MPM Pak: Package Insert / Prescribing Info
Package insert / product label
Generic name: mifepristone ondansetron, ibuprofen
Dosage form: tablets, kit
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
- Medication Guide
- Warnings
- Precautions
- Drug Abuse and Dependence
Highlights of Prescribing Information
Mifepristone tablets, 200mg for oral use
Initial U.S. Approval: 2000
WARNING: SERIOUS AND SOMETIMES FATAL INFECTIONS OR BLEEDING
See full prescribing information for complete boxed warning.
Serious and sometimes fatal infections and bleeding occur very rarely following spontaneous, surgical, and medical abortions, including following Mifepristone tablets, 200 mg use.
- Atypical Presentation of Infection. Patients with serious bacterial infections and sepsis can present without fever, bacteremia or significant findings on pelvic examination. A high index of suspicion is needed to rule out serious infection and sepsis. ( 5.1)
- Bleeding. Prolonged heavy bleeding may be a sign of incomplete abortion or other complications and prompt medical or surgical intervention may be needed. ( 5.2)
Mifepristone tablets, 200 mg is only available through a restricted program called the mifepristone REMS Program ( 5.3).
Before prescribing Mifepristone tablets, 200 mg, inform the patient about these risks. Ensure the patient knows whom to call and what to do if they experience sustained fever, severe abdominal pain, prolonged heavy bleeding, or syncope, or if they experience abdominal pain or discomfort or general malaise for more than 24 hours after taking misoprostol.
Indications and Usage for MPM Pak
Mifepristone tablets, 200 mg is a progestin antagonist indicated, in a regimen with misoprostol, for the medical termination of intrauterine pregnancy through 70 days gestation. ( 1)
MPM Pak Dosage and Administration
Dosage Forms and Strengths
Tablets containing 200 mg of mifepristone each, supplied as 1 tablet on one blister card (3)
Contraindications
- Confirmed/suspected ectopic pregnancy or undiagnosed adnexal mass ( 4)
- Chronic adrenal failure ( 4)
- Concurrent long-term corticosteroid therapy ( 4)
- History of allergy to mifepristone, misoprostol, or other prostaglandins ( 4)
- Hemorrhagic disorders or concurrent anticoagulant therapy ( 4)
- Inherited porphyria ( 4)
- Intrauterine device (IUD) in place ( 4)
Warnings and Precautions
Adverse Reactions/Side Effects
Most common adverse reactions (>15%) are nausea, weakness, fever/chills, vomiting, headache, diarrhea, and dizziness. ( 6)
To report SUSPECTED ADVERSE REACTIONS, contact GenBioPro, Inc. at 1-855-643-3463 or medical@genbiopro.com or www.MIFEINFO.com or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Use In Specific Populations
- Pregnancy: Risk of fetal malformations in ongoing pregnancy if not terminated is unknown. ( 8.1)
See 17 for PATIENT COUNSELING INFORMATION, Medication Guide
MPM Pak Dosage and Administration
The recommended adult oral dose of Misoprostol Tablets for reducing the risk of NSAID-induced gastric ulcers is 200 mcg four times daily with food. If this dose cannot be tolerated, a dose of 100 mcg can be used. See Clinical PharmacologyClinical studies . Misoprostol Tablets should be taken for the duration of NSAID therapy as prescribed by the physician. Misoprostol Tablets should be taken with a meal, and the last dose of the day should be at bedtime.
Indications and Usage for MPM Pak
Ondansetron orally disintegrating tablets are a 5-HT 3 receptor antagonist indicated for the prevention of:
- nausea and vomiting associated with highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m 2. (1)
- nausea and vomiting associated with initial and repeat courses of moderately emetogenic cancer chemotherapy. (1)
- nausea and vomiting associated with radiotherapy in patients receiving either total body irradiation, single high-dose fraction to the abdomen, or daily fractions to the abdomen. (1)
- postoperative nausea and/or vomiting. (1)
MPM Pak Dosage and Administration
MPM Pak Dosage and Administration
- Orally Disintegrating Tablets: 4 mg and 8 mg. ( 3)
Contraindications
Warnings and Precautions
- Hypersensitivity Reactions, Including Anaphylaxis and Bronchospasm: Discontinue ondansetron if suspected. Monitor and treat promptly per standard of care until signs and symptoms resolve. ( 5.1)
- QT Interval Prolongation and Torsade de Pointes: Avoid ondansetron in patients with congenital long QT syndrome; monitor with electrocardiograms (ECGs) if concomitant electrolyte abnormalities, cardiac failure or arrhythmias, or use of other QT prolonging drugs. ( 5.2)
- Serotonin Syndrome: Reported with 5-HT 3 receptor antagonists alone but particularly with concomitant use of serotonergic drugs. If such symptoms occur, discontinue ondansetron and initiate supportive treatment. If concomitant use of ondansetron with other serotonergic drugs is clinically warranted, patients should be made aware of a potential increased risk for serotonin syndrome. ( 5.3)
- Myocardial Ischemia: Monitor or advise patients for signs and symptoms of myocardial ischemia after oral administration. ( 5.4)
- Masking of Progressive Ileus and/or Gastric Distension Following Abdominal Surgery or Chemotherapy-Induced Nausea and Vomiting: Monitor for decreased bowel activity, particularly in patients with risk factors for gastrointestinal obstruction. ( 5.5)
- Phenylketonuria: Patients should be informed that ondansetron orally disintegrating tablets contain phenylalanine (a component of aspartame). Each 4 mg orally disintegrating tablet contains 1.68 mg phenylalanine and 8 mg orally disintegrating tablet contains 3.37 mg phenylalanine. ( 5.6)
Adverse Reactions/Side Effects
The most common adverse reactions in adults for the:
- prevention of chemotherapy-induced (≥ 5%) are: headache, malaise/fatigue, constipation, diarrhea. ( 6.1)
- prevention of radiation-induced nausea and vomiting (≥ 2%) are: headache, constipation, and diarrhea. ( 6.1)
- prevention of postoperative nausea and vomiting (≥ 9%) are: headache and hypoxia. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Rising Health, LLC at 1-833-395-6928 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION, Medication Guide and PATIENT COUNSELING INFORMATION.
Revised: 1/2023
Full Prescribing Information
WARNING: SERIOUS AND SOMETIMES FATAL INFECTIONS OR BLEEDING
Serious and sometimes fatal infections and bleeding occur very rarely following spontaneous, surgical, and medical abortions, including following Mifepristone tablets, 200 mg use. No causal relationship between the use of Mifepristone tablets, 200 mg and misoprostol and these events has been established.
- Atypical Presentation of Infection. Patients with serious bacterial infections (e.g., Clostridium sordellii) and sepsis can present without fever, bacteremia, or significant findings on pelvic examination following an abortion. Very rarely, deaths have been reported in patients who presented without fever, with or without abdominal pain, but with leukocytosis with a marked left shift, tachycardia, hemoconcentration, and general malaise. A high index of suspicion is needed to rule out serious infection and sepsis [see Warnings and Precautions ( 5.1)]
- Bleeding. Prolonged heavy bleeding may be a sign of incomplete abortion or other complications and prompt medical or surgical intervention may be needed. Advise patients to seek immediate medical attention if they experience prolonged heavy vaginal bleeding [see Warnings and Precautions ( 5.2)].
Because of the risks of serious complications described above, Mifepristone tablets, 200mg is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Mifepristone REMS Program [see Warnings and Precautions ( 5.3)].
Before prescribing mifepristone, inform the patient about the risk of these serious events. Ensure that the patient knows whom to call and what to do, including going to an Emergency Room if none of the provided contacts are reachable, if they experience sustained fever, severe abdominal pain, prolonged heavy bleeding, or syncope, or if they experience abdominal pain or discomfort, or general malaise (including weakness, nausea, vomiting or diarrhea) for more than 24 hours after taking misoprostol.
1. Indications and Usage for MPM Pak
Mifepristone tablets, 200 mg is indicated, in a regimen with misoprostol, for the medical termination of intrauterine pregnancy through 70 days gestation.
2. MPM Pak Dosage and Administration
2.1 Dosing Regimen
For purposes of this treatment, pregnancy is dated from the first day of the last menstrual period. The duration of pregnancy may be determined from menstrual history and clinical examination. Assess the pregnancy by ultrasonographic scan if the duration of pregnancy is uncertain or if ectopic pregnancy is suspected.
Remove any intrauterine device (“IUD”) before treatment with Mifepristone tablets, 200mg begins [see
Contraindications (
4)].
The dosing regimen for Mifepristone tablets, 200 mg and misoprostol is:
Mifepristone 200 mg orally + misoprostol 800 mcg buccally
• Day One: Mifepristone 200 mg Administration
One 200 mg tablet of Mifepristone is taken in a single oral dose.
• Day Two or Three: Misoprostol Administration (minimum 24-hour interval between, Mifepristone and misoprostol)
Four 200 mcg tablets (total dose 800 mcg) of misoprostol are taken by the buccal route.
Tell the patient to place two 200 mcg misoprostol tablets in each cheek pouch (the area between the cheek and gums) for 30 minutes and then swallow any remnants with water or another liquid (see Figure 1).
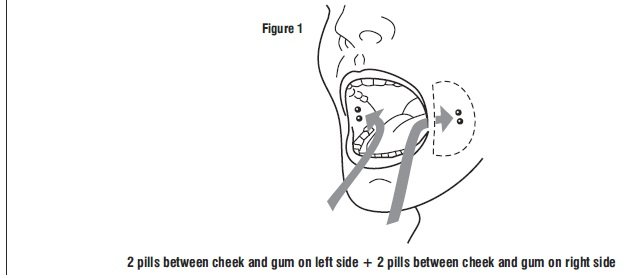
Patients taking Mifepristone tablets, 200 mg must take misoprostol within 24 to 48 hours after taking Mifepristone. The effectiveness of the regimen may be lower if misoprostol is administered less than 24 hours or more than 48 hours after mifepristone administration.
Because most women will expel the pregnancy within 2 to 24 hours of taking misoprostol
[see Clinical Studies (
14)]
, discuss with the patient an appropriate location for them to be when taking the misoprostol, taking into account that expulsion could begin within 2 hours of administration.
2.2 Patient Management Following Misoprostol Administration
During the period immediately following the administration of misoprostol, the patient may need medication for cramps or gastrointestinal symptoms [see Adverse Reactions ( 6)] .
Give the patient:
- Instructions on what to do if significant discomfort, excessive vaginal bleeding or other adverse reactions occur
- A phone number to call if the patient has questions following the administration of the misoprostol
- The name and phone number of the healthcare provider who will behandling emergencies.
2.3 Post-treatment Assessment: Day 7 to 14
Patients should follow-up with their healthcare provider approximately 7 to 14 days after the administration of Mifepristone. This assessment is very important to confirm that complete termination of pregnancy has occurred and to evaluate the degree of bleeding. Termination can be confirmed by medical history, clinical examination, human Chorionic Gonadotropin (hCG) testing, or ultrasonographic scan. Lack of bleeding following treatment usually indicates failure; however, prolonged or heavy bleeding is not proof of a complete abortion.
The existence of debris in the uterus (e.g., if seen on ultrasonography) following the treatment procedure will not necessarily require surgery for its removal.
Patients should expect to experience vaginal bleeding or spotting for an average of 9 to 16 days. Women report experiencing heavy bleeding for a median duration of 2 days. Up to 8% of women may experience some type of bleeding for more than 30 days. Persistence of heavy or moderate vaginal bleeding at the time of follow-up, however, could indicate an incomplete abortion.
If complete expulsion has not occurred, but the pregnancy is not ongoing, patients may be treated with another dose of misoprostol 800mcg buccally. There have been rare reports of uterine rupture in women who took mifepristone tablets, 200 mg and misoprostol, including women with prior uterine rupture or uterine scar and patients who received multiple doses of misoprostol within 24 hours. Patients who choose to use a repeat dose of misoprostol should have a follow-up visit with their healthcare provider in approximately 7 days to assess for complete termination.
Surgical evacuation is recommended to manage ongoing pregnancies after medical abortion
[see
Use in Specific Populations (
8.1)]
. Advise the patient whether you will provide such care or will refer her to another provider as part of counseling prior to prescribing Mifepristone tablets, 200 mg.
3. Dosage Forms and Strengths
Tablets containing 200 mg of mifepristone each, supplied as 1 tablet on one blister card. Mifepristone tablets are light yellow in color, circular, bio-convex tablets, approximately 11mm in diameter and imprinted on one side with “S.”
4. Contraindications
- Administration of Mifepristone tablets, 200 mg and misoprostol for the termination of pregnancy (the “treatment procedure”) is contraindicated in patients with any of the following conditions:
- Confirmed or suspected ectopic pregnancy or undiagnosed adnexal mass (the treatment procedure will not be effective to terminate an ectopic pregnancy) [see Warnings and Precautions ( 5.4)]
- Chronic adrenal failure (risk of acute adrenal insufficiency)
- Concurrent long-term corticosteroid therapy (risk of acute adrenal insufficiency)
- History of allergy to mifepristone, misoprostol, or other prostaglandins (allergic reactions including anaphylaxis, angioedema, rash, hives, and itching have been reported [see Adverse Reactions ( 6.2)])
- Hemorrhagic disorders or concurrent anticoagulant therapy (risk of heavy bleeding)
- Inherited porphyrias (risk of worsening or of precipitation of attacks)
- Use of Mifepristone tablets, 200 mg and misoprostol for termination of intrauterine pregnancy is contraindicated in patients with an intrauterine device (“IUD”) in place (the IUD might interfere with pregnancy termination). If the IUD is removed, Mifepristone tablets, 200 mg may be used.
5. Warnings and Precautions
5.1 Infection and Sepsis
As with other types of abortion, cases of serious bacterial infection, including very rare cases of fatal septic shock, have been reported following the use of Mifepristone tablets, 200 mg [see Boxed Warning]. Healthcare providers evaluating a patient who is undergoing a medical abortion should be alert to the possibility of this rare event. A sustained (>4 hours) fever of 100.4°F or higher, severe abdominal pain, or pelvic tenderness in the days after a medical abortion may be an indication of infection.
A high index of suspicion is needed to rule out sepsis (e.g., from Clostridium sordellii) if a patient reports abdominal pain or discomfort or general malaise (including weakness, nausea, vomiting or diarrhea) more than 24 hours after taking misoprostol. Very rarely, deaths have been reported in patients who presented without fever, with or without abdominal pain, but with leukocytosis with a marked left shift, tachycardia, hemoconcentration, and general malaise. No causal relationship between Mifepristone tablets, 200 mg and misoprostol use and an increased risk of infection or death has been established. Clostridium sordellii infections have also been reported very rarely following childbirth (vaginal delivery and caesarian section), and in other gynecologic and non-gynecologic conditions.
5.2 Uterine Bleeding
Uterine bleeding occurs in almost all patients during a medical abortion. Prolonged heavy bleeding (soaking through two thick full-size sanitary pads per hour for two consecutive hours) may be a sign of incomplete abortion or other complications and prompt medical or surgical intervention may be needed to prevent the development of hypovolemic shock. Counsel patients to seek immediate medical attention if they experience prolonged heavy vaginal bleeding following a medical abortion [see Boxed Warning].
Women should expect to experience vaginal bleeding or spotting for an average of 9 to 16 days. Women report experiencing heavy bleeding for a median duration of 2 days.
Up to 8% of all subjects may experience some type of bleeding for 30 days or more. In general, the duration of bleeding and spotting increased as the duration of the pregnancy increased.
Decreases in hemoglobin concentration, hematocrit, and red blood cell count may occur in patients who bleed heavily.
Excessive uterine bleeding usually requires treatment by uterotonics, vasoconstrictor drugs, surgical uterine evacuation, administration of saline infusions, and/or blood transfusions. Based on data from several large clinical trials, vasoconstrictor drugs were used in 4.3% of all subjects, there was a decrease in hemoglobin of more than 2 g/dL in 5.5% of subjects, and blood transfusions were administered to ≤0.1% of subjects. Because heavy bleeding requiring surgical uterine evacuation occurs in about 1% of patients, special care should be given to patients with hemostatic disorders, hypocoagulability, or severe anemia.
5.3 Mifepristone REMS Program
Mifepristone tablets, 200 mg is available only through a restricted program under a REMS called the mifepristone REMS Program, because of the risks of serious complications [see Warnings and Precautions ( 5.1, 5.2)]
Notable requirements of the mifepristone REMS Program include the following:
- Prescribers must be certified with the program by completing the Prescriber Agreement Form
- Patients must sign a Patient Agreement Form
- Mifepristone tablets, 200 mg must be dispensed to patients by or under the supervision of a certified prescriber, or by certified pharmacies on prescriptions issued by certified prescribers
Further information is available at 1-855-MIFEINFO (1-855-643-3463).
5.4 Ectopic Pregnancy
Mifepristone tablets, 200 mg is contraindicated in patients with a confirmed or suspected ectopic pregnancy because mifepristone is not effective for terminating ectopic pregnancies [see Contraindications ( 4)]. Healthcare providers should remain alert to the possibility that a patient who is undergoing a medical abortion could have an undiagnosed ectopic pregnancy because some of the expected symptoms experienced with a medical abortion (abdominal pain, uterine bleeding) may be similar to those of a ruptured ectopic pregnancy. The presence of an ectopic pregnancy may have been missed even if the patient underwent ultrasonography prior to being prescribed Mifepristone tablets, 200 mg.
Patients who became pregnant with an IUD in place should be assessed for ectopic pregnancy.
6. Adverse Reactions/Side Effects
The following adverse reactions are described in greater detail in other sections:
- Infection and sepsis [see Warnings and Precautions ( 5.1)]
- Uterine bleeding [see Warnings and Precautions ( 5.2)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
Information presented on common adverse reactions relies solely on data from US studies, because rates reported in non-US studies were markedly lower and are not likely generalizable to the US population. In three US clinical studies totaling 1,248 women through 70 days gestation who used mifepristone 200 mg orally followed 24-48 hours later by misoprostol 800mcg buccally, women reported adverse reactions in diaries and in interviews at the follow-up visit. These studies enrolled generally healthy women of reproductive age without contraindications to mifepristone or misoprostol use according to the Mifepristone tablets, 200 mg product label.Gestational age was assessed prior to study enrollment using the date of the woman's last menstrual period, clinical evaluation, and/or ultrasound examination.
About 85% of patients report at least one adverse reaction following administration of Mifepristone tablets, 200 mg and misoprostol, and many can be expected to report more than one such reaction. The most commonly reported adverse reactions (>15%) were nausea, weakness, fever/chills, vomiting, headache, diarrhea, and dizziness (see Table 1). The frequency of adverse reactions varies between studies and may be dependent on many factors including the patient population and gestational age.
Abdominal pain/cramping is expected in all medical abortion patients and its incidence is not reported in clinical studies. Treatment with Mifepristone tablets, 200 mg and misoprostol is designed to induce uterine bleeding and cramping to cause termination of an intrauterine pregnancy. Uterine bleeding and cramping are expected consequences of the action of Mifepristone tablets, 200 mg and misoprostol as used in the treatment procedure. Most patients can expect bleeding more heavily than they do during a heavy menstrual period [see Warnings and Precautions ( 5.2) ].
Table 1 lists the adverse reactions reported in US clinical studies with incidence >15% of women.
Table 1 Adverse Reactions Reported in Women Following Administration of Mifepristone (oral) and Misoprostol (buccal) in US Clinical Studies
| Adverse Reaction | # US
studies | Number of Evaluable
Women | Range of
frequency (%) | Upper Gestational Age of Studies
Reporting Outcome |
| Nausea | 3 | 1,248 | 51-75% | 70 days |
| Weakness | 2 | 630 | 55-58% | 63 days |
| Fever/chills | 1 | 414 | 48% | 63 days |
| Vomiting | 3 | 1,248 | 37-48% | 70 days |
| Headache | 2 | 630 | 41-44% | 63 days |
| Diarrhea | 3 | 1,248 | 18-43% | 70 days |
| Dizziness | 2 | 630 | 39-41% | 63 days |
One study provided gestational-age stratified adverse reaction rates for women who were 57-63 and 64-70 days; there was little difference in frequency of the reported common adverse reactions by gestational age.
Information on serious adverse reactions was reported in six US and four non-US clinical studies, totaling 30,966 women through 70 days gestation who used mifepristone 200mg orally followed 24-48 hours later by misoprostol 800mcg buccally. Serious adverse reaction rates were similar between US and non-US studies, so rates from both US and non-US studies are presented. In the US studies, one studied women through 56 days gestation, four through 63 days gestation, and one through 70 days gestation, while in the non-US studies, two studied women through 63 days gestation, and two through 70 days gestation. Serious adverse reactions were reported in <0.5% of women. Information from the US and non-US studies is presented in Table 2.
Table 2 Serious Adverse Reactions Reported in Women Following Administration of Mifepristone (oral) and Misoprostol (buccal) in US and Non-US Clinical Studies
|
NR= Not reported *This outcome represents a single patient who experienced death related to sepsis. |
||||||
| Adverse
Reaction | US | Non-US | ||||
| # of studies | Number of Evaluable Women | Range of
frequency (%) | # of
studies | Number of
Evaluable Women | Range of
frequency (%) |
|
| Transfusion | 4 | 17,774 | 0.03-0.5% | 3 | 12,134 | 0-0.1% |
| Sepsis | 1 | 629 | 0.2% | 1 | 11,155 | <0.01% * |
| ER visit | 2 | 1,043 | 2.9-4.6% | 1 | 95 | 0 |
| Hospitalization
Related to Medical Abortion | 3 | 14,339 | 0.04-0.6% | 3 | 1,286 | 0-0.7% |
| Infection without
sepsis | 1 | 216 | 0 | 1 | 11,155 | 0.2% |
| Hemorrhage | NR | NR | NR | 1 | 11,155 | 0.1% |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of Mifepristone tablets, 200 mg and misoprostol. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Infections and infestations: post-abortal infection (including endometritis, endomyometritis, parametritis, pelvic infection, pelvic inflammatory disease, salpingitis)
Blood and the lymphatic system disorders: anemia
Immune system disorders: allergic reaction (including anaphylaxis, angioedema, hives, rash, itching)
Psychiatric disorders: anxiety
Cardiac disorders: tachycardia (including racing pulse, heart palpitations, heart pounding)
Vascular disorders: syncope, fainting, loss of consciousness, hypotension (including orthostatic), light-headedness
Respiratory, thoracic and mediastinal disorders: shortness of breath
Gastrointestinal disorders: dyspepsia
Musculoskeletal, connective tissue and bone disorders: back pain, leg pain
Reproductive system and breast disorders: uterine rupture, ruptured ectopic pregnancy, hematometra, leukorrhea
General disorders and administration site conditions: pain
7. Drug Interactions
7.1 Drugs that May Reduce Mifepristone tablets, 200 mg Exposure (Effect of CYP 3A4 Inducers on Mifepristone tablets, 200mg)
CYP450 3A4 is primarily responsible for the metabolism of mifepristone. CYP3A4 inducers such as rifampin, dexamethasone, St. John's Wort, and certain anticonvulsants (such as phenytoin, phenobarbital, carbamazepine) may induce mifepristone metabolism (lowering serum concentrations of mifepristone). Whether this action has an impact on the efficacy of the dose regimen is unknown. Refer to the follow-up assessment [see Dosage and Administration ( 2.3)] to verify that treatment has been successful.
7.2 Drugs that May Increase Mifepristone tablets, 200 mg Exposure (Effect of CYP 3A4 Inhibitors on Mifepristone tablets, 200mg)
Although specific drug or food interactions with mifepristone have not been studied, on the basis of this drug's metabolism by CYP 3A4, it is possible that ketoconazole, itraconazole erythromycin, and grapefruit juice may inhibit its metabolism (increasing serum concentrations of mifepristone). Mifepristone tablets, 200mg should be used with caution in patients currently or recently treated with CYP 3A4 inhibitors.
7.3 Effects of Mifepristone tablets, 200 mg on Other Drugs (Effect of Mifepristone tablets, 200 mg on CYP 3A4Substrates)
Based on in vitro inhibition information, coadministration of mifepristone may lead to an increase in serum concentrations of drugs that are CYP 3A4 substrates. Due to the slow elimination of mifepristone from the body, such interaction may be observed for a prolonged period after its administration. Therefore, caution should be exercised when mifepristone is administered with drugs that are CYP 3A4 substrates and have a narrow therapeutic range.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Mifepristone is indicated, in a regimen with misoprostol, for the medical termination of intrauterine pregnancy through 70 days gestation. Risks to pregnant patients are discussed throughout the labeling.
Refer to misoprostol labeling for risks to pregnant patients with the use of misoprostol.
The risk of adverse developmental outcomes with a continued pregnancy after a failed pregnancy termination with Mifepristone tablets, 200 mg in a regimen with misoprostol is unknown; however, the process of a failed pregnancy termination could disrupt normal embryo-fetal development and result in adverse developmental effects. Birth defects have been reported with a continued pregnancy after a failed pregnancy termination with Mifepristone tablets, 200 mg in a regimen with misoprostol. In animal reproduction studies, increased fetal losses were observed in mice, rats, and rabbits and skull deformities were observed in rabbits with administration of mifepristone at doses lower than the human exposure level based on body surface area.
Data
Animal Data
In teratology studies in mice, rats and rabbits at doses of 0.25 to 4.0mg/kg (less than 1/100 to approximately 1/3 the human exposure based on body surface area), because of the antiprogestational activity of mifepristone, fetal losses were much higher than in control animals. Skull deformities were detected in rabbit studies at approximately 1/6 the human exposure, although no teratogenic effects of mifepristone have been observed to date in rats or mice.
These deformities were most likely due to the mechanical effects of uterine contractions resulting from inhibition of progesterone action.
8.2 Lactation
Mifepristone tablets, 200 mg is present in human milk. Limited data demonstrate undetectable to low levels of the drug in human milk with the relative (weight-adjusted) infant dose 0.5% or less as compared to maternal dosing. There is no information on the effects of Mifepristone tablets, 200 use of misoprostol. The developmental and health benefits of breast-feeding should be considered along with any potential adverse effects on the breast-fed child from Mifepristone tablets, 200 mg in a regimen with misoprostol.
8.4 Pediatric Use
Safety and efficacy of Mifepristone tablets, 200 mg have been established in pregnant females. Data from a clinical study of Mifepristone tablets, 200 mg that included a subset of 322 females under age 17 demonstrated a safety and efficacy profile similar to that observed in adults.
10. Overdosage
No serious adverse reactions were reported in tolerance studies in healthy non-pregnant female and healthy male subjects where mifepristone was administered in single doses greater than 1,800 mg (ninefold the recommended dose for medical abortion). If a patient ingests a massive overdose, the patient should be observed closely for signs of adrenal failure.
11. MPM Pak Description
Mifepristone tablets each contain 200 mg of mifepristone, a synthetic steroid with antiprogestational effects. The tablets are light yellow in color, circular, bi-convex, and are intended for oral administration only. The tablets include the inactive ingredients colloidal silicon dioxide, corn starch, povidone, microcrystalline cellulose, and magnesium stearate.
Mifepristone is a substituted 19-nor steroid compound chemically designated as 11ß-[ p- (Dimethylamino) phenyl]-17ß-hydroxy- 17-(1-propynyl) estra-4,9-dien-3-one. Its empirical formula is C 29H 35NO 2. Its structural formula is:
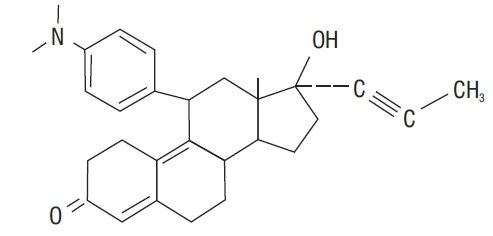
The compound is a yellow powder with a molecular weight of 429.6 and a melting point of 192- 196°C. It is freely soluble in methanol, chloroform and acetone and poorly soluble in water, hexane and isopropyl ether.
12. MPM Pak - Clinical Pharmacology
12.1 Mechanism of Action
The anti-progestational activity of mifepristone results from competitive interaction with progesterone at progesterone-receptor sites. Based on studies with various oral doses in several animal species (mouse, rat, rabbit, and monkey), the compound inhibits the activity of endogenous or exogenous progesterone, resulting in effects on the uterus and cervix that, when combined with misoprostol, result in termination of an intrauterine pregnancy.
During pregnancy, the compound sensitizes the myometrium to the contraction-inducing activity of prostaglandins.
12.2 Pharmacodynamics
Use of Mifepristone tablets, 200 mg in a regimen with misoprostol disrupts pregnancy by causing decidual necrosis, myometrial contractions, and cervical softening, leading to the expulsion of the products of conception.
Doses of 1mg/kg or greater of mifepristone have been shown to antagonize the endometrial and myometrial effects of progesterone in women.
Antiglucocorticoid and antiandrogenic activity: Mifepristone also exhibits antiglucocorticoid and weak antiandrogenic activity. The activity of the glucocorticoid dexamethasone in rats was inhibited following doses of 10 to 25 mg/kg of mifepristone. Doses of 4.5mg/kg or greater in human beings resulted in a compensatory elevation of adrenocorticotropic hormone (ACTH) and cortisol. Antiandrogenic activity was observed in rats following repeated administration of doses from 10 to 100 mg/kg.
12.3 Pharmacokinetics
Mifepristone is rapidly absorbed after oral ingestion with non-linear pharmacokinetics for C max after single oral doses of 200 mg and 600 mg in healthy subjects.
Absorption
The absolute bioavailability of a 20 mg mifepristone oral dose in females of childbearing age is 69%. Following oral administration of a single dose of 600mg, mifepristone is rapidly absorbed, with a peak plasma concentration of 1.98 ± 1.0mg/L occurring approximately 90 minutes after ingestion.
Following oral administration of a single dose of 200 mg in healthy men (n=8), mean C max was1.77 ± 0.7 mg/L occurring approximately 45 minutes after ingestion. Mean AUC0-∞ was 25.8± 6.2 mg*hr/L.
Distribution
Mifepristone is 98% bound to plasma proteins, albumin, and 1-acid glycoprotein. Binding to the latter protein is saturable, and the drug displays nonlinear kinetics with respect to plasma concentration and clearance.
Elimination
Following a distribution phase, elimination of mifepristone is slow at first (50% eliminated between 12 and 72 hours) and then becomes more rapid with a terminal elimination half-life of 18 hours.
Metabolism
Metabolism of mifepristone is primarily via pathways involving N-demethylation and terminal hydroxylation of the 17-propynyl chain.
In vitro studies have shown that CYP450 3A4 is primarily responsible for the metabolism. The three major metabolites identified in humans are: (1) RU 42 633, the most widely found in plasma, is the N-monodemethylated metabolite; (2) RU 42 848, which results from the loss of two methyl groups from the 4-dimethylaminophenyl in position 11ß; and (3) RU 42 698, which results from terminal hydroxylation of the 17-propynyl chain.
Excretion
By 11 days after a 600mg dose of tritiated compound, 83% of the drug has been accounted for by the feces and 9% by the urine. Serum concentrations are undetectable by 11 days.
Specific Populations
The effects of age, hepatic disease and renal disease on the safety, efficacy and pharmacokinetics of mifepristone have not been investigated.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No long-term studies to evaluate the carcinogenic potential of mifepristone have been performed.
Mutagenesis
Results from studies conducted
in vitro and in animals have revealed no genotoxic potential for mifepristone. Among the tests carried out were: Ames test with and without metabolic activation; gene conversion test in
Saccharomyces cerevisiae D4 cells; forward mutation in
Schizosaccharomyces pompe P1 cells; induction of unscheduled DNA synthesis in cultured HeLa cells; induction of chromosome aberrations in CHO cells;
in vitro test for gene mutation in V79 Chinese hamster lung cells; and micronucleus test in mice.
Impairment of Fertility
In rats, administration of 0.3mg/kg mifepristone per day caused severe disruption of the estrus cycles for the three weeks of the treatment period. Following resumption of the estrus cycle, animals were mated and no effects on reproductive performance were observed.
14. Clinical Studies
Safety and efficacy data from clinical studies of mifepristone 200 mg orally followed 24-48 hours later by misoprostol 800 mcg buccally through 70 days gestation are reported below. Success was defined as the complete expulsion of the products of conception without the need for surgical intervention. The overall rates of success and failure, shown by reason for failure based on 22 worldwide clinical studies (including 7 US studies) appear in Table 3.
The demographics of women who participated in the US clinical studies varied depending on study location and represent the racial and ethnic variety of American females. Females of all reproductive ages were represented, including females less than 18 and more than 40 years of age; most were 27 years or younger.
Table 3 Outcome Following Treatment with Mifepristone (oral) and Misoprostol (buccal) Through 70 Days Gestation
| US Trials | Non-US Trials | |
| N | 16,794 | 18,425 |
| Complete Medical Abortion | 97.4% | 96.2% |
| Surgical Intervention * | 2.6% | 3.8% |
| Ongoing Pregnancy ** | 0.7% | 0.9% |
| * Reasons for surgical intervention include ongoing pregnancy, medical necessity, persistent or heavy bleeding after treatment, patient request, or incomplete expulsion.
** Ongoing pregnancy is a subcategory of surgical intervention, indicating the percent of women who have surgical intervention due to an ongoing pregnancy. |
||
The results for clinical studies that reported outcomes, including failure rates for ongoing pregnancy, by gestational age are presented in Table 4.
Table 4 Outcome by Gestational Age Following Treatment with Mifepristone and Misoprostol (buccal) for US and Non-US Clinical Studies
| <49 days | 50-56 days | 57-63 days | 64-70 days | |||||||||
| N | % | Number of
Evaluable Studies | N | % | Number of
Evaluable Studies | N | % | Number of
Evaluable Studies | N | % | Number of
Evaluable Studies |
|
| Complete
medical abortion | 12,046 | 98.1 | 10 | 3,941 | 96.8 | 7 | 2,294 | 94.7 | 9 | 479 | 92.7 | 4 |
| Surgical
intervention for ongoing pregnancy | 10,272 | 0.3 | 6 | 3,788 | 0.8 | 6 | 2,211 | 2 | 8 | 453 | 3.1 | 3 |
One clinical study asked subjects through 70 days gestation to estimate when they expelled the pregnancy, with 70% providing data. Of these, 23-38% reported expulsion within 3 hours and over 90% within 24 hours of using misoprostol.
16. How is MPM Pak supplied
Mifepristone tablets, 200 mg is only available through a restricted program called the mifepristone REMS Program [see Warnings and Precautions ( 5.3)]
Mifepristone tablets, 200 mg is supplied as light yellow, circular, bi-convex, uncoated tablets debossed with “S” on one side and plain on other side. Each tablet contains 200 mg of mifepristone. One tablet is individually blistered on one blister card that is packaged in an individual package (National Drug Code 43393-001-01).
Store at 25°C (77°F); excursions permitted to 15 to 30°C (59 to 86°F) [see USP Controlled Room Temperature].
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide), included with each package of Mifepristone tablets, 200 mg. Additional copies of the Medication Guide are available by contacting GenBioPro, Inc., at 1-855-MIFEINFO (1-855-643-3463) or from www.MIFEINFO.com.
Serious Infections and Bleeding
- Inform the patient that uterine bleeding and uterine cramping will occur [see Warnings and Precautions ( 5.2)].
- Advise the patient that serious and sometimes fatal infections and bleeding can occur very rarely [see Warnings and Precautions ( 5.1, 5.2)].
- Mifepristone tablets, 200 mg are only available through a restricted program called the mifepristone REMS Program [see
Warnings and Precautions (
5.3)]. Under the mifepristone REMS Program:
- Patients must sign a Patient Agreement Form.
- Mifepristone tablets, 200mg is only available by or under the supervision of certified prescribers, or by certified pharmacies on prescriptions issued by certified prescribers.
Provider Contacts and Actions in Case of Complications
- Ensure that the patient knows whom to call and what to do, including going to an emergency room if none of the provided contacts are reachable, or if the patient experiences complications including prolonged heavy bleeding, severe abdominal pain, or sustained fever [see Boxed Warning].
Compliance with Treatment Schedule and Follow-up Assessment
- Advise the patient that it is necessary to complete the treatment schedule; including a follow-up assessment approximately 7 to14 days after taking Mifepristone tablets, 200mg [see Dosage and Administration ( 2.3)].
- Explain that:
- prolonged heavy vaginal bleeding is not proof of a complete abortion,
- if the treatment fails and the pregnancy continues, the risk of fetal malformation is unknown,
- it is recommended that ongoing pregnancy be managed by surgical termination [see Dosage and Administration ( 2.3)] . Advise the patient whether you will provide such care or will refer her to another provider.
Subsequent Fertility
- Inform the patient that another pregnancy can occur following medical abortion and before resumption of normal menses.
- Inform the patient that contraception can be initiated as soon as pregnancy expulsion has been confirmed, or before she resumes sexual intercourse.
Manufactured for:
GenBioPro, Inc.
P.O. Box 32011
Las Vegas, NV 89103
1-855-MIFEINFO (1-855-643-3463)
www.MIFEINFO.com
PI.MIF.R3 01/2023
MEDICATION GUIDE
Mifepristone (MIF-eh-pris-tone) tablets, 200 mg for oral use
Read this information carefully before taking Mifepristone tablets, 200 mg and misoprostol. It will help you understand how the treatment works. This Medication Guide does not take the place of talking with your healthcare provider.
What is the most important information I should know about Mifepristone tablets, 200 mg?
What symptoms should I be concerned with? Although cramping and bleeding are an expected part of ending a pregnancy, rarely, serious and potentially life-threatening bleeding, infections, or other problems can occur following a miscarriage, surgical abortion, medical abortion, or childbirth. Seeking medical attention as soon as possible is needed in these circumstances. Serious infection has resulted in death in a very small number of cases. There is no information that use of Mifepristone tablets, 200mg and misoprostol caused these deaths. If you have any questions, concerns, or problems, or if you are worried about any side effects or symptoms, you should contact your healthcare provider. You can write down your healthcare provider's telephone number here _______________________.
Be sure to contact your healthcare provider promptly if you have any of the following:
- Heavy Bleeding. Contact your healthcare provider right away if you bleed enough to soak through two thick full-size sanitary pads per hour for two consecutive hours or if you are concerned about heavy bleeding. In about 1 out of 100 women, bleeding can be so heavy that it requires a surgical procedure (surgical aspiration or D&C).
- Abdominal Pain or “Feeling Sick.” If you have abdominal pain or discomfort, or you are “feeling sick,” including weakness, nausea, vomiting, or diarrhea, with or without fever, more than 24 hours after taking misoprostol, you should contact your healthcare provider without delay. These symptoms may be a sign of a serious infection or another problem (including an ectopic pregnancy, a pregnancy outside the womb).
- Fever. In the days after treatment, if you have a fever of 100.4°F or higher that lasts for more than 4 hours, you should contact your healthcare provider right away. Fever may be a symptom of a serious infection or another problem.
If you cannot reach your healthcare provider, go to the nearest hospital emergency room.
What to do if you are still pregnant after Mifepristone tablets, 200 mg with misoprostol treatment. If you are still pregnant, your healthcare provider will talk with you about a surgical procedure to end your pregnancy. In many cases, this surgical procedure can be done in the office/clinic. The chance of birth defects if the pregnancy is not ended is unknown.
Talk with your healthcare provider. Before you take Mifepristone tablets, 200 mg you should read this Medication Guide and you and your healthcare provider should discuss the benefits and risks of your using Mifepristone tablets, 200 mg.
What is Mifepristone tablets, 200 mg?
Mifepristone tablets, 200 mg is used in a regimen with another prescription medicine called misoprostol, to end an early pregnancy. Early pregnancy means it is 70 days (10 weeks) or less since your last menstrual period began. Mifepristone tablets, 200 mg is not approved for ending pregnancies that are further along. Mifepristone tablets, 200 mg blocks a hormone needed for your pregnancy to continue. When you use Mifepristone tablets, 200 mg on Day 1, you also need to take another medicine called misoprostol 24 to 48 hours after you take Mifepristone tablets, 200 mg to cause the pregnancy to be passed from your uterus.
The pregnancy is likely to be passed from your uterus within 2 to 24 hours after taking Mifepristone tablets, 200 mg and misoprostol. When the pregnancy is passed from the uterus, you will have bleeding and cramping that will likely be heavier than your usual period. About 2 to 7 out of 100 women taking Mifepristone tablets, 200 mg will need a surgical procedure because the pregnancy did not completely pass from the uterus or to stop bleeding.
Who should not take Mifepristone tablets, 200 mg?
Some patients should not take Mifepristone tablets, 200 mg. Do not take Mifepristone tablets, 200 mg if you:
- Have a pregnancy that is more than 70 days (10 weeks). Your healthcare provider may do a clinical examination, an ultrasound examination, or other testing to determine how far along you are in pregnancy.
- Are using an IUD (intrauterine device or system). It must be taken out before you take Mifepristone tablets, 200 mg.
- Have been told by your healthcare provider that you have a pregnancy outside the uterus (ectopic pregnancy).
- Have problems with your adrenal glands (chronic adrenal failure).
- Take a medicine to thin your blood.
- Have a bleeding problem.
- Have porphyria.
- Take certain steroid medicines.
- Are allergic to mifepristone, misoprostol, or medicines that contain misoprostol, such as Cytotec or Arthrotec.
Ask your healthcare provider if you are not sure about all your medical conditions before taking this medicine to find out if you can take Mifepristone tablets, 200 mg.
What should I tell my healthcare provider before taking Mifepristone tablets, 200 mg? Before you take Mifepristone tablets, 200 mg, tell your healthcare provider if you:
- cannot follow-up within approximately 7 to 14 days of your first visit
- are breastfeeding. Mifepristone tablets, 200 mg can pass into your breast milk. The effect of the Mifepristone tablets, 200 mg and misoprostol regimen on the breastfed infant or on milk production is unknown.
- are taking medicines, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Mifepristone tablets, 200 mg and certain other medicines may affect each other if they are used together. This can cause side effects.
How should I take Mifepristone tablets, 200 mg?
- Mifepristone tablets, 200 mg will be given to you by a healthcare provider or pharmacy.
- You and your healthcare provider will plan the most appropriate location for you to take the misoprostol, because it may cause bleeding, cramps, nausea, diarrhea, and other symptoms that usually begin within 2 to 24 hours after taking it.
- Most women will pass the pregnancy within 2 to 24 hours after taking the misoprostol tablets.
Follow the instruction below on how to take Mifepristone tablets, 200 mg and misoprostol: Mifepristone tablets, 200 mg (1 tablet) orally + misoprostol (4 tablets) buccally
Day 1:
- Take 1 Mifepristone 200 mg tablet by mouth.
24 to 48 hours after taking Mifepristone tablets, 200 mg:
- Take 4 misoprostol tablets by placing 2 misoprostol tablets in each cheek pouch (the area between your teeth and cheek - see Figure A) for 30 minutes and then swallow anything left over with a drink of water or another liquid.
- The medicines may not work as well if you take misoprostol sooner than 24 hours after Mifepristone tablets, 200mg or later than 48 hours after Mifepristone tablets, 200mg.
- Misoprostol often causes cramps, nausea, diarrhea, and other symptoms. Your healthcare provider may send you home with medicines for these symptoms.
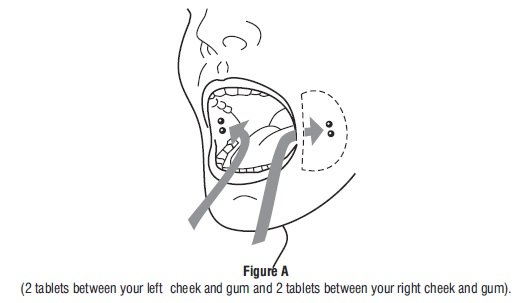
Follow-up Assessment at Day 7 to 14:
- This follow-up assessment is very important. You must follow-up with your healthcare provider about 7 to 14 days after you have taken Mifepristone tablets, 200 mg to be sure you are well and that you have had bleeding and the pregnancy has passed from your uterus.
- Your healthcare provider will assess whether your pregnancy has passed from your uterus. If your pregnancy continues, the chance that there may be birth defects is unknown. If you are still pregnant, your healthcare provider will talk with you about a surgical procedure to end your pregnancy.
- If your pregnancy has ended, but not yet completely passed from your uterus, your provider will talk with you about other choices you have, including waiting, taking another dose of misoprostol, or having surgical procedure to empty your uterus.
When should I begin birth control?
You can become pregnant again right after your pregnancy ends. If you do not want to become pregnant again, start using birth control as soon as your pregnancy ends or before you start having sexual intercourse again.
What should I avoid while taking Mifepristone tablets, 200 mg and misoprostol?
Do not take any other prescription or over-the-counter medicines (including herbal medicines or supplements) at any time during the treatment period without first asking your healthcare provider about them because they may interfere with the treatment. Ask your healthcare provider about what medicines you can take for pain and other side effects.
What are the possible side effects of Mifepristone tablets, 200 mg and misoprostol?
Mifepristone tablets, 200 mg may cause serious side effects. See “What is the most important information I should know about Mifepristone tablets, 200 mg?”
Cramping and bleeding. Cramping and vaginal bleeding are expected with this treatment. Usually, these symptoms mean that the treatment is working. But sometimes you can get cramping and bleeding and still be pregnant. This is why you must follow-up with your healthcare provider approximately 7 to 14 days after taking Mifepristone tablets, 200 mg. See “How should I take Mifepristone tablets, 200 mg?” for more information on your follow-up assessment. If you are not already bleeding after taking Mifepristone tablets, 200 mg you probably will begin to bleed once you take misoprostol, the medicine you take 24 to 48 hours after Mifepristone tablets, 200 mg. Bleeding or spotting can be expected for an average of 9 to16 days and may last for up to 30 days. Your bleeding may be similar to, or greater than, a normal heavy period. You may see blood clots and tissue. This is an expected part of passing the pregnancy.
The most common side effects of Mifepristone tablets, 200 mg treatment include: nausea, weakness, fever/chills, vomiting, headache, diarrhea and dizziness. Your provider will tell you how to manage any pain or other side effects. These are not all the possible side effects of Mifepristone tablets, 200 mg.
Call your healthcare provider for medical advice about any side effects that bother you or do not go away. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of Mifepristone tablets, 200 mg.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. This Medication Guide summarizes the most important information about Mifepristone tablets, 200 mg. If you would like more information, talk with your healthcare provider. You may ask your healthcare provider for information about Mifepristone tablets, 200 mg that is written for healthcare professionals. For more information about Mifepristone tablets, 200 mg, go to www.MIFEINFO.com or call 1- 855-MIFEINFO (1- 855-643-3463).
Manufactured for:
GenBioPro, Inc.
P.O. Box 32011
Las Vegas, NV 89103
1-855-MIFEINFO (1-855-643-3463)
www.MIFEINFO.com
This Medication Guide has been approved by the U.S. Food and Drug Administration.
MG.MIF.R3
Approved 01/2023
WARNING
MISOPROSTOL ADMINISTRATION TO WOMEN WHO ARE PREGNANT CAN CAUSE BIRTH DEFECTS, ABORTION, OR PREMATURE BIRTH. UTERINE RUPTURE HAS BEEN REPORTED WHEN MISOPROSTOL TABLETS WERE ADMINISTERED IN PREGNANT WOMEN TO INDUCE LABOR OR TO INDUCE ABORTION BEYOND THE EIGHTH WEEK OF PREGNANCY (see also PRECAUTIONS and LABOR AND DELIVERY). MISOPROSTOL TABLETS SHOULD NOT BE TAKEN BY PREGNANT WOMEN TO REDUCE THE RISK OF ULCERS INDUCED BY NONSTEROIDAL ANTI-INFLAMMATORY DRUGS (NSAIDs) (see CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS).
PATIENTS MUST BE ADVISED OF THE ABORTIFACIENT PROPERTY AND WARNED NOT TO GIVE THE DRUG TO OTHERS.
Misoprostol Tablets should not be used for reducing the risk of NSAID-induced ulcers in women of childbearing potential unless the patient is at high risk of complications from gastric ulcers associated with use of the NSAID, or is at high risk of developing gastric ulceration. In such patients, Misoprostol Tablets may be prescribed if the patient
- has had a negative serum pregnancy test within 2 weeks prior to beginning therapy.
- is capable of complying with effective contraceptive measures.
- has received both oral and written warnings of the hazards of Misoprostol Tablets, the risk of possible contraception failure, and the danger to other women of childbearing potential should the drug be taken by mistake.
- will begin Misoprostol Tablets only on the second or third day of the next normal menstrual period.
MPM Pak Description
Misoprostol oral tablets contain either 100 mcg or 200 mcg of misoprostol, a synthetic prostaglandin E1 analog.
Misoprostol contains approximately equal amounts of the two diastereomers presented below with their enantiomers indicated by (±):
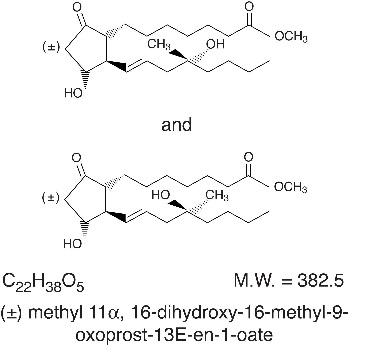
Misoprostol is a water-soluble, viscous liquid.
Inactive ingredients of tablets are hydrogenated castor oil, microcrystalline cellulose, and crospovidone
MPM Pak - Clinical Pharmacology
Pharmacokinetics
Misoprostol is extensively absorbed, and undergoes rapid de-esterification to its free acid, which is responsible for its clinical activity and, unlike the parent compound, is detectable in plasma. The alpha side chain undergoes beta oxidation and the beta side chain undergoes omega oxidation followed by reduction of the ketone to give prostaglandin F analogs.
In normal volunteers, misoprostol is rapidly absorbed after oral administration with a T max of misoprostol acid of 12 ± 3 minutes and a terminal half-life of 20–40 minutes.
There is high variability of plasma levels of misoprostol acid between and within studies but mean values after single doses show a linear relationship with dose over the range of 200–400 mcg. No accumulation of misoprostol acid was noted in multiple dose studies; plasma steady state was achieved within two days.
Maximum plasma concentrations of misoprostol acid are diminished when the dose is taken with food and total availability of misoprostol acid is reduced by use of concomitant antacid. Clinical trials were conducted with concomitant antacid, however, so this effect does not appear to be clinically important.
| Mean ± SD
| C
max (pg/ml)
| AUC(0–4) (pg·hr/ml)
| T
max (min)
|
| Fasting
| 811 ± 317
| 417 ± 135
| 14 ± 8
|
| With Antacid
| 689 ± 315
| 349 ± 108*
| 20 ± 14
|
| With High Fat Breakfast
| 303 ± 176*
| 373 ± 111
| 64 ± 79*
|
*Comparisons with fasting results statistically significant, p<0.05.
After oral administration of radiolabeled misoprostol, about 80% of detected radioactivity appears in urine. Pharmacokinetic studies in patients with varying degrees of renal impairment showed an approximate doubling of T 1/2, C max, and AUC compared to normals, but no clear correlation between the degree of impairment and AUC. In subjects over 64 years of age, the AUC for misoprostol acid is increased. No routine dosage adjustment is recommended in older patients or patients with renal impairment, but dosage may need to be reduced if the usual dose is not tolerated.
Drug interaction studies between misoprostol and several nonsteroidal anti-inflammatory drugs showed no effect on the kinetics of ibuprofen or diclofenac, and a 20% decrease in aspirin AUC, not thought to be clinically significant.
Pharmacokinetic studies also showed a lack of drug interaction with antipyrine and propranolol when these drugs were given with misoprostol. Misoprostol given for 1 week had no effect on the steady state pharmacokinetics of diazepam when the two drugs were administered 2 hours apart.
The serum protein binding of misoprostol acid is less than 90% and is concentration-independent in the therapeutic range.
After a single oral dose of misoprostol to nursing mothers, misoprostol acid was excreted in breast milk. The maximum concentration of misoprostol acid in expressed breast milk was achieved within 1 hour after dosing and was 7.6 pg/ml (CV 37%) and 20.9 pg/ml (CV 62%) after single 200 mcg and 600 mcg misoprostol administration, respectively. The misoprostol acid concentrations in breast milk declined to < 1 pg/ml at 5 hours post-dose.
Pharmacodynamics
Misoprostol has both antisecretory (inhibiting gastric acid secretion) and (in animals) mucosal protective properties. NSAIDs inhibit prostaglandin synthesis, and a deficiency of prostaglandins within the gastric mucosa may lead to diminishing bicarbonate and mucus secretion and may contribute to the mucosal damage caused by these agents. Misoprostol can increase bicarbonate and mucus production, but in man this has been shown at doses 200 mcg and above that are also antisecretory. It is therefore not possible to tell whether the ability of misoprostol to reduce the risk of gastric ulcer is the result of its antisecretory effect, its mucosal protective effect, or both.
In vitro studies on canine parietal cells using tritiated misoprostol acid as the ligand have led to the identification and characterization of specific prostaglandin receptors. Receptor binding is saturable, reversible, and stereospecific. The sites have a high affinity for misoprostol, for its acid metabolite, and for other E type prostaglandins, but not for F or I prostaglandins and other unrelated compounds, such as histamine or cimetidine. Receptor-site affinity for misoprostol correlates well with an indirect index of antisecretory activity. It is likely that these specific receptors allow misoprostol taken with food to be effective topically, despite the lower serum concentrations attained.
Misoprostol produces a moderate decrease in pepsin concentration during basal conditions, but not during histamine stimulation. It has no significant effect on fasting or postprandial gastrin nor on intrinsic factor output.
Effects on gastric acid secretion
Misoprostol, over the range of 50–200 mcg, inhibits basal and nocturnal gastric acid secretion, and acid secretion in response to a variety of stimuli, including meals, histamine, pentagastrin, and coffee. Activity is apparent 30 minutes after oral administration and persists for at least 3 hours. In general, the effects of 50 mcg were modest and shorter lived, and only the 200-mcg dose had substantial effects on nocturnal secretion or on histamine and meal-stimulated secretion.
Uterine effects
Misoprostol has been shown to produce uterine contractions that may endanger pregnancy. (See boxed WARNINGS.)
Other pharmacologic effects
Misoprostol does not produce clinically significant effects on serum levels of prolactin, gonadotropins, thyroid-stimulating hormone, growth hormone, thyroxine, cortisol, gastrointestinal hormones (somatostatin, gastrin, vasoactive intestinal polypeptide, and motilin), creatinine, or uric acid. Gastric emptying, immunologic competence, platelet aggregation, pulmonary function, or the cardiovascular system are not modified by recommended doses of misoprostol.
Clinical studies
In a series of small short-term (about 1 week) placebo-controlled studies in healthy human volunteers, doses of misoprostol were evaluated for their ability to reduce the risk of NSAID-induced mucosal injury. Studies of 200 mcg q.i.d. of misoprostol with tolmetin and naproxen, and of 100 and 200 mcg q.i.d. with ibuprofen, all showed reduction of the rate of significant endoscopic injury from about 70–75% on placebo to 10–30% on misoprostol. Doses of 25–200 mcg q.i.d. reduced aspirin-induced mucosal injury and bleeding.
Reducing the risk of gastric ulcers caused by nonsteroidal anti-inflammatory drugs (NSAIDs)
Two 12-week, randomized, double-blind trials in osteoarthritic patients who had gastrointestinal symptoms but no ulcer on endoscopy while taking an NSAID compared the ability of 200 mcg of misoprostol, 100 mcg of misoprostol, and placebo to reduce the risk of gastric ulcer (GU) formation. Patients were approximately equally divided between ibuprofen, piroxicam, and naproxen, and continued this treatment throughout the 12 weeks. The 200-mcg dose caused a marked, statistically significant reduction in gastric ulcers in both studies. The lower dose was somewhat less effective, with a significant result in only one of the studies.
Reduction of Risk of Gastric Ulcers Induced by Ibuprofen, Piroxicam, or Naproxen
[No. of patients with ulcer(s) (%)]
| Therapy
| Therapy Duration
|
|
||
| 4 weeks
| 8 weeks
| 12 weeks
|
||
| Study No. 1
|
||||
| Misoprostol 200 mcg
q.i.d. (n=74) | 1 (1.4)
| 0
| 0
| 1 (1.4)*
|
| Misoprostol 100 mcg
q.i.d. (n=77) | 3 (3.9)
| 1 (1.3)
| 1 (1.3)
| 5 (6.5)*
|
| Placebo (n=76)
| 11 (14.5)
| 4 (5.3)
| 4 (5.3)
| 19 (25.0)
|
| Study No. 2
|
||||
| Misoprostol 200 mcg
q.i.d. (n=65) | 1 (1.5)
| 1 (1.5)
| 0
| 2 (3.1)*
|
| Misoprostol 100 mcg
q.i.d. (n=66) | 2 (3.0)
| 2 (3.0)
| 1 (1.5)
| 5 (7.6)
|
| Placebo (n=62)
| 6 (9.7)
| 2 (3.2)
| 3 (4.8)
| 11 (17.7)
|
| Studies No. 1 & No. 2**
|
||||
| Misoprostol 200 mcg
q.i.d. (n=139) | 2 (1.4)
| 1 (0.7)
| 0
| 3 (2.2)*
|
| Misoprostol 100 mcg
q.i.d. (n=143) | 5 (3.5)
| 3 (2.1)
| 2 (1.4)
| 10 (7.0)*
|
| Placebo (n=138)
| 17 (12.3)
| 6 (4.3)
| 7 (5.1)
| 30 (21.7)
|
* Statistically significantly different from placebo at the 5% level.
** Combined data from Study No. 1 and Study No. 2.
In these trials there were no significant differences between misoprostol and placebo in relief of day or night abdominal pain. No effect of misoprostol in reducing the risk of duodenal ulcers was demonstrated, but relatively few duodenal lesions were seen.
In another clinical trial, 239 patients receiving aspirin 650–1300 mg q.i.d. for rheumatoid arthritis who had endoscopic evidence of duodenal and/or gastric inflammation were randomized to misoprostol 200 mcg q.i.d. or placebo for 8 weeks while continuing to receive aspirin. The study evaluated the possible interference of misoprostol on the efficacy of aspirin in these patients with rheumatoid arthritis by analyzing joint tenderness, joint swelling, physician's clinical assessment, patient's assessment, change in ARA classification, change in handgrip strength, change in duration of morning stiffness, patient's assessment of pain at rest, movement, interference with daily activity, and ESR. Misoprostol did not interfere with the efficacy of aspirin in these patients with rheumatoid arthritis.
Indications and Usage for MPM Pak
Misoprostol is indicated for reducing the risk of NSAID (nonsteroidal anti-inflammatory drugs, including aspirin)–induced gastric ulcers in patients at high risk of complications from gastric ulcer, e.g., the elderly and patients with concomitant debilitating disease, as well as patients at high risk of developing gastric ulceration, such as patients with a history of ulcer. Misoprostol Tablet has not been shown to reduce the risk of duodenal ulcers in patients taking NSAIDs. Misoprostol Tablets should be taken for the duration of NSAID therapy. Misoprostol Tablets has been shown to reduce the risk of gastric ulcers in controlled studies of 3 months' duration. It had no effect, compared to placebo, on gastrointestinal pain or discomfort associated with NSAID use.
Contraindications
See boxed WARNINGS.
Misoprostol Tablets should not be taken by pregnant women to reduce the risk of ulcers induced by nonsteroidal anti-inflammatory drugs (NSAIDs).
Misoprostol Tablets should not be taken by anyone with a history of allergy to prostaglandins.
Warnings
See boxed WARNINGS.
For hospital use only if misoprostol were to be used for cervical ripening, induction of labor, or for the treatment of serious post-partum hemorrhage, which are outside of the approved indication.
Precautions
Caution should be employed when administering misoprostol to patients with pre-existing cardiovascular disease.
Information for Patients
Women of childbearing potential using Misoprostol Tablets to decrease the risk of NSAID-induced ulcers should be told that they must not be pregnant when Misoprostol Tablets therapy is initiated, and that they must use an effective contraception method while taking Misoprostol Tablets.
See boxed WARNINGS.
Misoprostol Tablets is intended for administration along with nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin, to decrease the chance of developing an NSAID-induced gastric ulcer.
Misoprostol Tablets should be taken only according to the directions given by a physician.
If the patient has questions about or problems with Misoprostol Tablets, the physician should be contacted promptly.
THE PATIENT SHOULD NOT GIVE MISOPROSTOL TABLETS TO ANYONE ELSE. Misoprostol Tablets has been prescribed for the patient's specific condition, may not be the correct treatment for another person, and may be dangerous to the other person if she were to become pregnant.
The Misoprostol Tablets package the patient receives from the pharmacist will include a leaflet containing patient information. The patient should read the leaflet before taking Misoprostol Tablets and each time the prescription is renewed because the leaflet may have been revised.
Keep Misoprostol Tablets out of the reach of children.
SPECIAL NOTE FOR WOMEN: Misoprostol Tablets may cause birth defects, abortion (sometimes incomplete), or premature labor if given to pregnant women.
Misoprostol Tablets is available only as a unit-of-use package that includes a leaflet containing patient information. See Patient Information at the end of this labeling.
Drug Interactions
See Clinical Pharmacology. Misoprostol Tablets has not been shown to interfere with the beneficial effects of aspirin on signs and symptoms of rheumatoid arthritis. Misoprostol Tablets does not exert clinically significant effects on the absorption, blood levels, and antiplatelet effects of therapeutic doses of aspirin. Misoprostol Tablets has no clinically significant effect on the kinetics of diclofenac or ibuprofen.
Prostaglandins such as Misoprostol Tablets may augment the activity of oxytocic agents, especially when given less than 4 hours prior to initiating oxytocin treatment. Concomitant use is not recommended.
Animal toxicology
A reversible increase in the number of normal surface gastric epithelial cells occurred in the dog, rat, and mouse. No such increase has been observed in humans administered Misoprostol Tablets for up to 1 year.
An apparent response of the female mouse to Misoprostol Tablets in long-term studies at 100 to 1000 times the human dose was hyperostosis, mainly of the medulla of sternebrae. Hyperostosis did not occur in long-term studies in the dog and rat and has not been seen in humans treated with Misoprostol Tablets.
Carcinogenesis, Mutagenesis, Impairment of Fertility
There was no evidence of an effect of Misoprostol Tablets on tumor occurrence or incidence in rats receiving daily doses up to 150 times the human dose for 24 months. Similarly, there was no effect of Misoprostol Tablets on tumor occurrence or incidence in mice receiving daily doses up to 1000 times the human dose for 21 months. The mutagenic potential of Misoprostol Tablets was tested in several in vitro assays, all of which were negative.
Misoprostol, when administered to breeding male and female rats at doses 6.25 times to 625 times the maximum recommended human therapeutic dose, produced dose-related pre- and post-implantation losses and a significant decrease in the number of live pups born at the highest dose. These findings suggest the possibility of a general adverse effect on fertility in males and females.
Teratogenic Effects
See boxed WARNINGS.
Congenital anomalies sometimes associated with fetal death have been reported subsequent to the unsuccessful use of misoprostol as an abortifacient, but the drug's teratogenic mechanism has not been demonstrated. Several reports in the literature associate the use of misoprostol during the first trimester of pregnancy with skull defects, cranial nerve palsies, facial malformations, and limb defects.
Misoprostol Tablets is not fetotoxic or teratogenic in rats and rabbits at doses 625 and 63 times the human dose, respectively.
Nonteratogenic Effects
See boxed WARNINGS.
Misoprostol Tablets may endanger pregnancy (may cause abortion) and thereby cause harm to the fetus when administered to a pregnant woman. Misoprostol Tablets may produce uterine contractions, uterine bleeding, and expulsion of the products of conception. Abortions caused by Misoprostol Tablets may be incomplete. If a woman is or becomes pregnant while taking this drug to reduce the risk of NSAID-induced ulcers, the drug should be discontinued and the patient apprised of the potential hazard to the fetus.
Labor and Delivery
Misoprostol Tablets can induce or augment uterine contractions. Vaginal administration of Misoprostol Tablets, outside of its approved indication, has been used as a cervical ripening agent, for the induction of labor and for treatment of serious postpartum hemorrhage in the presence of uterine atony. A major adverse effect of the obstetrical use of Misoprostol Tablets is uterine tachysystole which may progress to uterine tetany with marked impairment of uteroplacental blood flow, uterine rupture (requiring surgical repair, hysterectomy, and/or salpingo-oophorectomy), or amniotic fluid embolism and lead to adverse fetal heart changes. Uterine activity and fetal status should be monitored by trained obstetrical personnel in a hospital setting.
The risk of uterine rupture increases with advancing gestational ages and prior uterine surgery, including Cesarean delivery. Grand multiparity also appears to be a risk factor for uterine rupture.
The use of Misoprostol Tablets outside of its approved indication may also be associated with meconium passage, meconium staining of amniotic fluid, and Cesarean delivery. Maternal shock, maternal death, fetal bradycardia, and fetal death have also been reported with the use of misoprostol.
Misoprostol Tablets should not be used in the third trimester in women with a history of Cesarean section or major uterine surgery because of an increased risk of uterine rupture. Misoprostol Tablets should not be used in cases where uterotonic drugs are generally contraindicated or where hyperstimulation of the uterus is considered inappropriate, such as cephalopelvic disproportion, grand multiparity, hypertonic or hyperactive uterine patterns, or fetal distress where delivery is not imminent, or when surgical intervention is more appropriate.
The effect of Misoprostol Tablets on later growth, development, and functional maturation of the child when Misoprostol Tablets is used for cervical ripening or induction of labor has not been established. Information on Misoprostol Tablet's effect on the need for forceps delivery or other intervention is unknown.
Nursing Mothers
Misoprostol is rapidly metabolized in the mother to misoprostol acid, which is biologically active and is excreted in breast milk. There are no published reports of adverse effects of misoprostol in breast-feeding infants of mothers taking misoprostol. Caution should be exercised when misoprostol is administered to a nursing woman.
Adverse Reactions/Side Effects
The following have been reported as adverse events in subjects receiving Misoprostol Tablets:
Gastrointestinal: In subjects receiving Misoprostol Tablets 400 or 800 mcg daily in clinical trials, the most frequent gastrointestinal adverse events were diarrhea and abdominal pain. The incidence of diarrhea at 800 mcg in controlled trials in patients on NSAIDs ranged from 14 to 40% and in all studies (over 5,000 patients) averaged 13%. Abdominal pain occurred in 13 to 20% of patients in NSAID trials and about 7% in all studies, but there was no consistent difference from placebo.
Diarrhea was dose related and usually developed early in the course of therapy (after 13 days), usually was self-limiting (often resolving after 8 days), but sometimes required discontinuation of Misoprostol Tablets (2% of the patients). Rare instances of profound diarrhea leading to severe dehydration have been reported. Patients with an underlying condition such as inflammatory bowel disease, or those in whom dehydration, were it to occur, would be dangerous, should be monitored carefully if Misoprostol Tablets is prescribed. The incidence of diarrhea can be minimized by administering after meals and at bedtime, and by avoiding coadministration of Misoprostol Tablets with magnesium-containing antacids.
Gynecological: Women who received Misoprostol Tablets during clinical trials reported the following gynecological disorders: spotting (0.7%), cramps (0.6%), hypermenorrhea (0.5%), menstrual disorder (0.3%) and dysmenorrhea (0.1%). Postmenopausal vaginal bleeding may be related to Misoprostol Tablets administration. If it occurs, diagnostic workup should be undertaken to rule out gynecological pathology. (See boxed WARNINGS.)
Elderly: There were no significant differences in the safety profile of Misoprostol Tablets in approximately 500 ulcer patients who were 65 years of age or older compared with younger patients.
Additional adverse events which were reported are categorized as follows:
Incidence greater than 1%: In clinical trials, the following adverse reactions were reported by more than 1% of the subjects receiving Misoprostol Tablets and may be causally related to the drug: nausea (3.2%), flatulence (2.9%), headache (2.4%), dyspepsia (2.0%), vomiting (1.3%), and constipation (1.1%). However, there were no significant differences between the incidences of these events for Misoprostol Tablets and placebo.
Causal relationship unknown: The following adverse events were infrequently reported. Causal relationships between Misoprostol Tablets and these events have not been established but cannot be excluded:
Body as a whole: aches/pains, asthenia, fatigue, fever, chills, rigors, weight changes.
Skin: rash, dermatitis, alopecia, pallor, breast pain.
Special senses: abnormal taste, abnormal vision, conjunctivitis, deafness, tinnitus, earache.
Respiratory: upper respiratory tract infection, bronchitis, bronchospasm, dyspnea, pneumonia, epistaxis.
Cardiovascular: chest pain, edema, diaphoresis, hypotension, hypertension, arrhythmia, phlebitis, increased cardiac enzymes, syncope, myocardial infarction (some fatal), thromboembolic events (e.g., pulmonary embolism, arterial thrombosis, and CVA).
Gastrointestinal: GI bleeding, GI inflammation/infection, rectal disorder, abnormal hepatobiliary function, gingivitis, reflux, dysphagia, amylase increase.
Hypersensitivity: anaphylactic reaction
Metabolic: glycosuria, gout, increased nitrogen, increased alkaline phosphatase.
Genitourinary: polyuria, dysuria, hematuria, urinary tract infection.
Nervous system/Psychiatric: anxiety, change in appetite, depression, drowsiness, dizziness, thirst, impotence, loss of libido, sweating increase, neuropathy, neurosis, confusion.
Musculoskeletal: arthralgia, myalgia, muscle cramps, stiffness, back pain.
Blood/Coagulation: anemia, abnormal differential, thrombocytopenia, purpura, ESR increased.
Overdosage
The toxic dose of Misoprostol Tablets in humans has not been determined. Cumulative total daily doses of 1600 mcg have been tolerated, with only symptoms of gastrointestinal discomfort being reported. In animals, the acute toxic effects are diarrhea, gastrointestinal lesions, focal cardiac necrosis, hepatic necrosis, renal tubular necrosis, testicular atrophy, respiratory difficulties, and depression of the central nervous system. Clinical signs that may indicate an overdose are sedation, tremor, convulsions, dyspnea, abdominal pain, diarrhea, fever, palpitations, hypotension, or bradycardia. Symptoms should be treated with supportive therapy.
It is not known if misoprostol acid is dialyzable. However, because misoprostol is metabolized like a fatty acid, it is unlikely that dialysis would be appropriate treatment for overdosage.
Renal impairment
Adjustment of the dosing schedule in renally impaired patients is not routinely needed, but dosage can be reduced if the 200-mcg dose is not tolerated. (See Clinical Pharmacology.)
How is MPM Pak supplied
Misoprostol Tablets, 200 mcg are available as white, round, scored, flat beveled-edged tablets, marked with “N” above the score-line and “444” below the score-line and plain on the reverse side.
NDC NumberSize
68071-3458-4 Bottles of 4
Pharmacist: Dispense in this unit-of-use, child-resistant container as defined in the USP.
Provide Patient Information Leaflet with each dispensing.
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [See USP Controlled Room Temperature]. Store in a dry area.
All trademarks are the property of their respective owners.
Issued: 02/2023
PATIENT INFORMATION
PATIENT INFORMATION
Read this leaflet before taking misoprostol and each time your prescription is renewed, because the leaflet may be changed.
Misoprostol is being prescribed by your doctor to decrease the chance of getting stomach ulcers related to the arthritis/pain medication that you take.
Do not take misoprostol to reduce the risk of NSAID-induced ulcers if you are pregnant. (See boxed WARNINGS.) Misoprostol can cause abortion (sometimes incomplete which could lead to dangerous bleeding and require hospitalization and surgery), premature birth, or birth defects. It is also important to avoid pregnancy while taking this medication and for at least one month or through one menstrual cycle after you stop taking it. Misoprostol may cause the uterus to tear (uterine rupture) during pregnancy. The risk of uterine rupture increases as your pregnancy advances and if you have had surgery on the uterus, such as a Cesarean delivery. Rupture (tearing) of the uterus can result in severe bleeding, hysterectomy, and/or maternal or fetal death.
If you become pregnant during misoprostol therapy, stop taking misoprostol and contact your physician immediately. Remember that even if you are on a means of birth control it is still possible to become pregnant. Should this occur, stop taking misoprostol and contact your physician immediately.
Misoprostol may cause diarrhea, abdominal cramping, and/or nausea in some people. In most cases these problems develop during the first few weeks of therapy and stop after about a week. You can minimize possible diarrhea by making sure you take misoprostol with food.
Because these side effects are usually mild to moderate and usually go away in a matter of days, most patients can continue to take misoprostol. If you have prolonged difficulty (more than 8 days), or if you have severe diarrhea, cramping and/or nausea, call your doctor.
Take misoprostol only according to the directions given by your physician.
Do not give misoprostol to anyone else. It has been prescribed for your specific condition, may not be the correct treatment for another person, and would be dangerous if the other person were pregnant.
This information sheet does not cover all possible side effects of misoprostol. This patient information leaflet does not address the side effects of your arthritis/pain medication. See your doctor if you have questions.
Keep out of reach of children.
All trademarks are the property of their respective owners.
Revised February 2023
Marketed By: GenBioPro, Inc.
P.O. Box 32011
Las Vegas, NV 89103
(1-855-643-3463)
1. Indications and Usage for MPM Pak
Ondansetron orally disintegrating tablets are indicated for the prevention of nausea and vomiting associated with:
- highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m 2
- initial and repeat courses of moderately emetogenic cancer chemotherapy
- radiotherapy in patients receiving either total body irradiation, single high-dose fraction to the abdomen, or daily fractions to the abdomen
Ondansetron orally disintegrating tablets also indicated for the prevention of postoperative nausea and/or vomiting.
2. MPM Pak Dosage and Administration
2.1 Dosage
The recommended dosage regimens for adult and pediatric patients are described in Table 1 and Table 2, respectively.
Corresponding doses of ondansetron tablets, ondansetron orally disintegrating tablets and ondansetron oral solution may be used interchangeably.
| Indication
| Dosage Regimen
|
| Highly Emetogenic Cancer Chemotherapy
| A single 24 mg dose administered 30 minutes before the start of single-day highly emetogenic chemotherapy, including cisplatin greater than or equal to 50 mg/m
2.
|
| Moderately Emetogenic Cancer Chemotherapy
| 8 mg administered 30 minutes before the start of chemotherapy, with a subsequent 8 mg dose 8 hours after the first dose.
Then administer 8 mg twice a day (every 12 hours) for 1 to 2 days after completion of chemotherapy. |
| Radiotherapy
| For total body irradiation: 8 mg administered 1 to 2 hours before each fraction of radiotherapy each day.
For single high-dose fraction radiotherapy to the abdomen: 8 mg administered 1 to 2 hours before radiotherapy, with subsequent 8 mg doses every 8 hours after the first dose for 1 to 2 days after completion of radiotherapy. For daily fractionated radiotherapy to the abdomen: 8 mg administered 1 to 2 hours before radiotherapy, with subsequent 8 mg doses every 8 hours after the first dose for each day radiotherapy is given. |
| Postoperative
| 16 mg administered 1 hour before induction of anesthesia.
|
| Indication
| Dosage Regimen
|
| Moderately Emetogenic Cancer Chemotherapy
| 12 to 17 years of age: 8 mg administered 30 minutes before the start of chemotherapy, with a subsequent 8 mg dose 8 hours after the first dose.
Then administer 8 mg twice a day (every 12 hours) for 1 to 2 days after completion of chemotherapy. 4 to 11 years of age: 4 mg administered 30 minutes before the start of chemotherapy, with a subsequent 4 mg dose 4 and 8 hours after the first dose. Then administer 4 mg three times a day for 1 to 2 days after completion of chemotherapy. |
2.2 Dosage in Hepatic Impairment
In patients with severe hepatic impairment (Child-Pugh score of 10 or greater), do not exceed a total daily dose of 8 mg [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)] .
2.3 Administration Instructions for Ondansetron Orally Disintegrating Tablets
Do not attempt to push ondansetron orally disintegrating tablets through the foil backing. With dry hands, remove the tablet from the bottle or PEEL BACK the foil backing of 1 blister and GENTLY remove the tablet. IMMEDIATELY place the ondansetron orally disintegrating tablet on top of the tongue where it will dissolve in seconds, then swallow with saliva. Administration with liquid is not necessary.
3. MPM Pak Dosage and Administration
Ondansetron Orally Disintegrating Tablets USP, 4 mg are white to off-white, round tablets debossed with ‘5’ on one side and ‘E’ on the other side with an embossed circular edge.
Ondansetron Orally Disintegrating Tablets USP, 8 mg are white to off-white, round tablets debossed with ‘7’ on one side and ‘E’ on the other side with an embossed circular edge.
4. Contraindications
Ondansetron orally disintegrating tablets are contraindicated in patients:
- known to have hypersensitivity (e.g., anaphylaxis) to ondansetron or any of the components of the formulation [see Adverse Reactions (6.2)]
- receiving concomitant apomorphine due to the risk of profound hypotension and loss of consciousness
5. Warnings and Precautions
5.1 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis and bronchospasm, have been reported in patients who have exhibited hypersensitivity to other selective 5-HT 3 receptor antagonists. If hypersensitivity reactions occur, discontinue use of ondansetron; treat promptly per standard of care and monitor until signs and symptoms resolve [see Contraindications (4)] .
5.2 QT Prolongation
Electrocardiogram (ECG) changes, including QT interval prolongation have been seen in patients receiving ondansetron. In addition, postmarketing cases of Torsade de Pointes have been reported in patients using ondansetron. Avoid ondansetron in patients with congenital long QT syndrome. ECG monitoring is recommended in patients with electrolyte abnormalities (e.g., hypokalemia or hypomagnesemia), congestive heart failure, bradyarrhythmias, or patients taking other medicinal products that lead to QT prolongation [see Clinical Pharmacology (12.2)] .
5.3 Serotonin Syndrome
The development of serotonin syndrome has been reported with 5-HT
3 receptor antagonists alone. Most reports have been associated with concomitant use of serotonergic drugs (e.g., selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), monoamine oxidase inhibitors, mirtazapine, fentanyl, lithium, tramadol, and intravenous methylene blue). Some of the reported cases were fatal. Serotonin syndrome occurring with overdose of ondansetron alone has also been reported. The majority of reports of serotonin syndrome related to 5-HT
3 receptor antagonist use occurred in a post-anesthesia care unit or an infusion center.
Symptoms associated with serotonin syndrome may include the following combination of signs and symptoms: mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, with or without gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Patients should be monitored for the emergence of serotonin syndrome, especially with concomitant use of ondansetron and other serotonergic drugs. If symptoms of serotonin syndrome occur, discontinue ondansetron and initiate supportive treatment. Patients should be informed of the increased risk of serotonin syndrome, especially if ondansetron is used concomitantly with other serotonergic drugs
[see
Drug Interactions (7.1),
Overdosage (10)]
.
5.4 Myocardial Ischemia
Myocardial ischemia has been reported in patients treated with ondansetron. In some cases, predominantly during intravenous administration, the symptoms appeared immediately after administration but resolved with prompt treatment. Coronary artery spasm appears to be the most common underlying cause. Therefore, monitor or advise patients for signs or symptoms of myocardial ischemia after oral administration of ondansetron [see Adverse Reactions (6.2)].
5.5 Masking of Progressive Ileus and Gastric Distension
The use of ondansetron in patients following abdominal surgery or in patients with chemotherapy-induced nausea and vomiting may mask a progressive ileus and/or gastric distension. Monitor for decreased bowel activity, particularly in patients with risk factors for gastrointestinal obstruction.
Ondansetron is not a drug that stimulates gastric or intestinal peristalsis. It should not be used instead of nasogastric suction.
5.6 Phenylketonuria
Phenylketonuric patients should be informed that ondansetron orally disintegrating tablets contain phenylalanine (a component of aspartame). Each 4 mg orally disintegrating tablet contains 1.68 mg phenylalanine and 8 mg orally disintegrating tablet contains 3.37 mg phenylalanine.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- QT Prolongation [see Warnings and Precautions (5.2)]
- Serotonin Syndrome [see Warnings and Precautions (5.3)]
- Myocardial Ischemia [see Warnings and Precautions (5.4)]
- Masking of Progressive Ileus and Gastric Distension [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The following adverse reactions have been reported in clinical trials of patients treated with ondansetron, the active ingredient of ondansetron orally disintegrating tablets. A causal relationship to therapy with ondansetron was unclear in many cases.
Prevention of Chemotherapy-Induced Nausea and Vomiting
The most common adverse reactions reported in greater than or equal to 4% of 300 adults receiving a single 24 mg dose of ondansetron orally in 2 trials for the prevention of nausea and vomiting associated with highly emetogenic chemotherapy (cisplatin greater than or equal to 50 mg/m
2) were: headache (11%) and diarrhea (4%).
The most common adverse reactions reported in 4 trials in adults for the prevention of nausea and vomiting associated with moderately emetogenic chemotherapy (primarily cyclophosphamide-based regimens) are shown in Table 3.
| a Reported in greater than or equal to 5% of patients treated with ondansetron orally disintegrating tablets and at a rate that exceeded placebo. | ||
| Adverse Reaction
| Ondansetron Orally
Disintegrating Tablets 8 mg Twice Daily (n = 242) | Placebo
(n = 262) |
| Headache
| 58 (24%)
| 34 (13%)
|
| Malaise/Fatigue
| 32 (13%)
| 6 (2%)
|
| Constipation
| 22 (9%)
| 1 (< 1%)
|
| Diarrhea
| 15 (6%)
| 10 (4%)
|
Less Common Adverse Reactions
Central Nervous System: Extrapyramidal reactions (less than 1% of patients).
Hepatic: Aspartate transaminase (AST) and/or alanine transaminase (ALT) values exceeded twice the upper limit of normal in approximately 1% to 2% of 723 patients receiving ondansetron and cyclophosphamide-based chemotherapy in U.S. clinical trials. The increases were transient and did not appear to be related to dose or duration of therapy. On repeat exposure, similar transient elevations in transaminase values occurred in some courses, but symptomatic hepatic disease did not occur. The role of cancer chemotherapy in these biochemical changes is unclear.
Liver failure and death has been reported in cancer patients receiving concurrent medications, including potentially hepatotoxic cytotoxic chemotherapy and antibiotics. The etiology of the liver failure is unclear.
Integumentary: Rash (approximately 1% of patients).
Other (less than 2%): Anaphylaxis, bronchospasm, tachycardia, angina, hypokalemia, electrocardiographic alterations, vascular occlusive events, and grand mal seizures. Except for bronchospasm and anaphylaxis, the relationship to ondansetron is unclear.
Prevention of Radiation-Induced Nausea and Vomiting
The most common adverse reactions (greater than or equal to 2%) reported in patients receiving ondansetron and concurrent radiotherapy were similar to those reported in patients receiving ondansetron and concurrent chemotherapy and were headache, constipation, and diarrhea.
Prevention of Postoperative Nausea and/or Vomiting
The most common adverse reactions reported in adults in trial(s) of prevention of postoperative nausea and vomiting are shown in Table 4. In these trial(s), patients were receiving multiple concomitant perioperative and postoperative medications in both treatment groups.
| a Reported in greater than or equal to 5% of patients treated with ondansetron orally disintegrating tablets and at a rate that exceeded placebo. | ||
| Adverse Reaction
| Ondansetron Orally
Disintegrating Tablets 16 mg as a Single Dose (n = 550) | Placebo
(n = 531) |
| Headache
| 49 (9%)
| 27 (5%)
|
| Hypoxia
| 49 (9%)
| 35 (7%)
|
| Pyrexia
| 45 (8%)
| 34 (6%)
|
| Dizziness
| 36 (7%)
| 34 (6%)
|
| Gynecological disorder
| 36 (7%)
| 33 (6%)
|
| Anxiety/Agitation
| 33 (6%)
| 29 (5%)
|
| Urinary retention
| 28 (5%)
| 18 (3%)
|
| Pruritus
| 27 (5%)
| 20 (4%)
|
In a crossover study with 25 subjects, headache was reported in 6 subjects administered ondansetron orally disintegrating tablets with water (24%) as compared with 2 subjects administered ondansetron orally disintegrating tablets without water (8%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of ondansetron. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiovascular
Arrhythmias (including ventricular and supraventricular tachycardia, premature ventricular contractions, and atrial fibrillation), bradycardia, electrocardiographic alterations (including second-degree heart block, QT/QTc interval prolongation, and ST segment depression), palpitations, and syncope. Rarely and predominantly with intravenous ondansetron, transient ECG changes, including QT interval prolongation have been reported.
Myocardial ischemia was reported predominately with intravenous administration
[see
Warnings and Precautions (5.4)].
General
Flushing: Rare cases of hypersensitivity reactions, sometimes severe (e.g., anaphylactic reactions, angioedema, bronchospasm, shortness of breath, hypotension, laryngeal edema, stridor) have also been reported.
Laryngospasm, shock, and cardiopulmonary arrest have occurred during allergic reactions in patients receiving injectable ondansetron.
Hepatobiliary
Liver enzyme abnormalities.
Lower Respiratory
Hiccups.
Neurology
Oculogyric crisis, appearing alone, as well as with other dystonic reactions.
Skin
Urticaria, Stevens-Johnson syndrome, and toxic epidermal necrolysis.
Eye Disorders
Cases of transient blindness, predominantly during intravenous administration, have been reported. These cases of transient blindness were reported to resolve within a few minutes up to 48 hours.
7. Drug Interactions
7.1 Serotonergic Drugs
Serotonin syndrome (including altered mental status, autonomic instability, and neuromuscular symptoms) has been described following the concomitant use of 5-HT 3 receptor antagonists and other serotonergic drugs, including SSRIs and SNRIs. Monitor for the emergence of serotonin syndrome. If symptoms occur, discontinue ondansetron and initiate supportive treatment [see Warnings and Precautions (5.3)] .
7.2 Drugs Affecting Cytochrome P-450 Enzymes
Ondansetron does not itself appear to induce or inhibit the cytochrome P-450 drug-metabolizing enzyme system of the liver [see Clinical Pharmacology (12.3)] . Because ondansetron is metabolized by hepatic cytochrome P-450 drug-metabolizing enzymes (CYP3A4, CYP2D6, CYP1A2), inducers or inhibitors of these enzymes may change the clearance and, hence, the half-life of ondansetron. In patients treated with potent inducers of CYP3A4 (i.e., phenytoin, carbamazepine, and rifampin), the clearance of ondansetron was significantly increased and ondansetron blood concentrations were decreased. However, on the basis of available data, no dosage adjustment for ondansetron is recommended for patients on these drugs [see Clinical Pharmacology (12.3)] .
7.3 Tramadol
Although no pharmacokinetic drug interaction between ondansetron and tramadol has been observed, data from 2 small trials indicate that when used together, ondansetron may increase patient-controlled administration of tramadol. Monitor patients to ensure adequate pain control when ondansetron is administered with tramadol.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Published epidemiological studies on the association between ondansetron use and major birth defects have reported inconsistent findings and have important methodological limitations that preclude conclusions about the safety of ondansetron use in pregnancy (see Data). Available postmarketing data have not identified a drug-associated risk of miscarriage or adverse maternal outcomes. Reproductive studies in rats and rabbits did not show evidence of harm to the fetus when ondansetron was administered during organogenesis at approximately 6 and 24 times the maximum recommended human oral dose of 24 mg/day, based on body surface area (BSA), respectively (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, miscarriages, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
Available data on ondansetron use in pregnant women from several published epidemiological studies preclude an assessment of a drug-associated risk of adverse fetal outcomes due to important methodological limitations, including the uncertainty of whether women who filled a prescription actually took the medication, the concomitant use of other medications or treatments, recall bias, and other unadjusted confounders.
Ondansetron exposure in utero has not been associated with overall major congenital malformations in aggregate analyses. One large retrospective cohort study examined 1970 women who received a prescription for ondansetron during pregnancy and reported no association between ondansetron exposure and major congenital malformations, miscarriage, stillbirth, preterm delivery, infants of low birth weight, or infants small for gestational age.
Two large retrospective cohort studies and one case-control study have assessed ondansetron exposure in the first trimester and risk of cardiovascular defects with inconsistent findings. Relative risks (RR) ranged from 0.97 (95% CI 0.86 to 1.10) to 1.62 (95% CI 1.04, 2.54). A subset analysis in one of the cohort studies observed that ondansetron was specifically associated with cardiac septal defects (RR 2.05, 95% CI 1.19, 3.28); however, this association was not confirmed in other studies.
Several studies have assessed ondansetron and the risk of oral clefts with inconsistent findings. A retrospective cohort study of 1.8 million pregnancies in the U.S. Medicaid Database showed an increased risk of oral clefts among 88,467 pregnancies in which oral ondansetron was prescribed in the first trimester (RR 1.24, 95% CI 1.03, 1.48), but no such association was reported with intravenous ondansetron in 23,866 pregnancies (RR 0.95, 95% CI 0.63, 1.43). In the subgroup of women who received both forms of administration, the RR was 1.07 (95% CI 0.59, 1.93). Two case-control studies, using data from birth defects surveillance programs, reported conflicting associations between maternal use of ondansetron and isolated cleft palate (OR 1.6 [95% CI 1.1, 2.3] and 0.5 [95% CI 0.3, 1.0]). It is unknown whether ondansetron exposure in utero in the cases of cleft palate occurred during the time of palate formation (the palate is formed between the 6th and 9th weeks of pregnancy).
Animal Data
In embryo-fetal development studies in rats and rabbits, pregnant animals received oral doses of ondansetron up to 15 mg/kg/day and 30 mg/kg/day, respectively, during the period of organogenesis. With the exception of a slight decrease in maternal body weight gain in the rabbits, there were no significant effects of ondansetron on the maternal animals or the development of the offspring. At doses of 15 mg/kg/day in rats and 30 mg/kg/day in rabbits, the maternal exposure margin was approximately 6 and 24 times the maximum recommended human oral dose of 24 mg/day, respectively, based on BSA.
In a pre- and postnatal developmental toxicity study, pregnant rats received oral doses of ondansetron up to 15 mg/kg/day from Day 17 of pregnancy to litter Day 21. With the exception of a slight reduction in maternal body weight gain, there were no effects upon the pregnant rats and the pre- and postnatal development of their offspring, including reproductive performance of the mated F1 generation. At a dose of 15 mg/kg/day in rats, the maternal exposure margin was approximately 6 times the maximum recommended human oral dose of 24 mg/day, based on BSA.
8.2 Lactation
Risk Summary
It is not known whether ondansetron is present in human milk. There are no data on the effects of ondansetron on the breastfed infant or the effects on milk production. However, it has been demonstrated that ondansetron is present in the milk of rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ondansetron and any potential adverse effects on the breastfed infant from ondansetron or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of orally administered ondansetron have been established in pediatric patients 4 years and older for the prevention of nausea and vomiting associated with moderately emetogenic cancer chemotherapy. Use of ondansetron in these age-groups is supported by evidence from adequate and well- controlled studies of ondansetron in adults with additional data from 3 open-label, uncontrolled, non-U.S. trials in 182 pediatric patients aged 4 to 18 years with cancer who were given a variety of cisplatin or noncisplatin regimens
[see
Dosage and Administration (2.2),
Clinical Studies (14.1)]
.
Additional information on the use of ondansetron in pediatric patients may be found in ondansetron Injection prescribing information.
The safety and effectiveness of orally administered ondansetron have not been established in pediatric patients for:
- prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy
- prevention of nausea and vomiting associated with radiotherapy
- prevention of postoperative nausea and/or vomiting
8.5 Geriatric Use
Of the total number of subjects enrolled in cancer chemotherapy-induced and postoperative nausea and vomiting in U.S.- and foreign-controlled clinical trials, for which there were subgroup analyses, 938 (19%) were aged 65 years and older.
No overall differences in safety or effectiveness were observed between subjects 65 years of age and older and younger subjects. A reduction in clearance and increase in elimination half-life were seen in patients older than 75 years compared with younger subjects
[see
Clinical Pharmacology (12.3)]
. There were an insufficient number of patients older than 75 years of age and older in the clinical trials to permit safety or efficacy conclusions in this age group. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. No dosage adjustment is needed in elderly patients.
8.6 Hepatic Impairment
No dosage adjustment is needed in patients with mild or moderate hepatic impairment.
In patients with severe hepatic impairment, clearance is reduced and the apparent volume of distribution is increased, resulting in a significant increase in the half-life of ondansetron. Therefore, do not exceed a total daily dose of 8 mg in patients with severe hepatic impairment (Child-Pugh score of 10 or greater)
[see
Dosage and Administration (2.2),
Clinical Pharmacology (12.3)]
.
8.7 Renal Impairment
No dosage adjustment is recommended for patients with any degree of renal impairment (mild, moderate, or severe). There is no experience beyond first-day administration of ondansetron [see Clinical Pharmacology (12.3)] .
9. Drug Abuse and Dependence
Animal studies have shown that ondansetron is not discriminated as a benzodiazepine nor does it substitute for benzodiazepines in direct addiction studies.
10. Overdosage
There is no specific antidote for ondansetron overdose. Patients should be managed with appropriate supportive therapy.
In addition to the adverse reactions listed above, the following adverse reactions have been described in the setting of ondansetron overdose: “Sudden blindness” (amaurosis) of 2 to 3 minutes’ duration plus severe constipation occurred in one patient that was administered 72 mg of ondansetron intravenously as a single dose. Hypotension (and faintness) occurred in a patient that took 48 mg of ondansetron tablets. Following infusion of 32 mg over only a 4-minute period, a vasovagal episode with transient second-degree heart block was observed. In all instances, the adverse reactions resolved completely.
Pediatric cases consistent with serotonin syndrome have been reported after inadvertent oral overdoses of ondansetron (exceeding estimated ingestion of 5 mg per kg) in young children. Reported symptoms included somnolence, agitation, tachycardia, tachypnea, hypertension, flushing, mydriasis, diaphoresis, myoclonic movements, horizontal nystagmus, hyperreflexia, and seizure. Patients required supportive care, including intubation in some cases, with complete recovery without sequelae within 1 to 2 days.
11. MPM Pak Description
The active ingredient in ondansetron orally disintegrating tablets, USP is ondansetron base, the racemic form of ondansetron and a selective blocking agent of the serotonin 5-HT
3 receptor type. Chemically it is (±) 1, 2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one. It has the following structural formula:
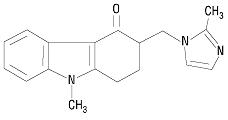
The molecular formula is C
18H
19N
3O representing a molecular weight of 293.4 g/mol. Ondansetron is a white to off-white powder.
Each 4 mg ondansetron orally disintegrating tablet, USP for oral administration contains 4 mg ondansetron base. Each 8 mg ondansetron orally disintegrating tablet, USP for oral administration contains 8 mg ondansetron base. Each ondansetron orally disintegrating tablet, USP also contains the inactive ingredients mannitol, crospovidone, lactose monohydrate, microcrystalline cellulose, aspartame, strawberry guarana flavor, colloidal silicon dioxide, and magnesium stearate. The strawberry guarana flavor contains maltodextrin, propylene glycol, artificial flavors, and acetic acid. Ondansetron orally disintegrating tablets, USP are orally administered formulation of ondansetron which disintegrates on the tongue and does not require water to aid dissolution or swallowing.
Meets USP Disintegration Test 2.
12. MPM Pak - Clinical Pharmacology
12.1 Mechanism of Action
Ondansetron is a selective 5-HT 3 receptor antagonist. While its mechanism of action has not been fully characterized, ondansetron is not a dopamine-receptor antagonist. Serotonin receptors of the 5-HT 3 type are present both peripherally on vagal nerve terminals and centrally in the chemoreceptor trigger zone of the area postrema. It is not certain whether ondansetron’s antiemetic action is mediated centrally, peripherally, or in both sites. However, cytotoxic chemotherapy appears to be associated with release of serotonin from the enterochromaffin cells of the small intestine. In humans, urinary 5-hydroxyindoleacetic acid (5-HIAA) excretion increases after cisplatin administration in parallel with the onset of emesis. The released serotonin may stimulate the vagal afferents through the 5-HT 3 receptors and initiate the vomiting reflex.
12.2 Pharmacodynamics
In healthy subjects, single intravenous doses of 0.15 mg/kg of ondansetron had no effect on esophageal motility, gastric motility, lower esophageal sphincter pressure, or small intestinal transit time. Multiday administration of ondansetron has been shown to slow colonic transit in healthy subjects. Ondansetron has no effect on plasma-prolactin concentrations.
Cardiac Electrophysiology
QTc interval prolongation was studied in a double-blind, single-intravenous dose, placebo- and positive- controlled, crossover trial in 58 healthy subjects. The maximum mean (95% upper confidence bound) difference in QTcF from placebo after baseline correction was 19.5 (21.8) milliseconds and 5.6 (7.4) milliseconds after 15-minute intravenous infusions of 32 mg and 8 mg of ondansetron injection, respectively. A significant exposure- response relationship was identified between ondansetron concentration and ΔΔQTcF. Using the established exposure-response relationship, 24 mg infused intravenously over 15 minutes had a mean predicted (95% upper prediction interval) ΔΔQTcF of 14.0 (16.3) milliseconds. In contrast, 16 mg infused intravenously over 15 minutes using the same model had a mean predicted (95% upper prediction interval) ΔΔQTcF of 9.1 (11.2) milliseconds. In this study, the 8 mg dose infused over 15 minutes did not prolong the QT interval to any clinically relevant extent.
12.3 Pharmacokinetics
Absorption
Ondansetron is absorbed from the gastrointestinal tract and undergoes some first-pass metabolism. Mean bioavailability in healthy subjects, following administration of a single 8 mg tablet, is approximately 56%.
Ondansetron systemic exposure does not increase proportionately to dose. The area under curve (AUC) from a 16 mg tablet was 24% greater than predicted from an 8 mg tablet dose. This may reflect some reduction of first-pass metabolism at higher oral doses.
Food Effects: Bioavailability is also slightly enhanced by the presence of food.
Distribution
Plasma protein binding of ondansetron as measured
in vitro was 70% to 76% over the concentration range of 10 to 500 ng/mL. Circulating drug also distributes into erythrocytes.
Elimination
Metabolism and Excretion: Ondansetron is extensively metabolized in humans, with approximately 5% of a radiolabeled dose recovered as the parent compound from the urine. The metabolites are observed in the urine. The primary metabolic pathway is hydroxylation on the indole ring followed by subsequent glucuronide or sulfate conjugation.
In vitro metabolism studies have shown that ondansetron is a substrate for human hepatic cytochrome P-450 enzymes, including CYP1A2, CYP2D6, and CYP3A4. In terms of overall ondansetron turnover, CYP3A4 played the predominant role. Because of the multiplicity of metabolic enzymes capable of metabolizing ondansetron, it is likely that inhibition or loss of one enzyme (e.g., CYP2D6 genetic deficiency) will be compensated by others and may result in little change in overall rates of ondansetron elimination.
Although some nonconjugated metabolites have pharmacologic activity, these are not found in plasma at concentrations likely to significantly contribute to the biological activity of ondansetron.
Specific Populations
Age: Geriatric Population: A reduction in clearance and increase in elimination half-life are seen in patients older than 75 years compared to younger subjects
[see
Use in Specific Populations (8.5)]
.
Sex: Gender differences were shown in the disposition of ondansetron given as a single dose. The extent and rate of absorption are greater in women than men. Slower clearance in women, a smaller apparent volume of distribution (adjusted for weight), and higher absolute bioavailability resulted in higher plasma ondansetron concentrations. These higher plasma concentrations may in part be explained by differences in body weight between men and women. It is not known whether these sex-related differences were clinically important. More detailed pharmacokinetic information is contained in Tables 5 and 6.
| Age-group (years) Sex (M/F)
| Mean
Weight (kg) | N
| Peak Plasma
Concentration (ng/mL) | Time of
Peak Plasma Concentration (h) | Mean
Elimination Half-life (h) | Systemic
Plasma Clearance L/h/kg | Absolute
Bioavailability |
| 18 to 40 M
F | 69.0
62.7 | 6
5 | 26.2
42.7 | 2.0
1.7 | 3.1
3.5 | 0.403
0.354 | 0.483
0.663 |
| 61 to 74 M
F | 77.5
60.2 | 6
6 | 24.1
52.4 | 2.1
1.9 | 4.1
4.9 | 0.384
0.255 | 0.585
0.643 |
| ≥ 75 M
F | 78.0
67.6 | 5
6 | 37.0
46.1 | 2.2
2.1 | 4.5
6.2 | 0.277
0.249 | 0.619
0.747 |
| Age-group (years)
Sex (M/F) |
Mean Weight (kg) | N
| Peak Plasma Concentration (ng/mL)
| Time of Peak Plasma Concentration
(h) | Mean Elimination Half-life
(h) |
| 18 to 43 M
F | 84.1
71.8 | 8
8 | 125.8
194.4 | 1.9
1.6 | 4.7
5.8 |
Renal Impairment: Renal impairment is not expected to significantly influence the total clearance of ondansetron as renal clearance represents only 5% of the overall clearance. However, the mean plasma clearance of ondansetron was reduced by about 50% in patients with severe renal impairment (creatinine clearance less than 30 mL/min). The reduction in clearance was variable and not consistent with an increase in half-life
[see
Use in Specific Populations (8.7)]
.
Hepatic Impairment: In patients with mild-to-moderate hepatic impairment, clearance is reduced 2-fold and mean half-life is increased to 11.6 hours compared with 5.7 hours in healthy subjects. In patients with severe hepatic impairment (Child-Pugh score of 10 or greater), clearance is reduced 2-fold to 3-fold and apparent volume of distribution is increased with a resultant increase in half-life to 20 hours
[see
Dosage and Administration (2.2),
Use in Specific Populations (8.6)]
.
Drug Interaction Studies
CYP 3A4 Inducers: Ondansetron elimination may be affected by cytochrome P-450 inducers. In a pharmacokinetic trial of 16 epileptic patients maintained chronically on CYP3A4 inducers, carbamazepine, or phenytoin, a reduction in AUC, C
max, and t
½ of ondansetron was observed. This resulted in a significant increase in the clearance of ondansetron. However, this increase is not thought to be clinically relevant
[see
Drug Interactions (7.2)]
.
Chemotherapeutic Agents: Carmustine, etoposide, and cisplatin do not affect the pharmacokinetics of ondansetron
[see
Drug Interactions (7.4)]
.
Antacids: Concomitant administration of antacids does not alter the absorption of ondansetron.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenic effects were not seen in 2-year studies in rats and mice with oral ondansetron doses up to 10 mg/kg per day and 30 mg/kg per day, respectively (approximately 4 and 6 times the maximum recommended human oral dose of 24 mg per day, based on BSA).
Ondansetron was not mutagenic in standard tests for mutagenicity.
Oral administration of ondansetron up to 15 mg/kg per day (approximately 6 times the maximum recommended human oral dose of 24 mg per day, based on BSA) did not affect fertility or general reproductive performance of male and female rats.
14. Clinical Studies
14.1 Prevention of Chemotherapy-Induced Nausea and Vomiting
p>
Highly Emetogenic Chemotherapy
In 2 randomized, double-blind, monotherapy trials, a single 24 mg oral dose of ondansetron was superior to a relevant historical placebo control in the prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m
2. Steroid administration was excluded from these clinical trials. More than 90% of patients receiving a cisplatin dose greater than or equal to 50 mg/m
2 in the historical-placebo comparator, experienced vomiting in the absence of antiemetic therapy.
The first trial compared oral doses of ondansetron 24 mg as a single dose, 8 mg every 8 hours for 2 doses, and 32 mg as a single dose in 357 adult cancer patients receiving chemotherapy regimens containing cisplatin greater than or equal to 50 mg/m
2. The first or single dose was administered 30 minutes prior to chemotherapy. A total of 66% of patients in the ondansetron 24 mg once-a-day group, 55% in the ondansetron 8 mg twice-a-day group, and 55% in the ondansetron 32 mg once-a-day group, completed the 24-hour trial period with 0 emetic episodes and no rescue antiemetic medications, the primary endpoint of efficacy. Each of the 3 treatment groups was shown to be statistically significantly superior to a historical placebo control.
In the same trial, 56% of patients receiving a single 24 mg oral dose of ondansetron experienced no nausea during the 24-hour trial period, compared with 36% of patients in the oral ondansetron 8 mg twice-a-day group (
P = 0.001) and 50% in the oral ondansetron 32 mg once-a-day group. Dosage regimens of ondansetron 8 mg twice daily and 32 mg once daily are not recommended for the prevention of nausea and vomiting associated with highly emetogenic chemotherapy
[see
Dosage and Administration (2.1)]
.
In a second trial, efficacy of a single 24 mg oral dose of ondansetron for the prevention of nausea and vomiting associated with highly emetogenic cancer chemotherapy, including cisplatin greater than or equal to 50 mg/m
2, was confirmed.
Moderately Emetogenic Chemotherapy
A randomized, placebo-controlled, double-blind trial was conducted in the U.S. in 67 patients receiving a cyclophosphamide-based chemotherapy regimen containing doxorubicin. The first 8 mg dose of ondansetron was administered 30 minutes before the start of chemotherapy, with a subsequent dose 8 hours after the first dose, followed by 8 mg of ondansetron twice a day for 2 days after the completion of chemotherapy. Ondansetron orally disintegrating tablets was significantly more effective than placebo in preventing vomiting. Treatment response was based on the total number of emetic episodes over the 3-day trial period. The results of this trial are summarized in Table 7.
| a Median undefined since at least 50% of the patients were withdrawn or had more than 2 emetic episodes.
b Median undefined since at least 50% of patients did not have any emetic episodes. |
|||
|
| Ondansetron Orally Disintegrating Tablets
(n = 33) | Placebo
(n = 34) | P-v
alue
|
| Treatment response
0 Emetic episodes 1 to 2 Emetic episodes More than 2 emetic episodes/withdrawn |
20 (61%) 6 (18%) 7 (21%) |
2 (6%) 8 (24%) 24 (71%) |
< 0.001 < 0.001 |
| Median number of emetic episodes
| 0.0
| Undefined
a
|
|
| Median time to first emetic episode (hours)
| Undefined
b
| 6.5
|
|
In a double-blind, U.S. trial in 336 patients receiving a cyclophosphamide-based chemotherapy regimen containing either methotrexate or doxorubicin, ondansetron 8 mg administered twice a day, was as effective as ondansetron 8 mg administered 3 times a day in preventing nausea and vomiting. Ondansetron 8 mg three times daily is not a recommended regimen for the treatment of moderately emetogenic chemotherapy
[see
Dosage and Administration (2.1)]
.
Treatment response was based on the total number of emetic episodes over the 3-day trial period. See Table 8 for the details of the dosage regimens studied and results of this trial.
| a The first 8 mg dose was administered 30 minutes before the start of emetogenic chemotherapy, with a subsequent 8 mg dose 8 hours after the first dose, followed by 8 mg administered twice a day for 2 days after the completion of chemotherapy.
b The first 8 mg dose was administered 30 minutes before the start of emetogenic chemotherapy, with subsequent 8 mg doses at 4 hours and 8 hours after the first dose, followed by 8 mg administered 3 times a day for 2 days after the completion of chemotherapy. c Median undefined since at least 50% of patients did not have any emetic episodes. d Visual analog scale assessment: 0 = no nausea, 100 = nausea as bad as it can be. |
||||||||||||||||
|
| Ondansetron Tablets
|
|||||||||||||||
| 8 mg Twice Daily
a
(n = 165) | 8 mg Three Times a Day
b
(n = 171) |
|||||||||||||||
| Treatment response
0 Emetic episodes 1 to 2 Emetic episodes More than 2 emetic episodes/withdrawn |
101 (61%) 16 (10%) 48 (29%) |
99 (58%) 17 (10%) 55 (32%) |
||||||||||||||
| Median number of emetic episodes
| 0.0
| 0.0
|
||||||||||||||
| Median time to first emetic episode (h)
| Undefined
c
| Undefined
c
|
||||||||||||||
| Median nausea scores (0 to 100)
d
| 6
| 6
|
||||||||||||||
Re-treatment
In single-arm trials, 148 patients receiving cyclophosphamide-based chemotherapy were re-treated with ondansetron 8 mg three times daily during subsequent chemotherapy for a total of 396 re-treatment courses. No emetic episodes occurred in 314 (79%) of the re-treatment courses, and only 1 to 2 emetic episodes occurred in 43 (11%) of the re-treatment courses.
Pediatric Trials
Three open-label, single-arm, non-U.S. trials have been performed with 182 pediatric patients aged 4 to 18 years with cancer who were given a variety of cisplatin or noncisplatin regimens. The initial dose of ondansetron injection ranged from 0.04 to 0.87 mg per kg (total dose of 2.16 mg to 12 mg) followed by the administration of oral doses of ondansetron ranging from 4 to 24 mg daily for 3 days. In these trials, 58% of the 170 evaluable patients had a complete response (no emetic episodes) on Day 1. In 2 trials, the response rates to ondansetron 4 mg three times a day in patients younger than 12 years was similar to ondansetron 8 mg three times daily in patients 12 to 18 years. Prevention of emesis in these pediatric patients was essentially the same as for adults.
14.2 Radiation-Induced Nausea and Vomiting
Total Body Irradiation
In a randomized, placebo-controlled, double-blind trial in 20 patients, 8 mg of ondansetron administered 1.5 hours before each fraction of radiotherapy for 4 days was significantly more effective than placebo in preventing vomiting induced by total body irradiation. Total body irradiation consisted of 11 fractions (120 cGy per fraction) over 4 days for a total of 1,320 cGy. Patients received 3 fractions for 3 days, then 2 fractions on Day 4.
Single High-Dose Fraction Radiotherapy
In an active-controlled, double-blind trial in 105 patients receiving single high-dose radiotherapy (800 to 1,000 cGy) over an anterior or posterior field size of greater than or equal to 80 cm
2 to the abdomen, ondansetron was significantly more effective than metoclopramide with respect to complete control of emesis (0 emetic episodes). Patients received the first dose of ondansetron (8 mg) or metoclopramide (10 mg) 1 to 2 hours before radiotherapy. If radiotherapy was given in the morning, 8 mg of ondansetron or 10 mg of metoclopramide was administered in the late afternoon and repeated again before bedtime. If radiotherapy was given in the afternoon, patients took 8 mg of ondansetron or 10 mg of metoclopramide only once before bedtime. Patients continued the doses of oral medication three times daily for 3 days.
Daily Fractionated Radiotherapy
In an active-controlled, double-blind trial in 135 patients receiving a 1- to 4- week course of fractionated radiotherapy (180 cGy doses) over a field size of greater than or equal to 100 cm
2 to the abdomen, ondansetron was significantly more effective than prochlorperazine with respect to complete control of emesis (0 emetic episodes). Patients received the first dose of ondansetron (8 mg) or prochlorperazine (10 mg) 1 to 2 hours before the first daily radiotherapy fraction, with subsequent 8 mg doses approximately every 8 hours on each day of radiotherapy.
14.3 Postoperative Nausea and/or Vomiting
In 2 placebo-controlled, double-blind trials (one conducted in the U.S. and the other outside the U.S.) in 865 females undergoing inpatient surgical procedures, ondansetron 16 mg as a single dose or placebo was administered one hour before the induction of general balanced anesthesia (barbiturate, opioid, nitrous oxide, neuromuscular blockade, and supplemental isoflurane or enflurane), ondansetron was significantly more effective than placebo in preventing postoperative nausea and vomiting.
No trials have been performed in males.
16. How is MPM Pak supplied
Ondansetron Orally Disintegrating Tablets USP, 8 mg are white to off-white, round tablets debossed with ‘7’ on one side and ‘E’ on the other side with an embossed circular edge.
Tablets of 15 NDC 68071-2865-5
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Dispense in a tight, light-resistant container as defined in the USP.
17. Patient Counseling Information
Hypersensitivity Reactions
Inform patients that ondansetron may cause hypersensitivity reactions, some as severe as anaphylaxis and bronchospasm. Instruct patients to immediately report any signs and symptoms of hypersensitivity reactions, including fever, chills, rash, or breathing problems to their healthcare provider [see Warnings and Precautions (5.1)].
QT Prolongation
Inform patients that ondansetron may cause serious cardiac arrhythmias, such as QT prolongation. Instruct patients to tell their healthcare provider right away if they perceive a change in their heart rate, if they feel lightheaded, or if they have a syncopal episode [see Warnings and Precautions (5.2)].
Drug Interactions
- Instruct the patient to report the use of all medications, especially apomorphine, to their healthcare provider. Concomitant use of apomorphine and ondansetron may cause a significant drop in blood pressure and loss of consciousness.
- Advise patients of the possibility of serotonin syndrome with concomitant use of ondansetron and another serotonergic agent, such as medications to treat depression and migraines. Advise patients to seek immediate medical attention if the following symptoms occur: changes in mental status, autonomic instability, neuromuscular symptoms with or without gastrointestinal symptoms [see Warnings and Precautions (5.3)].
Myocardial Ischemia
Inform patients that ondansetron may cause myocardial ischemia. Advise patients to seek immediate medical help if any symptoms suggestive of a myocardial ischemia occur, such as sudden chest pain or chest tightness [see Warnings and Precautions (5.4)].
Masking of Progressive Ileus and Gastric Distension
Inform patients following abdominal surgery or those with chemotherapy-induced nausea and vomiting that ondansetron may mask signs and symptoms of bowel obstruction. Instruct patients to immediately report any signs or symptoms consistent with a potential bowel obstruction to their healthcare provider [see Warnings and Precautions (5.5)].
Administration of Ondansetron Orally Disintegrating Tablets
Instruct patients not to remove ondansetron orally disintegrating tablets from the blister until just prior to dosing.
- Do not attempt to push ondansetron orally disintegrating tablets through the foil backing.
- With dry hands, remove the tablet from the bottle or peel back the foil backing of 1 blister and gently remove the tablet.
- Immediately place the ondansetron orally disintegrating tablet on top of the tongue where it will dissolve in seconds, then swallow with saliva.
- Administration with liquid is not necessary.
- Peelable illustrated stickers are affixed to the product carton that can be provided with the prescription to ensure proper use and handling of the product.
Distributed by:
Rising Health, LLC
Saddle Brook, NJ 07663
Made in India
Code: TS/DRUGS/19/1993
Revised: 12/2021
Cardiovascular Thrombotic Events
- Nonsteroidal anti-inflammatory drugs (NSAIDs) cause an increased risk of serious cardiovascular thrombotic events, including myocardial infarction and stroke, which can be fatal. This risk may occur early in treatment and may increase with duration of use [see Warnings and Precautions].
- Ibuprofen tablets are contraindicated in the setting of coronary artery bypass graft (CABG) surgery [see Contraindications and Warnings].
Gastrointestinal Risk
- NSAIDs cause an increased risk of serious gastrointestinal adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which can be fatal. These events can occur at any time during use and without warning symptoms. Elderly patients are at greater risk for serious gastrointestinal events (see WARNINGS).
MPM Pak Description
Ibuprofen tablets USP contain the active ingredient ibuprofen, which is (±) - 2 - ( p - isobutylphenyl) propionic acid. Ibuprofen is a white to off-white, crystalline powder with a melting point of 74 to 77°C and is very soluble in alcohol, in methanol, in acetone and in chloroform, slightly soluble in ethyl acetate, practically insoluble in water.
The structural formula is represented below:
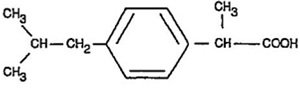
Ibuprofen tablets USP, a nonsteroidal anti-inflammatory drug (NSAID), is available in 400 mg, 600 mg, and 800 mg tablets for oral administration. Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol-part. hydrolyzed, povidone, stearic acid, talc and titanium dioxide.
MPM Pak - Clinical Pharmacology
Ibuprofen tablets contain ibuprofen which possesses analgesic and antipyretic activities. Its mode of action, like that of other NSAIDs, is not completely understood, but may be related to prostaglandin synthetase inhibition.
In clinical studies in patients with rheumatoid arthritis and osteoarthritis, ibuprofen tablets have been shown to be comparable to aspirin in controlling pain and inflammation and to be associated with a statistically significant reduction in the milder gastrointestinal side effects (see ADVERSE REACTIONS). Ibuprofen tablets may be well tolerated in some patients who have had gastrointestinal side effects with aspirin, but these patients when treated with ibuprofen tablets should be carefully followed for signs and symptoms of gastrointestinal ulceration and bleeding. Although it is not definitely known whether ibuprofen tablets causes less peptic ulceration than aspirin, in one study involving 885 patients with rheumatoid arthritis treated for up to one year, there were no reports of gastric ulceration with ibuprofen tablets whereas frank ulceration was reported in 13 patients in the aspirin group (statistically significant p<.001).
Gastroscopic studies at varying doses show an increased tendency toward gastric irritation at higher doses. However, at comparable doses, gastric irritation is approximately half that seen with aspirin. Studies using 51Cr-tagged red cells indicate that fecal blood loss associated with ibuprofen tablets in doses up to 2400 mg daily did not exceed the normal range, and was significantly less than that seen in aspirin-treated patients.
In clinical studies in patients with rheumatoid arthritis, ibuprofen tablets have been shown to be comparable to indomethacin in controlling the signs and symptoms of disease activity and to be associated with a statistically significant reduction of the milder gastrointestinal (see ADVERSE REACTIONS) and CNS side effects.
Ibuprofen tablets may be used in combination with gold salts and/or corticosteroids.
Controlled studies have demonstrated that ibuprofen tablets are a more effective analgesic than propoxyphene for the relief of episiotomy pain, pain following dental extraction procedures, and for the relief of the symptoms of primary dysmenorrhea.
In patients with primary dysmenorrhea, ibuprofen tablets have been shown to reduce elevated levels of prostaglandin activity in the menstrual fluid and to reduce resting and active intrauterine pressure, as well as the frequency of uterine contractions. The probable mechanism of action is to inhibit prostaglandin synthesis rather than simply to provide analgesia.
Pharmacodynamics
In a healthy volunteer study, ibuprofen 400 mg given once daily, administered 2 hours prior to immediate-release aspirin (81 mg) for 6 days, showed an interaction with the antiplatelet activity of aspirin as measured by % serum thromboxane B2 (TxB2) inhibition at 24 hours following the day-6 aspirin dose [53%]. An interaction was still observed, but minimized, when ibuprofen 400 mg given once-daily was administered as early as 8 hours prior to the immediate-release aspirin dose [90.7%]. However, there was no interaction with the antiplatelet activity of aspirin when ibuprofen 400 mg, given once daily, was administered 2 hours after (but not concomitantly, 15 min, or 30 min after) the immediate-release aspirin dose [99.2%].
In another study, where immediate-release aspirin 81 mg was administered once daily with ibuprofen 400 mg given three times daily (1, 7, and 13 hours post-aspirin dose) for 10 consecutive days, the mean % serum thromboxane B2 (TxB2) inhibition suggested no interaction with the antiplatelet activity of aspirin [98.3%]. However, there were individual subjects with serum TxB2 inhibition below 95%, with the lowest being 90.2%.
When a similarly designed study was conducted with enteric-coated aspirin, where healthy subjects were administered enteric-coated aspirin 81 mg once daily for 6 days and ibuprofen 400 mg three times daily (2, 7 and 12 h post-aspirin dose) for 6 days, there was an interaction with the antiplatelet activity at 24 hours following the day-6 aspirin dose [67%]. [ See Precautions/ Drug Interactions].
Pharmacokinetics
The ibuprofen in ibuprofen tablets is rapidly absorbed. Peak serum ibuprofen levels are generally attained one to two hours after administration. With single doses up to 800 mg, a linear relationship exists between amount of drug administered and the integrated area under the serum drug concentration vs time curve. Above 800 mg, however, the area under the curve increases less than proportional to increases in dose. There is no evidence of drug accumulation or enzyme induction.
The administration of ibuprofen tablets either under fasting conditions or immediately before meals yields quite similar serum ibuprofen concentration-time profiles. When ibuprofen tablets are administered immediately after a meal, there is a reduction in the rate of absorption but no appreciable decrease in the extent of absorption. The bioavailability of the drug is minimally altered by the presence of food.
A bioavailability study has shown that there was no interference with the absorption of ibuprofen when ibuprofen tablets were given in conjunction with an antacid containing both aluminum hydroxide and magnesium hydroxide.
Ibuprofen is rapidly metabolized and eliminated in the urine. The excretion of ibuprofen is virtually complete 24 hours after the last dose. The serum half-life is 1.8 to 2.0 hours.
Studies have shown that following ingestion of the drug, 45% to 79% of the dose was recovered in the urine within 24 hours as metabolite A (25%), (+)-2-[ p-(2hydroxymethyl-propyl) phenyl] propionic acid and metabolite B (37%), (+)-2-[ p-(2carboxypropyl)phenyl] propionic acid; the percentages of free and conjugated ibuprofen were approximately 1% and 14%, respectively.
Indications and Usage for MPM Pak
Carefully consider the potential benefits and risks of ibuprofen tablets and other treatment options before deciding to use ibuprofen. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS).
Ibuprofen tablets are indicated for relief of the signs and symptoms of rheumatoid arthritis and osteoarthritis.
Ibuprofen tablets are indicated for relief of mild to moderate pain.
Ibuprofen tablets are also indicated for the treatment of primary dysmenorrhea.
Controlled clinical trials to establish the safety and effectiveness of ibuprofen tablets in children have not been conducted.
Contraindications
Ibuprofen tablets are contraindicated in patients with known hypersensitivity to Ibuprofen.
Ibuprofen tablets should not be given to patients who have experienced asthma, urticaria, or allergic-type reactions after taking aspirin or other NSAIDs. Severe, rarely fatal, anaphylactic-like reactions to NSAIDs have been reported in such patients (see
WARNINGS,
Anaphylactoid Reactions, and
PRECAUTIONS,
Preexisting Asthma).
- In the setting of coronary artery bypass graft (CABG) surgery [see Warnings]
Warnings
Cardiovascular Effects
Cardiovascular Thrombotic Events
Clinical trials of several COX-2 selective and nonselective NSAIDs of up to three years duration have shown an increased risk of serious cardiovascular (CV) thrombotic events, including myocardial infarction (MI) and stroke, which can be fatal. Based on available data, it is unclear that the risk for CV thrombotic events is similar for all NSAIDs. The relative increase in serious CV thrombotic events over baseline conferred by NSAID use appears to be similar in those with and without known CV disease or risk factors for CV disease. However, patients with known CV disease or risk factors had a higher absolute incidence of excess serious CV thrombotic events, due to their increased baseline rate. Some observational studies found that this increased risk of serious CV thrombotic events began as early as the first weeks of treatment. The increase in CV thrombotic risk has been observed most consistently at higher doses.
To minimize the potential risk for an adverse CV event in NSAID-treated patients, use the lowest effective dose for the shortest duration possible. Physicians and patients should remain alert for the development of such events, throughout the entire treatment course, even in the absence of previous CV symptoms. Patients should be informed about the symptoms of serious CV events and the steps to take if they occur.
There is no consistent evidence that concurrent use of aspirin mitigates the increased risk of serious CV thrombotic events associated with NSAID use. The concurrent use of aspirin and an NSAID, such as ibuprofen, increases the risk of serious gastrointestinal (GI) events [ see Warnings].
Status Post Coronary Artery Bypass Graft (CABG) Surgery
Two large, controlled clinical trials of a COX-2 selective NSAID for the treatment of pain in the first 10 to 14 days following CABG surgery found an increased incidence of myocardial infarction and stroke. NSAIDs are contraindicated in the setting of CABG [ see Contraindications].
Post-MI Patients
Observational studies conducted in the Danish National Registry have demonstrated that patients treated with NSAIDs in the post-MI period were at increased risk of reinfarction, CV-related death, and all-cause mortality beginning in the first week of treatment. In this same cohort, the incidence of death in the first year post MI was 20 per 100 person years in NSAID-treated patients compared to 12 per 100 person years in non-NSAID exposed patients. Although the absolute rate of death declined somewhat after the first year post-MI, the increased relative risk of death in NSAID users persisted over at least the next four years of follow-up.
Avoid the use of ibuprofen tablets in patients with a recent MI unless the benefits are expected to outweigh the risk of recurrent CV thrombotic events. If ibuprofen tablets are used in patients with a recent MI, monitor patients for signs of cardiac ischemia.
Hypertension
NSAIDs including ibuprofen tablets, can lead to onset of new hypertension or worsening of pre-existing hypertension, either of which may contribute to the increased incidence of CV events. Patients taking thiazides or loop diuretics may have impaired response to these therapies when taking NSAIDs. NSAIDs, including ibuprofen tablets, should be used with caution in patients with hypertension. Blood pressure (BP) should be monitored closely during the initiation of NSAID treatment and throughout the course of therapy.
Heart Failure and Edema
The Coxib and traditional NSAID Trialists’ Collaboration meta-analysis of randomized controlled trials demonstrated an approximately two-fold increase in hospitalizations for heart failure in COX-2 selective-treated patients and nonselective NSAID-treated patients compared to placebo-treated patients. In a Danish National Registry study of patients with heart failure, NSAID use increased the risk of MI, hospitalization for heart failure, and death.
Additionally, fluid retention and edema have been observed in some patients treated with NSAIDs. Use of ibuprofen may blunt the CV effects of several therapeutic agents used to treat these medical conditions [e.g., diuretics, ACE inhibitors, or angiotensin receptor blockers (ARBs)] [see Drug Interactions].
Avoid the use of ibuprofen tablets in patients with severe heart failure unless the benefits are expected to outweigh the risk of worsening heart failure. If ibuprofen tablets are used in patients with severe heart failure, monitor patients for signs of worsening heart failure.
Gastrointestinal Effects - Risk of Ulceration, Bleeding, and Perforation
p>NSAIDs, including ibuprofen tablets, can cause serious gastrointestinal (GI) adverse events including inflammation, bleeding, ulceration, and perforation of the stomach, small intestine, or large intestine, which can be fatal. These serious adverse events can occur at any time, with or without warning symptoms, in patients treated with NSAIDs. Only one in five patients, who develop a serious upper GI adverse event on NSAID therapy, is symptomatic. Upper GI ulcers, gross bleeding, or perforation caused by NSAIDs occur in approximately 1% of patients treated for 3 to 6 months, and in about 2 to 4% of patients treated for one year. These trends continue with longer duration of use, increasing the likelihood of developing a serious GI event at some time during the course of therapy. However, even short-term therapy is not without risk. NSAIDs should be prescribed with extreme caution in those with a prior history of ulcer disease or gastrointestinal bleeding. Patients with a prior history of peptic ulcer disease and/or gastrointestinal bleeding who use NSAIDs have a greater than 10-fold increased risk for developing a GI bleed compared to patients treated with neither of these risk factors. Other factors that increase the risk of GI bleeding in patients treated with NSAIDs include concomitant use of oral corticosteroids or anticoagulants, longer duration of NSAID therapy, smoking, use of alcohol, older age, and poor general health status. Most spontaneous reports of fatal GI events are in elderly or debilitated patients and therefore, special care should be taken in treating this population.
To minimize the potential risk for an adverse GI event in patients treated with an NSAID, the lowest effective dose should be used for the shortest possible duration. Patients and physicians should remain alert for signs and symptoms of GI ulcerations and bleeding during NSAID therapy and promptly initiate additional evaluation and treatment if a serious GI event is suspected. This should include discontinuation of the NSAID until a serious GI adverse event is ruled out. For high-risk patients, alternate therapies that do not involve NSAIDs should be considered.
Renal Effects
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury. Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients, administration of a NSAID may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics and ACE inhibitors, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
Advanced Renal Disease
No information is available from controlled clinical studies regarding the use of ibuprofen tablets in patients with advanced renal disease. Therefore, treatment with ibuprofen tablets is not recommended in these patients with advanced renal disease. If ibuprofen tablet therapy must be initiated, close monitoring of the patients renal function is advisable.
Anaphylactoid Reactions
As with other NSAIDs, anaphylactoid reactions may occur in patients without known prior exposure to ibuprofen tablets. Ibuprofen tablets should not be given to patients with the aspirin triad. This symptom complex typically occurs in asthmatic patients who experience rhinitis with or without nasal polyps, or who exhibit severe, potentially fatal bronchospasm after taking aspirin or other NSAIDs (see CONTRAINDICATIONS and PRECAUTIONS, Preexisting Asthma). Emergency help should be sought in cases where an anaphylactoid reaction occurs.
Skin Reactions
NSAIDs, including ibuprofen tablets, can cause serious skin adverse events such as exfoliative dermatitis, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN), which can be fatal. These serious events may occur without warning. Patients should be informed about the signs and symptoms of serious skin manifestations and use of the drug should be discontinued at the first appearance of skin rash or any other sign of hypersensitivity.
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) has been reported in patients taking NSAIDs such as ibuprofen tablets. Some of these events have been fatal or life-threatening. DRESS typically, although not exclusively, presents with fever, rash, lymphadenopathy, and/or facial swelling. Other clinical manifestations may include hepatitis, nephritis, hematological abnormalities, myocarditis, or myositis. Sometimes symptoms of DRESS may resemble an acute viral infection. Eosinophilia is often present. Because this disorder is variable in its presentation, other organ systems not noted here may be involved. It is important to note that early manifestations of hypersensitivity, such as fever or lymphadenopathy, may be present even though rash is not evident. If such signs or symptoms are present, discontinue ibuprofen tablets and evaluate the patient immediately.
Fetal Toxicity
Premature Closure of Fetal Ductus Arteriosus:
Avoid use of NSAIDs, including ibuprofen tablets, in pregnant women at about 30 weeks gestation and later. NSAIDs including ibuprofen tablets, increase the risk of premature closure of the fetal ductus arteriosus at approximately this gestational age.
Oligohydramnios/Neonatal Renal Impairment:
Use of NSAIDs, including ibuprofen tablets, at about 20 weeks gestation or later in pregnancy may cause fetal renal dysfunction leading to oligohydramnios and, in some cases, neonatal renal impairment. These adverse outcomes are seen, on average, after days to weeks of treatment, although oligohydramnios has been infrequently reported as soon as 48 hours after NSAID initiation. Oligohydramnios is often, but not always, reversible with treatment discontinuation. Complications of prolonged oligohydramnios may, for example, include limb contractures and delayed lung maturation. In some postmarketing cases of impaired neonatal renal function, invasive procedures such as exchange transfusion or dialysis were required.
If NSAID treatment is necessary between about 20 weeks and 30 weeks gestation, limit ibuprofen tablets use to the lowest effective dose and shortest duration possible. Consider ultrasound monitoring of amniotic fluid if ibuprofen tablets treatment extends beyond 48 hours. Discontinue ibuprofen tablets if oligohydramnios occurs and follow up according to clinical practice [ see PRECAUTIONS; Pregnancy] .
Precautions
General
Ibuprofen tablets cannot be expected to substitute for corticosteroids or to treat corticosteroid insufficiency. Abrupt discontinuation of corticosteroids may lead to disease exacerbation. Patients on prolonged corticosteroid therapy should have their therapy tapered slowly if a decision is made to discontinue corticosteroids.
The pharmacological activity of ibuprofen tablets in reducing fever and inflammation may diminish the utility of these diagnostic signs in detecting complications of presumed noninfectious, painful conditions.
Hepatic effects
Borderline elevations of one or more liver tests may occur in up to 15% of patients taking NSAIDs, including ibuprofen tablets. These laboratory abnormalities may progress, may remain unchanged, or may be transient with continuing therapy. Notable elevations of ALT or AST (approximately three or more times the upper limit of normal) have been reported in approximately 1% of patients in clinical trials with NSAIDs. In addition, rare cases of severe hepatic reactions, including jaundice, fulminant hepatitis, liver necrosis, and hepatic failure, some of them with fatal outcomes have been reported.
A patient with symptoms and/or signs suggesting liver dysfunction, or with abnormal liver test values, should be evaluated for evidence of the development of a more severe hepatic reaction while on therapy with ibuprofen tablets. If clinical signs and symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash, etc.), ibuprofen tablets should be discontinued.
Hematological effects
Anemia is sometimes seen in patients receiving NSAIDs, including ibuprofen tablets. This may be due to fluid retention, occult or gross GI blood loss, or an incompletely described effect upon erythropoiesis. Patients on long-term treatment with NSAIDs, including ibuprofen tablets, should have their hemoglobin or hematocrit checked if they exhibit any signs or symptoms of anemia.
In two postmarketing clinical studies the incidence of a decreased hemoglobin level was greater than previously reported. Decrease in hemoglobin of 1 gram or more was observed in 17.1% of 193 patients on 1600 mg ibuprofen daily (osteoarthritis), and in 22.8% of 189 patients taking 2400 mg of ibuprofen daily (rheumatoid arthritis). Positive stool occult blood tests and elevated serum creatinine levels were also observed in these studies.
NSAIDs inhibit platelet aggregation and have been shown to prolong bleeding time in some patients. Unlike aspirin, their effect on platelet function is quantitatively less, of shorter duration, and reversible.
Patients receiving ibuprofen tablets who may be adversely affected by alterations in platelet function, such as those with coagulation disorders or patients receiving anticoagulants should be carefully monitored.
Preexisting asthma
Patients with asthma may have aspirin-sensitive asthma. The use of aspirin in patients with aspirin-sensitive asthma has been associated with severe bronchospasm, which can be fatal. Since cross reactivity, including bronchospasm, between aspirin and NSAIDs has been reported in such aspirin-sensitive patients, ibuprofen tablets should not be administered to patients with this form of aspirin sensitivity and should be used with caution in patients with preexisting asthma.
Ophthalmological effects
Blurred and/or diminished vision, scotomata, and/or changes in color vision have been reported. If a patient develops such complaints while receiving ibuprofen tablets, the drug should be discontinued, and the patient should have an ophthalmologic examination which includes central visual fields and color vision testing.
Aseptic Meningitis
Aseptic meningitis with fever and coma has been observed on rare occasions in patients on ibuprofen therapy. Although it is probably more likely to occur in patients with systemic lupus erythematosus and related connective tissue diseases, it has been reported in patients who do not have an underlying chronic disease. If signs or symptoms of meningitis develop in a patient on ibuprofen tablets, the possibility of its being related to ibuprofen tablets should be considered.
Information for Patients
Patients should be informed of the following information before initiating therapy with an NSAID and periodically during the course of ongoing therapy. Patients should also be encouraged to read the NSAID Medication Guide that accompanies each prescription dispensed.
- Cardiovascular Thrombotic Events
Advise patients to be alert for the symptoms of cardiovascular thrombotic events, including chest pain, shortness of breath, weakness, or slurring of speech, and to report any of these symptoms to their health care provider immediately [ see Warnings].
- Ibuprofen tablets, like other NSAIDs, can cause GI discomfort and, rarely, serious GI side effects, such as ulcers and bleeding, which may result in hospitalization and even death. Although serious GI tract ulcerations and bleeding can occur without warning symptoms, patients should be alert for the signs and symptoms of ulcerations and bleeding, and should ask for medical advice when observing any indicative signs or symptoms including epigastric pain, dyspepsia, melena, and hematemesis. Patients should be apprised of the importance of this follow-up (see WARNINGS, Gastrointestinal Effects- Risk of Ulceration, Bleeding and Perforation).
- Serious Skin Reactions, including DRESS
Advise patients to stop taking ibuprofen tablets immediately if they develop any type of rash or fever and to contact their healthcare provider as soon as possible [ see Warnings].
- Heart Failure And Edema
Advise patients to be alert for the symptoms of congestive heart failure including shortness of breath, unexplained weight gain, or edema and to contact their healthcare provider if such symptoms occur [ see Warnings] .
- Patients should be informed of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, jaundice, right upper quadrant tenderness and "flu-like" symptoms). If these occur, patients should be instructed to stop therapy and seek immediate medical therapy.
- Patients should be informed of the signs of an anaphylactoid reaction (e.g. difficulty breathing, swelling of the face or throat). If these occur, patients should be instructed to seek immediate emergency help (see WARNINGS).
- Fetal Toxicity Inform pregnant women to avoid use of ibuprofen tablets and other NSAIDs starting at 30 weeks gestation because of the risk of the premature closing of the fetal ductus arteriosus. If treatment with ibuprofen tablets is needed for a pregnant woman between about 20 to 30 weeks gestation, advise her that she may need to be monitored for oligohydramnios, if treatment continues for longer than 48 hours [ see WARNINGS; Fetal Toxicity, PRECAUTIONS; Pregnancy] .
Laboratory Tests
Because serious GI tract ulcerations and bleeding can occur without warning symptoms, physicians should monitor for signs or symptoms of GI bleeding. Patients on long-term treatment with NSAIDs should have their CBC and chemistry profile checked periodically. If clinical signs and symptoms consistent with liver or renal disease develop, systemic manifestations occur (e.g., eosinophilia, rash etc.), or abnormal liver tests persist or worsen, ibuprofen tablets should be discontinued.
Laboratory Tests
Because serious GI tract ulcerations and bleeding can occur without warning symptoms, physicians should monitor for signs or symptoms of GI bleeding. Patients on long-term treatment with NSAIDs should have their CBC and chemistry profile checked periodically. If clinical signs and symptoms consistent with liver or renal disease develop, systemic manifestations occur (e.g., eosinophilia, rash etc.), or abnormal liver tests persist or worsen, ibuprofen tablets should be discontinued.
ACE-inhibitors
Reports suggest that NSAIDs may diminish the antihypertensive effect of ACE-inhibitors. This interaction should be given consideration in patients taking NSAIDs concomitantly with ACE-inhibitors.
Aspirin
Pharmacodynamic studies have demonstrated interference with the antiplatelet activity of aspirin when ibuprofen 400 mg, given three times daily, is administered with enteric-coated low-dose aspirin. The interaction exists even following a once-daily regimen of ibuprofen 400 mg, particularly when ibuprofen is dosed prior to aspirin. The interaction is alleviated if immediate-release low-dose aspirin is dosed at least 2 hours prior to a once-daily regimen of ibuprofen; however, this finding cannot be extended to enteric-coated low-dose aspirin [see Clinical Pharmacology/Pharmacodynamics].
Because there may be an increased risk of cardiovascular events due to the interference of ibuprofen with the antiplatelet effect of aspirin, for patients taking low-dose aspirin for cardioprotection who require analgesics, consider use of an NSAID that does not interfere with the antiplatelet effect of aspirin, or non-NSAID analgesics, where appropriate.
When ibuprofen tablets are administered with aspirin, its protein binding is reduced, although the clearance of free ibuprofen tablets is not altered. The clinical significance of this interaction is not known; however, as with other NSAIDs, concomitant administration of ibuprofen and aspirin is not generally recommended because of the potential for increased adverse effects.
Diuretics
Clinical studies, as well as post marketing observations, have shown that ibuprofen tablets can reduce the natriuretic effect-of furosemide and thiazides in some patients. This response has been attributed to inhibition of renal prostaglandin synthesis. During concomitant therapy with NSAIDs, the patient should be observed closely for signs of renal failure (see WARNINGS, Renal Effects), as well as to assure diuretic efficacy.
Lithium
Ibuprofen produced an elevation of plasma lithium levels and a reduction in renal lithium clearance in a study of eleven normal volunteers. The mean minimum lithium concentration increased 15% and the renal clearance of lithium was decreased by 19% during this period of concomitant drug administration. This effect has been attributed to inhibition of renal prostaglandin synthesis by ibuprofen. Thus, when ibuprofen and lithium are administered concurrently, subjects should be observed carefully for signs of lithium toxicity. (Read circulars for lithium preparation before use of such concurrent therapy.)
Lithium
Ibuprofen produced an elevation of plasma lithium levels and a reduction in renal lithium clearance in a study of eleven normal volunteers. The mean minimum lithium concentration increased 15% and the renal clearance of lithium was decreased by 19% during this period of concomitant drug administration. This effect has been attributed to inhibition of renal prostaglandin synthesis by ibuprofen. Thus, when ibuprofen and lithium are administered concurrently, subjects should be observed carefully for signs of lithium toxicity. (Read circulars for lithium preparation before use of such concurrent therapy.)
Methotrexate
NSAIDs have been reported to competitively inhibit methotrexate accumulation in rabbit kidney slices. This may indicate that they could enhance the toxicity of methotrexate. Caution should be used when NSAIDs are administered concomitantly with methotrexate.
Warfarin-type anticoagulants
Several short-term controlled studies failed to show that ibuprofen tablets significantly affected prothrombin times or a variety of other clotting factors when administered to individuals on coumarin-type anticoagulants. However, because bleeding has been reported when ibuprofen tablets and other NSAIDs have been administered to patients on coumarin-type anticoagulants, the physician should be cautious when administering ibuprofen tablets to patients on anticoagulants. The effects of warfarin and NSAIDs on GI bleeding are synergistic, such that the users of both drugs together have a risk of serious GI bleeding higher than users of either drug alone.
H-2 Antagonists
In studies with human volunteers, co-administration of cimetidine or ranitidine with ibuprofen had no substantive effect on ibuprofen serum concentrations.
Pregnancy
Risk Summary
Use of NSAIDs, including ibuprofen tablets, can cause premature closure of the fetal ductus arteriosus and fetal renal dysfunction leading to oligohydramnios and, in some cases, neonatal renal impairment. Because of these risks, limit dose and duration of ibuprofen tablets use between about 20 and 30 weeks of gestation, and avoid ibuprofen tablets use at about 30 weeks of gestation and later in pregnancy [ see WARNINGS; Fetal Toxicity].
Premature Closure of Fetal Ductus Arteriosus
Use of NSAIDs, including ibuprofen tablets, at about 30 weeks gestation or later in pregnancy increases the risk of premature closure of the fetal ductus arteriosus.
Oligohydramnios/Neonatal Renal Impairment
Use of NSAIDs at about 20 weeks gestation or later in pregnancy has been associated with cases of fetal renal dysfunction leading to oligohydramnios, and in some cases, neonatal renal impairment.
Data from observational studies regarding other potential embryofetal risks of NSAID use in women in the first or second trimesters of pregnancy are inconclusive. Reproductive studies conducted in rats and rabbits have not demonstrated evidence of developmental abnormalities. However, animal reproduction studies are not always predictive of human response. Based on animal data, prostaglandins have been shown to have an important role in endometrial vascular permeability, blastocyst implantation, and decidualization. In animal studies, administration of prostaglandin synthesis inhibitors such as ibuprofen, resulted in increased pre- and post-implantation loss. Prostaglandins also have been shown to have an important role in fetal kidney development. In published animal studies, prostaglandin synthesis inhibitors have been reported to impair kidney development when administered at clinically relevant doses.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Premature Closure of Fetal Ductus Arteriosus:
Avoid use of NSAIDs in women at about 30 weeks gestation and later in pregnancy, because NSAIDs, including ibuprofen tablets, can cause premature closure of the fetal ductus arteriosus (see WARNINGS; Fetal Toxicity) .
Oligohydramnios/Neonatal Renal Impairment
If an NSAID is necessary at about 20 weeks gestation or later in pregnancy, limit the use to the lowest effective dose and shortest duration possible. If ibuprofen tablets treatment extends beyond 48 hours, consider monitoring with ultrasound for oligohydramnios. If oligohydramnios occurs, discontinue ibuprofen tablets and follow up according to clinical practice (see WARNINGS; Fetal Toxicity) .
Data
Human Data
There are no adequate, well-controlled studies in pregnant women. Ibuprofen tablets should be used in pregnancy only if the potential benefit justifies the potential risk to the fetus.
Premature Closure of Fetal Ductus Arteriosus:
Published literature reports that the use of NSAIDs at about 30 weeks of gestation and later in pregnancy may cause premature closure of the fetal ductus arteriosus.
Oligohydramnios/Neonatal Renal Impairment:
Published studies and postmarketing reports describe maternal NSAID use at about 20 weeks gestation or later in pregnancy associated with fetal renal dysfunction leading to oligohydramnios, and in some cases, neonatal renal impairment. These adverse outcomes are seen, on average, after days to weeks of treatment, although oligohydramnios has been infrequently reported as soon as 48 hours after NSAID initiation. In many cases, but not all, the decrease in amniotic fluid was transient and reversible with cessation of the drug. There have been a limited number of case reports of maternal NSAID use and neonatal renal dysfunction without oligohydramnios, some of which were irreversible. Some cases of neonatal renal dysfunction required treatment with invasive procedures, such as exchange transfusion or dialysis.
Methodological limitations of these postmarketing studies and reports include lack of a control group; limited information regarding dose, duration, and timing of drug exposure; and concomitant use of other medications. These limitations preclude establishing a reliable estimate of the risk of adverse fetal and neonatal outcomes with maternal NSAID use. Because the published safety data on neonatal outcomes involved mostly preterm infants, the generalizability of certain reported risks to the full-term infant exposed to NSAIDs through maternal use is uncertain.
Labor and Delivery
In rat studies with NSAIDs, as with other drugs known to inhibit prostaglandin synthesis, an increased incidence of dystocia, delayed parturition, and decreased pup survival occurred. The effects of ibuprofen tablets on labor and delivery in pregnant women are unknown.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human-milk and because of the potential for serious adverse reactions in nursing infants from ibuprofen tablets, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Adverse Reactions/Side Effects
The most frequent type of adverse reaction occurring with ibuprofen tablets is gastrointestinal. In controlled clinical trials the percentage of patients reporting one or more gastrointestinal complaints ranged from 4% to 16%.
In controlled studies when ibuprofen tablets were compared to aspirin and indomethacin in equally effective doses, the overall incidence of gastrointestinal complaints was about half that seen in either the aspirin- or indomethacin-treated patients.
Adverse reactions observed during controlled clinical trials at an incidence greater than 1% are listed in the table. Those reactions listed in Column one encompass observations in approximately 3,000 patients. More than 500 of these patients were treated for periods of at least 54 weeks.
Still other reactions occurring less frequently than 1 in 100 were reported in controlled clinical trials and from marketing experience. These reactions have been divided into two categories: Column two of the table lists reactions with therapy with ibuprofen tablets where the probability of a causal relationship exists: for the reactions in Column three, a causal relationship with ibuprofen tablets has not been established.
Reported side effects were higher at doses of 3200 mg/day than at doses of 2400 mg or less per day in clinical trials of patients with rheumatoid arthritis. The increases in incidence were slight and still within the ranges reported in the table.
| Incidence Greater than 1%
(but less than 3%) Probable Causal Relationship | Precise Incidence Unknown
(but less than 1%) Probable Causal Relationship* | Precise Incidence
Unknown (but less than 1%) Causal Relationship Unknown* |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| *Reactions are classified under
"Probable Causal Relationship (PCR)" if there has been one positive rechallenge or if three or more cases occur which might be causally related. Reactions are classified under
"Causal Relationship Unknown" if seven or more events have been reported but the criteria for PCR have not been met.
†Reactions occurring in 3% to 9% of patients treated with ibuprofen. (Those reactions occurring in less than 3% of the patients are unmarked). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GASTROINTESTINAL
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Nausea†, epigastric pain†, heartburn†, diarrhea, abdominal distress, nausea and vomiting, indigestion, constipation, abdominal cramps or Pain, fullness of GI tract (bloating and flatulence)
| Gastric or duodenal ulcer with
bleeding and/or perforation, gastrointestinal hemorrhage, melena, gastritis, hepatitis, jaundice, abnormal liver function tests; pancreatitis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CENTRAL NERVOUS SYSTEM
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Dizziness†, headache, nervousness
| Depression, insomnia, confusion,
emotional liability, somnolence, aseptic meningitis with fever and coma (see PRECAUTIONS) | Paresthesias, hallucinations,
dream abnormalities, pseudo-tumor cerebri |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DERMATOLOGIC
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Rash† (including maculopapular type), pruritus |
Vesiculobullous eruptions, urticaria, erythema multiforme, Stevens- Johnson syndrome, alopecia |
Toxic epidermal necrolysis, photoallergic skin reactions |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SPECIAL SENSES
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Tinnitus |
Hearing loss, amblyopia (blurred and/or diminished vision, scotomata and/or changes in color vision) (see PRECAUTIONS) |
Conjunctivitis, diplopia, optic neuritis, cataracts |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HEMATOLOGIC
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
| Neutropenia, agranulocytosis, aplastic anemia, hemolytic anemia
(sometimes Coombs positive), thrombocytopenia with or without purpura, eosinophilia, decreases in hemoglobin and hematocrit (see PRECAUTIONS) |
Bleeding episodes (eg epistaxis, menorrhagia) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| METABOLIC/ENDOCRINE
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Decreased appetite |
|
Gynecomastia, hypoglycemic reaction, acidosis |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CARDIOVASCULAR
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Edema, fluid retention (generally
responds promptly to drug discontinuation) (see PRECAUTIONS) | Congestive heart failure in patients
with marginal cardiac function, elevated blood pressure, palpitations | Arrhythmias (sinus
tachycardia, sinus bradycardia) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ALLERGIC
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
| Syndrome of abdominal pain, fever,
chills, nausea and vomiting; anaphylaxis; bronchospasm (see CONTRAINDICATIONS) | Serum sickness, lupus erythematosus syndrome. Henoch- Schonlein vasculitis,
angioedema |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RENAL
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
| Acute renal failure (see
PRECAUTIONS), decreased creatinine clearance, polyuria, azotemia, cystitis, Hematuria | Renal papillary
necrosis |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MISCELLANEOUS
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
| Dry eyes and mouth, gingival ulcer,
rhinitis |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Overdosage
Approximately 1½ hours after the reported ingestion of from 7 to 10 ibuprofen tablets (400 mg), a 19-month old child weighing 12 kg was seen in the hospital emergency room, apneic and cyanotic, responding only to painful stimuli. This type of stimulus, however, was sufficient to induce respiration. Oxygen and parenteral fluids were given; a greenish-yellow fluid was aspirated from the stomach with no evidence to indicate the presence of ibuprofen. Two hours after ingestion the child's condition seemed stable; she still responded only to painful stimuli and continued to have periods of apnea lasting from 5 to 10 seconds. She was admitted to intensive care and sodium bicarbonate was administered as well as infusions of dextrose and normal saline. By four hours post-ingestion she could be aroused easily, sit by herself and respond to spoken commands. Blood level of ibuprofen was 102.9 mcg/mL approximately 8½ hours after accidental ingestion. At 12 hours she appeared to be completely recovered.
In two other reported cases where children (each weighing approximately 10 kg) accidentally, acutely ingested approximately 120 mg/kg, there were no signs of acute intoxication or late sequelae. Blood level in one child 90 minutes after ingestion was 700 mcg/mL — about 10 times the peak levels seen in absorption-excretion studies.
A 19-year old male who had taken 8,000 mg of ibuprofen over a period of a few hours complained of dizziness, and nystagmus was noted. After hospitalization, parenteral hydration and three days bed rest, he recovered with no reported sequelae.
In cases of acute overdosage, the stomach should be emptied by vomiting or lavage, though little drug will likely be recovered if more than an hour has elapsed since ingestion. Because the drug is acidic and is excreted in the urine, it is theoretically beneficial to administer alkali and induce diuresis. In addition to supportive measures, the use of oral activated charcoal may help to reduce the absorption and reabsorption of ibuprofen tablets.
Overdosage
Approximately 1½ hours after the reported ingestion of from 7 to 10 ibuprofen tablets (400 mg), a 19-month old child weighing 12 kg was seen in the hospital emergency room, apneic and cyanotic, responding only to painful stimuli. This type of stimulus, however, was sufficient to induce respiration. Oxygen and parenteral fluids were given; a greenish-yellow fluid was aspirated from the stomach with no evidence to indicate the presence of ibuprofen. Two hours after ingestion the child's condition seemed stable; she still responded only to painful stimuli and continued to have periods of apnea lasting from 5 to 10 seconds. She was admitted to intensive care and sodium bicarbonate was administered as well as infusions of dextrose and normal saline. By four hours post-ingestion she could be aroused easily, sit by herself and respond to spoken commands. Blood level of ibuprofen was 102.9 mcg/mL approximately 8½ hours after accidental ingestion. At 12 hours she appeared to be completely recovered.
In two other reported cases where children (each weighing approximately 10 kg) accidentally, acutely ingested approximately 120 mg/kg, there were no signs of acute intoxication or late sequelae. Blood level in one child 90 minutes after ingestion was 700 mcg/mL — about 10 times the peak levels seen in absorption-excretion studies.
A 19-year old male who had taken 8,000 mg of ibuprofen over a period of a few hours complained of dizziness, and nystagmus was noted. After hospitalization, parenteral hydration and three days bed rest, he recovered with no reported sequelae.
In cases of acute overdosage, the stomach should be emptied by vomiting or lavage, though little drug will likely be recovered if more than an hour has elapsed since ingestion. Because the drug is acidic and is excreted in the urine, it is theoretically beneficial to administer alkali and induce diuresis. In addition to supportive measures, the use of oral activated charcoal may help to reduce the absorption and reabsorption of ibuprofen tablets.
MPM Pak Dosage and Administration
Carefully consider the potential benefits and risks of ibuprofen tablets and other treatment options before deciding to use ibuprofen tablets. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS).
After observing the response to initial therapy with ibuprofen tablets, the dose and frequency should be adjusted to suit an individual patient's needs.
Do not exceed 3200 mg total daily dose. If gastrointestinal complaints occur, administer ibuprofen tablets with meals or milk.
Rheumatoid arthritis and osteoarthritis, including flare-ups of chronic disease
Suggested Dosage: 1200 mg to 3200 mg daily (300 mg qid; 400 mg, 600 mg or 800 mg tid or qid). Individual patients may show a better response to 3200 mg daily, as compared with 2400 mg, although in well-controlled clinical trials patients on 3200 mg did not show a better mean response in terms of efficacy. Therefore, when treating patients with 3200 mg/day, the physician should observe sufficient increased clinical benefits to offset potential increased risk.
The dose should be tailored to each patient, and may be lowered or raised depending on the severity of symptoms either at time of initiating drug therapy or as the patient responds or fails to respond.
In general, patients with rheumatoid arthritis seem to require higher doses of ibuprofen tablets than do patients with osteoarthritis.
The smallest dose of ibuprofen tablets that yields acceptable control should be employed. A linear blood level doseresponse relationship exists with single doses up to 800 mg (See CLINICAL PHARMACOLOGY for effects of food on rate of absorption).
The availability of three tablet strengths facilitates dosage adjustment.
In chronic conditions, a therapeutic response to therapy with ibuprofen tablets is sometimes seen in a few days to a week but most often is observed by two weeks. After a satisfactory response has been achieved, the patient's dose should be reviewed and adjusted as required.
Mild to moderate pain: 400 mg every 4 to 6 hours as necessary for relief of pain.
In controlled analgesic clinical trials, doses of ibuprofen tablets greater than 400 mg were no more effective than the 400 mg dose.
How is MPM Pak supplied
Ibuprofen Tablets USP, 800 mg are white to off-white, film-coated, capsule shaped tablets debossed with ‘I8’ on one side and plain on the other side.
NDC 68071-2903-7 BOTTLES OF 12
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature]. Avoid excessive heat 40ºC (104ºF).
Dispense with Medication Guide available at: www.aurobindousa.com/medication-guides.
Distributed by:
Aurobindo Pharma USA, Inc.
279 Princeton-Hightstown Road
East Windsor, NJ 08520
Manufactured by:
Aurobindo Pharma Limited
Hyderabad–500 032, India
Revised: 05/2021
| Medication Guide for Nonsteroidal Anti-inflammatory Drugs (NSAIDs)
|
| What is the most important information I should know about medicines called Nonsteroidal Anti-inflammatory Drugs (NSAIDs)?
NSAIDs can cause serious side effects, including:
Do not take NSAIDs right before or after a heart surgery called a "coronary artery bypass graft (CABG)." Avoid taking NSAIDs after a recent heart attack, unless your healthcare provider tells you to. You may have an increased risk of another heart attack if you take NSAIDs after a recent heart attack.
|
| What are NSAIDs?
NSAIDs are used to treat pain and redness, swelling, and heat (inflammation) from medical conditions such as different types of arthritis, menstrual cramps, and other types of short-term pain. |
| Who should not take NSAIDs?
Do not take NSAIDs:
|
Before taking NSAIDs, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all of the medicines you take, including prescription or over-the-counter medicines, vitamins or herbal supplements. NSAIDs and some other medicines can interact with each other and cause serious side effects. Do not start taking any new medicine without talking to your healthcare provider first. |
| What are the possible side effects of NSAIDs?
NSAIDs can cause serious side effects, including: See “What is the most important information I should know about medicines called Nonsteroidal Anti-inflammatory Drugs (NSAIDs)?”
These are not all the possible side effects of NSAIDs. For more information, ask your healthcare provider or pharmacist about NSAIDs. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
Other information about NSAIDs
|
| General information about the safe and effective us e of NSAIDs
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use NSAIDs for a condition for which it was not prescribed. Do not give NSAIDs to other people, even if they have the same symptoms that you have. It may harm them. If you would like more information about NSAIDs, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about NSAIDs that is written for health professionals. |
| Dispense with Medication Guide available at:
www.aurobindousa.com/medication-guides.
Distributed by: Aurobindo Pharma USA, Inc. 279 Princeton-Hightstown Road East Windsor, NJ 08520 Manufactured by: Aurobindo Pharma Limited Hyderabad-500 032, India For more information, call Aurobindo Pharma USA, Inc. at 1-866-850-2876. |
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: 05/2021





| MPM PAK
mpm pak kit |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Labeler - NuCare Pharmaceuticals,Inc. (010632300) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| NuCare Pharmaceuticals,Inc. | 010632300 | manufacture(70859-060) | |