Quadramet: Package Insert / Prescribing Info
Package insert / product label
Generic name: samarium Sm 153 lexidronam
Dosage form: injection, solution
Drug class: Therapeutic radiopharmaceuticals
Medically reviewed by Drugs.com. Last updated on Mar 31, 2025.
On This Page
Quadramet Description
QUADRAMET® is a therapeutic agent consisting of radioactive samarium and a tetraphosphonate chelator, ethylenediaminetetramethylenephosphonic acid (EDTMP). QUADRAMET® is formulated as a sterile, non-pyrogenic, clear, colorless to light amber isotonic solution of samarium-153 lexidronam for intravenous administration. QUADRAMET® does not contain a preservative.
Each milliliter contains 35 mg EDTMP∙H2O, 5.3 mg Ca [as Ca(OH)2], 14.1 mg Na [as NaOH], equivalent to 44 mg Ca/Na EDTMP (anhydrous calc.), 5-46 µg samarium (specific activity of approximately 1.0-11.0 mCi/µg Sm), and 1850 ± 185 MBq (50 ± 5 mCi) of samarium-153 at calibration.
The structural formula of samarium lexidronam pentasodium is:
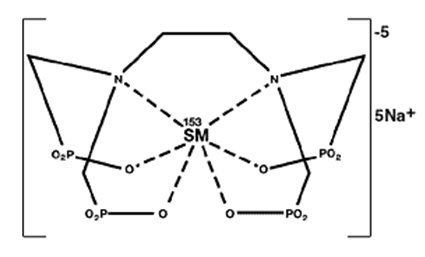
The ionic formula is 153Sm+3 [CH2N(CH2PO3-2)2]2 and the ionic formula weight is 581.1 daltons (pentasodium form, 696).
The pH of the solution is 7.0 to 8.5.
QUADRAMET® is supplied frozen in single-dose glass vials containing 3 mL with 5550 MBq (150 mCi) of samarium-153 at calibration.
Physical Characteristics
Samarium-153 is produced in high yield and purity by neutron irradiation of isotopically enriched samarium Sm 152 oxide (152Sm2O3). It emits both medium-energy beta particles and a gamma photon, and has a physical half-life of 46.3 hours (1.93 days). Samarium-153 has average and maximum beta particle ranges in water of 0.5 mm and 3.0 mm, respectively. The primary radiation emissions of samarium-153 are shown in Table 1.
External Radiation
The specific gamma-ray constant for samarium-153 is 0.46 R/mCi-hr at 1 cm (1.24×10-5 mSv/MBq- hr at 1 Meter). The half-value thickness of lead (Pb) for samarium-153 is approximately 0.10 mm. The use of 1 mm of lead will decrease the external radiation exposure by a factor of approximately 1,000. QUADRAMET® should be stored in a lead-shielded container and frozen until use.
Radioactive decay factors to be applied to the stated value for radioactive concentration at calibration are given in Table 2. All radioactivity is calibrated to the reference date and time on the vial.
| Time | Time | ||
|---|---|---|---|
| (hour)* | Factor | (hour)* | Factor |
|
|||
| -56.0 | 2.31 | +1.0 | 0.99 |
| -48.0 | 2.05 | +2.0 | 0.97 |
| -36.0 | 1.71 | +3.0 | 0.96 |
| -24.0 | 1.43 | +4.0 | 0.94 |
| -20.0 | 1.35 | +6.0 | 0.91 |
| -16.0 | 1.27 | +8.0 | 0.89 |
| -12.0 | 1.20 | +12.0 | 0.84 |
| -8.0 | 1.13 | +16.0 | 0.80 |
| -6.0 | 1.09 | +20.0 | 0.74 |
| -4.0 | 1.06 | +24.0 | 0.70 |
| -3.0 | 1.05 | +36.0 | 0.58 |
| -2.0 | 1.03 | +48.0 | 0.49 |
| -1.0 | 1.02 | +56.0 | 0.43 |
Quadramet - Clinical Pharmacology
QUADRAMET® (samarium Sm-153 EDTMP) has an affinity for bone and concentrates in areas of bone turnover in association with hydroxyapatite. In clinical studies employing planar imaging techniques, more QUADRAMET® accumulates in osteoblastic lesions than in normal bone with a lesion-to-normal bone ratio of approximately 5. The mechanism of action of QUADRAMET® in relieving the pain of bone metastases is not known.
Distribution
Human protein binding has not been studied; however, in dog, rat and bovine studies, less than 0.5% of samarium-153 EDTMP is bound to protein. At physiologic pH, >90% of the complex is present as 153Sm[EDTMP]-5, and <10% as 153SmH[EDTMP]-4. The octanol/ water partition coefficient is <10-5.
Skeletal Uptake
The greater the number of metastatic lesions, the more skeletal uptake of Sm-153 radioactivity. The relationship between skeletal uptake and the size of the metastatic lesions has not been studied. The total skeletal uptake of radioactivity was 65.5% ± 15.5% of the injected dose in 453 patients with metastatic lesions from a variety of primary malignancies. In a study of 22 patients with a wide range in the number of metastatic sites, the % of the injected dose (% ID) taken up by bone ranged from 56.3% in a patient with 5 metastatic lesions to 76.7% in a patient with 52 metastatic lesions. If the number of metastatic lesions is fixed, over the range 0.1 to 3.0 mCi/kg, the % ID taken up by bone is the same regardless of the dose.
Metabolism
The complex formed by samarium and EDTMP is excreted as an intact, single species that consists of one atom of the Sm-153 and one molecule of the EDTMP, as shown by an analysis of urine samples from patients (n=5) administered samarium Sm-153 EDTMP. Metabolic products of samarium Sm-153 EDTMP were not detected in humans.
Elimination
For QUADRAMET®, calculations of the % ID detected in the whole body, urine and blood were corrected for radionuclide decay. The clearance of activity through the urine is expressed as the cumulated activity excreted. The whole body retention is the simple reciprocal of the cumulated urine activity. (See Skeletal Uptake Section).
Blood
Clearance of radioactivity from the blood demonstrated biexponential kinetics after intravenous injection in 19 patients (10 men, 9 women) with a variety of primary cancers that were metastatic to bone. Over the first 30 minutes, the radioactivity (mean ± SD) in the blood decreased to 15% (±8%) of the injected dose with a t 1/2 of 5.5 min (±1.1 min). After 30 minutes, the radioactivity cleared from the blood more slowly with a t1/2 of 65.4 min (± 9.6 min). Less than 1% of the dose injected remained in the blood 5 hr after injection.
Gender Differences
Gender did not affect the samarium Sm-153 EDTMP blood pharmacokinetics, the cumulative % of radioactivity excreted in urine, or the % radioactivity retained in the skeleton when the number of metastatic lesions is taken into account.
Specific Populations
Elderly
The pharmacokinetics of samarium Sm-153 EDTMP did not change with age as seen from comparison of values from people in the age range of 22 to 64 compared to the range 65 to 86 years.
Pharmacodynamics
The beta particle of 153Sm-EDTMP travels a maximum distance of 3.0 mm in soft tissue and 1.7 mm in bone. In clinical trials of 78 patients with metastatic bone lesions who had 13 specific bone scan sites evaluated, the presence or absence of 153Sm-EDTMP uptake is similar to the presence or absence of 99mTc diphosphonate uptake (range 67 to 96% agreement depending upon the blinded reader and the site of the body). Whether the amount of 153Sm-EDTMP uptake varies with the size of the lesion or to the presence of osteolytic components has not been studied. The clinical benefit of Sm-153-EDTMP in patients with osteolytic lesions is not known. The relationship of different tumor cell types to clinical response has not been studied.
Clinical Studies
Overall QUADRAMET® was evaluated in 580 patients (see Adverse Events Section for demographic description). Of these patients, 270 (244 men, 26 women) were studied in two randomized, blinded, placebo controlled clinical trials. These patients had a mean age of 67, and a range 22 to 87 years. Eligible patients had painful metastatic bone lesions that had failed other treatments, had at least a 6 month expected survival and had a positive radionuclide bone scan. Routine x-rays to evaluate the metastatic lesions were not part of the protocol.
In study A, 118 patients were randomized to receive 0.5 mCi/kg QUADRAMET®, 1.0 mCi/kg QUADRAMET®, or a placebo intravenous injection. In study B, 152 patients were randomized to receive either 1.0 mCi/kg QUADRAMET® or a placebo intravenous injection. Both studies were double blind over a 4 week period. Patients scored their daily pain intensity on a visual analogue scale rated from 0 (no or low pain) to 10 (excruciating pain). The area under the pain curve (AUPC) was obtained by integrating the daily pain scores by week. Opioid analgesic use was recorded daily and averaged over each week and expressed in oral morphine milligram equivalents.
Of the 270 patients studied, 232 (86%) had prostate cancer and 38 (14%) had other primary cancers. In study A, 80 (68%) of the patients had prostate cancer and 38 (32%) had a variety of other primary tumors. In study B, all (100%) patients had prostate cancer.
The results of the patients' AUPC scores are shown in Table 3. In both trials for each of the 4 weeks of study, the mean AUPC scores decreased in patients who received QUADRAMET® (1.0 mCi/kg). In study A, pain (the AUPC) decrease from baseline was significantly different in QUADRAMET® 1.0 mCi/kg and placebo groups at weeks 3 and 4. In study B, pain (the AUPC) decrease from baseline was significantly different in QUADRAMET® 1.0 mCi/kg and placebo groups at weeks 2, 3 and 4.
| STUDY A (n = 73) † | STUDY B (n = 150) ‡ | |||
|---|---|---|---|---|
| WEEK | Placebo N=36 | 1.0 mCi/kg N=37 | Placebo N=50 | 1.0 mCi/kg N=100 |
|
||||
| Baseline | 26.5 (11.8) | 28.7 (12.3) | 28.5 (14.1) | 28.1 (12.9) |
| 1 | 26.1 (10.3) | 27.6 (14.1) | 27.9 (14.6) | 25.8 (13.1) |
| 2 | 24.4 (10.4) | 23.8 (13.7) | 28.1 (15.4) | 20.6 (13.9)§ |
| 3 | 24.3 (11.0) | 20.5 (11.5)§ | 25.8 (16.1) | 20.1 (13.3)§ |
| 4 | 24.7 (12.1) | 18.8 (10.8)§ | 24.7 (15.3) | 19.9 (13.7)§ |
In the two clinical trials, the patient use of analgesics differed. In Study A, the patients did not receive specific instructions on analgesic reduction. In Study B, patients were encouraged to adjust their pain medication as needed. As shown in Table 4, the morphine equivalent analgesic use in study A generally increased from baseline in both the QUADRAMET® and placebo treatment groups; however, the difference between the QUADRAMET® and placebo group change from baseline is not statistically significant. In study B, the placebo treated patients increased their use of opioid analgesics, while the QUADRAMET® treated patients decreased their use of opioid analgesics.
| STUDY A (n = 73) † | STUDY B (n = 150) ‡ | |||
|---|---|---|---|---|
| WEEK | Placebo N=36 | 1.0 mCi/kg N=37 | Placebo N=50 | 1.0 mCi/kg N=100 |
|
||||
| Baseline | 93.5 (154.0) * | 127.1 (189.9) | 78.4 (83.1) | 96.5 (166.6) |
| 1 | 106.8 (173.8) | 125.7 (192.6) | 84.5 (91.1) | 93.5 (165.5) |
| 2 | 127.1 (238.4) | 144.8 (276.7) | 85.6 (90.9) | 82.9 (122.9) |
| 3 | 133.9 (254.0) | 146.6 (278.2) | 100.1 (119.4) | 79.6 (131.2)§ |
| 4 | 135.6 (222.0) | 135.1 (274.0) | 106.3 (161.0) | 76.8 (132.3)§ |
In both studies, the numbers of patients who experienced any decrease in AUPC score without any increase in analgesic use at weeks 3 and 4 were also evaluated. In study A, this occurred in 20/37 (54%) of the patients who received QUADRAMET® 1.0 mCi/kg and 9/36 (25%) of the placebo treated patients. In study B, this occurred in 48/100 (48%) of the QUADRAMET® treated patients and 11/51 (22%) of the placebo treated patients.
Indications and Usage for Quadramet
QUADRAMET® is indicated for relief of pain in patients with confirmed osteoblastic metastatic bone lesions that enhance on radionuclide bone scan.
Contraindications
QUADRAMET® is contraindicated in patients who have known hypersensitivity to EDTMP or similar phosphonate compounds.
Warnings
QUADRAMET® causes bone marrow suppression. In clinical trials, white blood cell counts and platelet counts decreased to a nadir of approximately 40% to 50% of baseline in 123 (95%) of patients within 3 to 5 weeks after QUADRAMET®, and tended to return to pretreatment levels by 8 weeks. The grade of marrow toxicity is shown in Table 5 below.
| Hemoglobin | Leucocytes | Platelets | ||||
|---|---|---|---|---|---|---|
| Toxicity Grade* | Placebo N=85 | 1.0 mCi/kg N=185 | Placebo N=85 | 1.0 mCi/kg N=184 | Placebo N=85 | 1.0 mCi/kg N-185 |
|
||||||
| 0-2 | 78 (92%) | 162 (88%) | 85 (100%) | 169 (92%) | 85 (100%) | 173 (94%) |
| 3 | 6 (7%) | 20 (11%) | 0 (0%) | 15 (8%) | 0 (0%) | 10 (5%) |
| 4 | 1 (1%) | 3 (2%) | 0 (0%) | 0 (0%) | 0 (0%) | 2 (1%) |
Before QUADRAMET® is administered, consideration should be given to the patient's current clinical and hematologic status and bone marrow response history to treatment with myelotoxic agents. Metastatic prostate and other cancers can be associated with disseminated intravascular coagulation (DIC); caution should be exercised in treating cancer patients whose platelet counts are falling or who have other clinical or laboratory findings suggesting DIC. Because of the unknown potential for additive effects on bone marrow, QUADRAMET® should not be given concurrently with chemotherapy or external beam radiation therapy unless the clinical benefits outweigh the risks. Use of QUADRAMET® in patients with evidence of compromised bone marrow reserve from previous therapy or disease involvement is not recommended unless the potential benefits of the treatment outweigh the risks. Blood counts should be monitored weekly for at least 8 weeks, or until recovery of adequate bone marrow function.
Pregnancy
As with other radiopharmaceutical drugs, QUADRAMET® can cause fetal harm when administered to a pregnant woman. Adequate and well controlled studies have not been conducted in animals or pregnant women. Women of child-bearing age should have a negative pregnancy test before administration of QUADRAMET®. If this drug is used during pregnancy, or if a patient becomes pregnant after taking this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant soon after receiving QUADRAMET®. Men and women patients should be advised to use an effective method of contraception after the administration of QUADRAMET®.
The vial stopper contains dry natural rubber latex and may cause allergic reactions in providers or patients who are sensitive to latex
Precautions
EDTMP is a chelating agent. Although the chelating effects have not been evaluated thoroughly in humans, dogs that received non-radioactive samarium EDTMP (6 times the human dose based on body weight, 3 times based on surface area) developed a variety of electrocardiographic (ECG) changes (with or without the presence of hypocalcemia). The causal relationship between the hypocalcemia and ECG changes has not been studied. Whether QUADRAMET® causes electrocardiographic changes or arrhythmias in humans has not been studied. Caution and appropriate monitoring should be given when administering QUADRAMET® to patients (See Laboratory Tests).
Because concomitant hydration is recommended to promote the urinary excretion of QUADRAMET®, appropriate monitoring and consideration of additional supportive treatment should be used in patients with a history of congestive heart failure or renal impairment.
Renal Impairment: Because approximately one third of Samarium Sm-153 EDTMP is excreted in the urine, clearance may be reduced in patients with renal impairment; it is not known if dose adjustment is needed.
This drug should be used with caution in patients with compromised bone marrow reserves. See Warnings.
Skeletal
Spinal cord compression frequently occurs in patients with known metastases to the cervical, thoracic or lumbar spine. In clinical studies of QUADRAMET®, spinal cord compression was reported in 7% of patients who received placebo and in 8.3% of patients who received 1.0 mCi/kg QUADRAMET®. QUADRAMET® is not indicated for treatment of spinal cord compression. QUADRAMET® administration for pain relief of metastatic bone cancer does not prevent the development of spinal cord compression. When there is a clinical suspicion of spinal cord compression, appropriate diagnostic and therapeutic measures must be taken immediately to avoid permanent disability.
Radiopharmaceutical agents should be used only by physicians who are qualified by training and experience in the safe use and handling of radionuclides and whose experience and training have been approved by the appropriate government agency authorized to license the use of radionuclides.
QUADRAMET®, like other radioactive drugs, must be handled with care, and appropriate safety measures must be taken to minimize radiation exposure of clinical personnel and others in the patient environment.
Special precautions, such as bladder catheterization, should be taken with incontinent patients to minimize the risk of radioactive contamination of clothing, bed linen, and the patient's environment. Urinary excretion of radioactivity occurs over about 12 hours (with 35% occurring during the first 6 hours). Studies have not been done on the use of QUADRAMET® in patients with renal impairment.
INFORMATION FOR PATIENTS
Patients who receive QUADRAMET® should be advised that for several hours following administration, radioactivity will be present in excreted urine. To help protect themselves and others in their environment, precautions need to be taken for 12 hours following administration. Whenever possible, a toilet should be used, rather than a urinal, and the toilet should be flushed several times after each use. Spilled urine should be cleaned up completely and patients should wash their hands thoroughly. If blood or urine gets onto clothing, the clothing should be washed separately, or stored for 1-2 weeks to allow for decay of the Sm-153.
Some patients have reported a transient increase in bone pain shortly after injection (flare reaction). This is usually mild and self-limiting and occurs within 72 hours of injection. Such reactions are usually responsive to analgesics.
Patients who respond to QUADRAMET® might begin to notice the onset of pain relief one week after QUADRAMET®. Maximal pain relief generally occurs at 3-4 weeks after injection of QUADRAMET®. Patients who experience a reduction in pain may be encouraged to decrease their use of opioid analgesics.
LABORATORY TESTS
Because of the potential for bone marrow suppression, beginning 2 weeks after QUADRAMET® administration, blood counts should be monitored weekly for at least 8 weeks, or until recovery of adequate bone marrow function.
In a subset of 31 patients who had serum calcium monitored during the first 2 hours after QUADRAMET® infusion, a clear pattern of calcium change was not identified. However, 10 (32%) patients had at least one serum calcium level that was below normal (7.16 to 8.28). The extent to which samarium-153-EDTMP is related to this hypocalcemia is not known. Caution should be exercised when administering QUADRAMET® to patients at risk for developing hypocalcemia.
DRUG INTERACTIONS
The potential for additive bone marrow toxicity of QUADRAMET® with chemotherapy or external beam radiation has not been studied. QUADRAMET® should not be given concurrently with chemotherapy or external beam radiation therapy unless the benefit outweighs the risks. QUADRAMET® should not be given after either of these treatments until there has been time for adequate marrow recovery. (See Warnings Section).
CARCINOGENESIS, MUTAGENESIS, IMPAIRMENT OF FERTILITY
Carcinogenesis in humans given EDTMP, in QUADRAMET®, is not likely. Osteosarcomas occurred in a 2-year toxicity/carcinogenicity study of EDTMP administered by gastric intubation to Sprague-Dawley rats, in male rats at 50 mg/kg/day and in male and female rats at 150 mg/kg/day (the dosage was increased to 333 mg/kg/day on day 329 of treatment). Osteosarcomas were not reported in a published chronic dietary study of up to 130 weeks of EDTMP in Fisher 344 rats, at dietary doses up to 100 mg/kg/day (not the maximum tolerated dose). However, at study termination in female Fisher 344 rats, this dose was associated with statistically significantly higher rate of pancreatic islet-cell adenomas and carcinomas.
The results of the following genotoxicity assays with non-radioactive samarium- EDTMP were negative: Salmonella reverse mutation (AMES) assay, unscheduled DNA synthesis in rat liver primary cell culture, chromosomal aberration assay in rat lymphocytes, CHO/HGPRT forward mutation assay, and mouse bone marrow micronucleus test.
Studies have not been performed to assess the effect of QUADRAMET® on fertility.
NURSING MOTHERS
It is not known whether QUADRAMET® is excreted in human milk. Because of the potential for serious adverse reactions in nursing infants from QUADRAMET®, a decision should be made whether to continue nursing or to administer the drug. If QUADRAMET® is administered, formula feedings should be substituted for breast feedings.
Adverse Reactions/Side Effects
Adverse events were evaluated in a total of 580 patients who received QUADRAMET® in clinical trials. Of the 580 patients, there were 472 men and 108 women with a mean age of 66 (range 20 to 87).
Of these patients, 472 (81%) had at least one adverse event. In a subgroup of 399 patients who received QUADRAMET® 1.0 mCi/kg, there were 23 deaths and 46 serious adverse events. The deaths occurred an average of 67 days (9 to 130) after QUADRAMET®. Serious events occurred an average of 46 days (1 - 118) after QUADRAMET®. Although most of the patient deaths and serious adverse events appear to be related to the underlying disease, the relationship of end stage disease, marrow invasion by cancer cells, previous myelotoxic treatment and QUADRAMET® toxicity can not be easily distinguished. In clinical studies, two patients with rapidly progressive prostate cancer developed thrombocytopenia and died 4 weeks after receiving QUADRAMET®. One of the patients showed evidence of disseminated intravascular coagulation (DIC); the other patient experienced a fatal cerebrovascular accident, with a suspicion of DIC. The relationship of the DIC to the bone marrow suppressive effect of Samarium is not known. Marrow toxicity occurred in 277 (48%) patients (See Warnings section).
In controlled studies, 7% of patients receiving 1.0 mCi/kg QUADRAMET® (as compared to 6% of patients receiving placebo) reported a transient increase in bone pain shortly after injection (flare reaction). This was usually mild, self-limiting, and responded to analgesics.
The most common adverse events observed in controlled clinical studies of QUADRAMET®, are given in Table 6.
| ADVERSE EVENT | Placebo N=90 | QUADRAMET®
1.0 mCi/kg N=199 |
|---|---|---|
|
||
| # Patients with Any Adverse Event | 72 (80%) | 169 (85%) |
| Body As A Whole | 56 (62%) | 100 (50%) |
| Pain Flare Reaction | 5 (5.6%) | 14 (7.0%) |
| Cardiovascular | 19 (21%) | 32 (16%) |
| Arrhythmias | 2 (2.2%) | 10 (5.0%) |
| Chest Pain | 4 (4.4%) | 8 (4.0%) |
| Hypertension | 0 | 6 (3.0%) |
| Hypotension | 2 (2.2%) | 4 (2.0%) |
| Digestive | 44 (49%) | 82 (41%) |
| Abdominal Pain | 7 (7.8%) | 12 (6.0%) |
| Diarrhea | 3 (3.3%) | 12 (6.0%) |
| Nausea &/or Vomiting | 37 (41.1%) | 65 (32.7%) |
| Hematologic & Lymphatic | 12 (13%) | 54 (27%) |
| Coagulation Disorder | 0 | 3 (1.5%) |
| Hemoglobin Decreased | 21 (23.3%) | 81 (40.7%) |
| Leukopenia | 6 (6.7%) | 118(59.3%) |
| Lymphadenopathy | 0 | 4 (2.0%) |
| Thrombocytopenia | 8 (8.9%) | 138(69.3%) |
| Any Bleeding Manifestations* | 8 (8.9%) | 32 (16.1%) |
| Ecchymosis | 1 (1.1%) | 3 (3.0%) |
| Epistaxis | 1 (1.1%) | 4 (2.0%) |
| Hematuria | 3 (3.3%) | 10 (5%) |
| Infection | 10 (11.1%) | 34 (17.1%) |
| Fever and/or Chills | 10 (11.1%) | 17 (8.5%) |
| Infection, Not Specified | 4 (4.4%) | 14 (7.0%) |
| Oral Moniliasis | 1 (1.1%) | 4 (2.0%) |
| Pneumonia | 1 (1.1%) | 3 (1.5%) |
| Musculoskeletal | 28 (31%) | 55 (27%) |
| Myasthenia | 8 (8.9%) | 13 (6.5%) |
| Pathologic Fracture | 2 (2.2%) | 5 (2.5%) |
| Nervous | 39 (43%) | 59 (30%) |
| Dizziness | 1 (1.1%) | 8 (4.0%) |
| Paresthesia | 7 (7.8%) | 4 (2.0%) |
| Spinal Cord Compression | 5 (5.5%) | 13 (6.5%) |
| Cerebrovascular Accident/Stroke | 0 | 2 (1.0%) |
| Respiratory | 24 (27%) | 35 (18%) |
| Bronchitis/Cough Increased | 2 (2.2%) | 8 (4.0%) |
| Special Senses | 11 (12%) | 11 (6%) |
| Skin & Appendages | 17 (19%) | 13 (7%) |
| Purpura | 0 | 2 (1%) |
| Rash | 2 (2.2%) | 2 (1%) |
In an additional 200 patients who received QUADRAMET® in uncontrolled clinical trials, adverse events that were reported at a rate of greater than or equal to 1.0% were similar except for 9 (4.5%) patients who had agranulocytosis. Other selected adverse events that were reported in <1% of the patients who received QUADRAMET® 1.0 mCi/kg in any clinical trial include: alopecia, angina, congestive heart failure, sinus bradycardia, and vasodilation.
Related/similar drugs
Lutathera
Lutathera is a targeted radiotherapy used for gastroenteropancreatic neuroendocrine tumors ...
Botox
Botox is used cosmetically to reduce facial lines and wrinkles and for medical purposes for ...
Pluvicto
Pluvicto (lutetium lu 177 vipivotide tetraxetan) is used for PSMA-positive metastatic ...
Xofigo
XOFIGO radium 223 treatment is a radioactive substance used to treat advanced prostate cancer ...
Sodium iodide-i-131
Sodium iodide-i-131 is used for diagnosis and investigation, hyperthyroidism, thyroid cancer
Lutetium lu 177 vipivotide tetraxetan
Lutetium lu 177 vipivotide tetraxetan is used for prostate cancer
Overdosage
Overdosage with QUADRAMET® has not been reported. An antidote for QUADRAMET® overdosage is not known. The anticipated complications of overdosage would likely be secondary to bone marrow suppression from the radioactivity of 153Sm, or secondary to hypocalcemia and cardiac arrhythmias related to the EDTMP.
Quadramet Dosage and Administration
The recommended dose of QUADRAMET® is 1.0 mCi/kg, administered intravenously over a period of one minute through a secure in-dwelling catheter and followed with a saline flush. Dose adjustment in patients at the extremes of weight have not been studied. Caution should be exercised when determining the dose in very thin or very obese patients.
The dose should be measured by a suitable radioactivity calibration system, such as a radioisotope dose calibrator, immediately before administration.
The dose of radioactivity to be administered and the patient should be verified before administering QUADRAMET®. Patients should not be released until their radioactivity levels and exposure rates comply with federal and local regulations.
The patient should ingest (or receive by i.v. administration) a minimum of 500 mL (2 cups) of fluids prior to injection and should void as often as possible after injection to minimize radiation exposure to the bladder.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit. The solution should not be used if it is cloudy or if it contains particulate matter.
QUADRAMET® contains calcium and may be incompatible with solutions that contain molecules that can complex with and form calcium precipitates.
QUADRAMET® should not be diluted or mixed with other solutions.
Thaw at room temperature before administration and use within 8 hours of thawing.
Radiation Dosimetry
The estimated absorbed radiation doses to an average 70 kg adult patient from an i.v. injection of QUADRAMET® are shown in Table 7. The dosimetry estimates were based on clinical biodistribution studies using methods developed for radiation dose calculations by the Medical Internal Radiation Dose (MIRD) Committee of the Society of Nuclear Medicine.
Radiation exposure is based on a urinary voiding interval of 4.8 hours. Radiation dose estimates for bone and marrow assume that radioactivity is deposited on bone surfaces, as noted in autoradiograms of biopsy bone samples in 7 patients who received QUADRAMET®. Although electron emissions from 153Sm are abundant, with energies up to 810 keV, rapid blood clearance of QUADRAMET® and low energy and abundant photon emissions generally result in low radiation doses to those parts of the body where the complex does not localize.
When blastic osseous lesions are present, significantly enhanced localization of the radiopharmaceutical will occur, with correspondingly higher doses to the lesions compared with normal bones and other organs. (See Clinical Pharmacology, Skeletal Uptake and Pharmacodynamics Sections).
| RADIATION ABSORBED DOSES 70 kg ADULT |
||
|---|---|---|
| Target Organ | Rad/mCi | mGy/MBq |
| Bone Surfaces | 25.0 | 6.76 |
| Red Marrow | 5.70 | 1.54 |
| Urinary Bladder Wall | 3.60 | 0.097 |
| Kidneys | 0.065 | 0.018 |
| Whole Body | 0.040 | 0.011 |
| Lower large intestine | 0.037 | 0.010 |
| Ovaries | 0.032 | 0.0086 |
| Muscle | 0.028 | 0.0076 |
| Small Intestine | 0.023 | 0.0062 |
| Upper Large Intestine | 0.020 | 0.0054 |
| Testes | 0.020 | 0.0054 |
| Liver | 0.019 | 0.0051 |
| Spleen | 0.018 | 0.0049 |
| Stomach | 0.015 | 0.0041 |
How is Quadramet supplied
QUADRAMET® is supplied frozen in a single-dose 10 mL glass vial containing 1850 ± 185 MBq/mL (50 ± 5 mCi/mL) of samarium-153, at calibration.
QUADRAMET® is available in the following size:
| NDC# 11994-016-01 | 3mL fill size with total activity of 5550 MBq (150mCi). |
The vial is shipped in a lead shield; a package insert is included.
The drug product expires 56 hours after the time of calibration noted on the label, or 8 hours after thawing, whichever is earlier.
This radioactive drug is approved for distribution to persons licensed pursuant to the Code of Massachusetts Regulations 105 CMR 120.100 for the uses listed in 105 CMR 120.589 or under equivalent regulations of the U.S. Nuclear Regulatory Commission, an Agreement State or a Licensing State.
THIS PRODUCT INFORMATION ISSUED March 2018.
Lantheus Medical Imaging, Inc.
331 Treble Cove Road
N. Billerica, Massachusetts 01862
USA
For Ordering Call Toll Free: 800-299-3431
All other business: 800-362-2668
For Massachusetts and International call: 978-667-9531
Printed in U.S.A.
Logo
513145-0318
March 2018
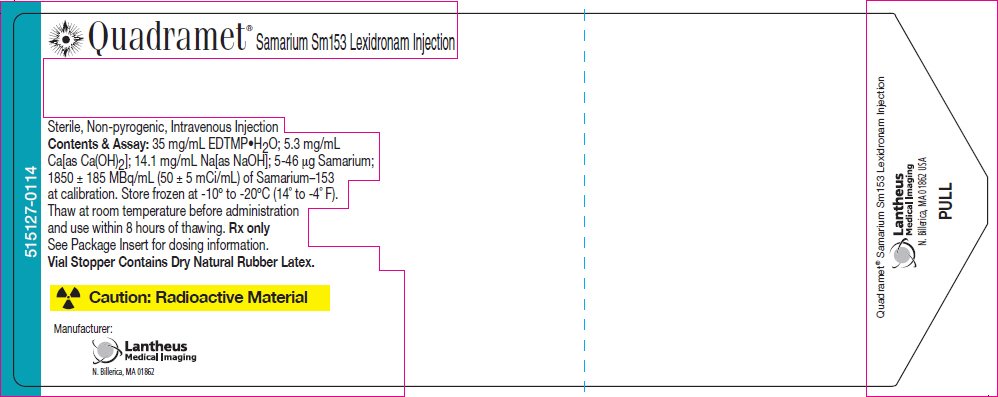
PRINCIPAL DISPLAY PANEL - 3 mL Vial Label
Quadramet® Samarium Sm153 Lexidronam Injection
Sterile, Non-pyrogenic, Intravenous Injection
Contents & Assay: 35 mg/mL EDTMP•H2O; 5.3 mg/mL
Ca[as Ca(OH)2]; 14.1 mg/mL Na[as NaOH]; 5-46 µg Samarium;
1850 ± 185 MBq/mL (50 ± 5 mCi/mL) of Samarium–153
at calibration. Store frozen at -10° to -20°C (14° to -4° F).
Thaw at room temperature before administration
and use within 8 hours of thawing. Rx only
See Package Insert for dosing information.
Vial Stopper Contains Dry Natural Rubber Latex.
Caution: Radioactive Material
Manufacturer:
Lantheus
Medical Imaging
N. Billerica, MA 01862
515127-0114
| QUADRAMET
samarium sm 153 lexidronam injection, solution |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - Lantheus Medical Imaging, Inc. (176786812) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Lantheus Medical Imaging, Inc. | 176786812 | MANUFACTURE(11994-016) , PACK(11994-016) , LABEL(11994-016) | |
More about Quadramet (samarium sm 153 lexidronam)
- Check interactions
- Compare alternatives
- Side effects
- Dosage information
- Drug class: therapeutic radiopharmaceuticals
- Breastfeeding