All other endpoints are derived similarly. |
|||
|
Variable |
Subjects | Mean number of events, days or visits per subject per yeara | |
| N | % | ||
| Work/school days missed | 23 | 50.0 | 12.95 |
| Days of normal activities missed | 18 | 39.1 | 7.28 |
| Days in hospital | 4 | 8.7 | 0.77 |
| Visits to physician/ER | 29 | 63.0 | 4.31 |
| Number of other documented infectious episodes | 33 | 71.7 | 1.96 |
| Days of therapeutic oral antibiotic use | 35 | 76.1 | 55.52 |
| Days of therapeutic parenteral antibiotic use | 2 | 4.3 | 0.14 |
| Days of other therapeutic antibiotic use | 16 | 34.8 | 44.30 |
| Days of prophylactic oral antibiotic use | 19 | 41.3 | 81.08 |
| Days of prophylactic parenteral antibiotic use | 1 | 2.3 | 0.02 |
| Days of other prophylactic antibiotic use | 0 | 0.0 | 0.00 |
A multicenter, open-label, historically controlled study was conducted in the United States to assess the efficacy of Flebogamma 5% DIF in pediatric subjects with PI. A total of 24 subjects aged 2-16 years (79% male, 21% female) were enrolled and treated with Flebogamma 5% DIF at a dose of 262-625 mg per kg per infusion every 3-4 weeks for 12 months.
The annual rate of acute serious bacterial infections, defined as bacterial pneumonia, bacteremia or sepsis, osteomyelitis/septic arthritis, visceral abscess, and bacterial meningitis per subject per year, was 0.051 (with an upper 1-sided 99% confidence limit of 0.53). One subject had one episode of bacterial pneumonia and there were no other episodes of serious bacterial infections reported.
15. References
- Cayco AV, Perazella MA, Hayslett JP. Renal insufficiency after intravenous immune globulin therapy: a report of two cases and an analysis of the literature. J Am Soc Nephrol 1997; 8:1788-94.
- Winward DB, Brophy MT. Acute renal failure after administration of intravenous immunoglobulin: review of the literature and case report. Pharmacotherapy 1995; 15:765-72.
- Tan E, Hajinazarian M, Bay W, et al. Acute renal failure resulting from intravenous immunoglobulin therapy. Arch Neurol 1993; 50:137-9.
- Sekul EA, Cupler EJ, Dalakas MC. Aseptic meningitis associated with high-dose intravenous immunoglobulin therapy: frequency and risk factors. Ann Intern Med 1994; 121:259-62.
- Kato E, Shindo S, Eto Y, et al. Administration of immune globulin associated with aseptic meningitis. JAMA 1988; 259:3269-71.
- Casteels-Van Daele M, Wijndaele L, Hanninck K, et al. Intravenous immune globulin and acute aseptic meningitis. N Engl J Med 1990; 323:614-5.
- Scribner CL, Kapit RM, Phillips ET, et al. Aseptic meningitis and intravenous immunoglobulin therapy. Ann Intern Med 1994; 121:305-6.
- Copelan EA, Strohm PL, Kennedy MS, et al. Hemolysis following intravenous immune globulin therapy. Transfusion 1986; 26:410-2.
- Thomas MJ, Misbah SA, Chapel HM, et al. Hemolysis after high-dose intravenous Ig. Blood 1993; 15:3789.
- Reinhart WH, Berchtold PE. Effect of high-dose intravenous immunoglobulin therapy on blood rheology. Lancet 1992; 339:662-4.
- Wilson JR, Bhoopalam N, Fisher M. Hemolytic anemia associated with intravenous immunoglobulin. Muscle Nerve 1997; 20:1142-1145.
- Kessary-Shoham H, Levy Y, Shoenfeld Y, et al. In vivo administration of intravenous immunoglobulin (IVIG) can lead to enhanced erythrocyte sequestration. J Autoimmune 1999; 13:129-35.
- Kahwaji J, Barker E, Pepkowitz S, et al. Acute hemolysis after high-dose intravenous immunoglobulin therapy in highly HLA sensitized patients. Clin J Am Soc Nephrol 2009; 4:1993-1997.
- Daw Z, Padmore R, Neurath D, et al. Hemolytic transfusion reactions after administration of intravenous immune (gamma) globulin: A case series analysis. Transfusion 2008; 48:1598-1601.
- Rizk A, Gorson KC, Kenney L, et al. Transfusion-related acute lung injury after the infusion of IVIG. Transfusion 2001; 41:264-8.
- Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin. Transfus Med Rev 2003; 17:241-51.
- Orbach H, Katz U, Sherer Y, Shoenfeld Y. Intravenous immunoglobulin: adverse effects and safe administration. Clin Rev Allergy Immunol, 2005; 29:173-184.
16. How is Flebogamma DIF supplied
Flebogamma 5% DIF is supplied in single-use, individually laser-etched vials containing the labeled amount of functionally active IgG. The following presentations of Flebogamma 5% DIF are available:
| NDC Number | Size | Grams Protein |
| 61953-0004-1 | 10 mL | 0.5 g |
| 61953-0004-2 | 50 mL | 2.5 g |
| 61953-0004-3 | 100 mL | 5.0 g |
| 61953-0004-4 | 200 mL | 10.0 g |
| 61953-0004-5 | 400 mL | 20.0 g |
Each vial has an integral suspension band and a label with two peel-off strips showing the product name and lot number.
Flebogamma 5% DIF may be stored at room temperature at 2 to 25 ºC (36 to 77 ºF) for 24 months, as indicated by the expiration date printed on the outer carton and container label. Discard after expiration date. Do not freeze.
Keep Flebogamma 5% DIF in its original carton to protect it from light.
Not made with natural rubber latex.
17. Patient Counseling Information
Instruct patients to immediately report the following signs and symptoms to their physician:
- Decreased urine output, sudden weight gain, fluid retention/edema, and/or shortness of breath (see Renal Failure [5.2])
- Symptoms of thrombosis which may include: pain and/or swelling of an arm or leg with warmth over the affected area, discoloration of an arm or leg, unexplained shortness of breath, chest pain or discomfort that worsens on deep breathing, unexplained rapid pulse, numbness or weakness on one side of the body (see Thrombosis [5.4])
- Severe headache, neck stiffness, drowsiness, fever, sensitivity to light, painful eye movements, nausea, and vomiting (see Aseptic Meningitis Syndrome [5.5])
- Fatigue, increased heart rate, yellowing of the skin or eyes, and dark-colored urine (see Hemolysis [5.6])
- Trouble breathing, chest pain, blue lips or extremities, fever (see TRALI [5.7])
Inform patients that Flebogamma 5% DIF is made from human plasma and may contain infectious agents that can cause disease (e.g., viruses, the vCJD agent and, theoretically, the CJD agent). The risk of Flebogamma 5% DIF transmitting an infectious agent has been reduced by screening plasma donors for prior exposure, testing the donated plasma, and inactivating and/or removing certain viruses during manufacturing. (see Warnings and Precautions [5.8]) Instruct patients to report any symptoms that concern them and might be caused by infections.
Inform patients that Flebogamma 5% DIF may interfere with their immune response to live viral vaccines such as measles, mumps, and rubella. Inform patients to notify their health care professional of this potential interaction when they are receiving vaccinations. (see Drug Interactions [7])
Manufactured by:
INSTITUTO GRIFOLS, S.A.
BARCELONA – SPAIN
U.S. License No. 1181
Principal Display Panel - Vial Label
NDC 61953-0004-8
Immune Globulin Intravenous (Human)
Flebogamma® 5% DIF
5 g in 100 mL Rx only.
5%
Dosage and directions for administration, see package insert.
Store at 2 - 25 °C (36 - 77 °F). DO NOT USE IF TURBID, ONCE THE
CONTAINER HAS BEEN ENTERED IT SHOULD BE USED PROMPTLY.
Manufactured by Instituto Grifols, S.A. Barcelona - SPAIN
U.S. License No. 1181
3039367
Lot No.:
Expires

Principal Display Panel - Carton Label
GRIFOLS
NDC 61953-0004-3
Immune Globulin
Intravenous (Human)
Flebogamma® 5% DIF
Solution for infusion
5 g in 100 mL
5%
Nanofiltered (20 nm)
DO NOT USE IF TURBID. ONCE THE CONTAINER HAS BEEN ENTERED IT SHOULD BE USED PROMPTLY.
CONTENTS
One each: 100 mL vial Immune Globulin Intravenous (Human)
Each 100 mL contains 5 g Immune Globulin (Human), 5 g D-sorbitol.
Protein content is 50 g/L
Contains no preservatives.
The patient and physician should discuss the risks and benefits of this product.
INSTRUCTIONS
For information on dosage and directions for administration, see enclosed package insert.
STORE AT 2 - 25 °C (36 - 77 °F)
Rx only.
PRECAUTION
Do not allow to freeze.
Single dose container for intravenous administration.
Discard any unused contents and administration devices after use.
Manufactured by:
Instituto Grifols, S.A.
Barcelona - SPAIN
U.S. License No. 1181
GTIN: 00361953000432
SN: XXXXXXXXXXXXXXXX
Lot: XXXXXXXXXX
Exp.: DD-MMM-YY
3048655

Principal Display Panel - Vial Label
NDC 61953-0004-9
Immune Globulin Intravenous (Human)
Flebogamma® 5% DIF
10 g in 200 mL
Rx only.
5%
CONTENTS: Each 200 mL contains
Immune Globulin (Human) 10 g; D-sorbitol 10 g; Protein content is 50 g/L
Dosage and directions for administration, see package insert.
Store at 2 - 25 °C (36 - 77 °F).
DO NOT USE IF TURBID, ONCE THE CONTAINER HAS BEEN ENTERED IT SHOULD BE USED PROMPTLY.
The patient and physician should discuss the risks and benefits of this product.
Manufactured by Instituto Grifols, S.A. Barcelona - SPAIN
U.S. License No. 1181
3039369
Lot No.:
Expires:

Principal Display Panel - Carton Label
GRIFOLS
NDC 61953-0004-4
Immune Globulin
Intravenous (Human)
Flebogamma® 5% DIF
Solution for infusion
10 g in 200 mL
5%
Nanofiltered (20 nm)
DO NOT USE IF TURBID. ONCE THE CONTAINER HAS BEEN ENTERED IT SHOULD BE USED PROMPTLY.
CONTENTS
One each: 200 mL vial Immune Globulin Intravenous (Human)
Each 200 mL contains 10 g Immune Globulin (Human), 10 g D-sorbitol.
Protein content is 50 g/L
Contains no preservatives.
The patient and physician should discuss the risks and benefits of this product.
INSTRUCTIONS
For information on dosage and directions for administration, see enclosed package insert.
STORE AT 2 - 25 °C (36 - 77 °F)
Rx only.
PRECAUTION
Do not allow to freeze.
Single dose container for intravenous administration.
Discard any unused contents and administration devices after use.
Manufactured by:
Instituto Grifols, S.A.
Barcelona - SPAIN
U.S. License No. 1181
GTIN: 00361953000449
SN: XXXXXXXXXXXXXXXX
Lot: XXXXXXXXXX
Exp.: DD-MMM-YY
3048656

Principal Display Panel - Vial Label
NDC 61953-0004-0
Immune Globulin Intravenous (Human)
Flebogamma® 5% DIF
20 g in 400 mL
Rx only.
5%
CONTENTS: Each 400 mL contains Immune Globulin (Humin) 20 g;
D-sorbitol 20 g; Protein content is 50 g/L
Dosage and directions for administration, see package insert.
Store at 2 - 25 °C (36 - 77 °F).
DO NOT USE IF TURBID, ONCE THE CONTAINER HAS BEEN ENTERED IT SHOULD BE USED PROMPTLY.
The patient and physician should discuss the risks and benefits of this product.
Manufactured by Instituto Grifols, S.A. Barcelona - SPAIN
U.S. License No. 1181
3039371
Lot No.:
Expires:
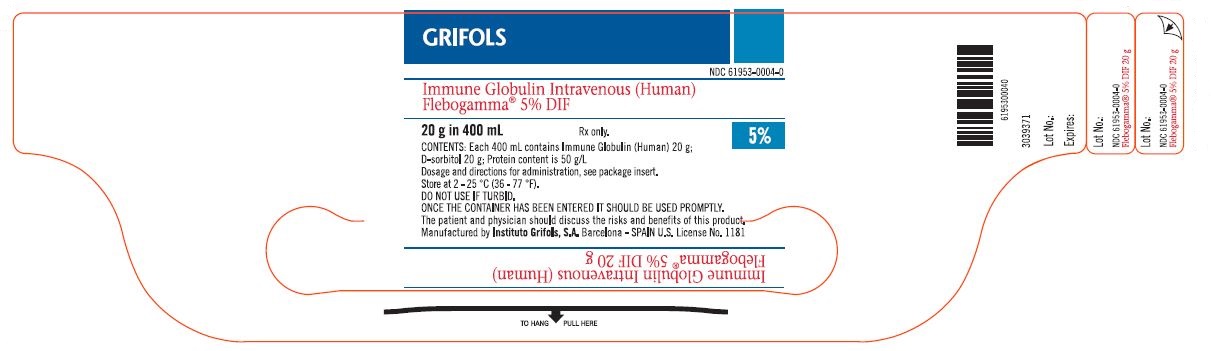
Principal Display Panel - Carton Label
GRIFOLS
NDC 61953-0004-5
Immune Globulin
Intravenous (Human)
Flebogamma® 5% DIF
Solution for infusion
20 g in 400 mL
5%
Nanofiltered (20 nm)
DO NOT USE IF TURBID. ONCE THE CONTAINER HAS BEEN ENTERED IT SHOULD BE USED PROMPTLY.
CONTENTS
One each: 400 mL vial Immune Globulin Intravenous (Human)
Each 400 mL contains 20 g Immune Globulin (Human), 20 g D-sorbitol.
Protein content is 50 g/L
Contains no preservatives.
The patient and physician should discuss the risks and benefits of this product.
INSTRUCTIONS
For information on dosage and directions for administration, see enclosed package insert.
STORE AT 2 - 25 °C (36 - 77 °F)
Rx only.
PRECAUTION
Do not allow to freeze.
Single dose container for intravenous administration.
Discard any unused contents and administration devices after use.
Manufactured by:
Instituto Grifols, S.A.
Barcelona - SPAIN
U.S. License No. 1181
GTIN: 00361953000456
SN: XXXXXXXXXXXXXXXX
Lot: XXXXXXXXXX
Exp.: DD-MMM-YY
3048658

| FLEBOGAMMA
DIF
immune globulin (human) injection, solution |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - GRIFOLS USA, LLC (048987452) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Instituto Grifols, S.A. | 465562213 | MANUFACTURE(61953-0004) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| GRIFOLS WORLDWIDE OPERATIONS LIMITED | 985528524 | label(61953-0004) , pack(61953-0004) | |
More about Flebogamma (immune globulin intravenous)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Latest FDA alerts (1)
- Side effects
- Dosage information
- During pregnancy
- Drug class: immune globulins
- En español
Patient resources
Professional resources
Other brands
Privigen, Octagam, Panzyga, Gammagard S/D, ... +6 more