Leflunomide Prescribing Information
Package insert / product label
Dosage form: tablet
Drug classes: Antirheumatics, Selective immunosuppressants
Medically reviewed by Drugs.com. Last updated on Aug 18, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
LEFLUNOMIDE tablets, for oral use
Initial U.S. Approval: 1998
See full prescribing information for complete boxed warning.
Embryo-Fetal Toxicity
- Teratogenicity and embryo-lethality occurred in animals administeredleflunomide. (5.1, 8.1)
- Exclude pregnancy prior to initiating leflunomide therapy. (5.1, 8.3)
- Advise use of effective contraception in females of reproductive potential during treatment and during a drug elimination procedure. (5.1, 5.3, 8.3)
- Stop leflunomide and use an accelerated drug elimination procedure if the patient becomes pregnant. (5.1, 5.3, 8.1)
Hepatotoxicity
- Severe liver injury and fatal liver failure have been reported. (5.2)
- Avoid leflunomide use in patients with pre-existing liver disease, or those with serum alanine aminotransferase (ALT) >2 x ULN. (5.2, 8.6)
- Use caution when leflunomide is given with other potentially hepatotoxic drugs. (5.2)
- Monitor ALT levels. Interrupt leflunomide treatment if ALT elevation >3-fold ULN. If likely leflunomide-induced, start accelerated drug elimination procedure and monitor liver tests weekly until normalized. (5.2, 5.3)
Indications and Usage for Leflunomide
Leflunomide is a pyrimidine synthesis inhibitor indicated for the treatment of adults with active rheumatoid arthritis. (1)
Leflunomide Dosage and Administration
- Loading dosage for patients at low risk for leflunomide-associated hepatotoxicity and leflunomide-associated myelosuppression: 100 mg daily for 3 days. (2.1)
- Maintenance dosage: 20 mg daily. (2.1)
- Maximum recommended daily dosage: 20 mg once daily. (2.1)
- If 20 mg once daily is not tolerated, may decrease dosage to 10 mg once daily. (2.1)
- Screen patients for active and latent tuberculosis, pregnancy test (females), blood pressure, and laboratory tests before starting leflunomide. (2.2)
Dosage Forms and Strengths
Tablets: 10 mg, 20 mg. (3)
Contraindications
Warnings and Precautions
- After stopping leflunomide, it is recommended that an accelerated drug elimination procedure be used to reduce the plasma concentrations of the active metabolite, teriflunomide. (5.3)
- Severe infections (including sepsis), pancytopenia, agranulocytosis and thrombocytopenia: Stop leflunomide and use accelerated elimination procedure. Do not start leflunomide in patients with active infection. Monitor CBCs during treatment with leflunomide. (5.4)
- Stevens-Johnson syndrome and toxic epidermal necrolysis, drug reaction with eosinophilia and systemic symptoms (DRESS): Stop leflunomide and use accelerated elimination procedure. (5.5)
- Skin ulcers: If skin ulcer is suspected, discontinue leflunomide treatment and consider an accelerated drug elimination procedure. (5.6)
- Peripheral neuropathy: If patient develops symptoms consistent with peripheral neuropathy, evaluate patient and consider discontinuing leflunomide. (5.8)
- Interstitial lung disease: May be fatal. New onset or worsening symptoms may necessitate discontinuation of leflunomide and initiation of accelerated elimination procedure. (5.9)
- Increased blood pressure: Monitor and treat. (5.11)
Adverse Reactions/Side Effects
- The most commonly reported adverse reactions (≥10%) regardless of relation to leflunomide treatment were diarrhea, respiratory infection, nausea, headache, rash, abnormal liver enzymes, and dyspepsia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Zydus Pharmaceuticals (USA) Inc. at 1-877-993-8779 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Drugs metabolized by CYP2C8 and OAT3 transporters: Monitor patients because teriflunomide may increase exposure of these drugs. (7)
- Teriflunomide may increase exposure of ethinylestradiol and levonorgestrel. Choose an appropriate oral contraceptive. (7)
- Drugs metabolized by CYP1A2: Monitor patients because teriflunomide may decrease exposure of these drugs. (7)
- Warfarin: Monitor INR as teriflunomide may decrease INR. (7)
- Drugs metabolized by BCRP and OATP1B1/B3 transporters: Monitor patients because teriflunomide may increase exposure of these drugs. (7)
- Rosuvastatin: The dose of rosuvastatin should not exceed 10 mg once daily in patients taking leflunomide. (7)
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2024
Full Prescribing Information
WARNING: EMBRYO-FETAL TOXICITY and HEPATOTOXICITY
WARNING: EMBRYO-FETAL TOXICITY and HEPATOTOXICITY
Embryo-Fetal Toxicity
Leflunomide is contraindicated for use in pregnant women because of the potential for fetal harm. Teratogenicity and embryo-lethality occurred in animals administered leflunomide at doses lower than the human exposure level. Exclude pregnancy before the start of treatment with leflunomide in females of reproductive potential. Advise females of reproductive potential to use effective contraception during leflunomide treatment and during an accelerated drug elimination procedure after leflunomide treatment. Stop leflunomide and use an accelerated drug elimination procedure if the patient becomes pregnant. [see Contraindications (4), Warnings and Precautions (5.1, 5.3), Use in Specific Populations (8.1, 8.3)], and Clinical Pharmacology (12.3)].
Hepatotoxicity
Severe liver injury, including fatal liver failure, has been reported in patients treated with leflunomide. Leflunomide is contraindicated in patients with severe hepatic impairment. Concomitant use of leflunomide with other potentially hepatotoxic drugs may increase the risk of liver injury. Patients with pre-existing acute or chronic liver disease, or those with serum alanine aminotransferase (ALT) >2 x ULN before initiating treatment, are at increased risk and should not be treated with leflunomide. Monitor ALT levels at least monthly for six months after starting leflunomide, and thereafter every 6 to 8 weeks. If leflunomide-induced liver injury is suspected, stop leflunomide treatment, start an accelerated drug elimination procedure, and monitor liver tests weekly until normalized [see Contraindications (4), Warnings and Precautions (5.2, 5.3), Use in Specific Populations (8.6)].
1. Indications and Usage for Leflunomide
Leflunomide tablets are indicated for the treatment of adults with active rheumatoid arthritis (RA).
2. Leflunomide Dosage and Administration
2.1 Recommended Dosage
The recommended dosage of leflunomide is 20 mg once daily. Treatment may be initiated with or without a loading dose, depending upon the patient's risk of leflunomide-associated hepatotoxicity and leflunomide-associated myelosuppression. The loading dose provides steady-state concentrations more rapidly.
- For patients who are at low risk for leflunomide-associated hepatotoxicity and leflunomide-associated myelosuppression the recommended leflunomide loading dose is 100 mg once daily for 3 days. Subsequently administer 20 mg once daily.
- For patients at high risk for leflunomide-associated hepatotoxicity (e.g., those taking concomitant methotrexate) or leflunomide-associated myelosuppression (e.g., patients taking concomitant immunosuppressants), the recommended leflunomide dosage is 20 mg once daily without a loading dose [see Warnings and Precautions (5.2, 5.4)].
The maximum recommended daily dosage is 20 mg once per day. Consider dosage reduction to 10 mg once daily for patients who are not able to tolerate 20 mg daily (i.e., for patients who experience any adverse events listed in Table 1).
Monitor patients carefully after dose reduction and after stopping therapy with leflunomide, since the active metabolite of leflunomide, teriflunomide, is slowly eliminated from the plasma [see Clinical Pharmacology (12.3)]. After stopping leflunomide treatment, an accelerated drug elimination procedure is recommended to reduce the plasma concentrations of the active metabolite, teriflunomide [see Warnings and Precautions (5.3)]. Without use of an accelerated drug elimination procedure, it may take up to 2 years to reach undetectable plasma teriflunomide concentrations after stopping leflunomide [see Clinical Pharmacology (12.3)].
2.2 Evaluation and Testing Prior to Starting Leflunomide
Prior to starting leflunomide treatment the following evaluations and tests are recommended:
- Evaluate patients for active tuberculosis and screen patients for latent tuberculosis infection [see Warnings and Precautions (5.4)]
- Laboratory tests including serum alanine aminotransferase (ALT); and white blood cell, hemoglobin or hematocrit, and platelet counts [see Warnings and Precautions (5.2, 5.4)]
- For females of reproductive potential, pregnancy testing [see Warnings and Precautions (5.1)]
- Check blood pressure [see Warnings and Precautions (5.11]
3. Dosage Forms and Strengths
Leflunomide tablets, USP are available in two strengths:
- Tablets: 10 mg, white to off white, round, film-coated tablets debossed with "57" on one side and "11" on the other side.
- Tablets: 20 mg, white to off white, triangular, film-coated tablets debossed with "58" on one side and "11" on the other side.
4. Contraindications
Leflunomide is contraindicated in:
- Pregnant women. Leflunomide may cause fetal harm. If a woman becomes pregnant while taking this drug, stop leflunomide, apprise the patient of the potential hazard to the fetus, and begin a drug elimination procedure [see Warnings and Precautions (5.1 , 5.3) and Use in Specific Populations (8.1)].
- Patients with severe hepatic impairment [see Warnings and Precautions (5.2)].
- Patients with known hypersensitivity to leflunomide or any of the other components of leflunomide tablets. Known reactions include anaphylaxis [see Adverse Reactions (6.1)].
- Patients being treated with teriflunomide [see Drug Interactions (7)].
5. Warnings and Precautions
5.1 Embryo-Fetal Toxicity
Leflunomide may cause fetal harm when administered to a pregnant woman. Teratogenicity and embryo-lethality occurred in animal reproduction studies with leflunomide at doses lower than the human exposure level [see Use in Specific Populations (8.1)].
Leflunomide is contraindicated for use in pregnant women [see Contraindications (4)]. Exclude pregnancy before starting treatment with leflunomide in females of reproductive potential [see Dosage and Administration (2.2)]. Advise females of reproductive potential to use effective contraception during leflunomide treatment and during an accelerated drug elimination procedure after leflunomide treatment [see Use in Specific Populations (8.3)]. If a woman becomes pregnant while taking leflunomide, stop treatment with leflunomide, apprise the patient of the potential risk to a fetus, and perform an accelerated drug elimination procedure to achieve nondetectable plasma concentrations of teriflunomide, the active metabolite of leflunomide [see Warnings and Precautions (5.3)].
Upon discontinuing leflunomide, it is recommended that all females of reproductive potential undergo an accelerated drug elimination procedure. Women receiving leflunomide treatment who wish to become pregnant must discontinue leflunomide and undergo an accelerated drug elimination procedure, which includes verification that plasma concentrations of the active metabolite of leflunomide, teriflunomide, are less than 0.02 mg/L (0.02 mcg/mL). Based on animal data, human plasma concentrations of teriflunomide of less than 0.02 mg/L (0.02 mcg/mL) are expected to have minimal embryo-fetal risk [see Contraindications (4), Warnings and Precautions (5.3), and Use in Specific Populations (8.1)].
5.2 Hepatotoxicity
Severe liver injury, including fatal liver failure, has been reported in some patients treated with leflunomide. Patients with pre-existing acute or chronic liver disease, or those with serum alanine aminotransferase (ALT) of greater than twice the upper limits of normal (>2xULN) before initiating treatment, should not be treated with leflunomide. Use caution when leflunomide is given with other potentially hepatotoxic drugs. Monitoring of ALT levels is recommended at least monthly for six months after starting leflunomide, and thereafter every 6 to 8 weeks. If ALT elevation >3-fold ULN occurs, interrupt leflunomide therapy and investigate the cause. If likely leflunomide-induced, perform the accelerated drug elimination procedure and monitor liver tests weekly until normalized [see Warnings and Precautions (5.3)].
If leflunomide-induced liver injury is unlikely because some other cause has been found, resumption of leflunomide therapy may be considered. If leflunomide and methotrexate are given concomitantly, follow the American College of Rheumatology (ACR) guidelines for monitoring methotrexate liver toxicity with ALT, AST, and serum albumin testing.
5.3 Procedure for Accelerated Elimination of Leflunomide and its Active Metabolite
The active metabolite of leflunomide, teriflunomide, is eliminated slowly from the plasma [see Clinical Pharmacology (12.3)].
Use of an accelerated drug elimination procedure will rapidly reduce plasma concentrations of leflunomide and its active metabolite, teriflunomide. Therefore, an accelerated elimination procedure should be considered at any time after discontinuation of leflunomide, and in particular, when a patient has experienced a severe adverse reaction (e.g., hepatotoxicity, serious infection, bone marrow suppression, Stevens-Johnson Syndrome, toxic epidermal necrolysis, peripheral neuropathy, interstitial lung disease), suspected hypersensitivity, or has become pregnant. It is recommended that all women of childbearing potential undergo an accelerated elimination procedure after stopping leflunomide treatment.
Without use of an accelerated drug elimination procedure, it may take up to 2 years to reach plasma teriflunomide concentrations of less than 0.02 mg/L, the plasma concentration not associated with embryo-fetal toxicity in animals.
Elimination can be accelerated by the following procedures:
1. Administer cholestyramine 8 grams orally 3 times daily for 11 days.
2. Alternatively, administer 50 grams of activated charcoal powder (made into a suspension) orally every 12 hours for 11 days.
Verify plasma teriflunomide concentrations of less than 0.02 mg/L (0.02 mcg/mL) by two separate tests at least 14 days apart. If plasma teriflunomide concentrations are higher than 0.02 mg/L, repeat cholestyramine and/or activated charcoal treatment.
The duration of accelerated drug elimination treatment may be modified based on the clinical status and tolerability of the elimination procedure. The procedure may be repeated as needed, based on teriflunomide concentrations and clinical status.
Use of the accelerated drug elimination procedure may potentially result in return of disease activity if the patient had been responding to leflunomide treatment.
5.4 Immunosuppression, Bone Marrow Suppression, and Risk of Serious Infections
Leflunomide is not recommended for patients with severe immunodeficiency, bone marrow dysplasia, or severe, uncontrolled infections. If a serious infection occurs, consider interrupting leflunomide therapy and initiating the accelerated drug elimination procedure [see Warnings and Precautions (5.3)]. Medications like leflunomide that have immunosuppression potential may cause patients to be more susceptible to infections, including opportunistic infections, especially Pneumocystis jirovecii pneumonia, tuberculosis (including extra-pulmonary tuberculosis), and aspergillosis. Severe infections including sepsis, which may be fatal, have been reported in patients receiving leflunomide, especially Pneumocystis jirovecii pneumonia and aspergillosis. Most of the reports were confounded by concomitant immunosuppressant therapy and/or comorbid illness which, in addition to rheumatoid arthritis, may predispose patients to infection.
Cases of tuberculosis were observed in clinical studies with teriflunomide, the metabolite of leflunomide. Prior to initiating leflunomide, all patients should be screened for active and inactive ("latent") tuberculosis infection as per commonly used diagnostic tests. Leflunomide has not been studied in patients with a positive tuberculosis screen, and the safety of leflunomide in individuals with latent tuberculosis infection is unknown. Patients testing positive in tuberculosis screening should be treated by standard medical practice prior to therapy with leflunomide and monitored carefully during leflunomide treatment for possible reactivation of the infection.
Pancytopenia, agranulocytosis and thrombocytopenia have been reported in patients receiving leflunomide alone. These events have been reported most frequently in patients who received concomitant treatment with methotrexate or other immunosuppressive agents, or who had recently discontinued these therapies; in some cases, patients had a prior history of a significant hematologic abnormality.
Patients taking leflunomide should have platelet, white blood cell count and hemoglobin or hematocrit monitored at baseline and monthly for six months following initiation of therapy and every 6 to 8 weeks thereafter. If used with concomitant methotrexate and/or other potential immunosuppressive agents, chronic monitoring should be monthly. If evidence of bone marrow suppression occurs in a patient taking leflunomide, stop treatment with leflunomide and perform an accelerated drug elimination procedure to reduce the plasma concentration of the leflunomide active metabolite, teriflunomide [see Warnings and Precautions (5.3)].
In any situation in which the decision is made to switch from leflunomide to another antirheumatic agent with a known potential for hematologic suppression, it would be prudent to monitor for hematologic toxicity, because there will be overlap of systemic exposure to both compounds.
5.5 Stevens-Johnson Syndrome, Toxic Epidermal Necrolysis, and Drug Reactions with Eosinophilia and Systemic Symptoms
Rare cases of Stevens-Johnson syndrome and toxic epidermal necrolysis, and drug reaction with eosinophilia and systemic symptoms (DRESS) have been reported in patients receiving leflunomide. If a patient taking leflunomide develops any of these conditions, stop leflunomide treatment and perform an accelerated drug elimination procedure [see Warnings and Precautions (5.3)].
5.6 Skin Ulcers
Skin ulcers may occur in patients during therapy with leflunomide. If leflunomide-associated skin ulcer is suspected or if skin ulcers persist despite appropriate therapy, leflunomide discontinuation and an accelerated drug elimination procedure should be considered [see Warnings and Precautions (5.3)]. The decision to resume leflunomide following skin ulcers should be based on clinical judgment of adequate wound healing.
5.7 Malignancy and Lymphoproliferative Disorders
The risk of malignancy, particularly lymphoproliferative disorders, is increased with the use of some immunosuppression medications. There is a potential for immunosuppression with leflunomide. No apparent increase in the incidence of malignancies and lymphoproliferative disorders was reported in the clinical trials of leflunomide, but larger dosages and longer-term studies would be needed to determine whether there is an increased risk of malignancy or lymphoproliferative disorders with leflunomide.
5.8 Peripheral Neuropathy
Cases of peripheral neuropathy have been reported in patients receiving leflunomide and in clinical studies with teriflunomide, the active metabolite of leflunomide. Most patients recovered after discontinuation of treatment, but some patients had persistent symptoms. Age older than 60 years, concomitant neurotoxic medications, and diabetes may increase the risk for peripheral neuropathy. If a patient taking leflunomide develops a peripheral neuropathy, consider discontinuing leflunomide therapy and performing an accelerated drug elimination procedure [see Warnings and Precautions (5.3)].
5.9 Interstitial Lung Disease
Interstitial lung disease and worsening of pre-existing interstitial lung disease have been reported during treatment with leflunomide and has been associated with fatal outcomes [see Adverse Reactions (6.2)]. The risk of leflunomide-associated interstitial lung disease is increased in patients with a history of interstitial lung disease. Interstitial lung disease is a potentially fatal disorder that may occur acutely at any time during therapy and has a variable clinical presentation. New onset or worsening pulmonary symptoms, such as cough and dyspnea, with or without associated fever, may be a reason for discontinuation of leflunomide therapy and for further investigation as appropriate. If discontinuation of leflunomide is necessary, consider performing an accelerated drug elimination procedure [see Warnings and Precautions (5.3)].
5.10 Vaccinations
No clinical data are available on the efficacy and safety of vaccinations during leflunomide treatment. Vaccination with live vaccines is, however, not recommended. The long half-life of the active metabolite of leflunomide should be considered when contemplating administration of a live vaccine after stopping leflunomide.
5.11 Blood Pressure Monitoring
In placebo-controlled studies with the active metabolite of leflunomide, teriflunomide, elevations in blood pressure were observed in some subjects. Blood pressure should be checked before starting treatment with leflunomide and monitored periodically thereafter [see Adverse Reactions (6.1)].
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described elsewhere in the labeling:
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Immunosuppression [see Warnings and Precautions (5.4)]
- Bone marrow suppression [see Warnings and Precautions (5.4)]
- Serious infections [see Warnings and Precautions (5.4)]
- Stevens-Johnson syndrome, toxic epidermal necrolysis, and drug reactions with eosinophilia and systemic symptoms [see Warnings and Precautions (5.5)]
- Skin ulcers [see Warnings and Precautions (5.6)]
- Peripheral neuropathy [see Warnings and Precautions (5.8)]
- Interstitial lung disease [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
In clinical studies (Trials 1, 2, and 3), 1,865 patients were treated with leflunomide administered as either monotherapy or in combination with methotrexate or sulfasalazine. Patients ranged in age from 19 to 85 years, with an overall median age of 58 years. The mean duration of RA was 6 years ranging from 0 to 45 years.
Elevation of Liver Enzymes
Treatment with leflunomide was associated with elevations of liver enzymes, primarily ALT and AST, in a significant number of patients; these effects were generally reversible. Most transaminase elevations were mild (≤2-fold ULN) and usually resolved while continuing treatment. Marked elevations (>3-fold ULN) occurred infrequently and reversed with dose reduction or discontinuation of treatment. Table 1 shows liver enzyme elevations seen with monthly monitoring in clinical trials Trial 1 and Trial 2. It was notable that the absence of folate use in Trial 3 was associated with a considerably greater incidence of liver enzyme elevation on methotrexate.
|
MTX = methotrexate, PL = placebo, SSZ = sulfasalazine, ULN = Upper limit of normal |
||||||||
|
*Only 10% of patients in Trial 3 received folate. All patients in Trial 1 received folate. |
||||||||
|
| Trial 1
| Trial 2
| Trial 3*
|
|||||
|
| Leflunomide
20 mg/day (n= 182) | PL
(n=118) | MTX
7.5 to 15 mg/wk (n=182) | Leflunomide
20 mg/day (n= 133) | PL
(n=92) | SSZ
2 g/day (n=133) | Leflunomide 20 mg/day
(n= 501) | MTX
7.5 to 15 mg/wk (n=498) |
| ALT (SGPT) >3-fold ULN (n %) | 8(4.4) | 3(2.5) | 5(2.7) | 2(1.5) | 1(1.1) | 2(1.5) | 13(2.6) | 83 (16.7) |
| Reversed to ≤2-fold ULN: | 8 | 3 | 5 | 2 | 1 | 2 | 12 | 82 |
| Timing of Elevation | ||||||||
| 0 to 3 Months | 6 | 1 | 1 | 2 | 1 | 2 | 7 | 27 |
| 4 to 6 Months | 1 | 1 | 3 | - | - | - | 1 | 34 |
| 7 to 9 Months | 1 | 1 | 1 | - | - | - | - | 16 |
| 10 to 12 Months | - | - | - | - | - | - | 5 | 6 |
In a 6-month study of 263 patients with persistent active rheumatoid arthritis despite methotrexate therapy, and with normal LFTs, leflunomide was administered to a group of 130 patients starting at 10 mg per day and increased to 20 mg as needed. An increase in ALT greater than or equal to three times the ULN was observed in 3.8% of patients compared to 0.8% in 133 patients continued on methotrexate with placebo.
Most Common Adverse Reactions
The most common adverse reactions in leflunomide-treated patients with RA include diarrhea, elevated liver enzymes (ALT and AST), alopecia, and rash. Table 2 displays the most common adverse reactions in the controlled studies in patients with RA at one year (≥5% in any leflunomide treatment group).
|
MTX = methotrexate, PL = placebo, SSZ = sulfasalazine |
|||||||
|
* Only 10% of patients in Trial 3 received folate. All patients in Trial 1 received folate; none in Trial 2 received folate. |
|||||||
|
† Includes all controlled and uncontrolled trials with leflunomide (duration up to 12 months). |
|||||||
|
‡ Hypertension as a preexisting condition was overrepresented in all leflunomide treatment groups in phase III trials. |
|||||||
|
| Placebo-Controlled Trials
| Active-Controlled Trials
| All RA Studies
|
||||
|
| Trial 1 and 2
| Trial 3 *
|
|
||||
|
| Leflunomide 20 mg/day (n=315)
| PL (n=210)
| SSZ
2 g/day (n=133) | MTX 7.5 to 15 mg/wk (n=182)
| Leflunomide 20 mg/day (n=501)
| MTX 7.5 to 15 mg/wk (n=498)
| Leflunomide (n=1339) †
|
| Diarrhea | 27% | 12% | 10% | 20% | 22% | 10% | 17% |
| Headache | 13% | 11% | 12% | 21% | 10% | 8% | 7% |
| Nausea | 13% | 11% | 19% | 18% | 13% | 18% | 9% |
| Rash | 12% | 7% | 11% | 9% | 11% | 10% | 10% |
| Abnormal Liver Enzymes | 10% | 2% | 4% | 10% | 6% | 17% | 5% |
| Alopecia | 9% | 1% | 6% | 6% | 17% | 10% | 10% |
| Hypertension‡
| 9% | 4% | 4% | 3% | 10% | 4% | 10% |
| Asthenia | 6% | 4% | 5% | 6% | 3% | 3% | 3% |
| Back Pain | 6% | 3% | 4% | 9% | 8% | 7% | 5% |
| GI/Abdominal Pain | 6% | 4% | 7% | 8% | 8% | 8% | 5% |
| Abdominal Pain | 5% | 4% | 4% | 8% | 6% | 4% | 6% |
| Allergic Reaction | 5% | 2% | 0% | 6% | 1% | 2% | 2% |
| Bronchitis | 5% | 2% | 4% | 7% | 8% | 7% | 7% |
| Dizziness | 5% | 3% | 6% | 5% | 7% | 6% | 4% |
| Mouth Ulcer | 5% | 4% | 3% | 10% | 3% | 6% | 3% |
| Pruritus | 5% | 2% | 3% | 2% | 6% | 2% | 4% |
| Rhinitis | 5% | 2% | 4% | 3% | 2% | 2% | 2% |
| Vomiting | 5% | 4% | 4% | 3% | 3% | 3% | 3% |
| Tenosynovitis | 2% | 0% | 1% | 2% | 5% | 1% | 3% |
Adverse events during a second year of treatment with leflunomide in clinical trials were consistent with those observed during the first year of treatment and occurred at a similar or lower incidence.
Less Common Adverse Reactions
In addition, in controlled clinical trials, the following adverse events in the leflunomide treatment group occurred at a higher incidence than in the placebo group. These adverse events were deemed possibly related to the study drug.
Blood and Lymphatic System: leukocytosis, thrombocytopenia
Cardiovascular: chest pain, palpitation, thrombophlebitis of the leg, varicose vein
Eye: blurred vision, eye disorder, papilledema, retinal disorder, retinal hemorrhage
Gastrointestinal: alkaline phosphatase increased, anorexia, bilirubinemia, flatulence, gamma-GT increased, salivary gland enlarged, sore throat, vomiting, dry mouth
General Disorders: malaise
Immune System: anaphylactic reaction
Infection: abscess, flu syndrome, vaginal moniliasis
Nervous System: dizziness, headache, somnolence
Respiratory System: dyspnea
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during postapproval use of leflunomide. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System: agranulocytosis, leukopenia, neutropenia, pancytopenia
Infection: opportunistic infections, severe infections including sepsis
Gastrointestinal: acute hepatic necrosis, colitis, including microscopic colitis, hepatitis, jaundice/cholestasis, pancreatitis, severe liver injury such as hepatic failure
Immune System: angioedema
Nervous System: peripheral neuropathy
Respiratory: interstitial lung disease, including interstitial pneumonitis and pulmonary fibrosis, which may be fatal, pulmonary hypertension
Skin and Appendages: erythema multiforme, vasculitis including cutaneous necrotizing vasculitis, cutaneous lupus erythematosus, pustular psoriasis or worsening psoriasis
7. Drug Interactions
Following oral administration, leflunomide is metabolized to an active metabolite, teriflunomide, which is responsible for essentially all of leflunomide's in vivo activity. Drug interaction studies have been conducted with both leflunomide and with its active metabolite, teriflunomide, where the metabolite was directly administered to the test subjects.
Effect of Potent CYP and Transporter Inducers
Leflunomide is metabolized by CYP450 metabolizing enzymes. Concomitant use of leflunomide and rifampin, a potent inducer of CYP and transporters, increased the plasma concentration of teriflunomide by 40%. However, when coadministered with the metabolite, teriflunomide, rifampin did not affect its pharmacokinetics. No dosage adjustment is recommended for leflunomide when coadministered with rifampin. Because of the potential for leflunomide concentrations to continue to increase with multiple dosing, caution should be used if patients are to be receiving both leflunomide and rifampin [see Clinical Pharmacology (12.3)].
Effect on CYP2C8 Substrates
Teriflunomide is an inhibitor of CYP2C8 in vivo. In patients taking leflunomide, exposure of drugs metabolized by CYP2C8 (e.g., paclitaxel, pioglitazone, repaglinide, rosiglitazone) may be increased. Monitor these patients and adjust the dose of the concomitant drug(s) metabolized by CYP2C8 as required [see Clinical Pharmacology (12.3)].
Effect on Warfarin
Coadministration of leflunomide with warfarin requires close monitoring of the international normalized ratio (INR) because teriflunomide, the active metabolite of leflunomide, may decrease peak INR by approximately 25%.
Effect on Oral Contraceptives
Teriflunomide may increase the systemic exposures of ethinylestradiol and levonorgestrel. Consideration should be given to the type or dose of contraceptives used in combination with leflunomide [see Clinical Pharmacology (12.3)].
Effect on CYP1A2 Substrates
Teriflunomide, the active metabolite of leflunomide, may be a weak inducer of CYP1A2 in vivo. In patients taking leflunomide, exposure of drugs metabolized by CYP1A2 (e.g., alosetron, duloxetine, theophylline, tizanidine) may be reduced. Monitor these patients and adjust the dose of the concomitant drug(s) metabolized by CYP1A2 as required [see Clinical Pharmacology (12.3)].
Effect on Organic Anion Transporter 3 (OAT3) Substrates
Teriflunomide inhibits the activity of OAT3 in vivo. In patients taking leflunomide, exposure of drugs which are OAT3 substrates (e.g., cefaclor, cimetidine, ciprofloxacin, penicillin G, ketoprofen, furosemide, methotrexate, zidovudine) may be increased. Monitor these patients and adjust the dose of the concomitant drug(s) which are OAT3 substrates as required [see Clinical Pharmacology (12.3)].
Effect on BCRP and Organic Anion Transporting Polypeptide B1 and B3 (OATP1B1/1B3) Substrates
Teriflunomide inhibits the activity of BCRP and OATP1B1/1B3 in vivo. For a patient taking leflunomide, the dose of rosuvastatin should not exceed 10 mg once daily. For other substrates of BCRP (e.g., mitoxantrone) and drugs in the OATP family (e.g., methotrexate, rifampin), especially HMG-Co reductase inhibitors (e.g., atorvastatin, nateglinide, pravastatin, repaglinide, and simvastatin), consider reducing the dose of these drugs and monitor patients closely for signs and symptoms of increased exposures to the drugs while patients are taking leflunomide [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to leflunomide during pregnancy. Health care providers and patients are encouraged to report pregnancies by calling 1-877-311-8972 or visit http://www.pregnancystudies.org/participate-in-a-study/.
Risk Summary
Leflunomide is contraindicated for use in pregnant women because of the potential for fetal harm. In animal reproduction studies, oral administration of leflunomide during organogenesis at a dose of 1/10 of, and equivalent to, the maximum recommended human dose (MRHD) based on AUC, respectively, in rats and rabbits, caused teratogenicity (rats and rabbits), and embryo-lethality (rats) [see Data]. Pregnancy exposure registry data are not available at this time to inform the presence or absence of drug-associated risk with the use of leflunomide during pregnancy. The background risk of major birth defects and miscarriage for the indicated populations is unknown. The background risk in the U.S. general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, stop treatment with leflunomide, apprise the patient of the potential hazard to a fetus, and perform the accelerated drug elimination procedure to achieve teriflunomide concentrations of less than 0.02 mg/L (0.02 mcg/mL) [see Warnings and Precautions (5.3)].
Clinical Considerations
Fetal/Neonatal adverse reactions
Lowering the plasma concentration of the active metabolite, teriflunomide, by instituting an accelerated drug elimination procedure as soon as pregnancy is detected may decrease the risk to the fetus from leflunomide. The accelerated drug elimination procedure includes verification that the plasma teriflunomide concentration is less than 0.02 mg/L [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
Data
Animal Data
In an embryo-fetal development study, pregnant rats administered leflunomide during organogenesis from gestation days 7 to 19 at a dose approximately 1/10 of the MRHD (on an AUC basis at a maternal oral dose of 15 mg/kg), teratogenic effects, most notably anophthalmia or microphthalmia and internal hydrocephalus, were observed. Under these exposure conditions, leflunomide also caused a decrease in the maternal body weight and an increase in embryolethality with a decrease in fetal body weight for surviving fetuses. In an embryo-fetal development study, pregnant rabbits administered leflunomide during organogenesis from gestation days 6 to 18 at a dose approximately equivalent to the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg), a teratogenic finding of fused, dysplastic sternebrae was observed. Leflunomide was not teratogenic in rats and rabbits at doses approximately 1/150 and 1/10 of the MRHD, respectively (on an AUC basis at maternal oral dose of 1 mg/kg in both rats and rabbits).
In a pre and post natal development study, when female rats were treated with leflunomide at a dose that was approximately 1/100 of the MRHD (on an AUC basis at a maternal dose of 1.25 mg/kg) beginning 14 days before mating and continuing until the end of lactation, the offspring exhibited marked (greater than 90%) decreases in postnatal survival.
8.2 Lactation
Clinical lactation studies have not been conducted to assess the presence of leflunomide in human milk, the effects of leflunomide on the breastfed child, or the effects of leflunomide on milk production. Because of the potential for serious adverse reactions in a breastfed infant from leflunomide, advise a nursing woman to discontinue breastfeeding during treatment with leflunomide.
8.3 Females and Males of Reproductive Potential
Leflunomide may cause fetal harm if administered during pregnancy [see Use in Specific Populations (8.1)].
Pregnancy Testing
Exclude pregnancy prior to initiation of treatment with leflunomide in females of reproductive potential. Advise females to notify their healthcare provider immediately if pregnancy occurs or is suspected during treatment [see Warnings and Precautions (5.1, 5.3) and Use in Specific Populations (8.1)].
Contraception
Females
Females of reproductive potential should use effective contraception while taking leflunomide. If leflunomide is discontinued, use of contraception should be continued until it is verified that plasma concentrations of teriflunomide are less than 0.02 mg/L (0.02 mcg/mL, the level expected to have minimal fetal risk, based on animal data).
Females of reproductive potential who wish to become pregnant should discontinue leflunomide and undergo an accelerated elimination procedure. Effective contraception should be used until it is verified that plasma concentrations of teriflunomide are less than 0.02 mg/L (0.02 mcg/mL) [see Warnings and Precautions (5.1, 5.3) and Use in Specific Populations (8.1)].
Males
Teriflunomide, the active metabolite of leflunomide, is detected in human semen. Animal studies to specifically evaluate the risk of male mediated fetal toxicity have not been conducted. To minimize any possible risk, men not wishing to father a child and their female partners should use effective contraception. Men wishing to father a child should discontinue use of leflunomide and either undergo an accelerated elimination procedure or wait until verification that the plasma teriflunomide concentration is less than 0.02 mg/L (0.02 mcg/mL) [see Warnings and Precautions (5.3)].
8.4 Pediatric Use
The safety and effectiveness of leflunomide in pediatric patients have not been established.
The safety and effectiveness of leflunomide in the treatment of polyarticular course juvenile idiopathic arthritis (JIA) was evaluated in a single multicenter, double-blind, active-controlled trial in 94 pediatric patients (1:1 randomization) with polyarticular course juvenile idiopathic arthritis (JIA) as defined by the American College of Rheumatology (ACR). In this population, leflunomide treatment was found not to be effective.
The safety of leflunomide was studied in 74 patients with polyarticular course JIA ranging in age from 3 to 17 years (47 patients from the active-controlled study and 27 from an open-label safety and pharmacokinetic study). The most common adverse events included abdominal pain, diarrhea, nausea, vomiting, oral ulcers, upper respiratory tract infections, alopecia, rash, headache, and dizziness. Less common adverse events included anemia, hypertension, and weight loss. Fourteen pediatric patients experienced ALT and/or AST elevations, nine between 1.2 and 3-fold the upper limit of normal, and five between 3 and 8-fold the upper limit of normal.
8.5 Geriatric Use
Of the total number of subjects in controlled clinical trials (Trials 1, 2, and 3) of leflunomide, 234 subjects were 65 years and over [see Clinical Studies (14)]. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. No dosage adjustment is needed in patients over 65.
8.6 Hepatic Impairment
Dedicated studies of the effect of hepatic impairment on leflunomide pharmacokinetics have not been conducted. Given the need to metabolize leflunomide into the active species, the role of the liver in drug elimination/recycling, and the possible risk of increased hepatic toxicity, the use of leflunomide in patients with hepatic impairment is not recommended.
10. Overdosage
There have been reports of chronic overdose in patients taking leflunomide at daily dose up to five times the recommended daily dose and reports of acute overdose in adults and children. Adverse events were consistent with the safety profile for leflunomide [see Adverse Reactions (6)]. The most frequent adverse events observed were diarrhea, abdominal pain, leukopenia, anemia, and elevated liver function tests.
In the event of a significant overdose or toxicity, perform an accelerated drug elimination procedure to accelerate elimination [see Warnings and Precautions (5.3)].
Studies with both hemodialysis and CAPD (chronic ambulatory peritoneal dialysis) indicate that teriflunomide, the primary metabolite of leflunomide, is not dialyzable [see Clinical Pharmacology (12.3)].
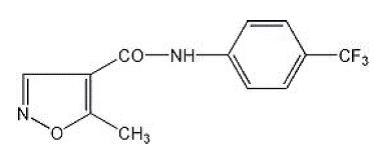
11. Leflunomide Description
Leflunomide, USP is a pyrimidine synthesis inhibitor. The chemical name for leflunomide is N-(4´-trifluoromethylphenyl)-5-methylisoxazole-4-carboxamide. It has a molecular formula C12H9F3N2O2, a molecular weight of 270.2 and the following structural formula:
Leflunomide tablets USP are available for oral administration as tablets containing 10 mg and 20 mg of active drug. Combined with leflunomide are the following inactive ingredients: colloidal silicon dioxide, corn starch, crospovidone, hypromellose, lactose monohydrate, magnesium stearate, polyethylene glycol, povidone, talc and titanium dioxide.
12. Leflunomide - Clinical Pharmacology
12.1 Mechanism of Action
Leflunomide is an isoxazole immunomodulatory agent that inhibits dihydroorotate dehydrogenase (a mitochondrial enzyme involved in de novo pyrimidine synthesis) and has antiproliferative activity. Several in vivo and in vitro experimental models have demonstrated an anti-inflammatory effect.
12.3 Pharmacokinetics
Following oral administration, leflunomide is metabolized to an active metabolite, teriflunomide, which is responsible for essentially all of leflunomide's in vivo activity. Plasma concentrations of the parent drug, leflunomide, have been occasionally seen at very low concentrations. Studies of the pharmacokinetics of leflunomide have primarily examined the plasma concentrations of the active metabolite, teriflunomide.
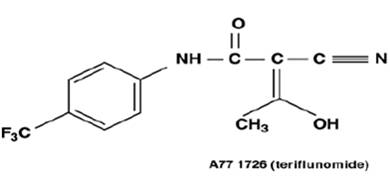
Following oral administration, peak teriflunomide concentrations occurred between 6 to 12 hours after dosing. Due to the very long half-life of teriflunomide (18 to 19 days), a loading dose of 100 mg for 3 days was used in clinical studies to facilitate the rapid attainment of steady-state teriflunomide concentrations. Without a loading dose, it is estimated that attainment of steady-state plasma concentrations would require about two months of dosing. The resulting plasma concentrations following both loading doses and continued clinical dosing indicate that plasma teriflunomide concentrations are dose proportional.
Effect of Food
Coadministration of leflunomide tablets with a high fat meal did not have a significant impact on teriflunomide plasma concentrations.
Distribution
Teriflunomide is extensively bound to plasma protein (>99%) and is mainly distributed in plasma. The volume of distribution is 11 L after a single intravenous (IV) administration.
Elimination
Teriflunomide, the active metabolite of leflunomide, has a median half-life of 18 to 19 days in healthy volunteers. The elimination of teriflunomide can be accelerated by administration of cholestyramine or activated charcoal. Without use of an accelerated drug elimination procedure, it may take up to 2 years to reach plasma teriflunomide concentrations of less than 0.02 mg/L, due to individual variation in drug clearance [see Warnings and Precautions (5.3)]. After a single IV administration of the metabolite (teriflunomide), the total body clearance of teriflunomide was 30.5 mL/h.
Metabolism
In vitro inhibition studies in human liver microsomes suggest that cytochrome P450 (CYP) 1A2, 2C19 and 3A4 are involved in leflunomide metabolism. In vivo, leflunomide is metabolized to one primary (teriflunomide) and many minor metabolites. In vitro, teriflunomide is not metabolized by CYP450 or flavin monoamine oxidase enzymes. The parent compound is rarely detectable in plasma.
Excretion
Teriflunomide, the active metabolite of leflunomide, is eliminated by direct biliary excretion of unchanged drug as well as renal excretion of metabolites. Over 21 days, 60.1% of the administered dose is excreted via feces (37.5%) and urine (22.6%). After an accelerated elimination procedure with cholestyramine, an additional 23.1% was recovered (mostly in feces).
Studies with both hemodialysis and CAPD (chronic ambulatory peritoneal dialysis) indicate that teriflunomide is not dialyzable.
Specific Populations
Gender. Gender has not been shown to cause a consistent change in the in vivo pharmacokinetics of teriflunomide.
Smoking. A population based pharmacokinetic analysis of the clinical trial data indicates that smokers have a 38% increase in clearance over non-smokers; however, no difference in clinical efficacy was seen between smokers and nonsmokers.
Drug Interaction Studies
Drug interaction studies have been conducted with both leflunomide and with its active metabolite, teriflunomide, where the metabolite was directly administered to the test subjects.
The potential effect of other drugs on leflunomide
- Potent CYP and transporter inducers:
Following concomitant administration of a single dose of leflunomide to subjects receiving multiple doses of rifampin, teriflunomide peak concentrations were increased (~40%) over those seen when leflunomide was given alone [see Drug Interactions (7)].
- An in vivointeraction study with leflunomide and cimetidine (nonspecific weak CYP inhibitor) has demonstrated a lack of a significant impact on teriflunomide exposure.
The potential effect of leflunomide on other drugs
- CYP2C8 Substrates
There was an increase in mean repaglinide Cmax and AUC (1.7 and 2.4-fold, respectively), following repeated doses of teriflunomide and a single dose of 0.25 mg repaglinide, suggesting that teriflunomide is an inhibitor of CYP2C8 in vivo. The magnitude of interaction could be higher at the recommended repaglinide dose [see Drug Interactions (7)].
- CYP1A2 Substrates
Repeated doses of teriflunomide decreased mean Cmax and AUC of caffeine by 18% and 55%, respectively, suggesting that teriflunomide may be a weak inducer of CYP1A2 in vivo.
- OAT3 Substrates
There was an increase in mean cefaclor Cmax and AUC (1.43 and 1.54-fold, respectively), following repeated doses of teriflunomide, suggesting that teriflunomide is an inhibitor of organic anion transporter 3 (OAT3) in vivo [see Drug Interactions (7)].
- BCRP and OATP1B1/1B3 Substrates
There was an increase in mean rosuvastatin Cmax and AUC (2.65 and 2.51-fold, respectively), following repeated doses of teriflunomide, suggesting that teriflunomide is an inhibitor of BCRP transporter and organic anion transporting polypeptide 1B1 and 1B3 (OATP1B1/1B3) [see Drug Interactions (7)].
- Oral Contraceptives
There was an increase in mean ethinylestradiol Cmax and AUC0-24 (1.58 and 1.54-fold, respectively) and levonorgestrel Cmax and AUC0-24 (1.33 and 1.41-fold, respectively) following repeated doses of teriflunomide [see Drug Interactions (7)].
- Teriflunomide did not affect the pharmacokinetics of bupropion (a CYP2B6 substrate), midazolam (a CYP3A4 substrate), S-warfarin (a CYP2C9 substrate), omeprazole (a CYP2C19 substrate), and metoprolol (a CYP2D6 substrate).
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No evidence of carcinogenicity was observed in a 2-year bioassay in rats at oral doses of leflunomide up to the maximally tolerated dose of 6 mg/kg (approximately 1/40 the maximum human teriflunomide systemic exposure based on AUC). However, male mice in a 2-year bioassay exhibited an increased incidence in lymphoma at an oral dose of 15 mg/kg, the highest dose studied (1.7 times the human teriflunomide exposure based on AUC). Female mice, in the same study, exhibited a dose-related increased incidence of bronchoalveolar adenomas and carcinomas combined beginning at 1.5 mg/kg (approximately 1/10 the human teriflunomide exposure based on AUC). The significance of the findings in mice relative to the clinical use of leflunomide is not known.
Leflunomide was not mutagenic in the Ames assay, the unscheduled DNA synthesis assay, or in the HGPRT gene mutation assay. In addition, leflunomide was not clastogenic in the in vivo mouse micronucleus assay or in the in vivo Chinese hamster bone marrow cell cytogenic test. However, 4-trifluoromethylaniline (TFMA), a minor metabolite of leflunomide, was mutagenic in the Ames assay and in the HGPRT gene mutation assay, and was clastogenic in the in vitro Chinese hamster cell chromosomal aberration assay. TFMA was not clastogenic in the in vivo mouse micronucleus assay or in the in vivo Chinese hamster bone marrow cell cytogenic test.
Leflunomide had no effect on fertility or reproductive performance in either male or female rats at oral doses up to 4 mg/kg (approximately 1/30 the human teriflunomide exposure based on AUC) [see Use in Specific Populations (8.1, 8.6)].
14. Clinical Studies
The efficacy of leflunomide in the treatment of rheumatoid arthritis (RA) was demonstrated in three controlled trials showing reduction in signs and symptoms, and inhibition of structural damage. In two placebo controlled trials, efficacy was demonstrated for improvement in physical function. In these trials, efficacy was evaluated by:
1. Reduction of signs and symptoms
Relief of signs and symptoms was assessed using the American College of Rheumatology (ACR) 20 Responder Index, a composite of clinical, laboratory, and functional measures in rheumatoid arthritis. An "ACR20 Responder" is a patient who had ≥ 20% improvement in both tender and swollen joint counts and in 3 of the following 5 criteria: physician global assessment, patient global assessment, functional ability measure (Modified Health Assessment Questionnaire [MHAQ]), visual analog pain scale, and erythrocyte sedimentation rate or C-reactive protein. An "ACR20 Responder at Endpoint" is a patient who completed the study and was an ACR20 Responder at the completion of the study.
2. Inhibition of structural damage
Inhibition of structural damage compared to control was assessed using the Sharp score, a composite score of x-ray erosions and joint space narrowing in hands/wrists and forefeet.
3. Improvement in physical function
Improvement in physical function was assessed using the Health Assessment Questionnaire (HAQ) and the Medical Outcomes Survey Short Form (SF-36).
In all leflunomide trials, participants of at least 18 years of age and in ARA functional class of I, II or III received an initial loading dose of 100 mg leflunomide per day for three days, followed by 20 mg per day thereafter.
Exclusion criteria included patients with a history of hypersensitivity to the study medication; women who were pregnant or breastfeeding and men or women of child bearing age and potential who had not received contraceptives for at least 4 weeks before entering the study and to be maintained throughout the study and for at least 6 months after discontinuing treatment; patients with a history of inflammatory disease, impaired renal function or liver impairment, cardiac failure, congenital or acquired immunodeficiency, impaired coagulation, or a history of recent major traumatic injury; and patients taking intra-articular or systemic concomitant medications which could affect the safety and/or efficacy of the study medication.
Trial 1
Trial 1, a 2-year study, randomized 482 patients with active RA of at least 6 months duration to leflunomide 20 mg/day (n=182), methotrexate 7.5 mg/week increasing to 15 mg/week (n=182), or placebo (n=118). All patients received folate 1 mg BID. The primary analysis was at 52 weeks with blinded treatment to 104 weeks.
Overall, 235 of the 508 randomized treated patients (482 in primary data analysis and an additional 26 patients), continued into a second 12 months of double-blind treatment (98 leflunomide, 101 methotrexate, 36 placebo). Leflunomide dose continued at 20 mg/day and the methotrexate dose could be increased to a maximum of 20 mg/week. In total, 190 patients (83 leflunomide, 80 methotrexate, 27 placebo) completed 2 years of double-blind treatment.
Trial 2
Trial 2 randomized 358 patients with active RA to leflunomide 20 mg/day (n=133), sulfasalazine 2 g/day (n=133), or placebo (n=92). Treatment duration was 24 weeks. An extension of the study was an optional 6-month blinded continuation of Trial 2 without the placebo arm, resulting in a 12-month comparison of leflunomide and sulfasalazine.
Of the 168 patients who completed 12 months of treatment, 146 patients (87%) entered a 1-year extension study of double-blind active treatment; (60 leflunomide, 60 sulfasalazine, 26 placebo/ sulfasalazine). Patients continued on the same daily dosage of leflunomide or sulfasalazine that they had been taking at the completion of Trial 2. A total of 121 patients (53 leflunomide, 47 sulfasalazine, 21 placebo/sulfasalazine) completed the 2 years of double-blind treatment.
Trial 3
Trial 3 randomized 999 patients with active RA to leflunomide 20 mg/day (n=501) or methotrexate at 7.5 mg/week increasing to 15 mg/week (n=498). Folate supplementation was used in 10% of patients. Treatment duration was 52 weeks.
Of the 736 patients who completed 52 weeks of treatment in study Trial 3, 612 (83%) entered the double-blind, 1-year extension study (292 leflunomide, 320 methotrexate). Patients continued on the same daily dosage of leflunomide or methotrexate that they had been taking at the completion of Trial 3. There were 533 patients (256 leflunomide, 277 methotrexate) who completed 2 years of double-blind treatment.
Clinical Trial Results
Clinical response
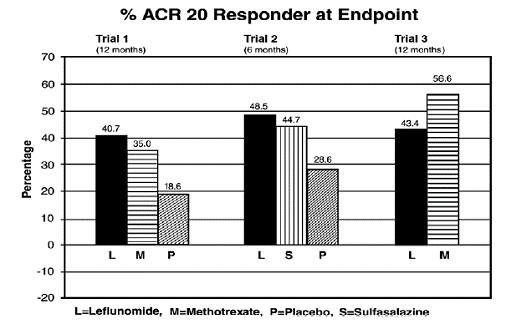
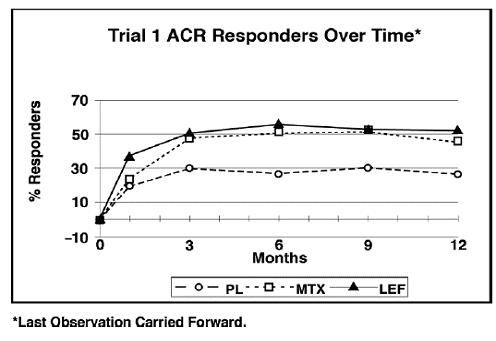
The ACR20 Responder at Endpoint rates are shown in Figure 1. Leflunomide was statistically significantly superior to placebo in reducing the signs and symptoms of RA by the primary efficacy analysis, ACR20 Responder at Endpoint, in study Trial 1 (at the primary 12 months endpoint) and Trial 2 (at 6-month endpoint). ACR20 Responder at Endpoint rates with leflunomide treatment were consistent across the 6 and 12-month studies (41% to 49%). No consistent differences were demonstrated between leflunomide and methotrexate or between leflunomide and sulfasalazine. Leflunomide treatment effect was evident by 1 month, stabilized by 3 to 6 months, and continued throughout the course of treatment as shown in Figure 1.
Figure 1: Percentage of ACR20 Responders at Endpoint in Patients with Active RA in Trials 1, 2, and 3
| Comparisons
| 95%Confidence Interval
| p Value
|
|
| Trial 1 | Leflunomide vs Placebo | (12, 32) | <0.0001 |
| Methotrexate vs Placebo | (8, 30) | <0.0001 |
|
| Leflunomide vs Methotrexate | (-4, 16) | NS |
|
| Trial 2 | Leflunomide vs Placebo | (7, 33) | 0.0026 |
| Sulfasalazine vs Placebo | (4, 29) | 0.0121 |
|
| Leflunomide vs Sulfasalazine | (-8, 16) | NS |
|
| Trial 3 | Leflunomide vs Methotrexate | (-19, -7) | <0.0001 |
Figure 2: ACR20 Responders over Time in Patients with Active RA in Trial 1*
ACR50 and ACR70 Responders are defined in an analogous manner to the ACR 20 Responder, but use improvements of 50% or 70%, respectively (Table 3). Mean change for the individual components of the ACR Responder Index are shown in Table 4.
| Study and Treatment Group | ACR20 | ACR50 | ACR70 |
| Placebo-Controlled Studies | |||
| Trial 1 (12 months) | |||
| Leflunomide (n=178)†
| 52‡
| 34‡
| 20‡
|
| Placebo (n=118)†
| 26 | 8 | 4 |
| Methotrexate (n=180)†
| 46 | 23 | 9 |
| Trial 2 (6 months) | |||
| Leflunomide (n=130)†
| 55‡
| 33‡
| 10§
|
| Placebo (n=91)†
| 29 | 14 | 2 |
| Sulfasalazine (n=132)†
| 57 | 30 | 8 |
| Non-Placebo Active-Controlled Studies | |||
| Trial 3 (12 months) | |||
| Leflunomide (n=495)†
| 51 | 31 | 10 |
| Methotrexate (n=489)†
| 65 | 44 | 16 |
| * Intent to treat (ITT) analysis using last observation carried forward (LOCF) technique for patients who discontinued early. † n is the number of ITT patients for whom adequate data were available to calculate the indicated rates. ‡ p<0.001 leflunomide vs placebo § p<0.02 leflunomide vs placebo |
|||
Table 4 shows the results of the components of the ACR response criteria for Trial 1, Trial 2 and Trial 3. Leflunomide was significantly superior to placebo in all components of the ACR Response criteria in study Trial 1 and Trial 2. In addition, leflunomide was significantly superior to placebo in improving morning stiffness, a measure of RA disease activity, not included in the ACR Response criteria. No consistent differences were demonstrated between leflunomide and the active comparators.
| Components
| Placebo-Controlled Studies
| Non-Placebo Controlled Study
|
||||||||||
| Trial 1
(12 months) | Trial 2 Non-US
(6 months) | Trial 3 Non-US
(12 months) |
||||||||||
| Leflunomide
| Methotrexate
| Placebo
| Leflunomide
| Sulfasalazine
| Placebo
| Leflunomide
| Methotrexate
|
|||||
| Tender joint count†
| -7.7 | -6.6 | -3 | -9.7 | -8.1 | -4.3 | -8.3 | -9.7 |
||||
| Swollen joint count†
| -5.7 | -5.4 | -2.9 | -7.2 | -6.2 | -3.4 | -6.8 | -9 |
||||
| Patient global assessment†
| -2.1 | -1.5 | 0.1 | -2.8 | -2.6 | -0.9 | -2.3 | -3 |
||||
| Physician global assessment‡
| -2.8 | -2.4 | -1 | -2.7 | -2.5 | -0.8 | -2.3 | -3.1 |
||||
| Physical function/disability (MHAQ/HAQ) | -0.29 | -0.15 | 0.07 | -0.5 | -0.29 | -0.04 | -0.37 | -0.44 |
||||
| Pain intensity‡
| -2.2 | -1.7 | -0.5 | -2.7 | -2 | -0.9 | -2.1 | -2.9 |
||||
| Erythrocyte Sedimentation rate | -6.26 | -6.48 | 2.56 | -7.48 | -16.56 | 3.44 | -10.12 | -22.18 |
||||
| C-reactive protein | -0.62 | -0.5 | 0.47 | -2.26 | -1.19 | 0.16 | -1.86 | -2.45 |
||||
| Not included in the ACR Responder Index
|
||||||||||||
| Morning Stiffness (min) | -101.4 | -88.7 | 14.7 | -93 | -42.4 | -6.8 | -63.7 | -86.6 |
||||
| * Last Observation Carried Forward; Negative Change Indicates Improvement † Based on 28 joint count ‡ Visual Analog Scale -0=Best; 10=Worst |
||||||||||||
After completing 12 months of treatment, patients continuing on study treatment were evaluated for an additional 12 months of double-blind treatment (total treatment period of 2 years). ACR Responder rates at 12 months were maintained over 2 years in most patients continuing a second year of treatment.
Improvement from baseline in the individual components of the ACR responder criteria was also sustained in most patients during the second year of leflunomide treatment in all three trials.
Radiographic Response
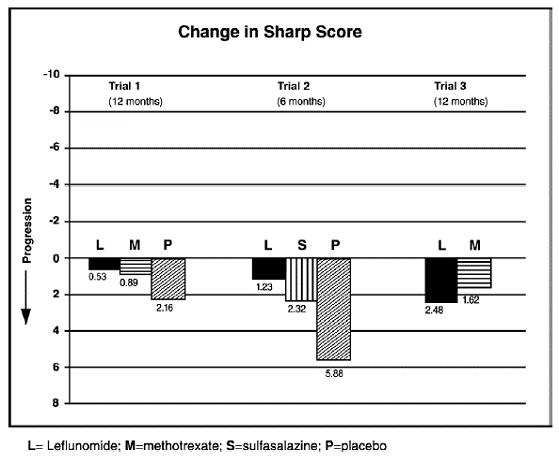
The change from baseline to endpoint in progression of structural disease, as measured by the Sharp x-ray score, is displayed in Figure 3. Leflunomide was statistically significantly superior to placebo in inhibiting the progression of disease by the Sharp score. No consistent differences were demonstrated between leflunomide and methotrexate or between leflunomide and sulfasalazine.
Figure 3: Change in Sharp Score in Patients with Active RA in Trials 1, 2, and 3
| Comparisons
| 95%Confidence Interval
| p Value
|
|
| Trial 1 | Leflunomide vs Placebo | (-4, -1.1) | 0.0007 |
| Methotrexate vs Placebo | (-2.6, -0.2) | 0.0196 |
|
| Leflunomide vs Methotrexate | (-2.3, 0) | 0.0499 |
|
| Trial 2 | Leflunomide vs Placebo | (-6.2, -1.8) | 0.0004 |
| Sulfasalazine vs Placebo | (-6.9, 0) | 0.0484 |
|
| Leflunomide vs Sulfasalazine | (-3.3, 1.2) | NS |
|
| Trial 3 | Leflunomide vs Methotrexate | (-2.2, 7.4) | NS |
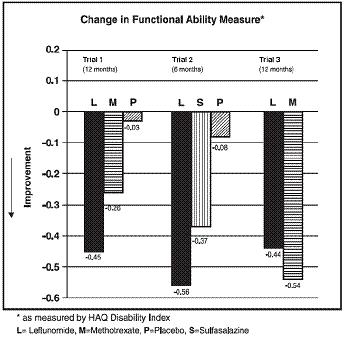
The Health Assessment Questionnaire (HAQ) assesses a patient's physical function and degree of disability. The mean change from baseline in functional ability as measured by the HAQ Disability Index (HAQ DI) in the 6 and 12 month placebo and active-controlled trials is shown in Figure 4. Leflunomide was statistically significantly superior to placebo in improving physical function. Superiority to placebo was demonstrated consistently across all eight HAQ DI subscales (dressing, arising, eating, walking, hygiene, reach, grip and activities) in both placebo controlled studies.
The Medical Outcomes Survey Short Form 36 (SF-36), a generic health-related quality of life questionnaire, further addresses physical function. In Trial 1, at 12 months, leflunomide provided statistically significant improvements compared to placebo in the Physical Component Summary (PCS) Score.
Figure 4: Change in Functional Ability Measure in Patients with Active RA in Trials 1, 2, and 3*
| Comparison
| 95% Confidence Interval
| p Value
|
|
| Trial 1 | Leflunomide vs Placebo | (-0.58, -0.29) | 0.0001 |
| Leflunomide vs Methotrexate | (-0.34, -0.07) | 0.0026 |
|
| Trial 2 | Leflunomide vs Placebo | (-0.67, -0.36) | <0.0001 |
| Leflunomide vs Sulfasalazine | (-0.33, -0.03) | 0.0163 |
|
| Trial 3 | Leflunomide vs Methotrexate | (0.01, 0.16) | 0.0221 |
The improvement in physical function demonstrated at 6 and 12 months was maintained over two years. In those patients continuing therapy for a second year, this improvement in physical function as measured by HAQ and SF-36 (PCS) was maintained.
16. How is Leflunomide supplied
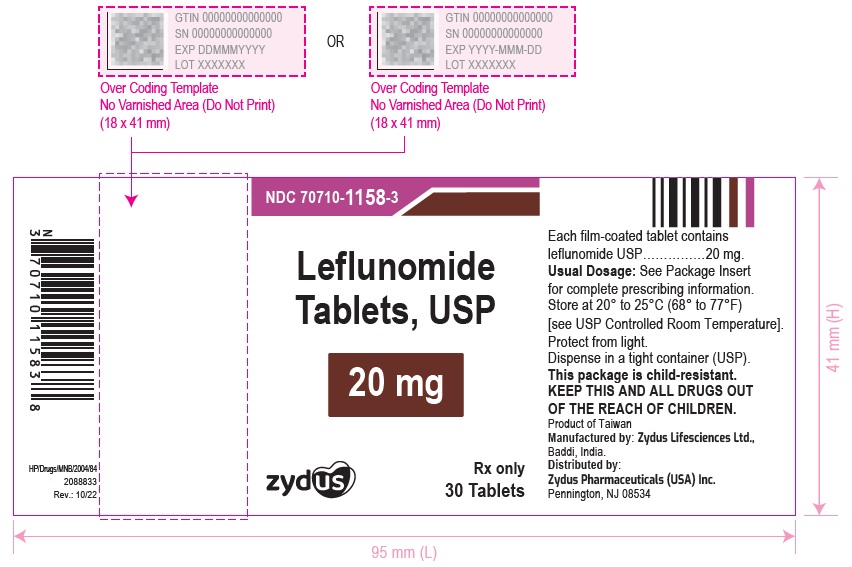
Leflunomide tablets USP, 10 mg, white to off white, round, film-coated tablets debossed with "57" on one side and "11" on the other side, and are supplied as follow:
NDC 70710-1157-3 in bottle of 30 tablets with child-resistant closure
NDC 70710-1157-1 in bottle of 100 tablets with child-resistant closure
NDC 70710-1157-5 in bottle of 500 tablets
Leflunomide tablets USP, 20 mg, white to off white, triangular, film-coated tablets debossed with "58" on one side and "11" on the other side, and are supplied as follow:
NDC 70710-1158-3 in bottle of 30 tablets with child-resistant closure
NDC 70710-1158-1 in bottle of 100 tablets with child-resistant closure
NDC 70710-1158-5 in bottle of 500 tablets
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature]. Protect from light.
17. Patient Counseling Information
Advise females of reproductive potential:
- Of the potential for fetal harm if leflunomide is taken during pregnancy.
- To notify their healthcare provider immediately if a pregnancy occurs or is suspected.
- To use effective contraception during treatment with leflunomide and until the active metabolite (teriflunomide) plasma concentration is verified to be less than 0.02 mg/L [see Warnings and Precautions (5.1, 5.3), Use in Specific Populations (8.1, 8.3), and Clinical Pharmacology (12.3)].
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to leflunomide during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Advise nursing women to discontinue breastfeeding during treatment with leflunomide [see Use in Specific Populations (8.2)].
Serious Skin Reactions
Advise patients of the possibility of rare, serious skin reactions. Instruct patients to promptly seek medical attention if they develop a skin rash, mucous membrane lesions, or skin ulcers.
Investigations
• Advise patients of the potential hepatotoxic effects of leflunomide and of the need for monitoring liver enzymes. Instruct patients to report if they develop symptoms such as unusual tiredness, abdominal pain or jaundice.
• Advise patients that they may develop a lowering of their blood counts and should have frequent hematologic monitoring. This is particularly important for patients who are receiving other immunosuppressive therapy concurrently with leflunomide, who have recently discontinued such therapy before starting treatment with leflunomide, or who have had a history of a significant hematologic abnormality. Instruct patients to promptly report if they notice symptoms consistent with pancytopenia, such as easy bruising or bleeding, recurrent infections, fever, paleness or unusual tiredness.
• Inform patients about the early warning signs of interstitial lung disease and ask them to contact their physician promptly if these symptoms appear or worsen during therapy.
Manufactured by:
Zydus Lifesciences Ltd., Baddi, India
Distributed by:
Zydus Pharmaceuticals (USA) Inc.
Pennington, NJ 08534
Rev.: 07/24
| LEFLUNOMIDE
leflunomide tablet, film coated |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| LEFLUNOMIDE
leflunomide tablet, film coated |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Zydus Pharmaceuticals (USA) Inc. (156861945) |
| Registrant - Zydus Lifesciences Global FZE (850107010) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Zydus Lifesciences Limited | 677605858 | ANALYSIS(70710-1157, 70710-1158) , MANUFACTURE(70710-1157, 70710-1158) | |
Frequently asked questions
More about leflunomide
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (113)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: antirheumatics
- Breastfeeding
- En español