Arzerra Prescribing Information
Package insert / product label
Generic name: ofatumumab
Dosage form: injection, solution
Drug classes: CD20 monoclonal antibodies, Selective immunosuppressants
J Code (medical billing code): J9302 (10 mg, injection)
Medically reviewed by Drugs.com. Last updated on Aug 4, 2023.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
ARZERRA® (ofatumumab) injection, for intravenous use
Initial U.S. Approval: 2009
WARNING: HEPATITIS B VIRUS REACTIVATION AND PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
See full prescribing information for complete boxed warning.
Recent Major Changes
Indications and Usage for Arzerra
ARZERRA (ofatumumab) is a CD20-directed cytolytic monoclonal antibody indicated for the treatment of chronic lymphocytic leukemia (CLL) (1):
- in combination with chlorambucil, for the treatment of previously untreated patients with CLL for whom fludarabine-based therapy is considered inappropriate.
- in combination with fludarabine and cyclophosphamide for the treatment of patients with relapsed CLL.
- for extended treatment of patients who are in complete or partial response after at least two lines of therapy for recurrent or progressive CLL.
- for the treatment of patients with CLL refractory to fludarabine and alemtuzumab.
Arzerra Dosage and Administration
- Dilute and administer as an intravenous infusion. Do not administer subcutaneously or as an intravenous push or bolus. (2.1)
- Previously untreated CLL in combination with chlorambucil recommended dosage and schedule is:
- 300 mg on Day 1 followed by 1,000 mg on Day 8 (Cycle 1)
- 1,000 mg on Day 1 of subsequent 28-day cycles for a minimum of 3 cycles until best response or a maximum of 12 cycles. (2.1)
- Relapsed CLL in combination with fludarabine and cyclophosphamide recommended dosage and schedule is:
- 300 mg on Day 1 followed by 1,000 mg on Day 8 (Cycle 1)
- 1,000 mg on Day 1 of subsequent 28-day cycles for a maximum of 6 cycles (2.1)
- Extended treatment in CLL recommended dosage and schedule is:
- 300 mg on Day 1 followed by
- 1,000 mg 1 week later on Day 8, followed by
- 1,000 mg 7 weeks later and every 8 weeks thereafter for up to a maximum of 2 years. (2.1)
- Refractory CLL recommended dosage and schedule is:
- 300 mg initial dose, followed 1 week later by
- 2,000 mg weekly for 7 doses, followed 4 weeks later by
- 2,000 mg every 4 weeks for 4 doses. (2.1)
- Administer where facilities to adequately monitor and treat infusion reactions are available. (2.2)
- Pre-medicate with acetaminophen, antihistamine, and corticosteroid. (2.4)
Dosage Forms and Strengths
Contraindications
None. (4)
Warnings and Precautions
- Infusion Reactions: Pre-medicate with corticosteroid, acetaminophen, and an antihistamine. Monitor patients during infusions. Interrupt infusion if infusion reactions occur. (2.3, 2.4, 5.1)
- Tumor Lysis Syndrome: Anticipate TLS in high-risk patients; pre-medicate with anti-hyperuricemics and hydration. (5.5)
- Cytopenias: Neutropenia, anemia, and thrombocytopenia occur. Late-onset and prolonged neutropenia can also occur. Monitor complete blood counts at regular intervals. (5.6)
Adverse Reactions/Side Effects
- Previously Untreated CLL: Common adverse reactions (≥10%) were infusion reactions and neutropenia. (6)
- Relapsed CLL: Common adverse reactions (>10%) were infusion reactions, neutropenia, leukopenia and febrile neutropenia. (6)
- Extended Treatment in CLL: Common adverse reactions (≥10%) were infusion reactions, neutropenia, and upper respiratory tract infection. (6)
- Refractory CLL: Common adverse reactions (≥10%) were neutropenia, pneumonia, pyrexia, cough, diarrhea, anemia, fatigue, dyspnea, rash, nausea, bronchitis, and upper respiratory tract infections. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
- Pregnancy: May cause fetal B-cell depletion. (8.1)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2016
Full Prescribing Information
WARNING: HEPATITIS B VIRUS REACTIVATION AND PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
- Hepatitis B Virus (HBV) reactivation can occur in patients receiving CD20-directed cytolytic antibodies, including ARZERRA®, in some cases resulting in fulminant hepatitis, hepatic failure, and death [see Warnings and Precautions (5.2)].
- Progressive Multifocal Leukoencephalopathy (PML) resulting in death can occur in patients receiving CD20-directed cytolytic antibodies, including ARZERRA [see Warnings and Precautions (5.4)].
1. Indications and Usage for Arzerra
Chronic Lymphocytic Leukemia (CLL)
ARZERRA (ofatumumab) is indicated:
- in combination with chlorambucil, for the treatment of previously untreated patients with CLL for whom fludarabine-based therapy is considered inappropriate [see Clinical Studies (14.1)]
- in combination with fludarabine and cyclophosphamide for the treatment of patients with relapsed CLL [see Clinical Studies (14.2)]
- for extended treatment of patients who are in complete or partial response after at least two lines of therapy for recurrent or progressive CLL [see Clinical Studies (14.3)]
- for the treatment of patients with CLL refractory to fludarabine and alemtuzumab [see Clinical Studies (14.4)]
2. Arzerra Dosage and Administration
2.1 Recommended Dosage Regimen
- Dilute and administer as an intravenous infusion according to the following schedules.
- Do not administer as an intravenous push or bolus or as a subcutaneous injection.
- Pre-medicate before each infusion [see Dosage and Administration (2.4)].
Previously Untreated CLL:
The recommended dosage and schedule in combination with chlorambucil is:
- 300 mg on Day 1, followed 1 week later by 1,000 mg on Day 8 (Cycle 1), followed by
- 1,000 mg on Day 1 of subsequent 28-day cycles for a minimum of 3 cycles until best response or a maximum of 12 cycles.
Relapsed CLL:
The recommended dosage and schedule in combination with fludarabine and cyclophosphamide is:
- 300 mg on Day 1, followed 1 week later by 1,000 mg on Day 8 (Cycle 1), followed by
- 1,000 mg on Day 1 of subsequent 28-day cycles for a maximum of 6 cycles.
Extended Treatment in CLL: The recommended dosage and schedule as single-agent extended treatment in CLL is:
- 300 mg on Day 1, followed by
- 1,000 mg 1 week later on Day 8, followed by
- 1,000 mg 7 weeks later and every 8 weeks thereafter for up to a maximum of 2 years.
Refractory CLL: The recommended dosage and schedule is 12 doses administered as follows:
- 300 mg initial dose on Day 1, followed 1 week later by
- 2,000 mg weekly for 7 doses (Infusions 2 through 8), followed 4 weeks later by
- 2,000 mg every 4 weeks for 4 doses (Infusions 9 through 12).
2.2 Administration
Administer ARZERRA in an environment where facilities to adequately monitor and treat infusion reactions are available [see Warnings and Precautions (5.1)].
Prepare all doses in 1,000 mL of 0.9% Sodium Chloride Injection, USP [see Dosage and Administration (2.5)].
Previously Untreated CLL, Relapsed CLL, and Extended Treatment in CLL:
- For initial 300-mg dose: Initiate infusion at a rate of 3.6 mg/hour (12 mL/hour).
- For subsequent infusions of 1,000 mg: Initiate infusion at a rate of 25 mg/hour (25 mL/hour). Initiate infusion at a rate of 12 mg/hour if a Grade 3 or greater infusion-related adverse event was experienced during the previous infusion.
In the absence of an infusion-related adverse event, the rate of infusion may be increased every 30 minutes (Table 1). Do not exceed the infusion rates in Table 1.
| a Initial 300 mg: median durations of infusions = 4.8 to 5.2 hours.
b Subsequent infusions of 1,000 mg: median durations of infusions = 4.2 to 4.4 hours. |
||
|
Interval After Start of Infusion (min) |
Initial 300 mg Dosea (mL/hour) |
Subsequent Infusionsb (mL/hour) |
|
0-30 |
12 |
25 |
|
31-60 |
25 |
50 |
|
61-90 |
50 |
100 |
|
91-120 |
100 |
200 |
|
121-150 |
200 |
400 |
|
151-180 |
300 |
400 |
|
>180 |
400 |
400 |
Refractory CLL:
- Infusion 1 (300-mg dose): Initiate infusion at a rate of 3.6 mg/hour (12 mL/hour).
- Infusion 2 (2,000-mg dose): Initiate infusion at a rate of 24 mg/hour (12 mL/hour).
- Infusions 3 through 12 (2,000-mg doses): Initiate infusion at a rate of 50 mg/hour (25 mL/hour).
In the absence of an infusion-related adverse event, the rate of infusion may be increased every 30 minutes (Table 2). Do not exceed the infusion rates in Table 2.
| a Infusions 1 and 2 (300 mg and 2,000 mg): median duration of infusions = 6.8 hours.
b Subsequent infusions of 2,000 mg: median durations of infusions = 4.2 to 4.4 hours. |
||
|
Interval after Start of Infusion (min) |
Infusions 1 and 2a (mL/hour) |
Subsequent Infusions b (mL/hour) |
|
0-30 |
12 |
25 |
|
31-60 |
25 |
50 |
|
61-90 |
50 |
100 |
|
91-120 |
100 |
200 |
|
>120 |
200 |
400 |
2.3 Infusion Rate Dose Modification for Infusion Reactions
- Interrupt infusion for infusion reactions of any severity [see Warnings and Precautions (5.1)]. Treatment can be resumed at the discretion of the treating physician. The following infusion rate modifications can be used as a guide.
- If the infusion reaction resolves or remains less than or equal to Grade 2, resume infusion with the following modifications according to the initial Grade of the infusion reaction.
- Grade 1 or 2: Infuse at one‑half of the previous infusion rate.
- Grade 3 or 4: Infuse at a rate of 12 mL/hour.
- After resuming the infusion, the infusion rate may be increased according to Tables 1 and 2 above, based on patient tolerance.
- Consider permanent discontinuation of ARZERRA if the severity of the infusion reaction does not resolve to less than or equal to Grade 2 despite adequate clinical intervention.
- Permanently discontinue therapy for patients who develop an anaphylactic reaction to ARZERRA.
2.4 Premedication
Patients should receive all of the following premedication agents 30 minutes to 2 hours prior to each infusion of ARZERRA. See Table 3 for pre-medication schedule prior to each infusion.
| a Up to 13 infusions in previously untreated CLL; up to 7 infusions in relapsed CLL, up to 14 infusions in extended treatment in CLL. b Corticosteroid may be reduced or omitted for subsequent infusions if a Grade 3 or greater infusion-related adverse event did not occur with the preceding infusion(s). c Prednisolone may be given at reduced dose of 50 mg to 100 mg (or equivalent) if a Grade 3 or greater infusion-related adverse event did not occur with Infusion 9. |
|||||
| Previously Untreated CLL, Relapsed CLL or Extended Treatment in CLL | Refractory CLL | ||||
| Infusion Number | 1 and 2 | 3 and beyonda | 1, 2, and 9 | 3 to 8 | 10 to 12 |
| Intravenous corticosteroid (prednisolone or equivalent) | 50 mg | 0-50 mgb | 100 mg | 0-100 mgb | 50-100 mgc |
| Oral acetaminophen | 1,000 mg | ||||
| Oral or intravenous antihistamine | Diphenhydramine 50 mg or cetirizine 10 mg (or equivalent) | ||||
2.5 Preparation and Administration
- Do not shake product.
- Inspect parenteral drug products visually for particulate matter and discoloration prior to administration. ARZERRA should be a clear to opalescent, colorless solution. The solution should not be used if discolored or cloudy, or if foreign particulate matter is present.
Preparation of Solution:
- 300-mg dose: Withdraw and discard 15 mL from a 1,000-mL bag of 0.9% Sodium Chloride Injection, USP. Withdraw 5 mL from each of 3 single-use 100-mg vials of ARZERRA and add to the bag. Mix diluted solution by gentle inversion.
- 1,000-mg dose: Withdraw and discard 50 mL from a 1,000-mL bag of 0.9% Sodium Chloride Injection, USP. Withdraw 50 mL from 1 single-use 1,000-mg vial of ARZERRA and add to the bag. Mix diluted solution by gentle inversion.
- 2,000-mg dose: Withdraw and discard 100 mL from a 1,000-mL bag of 0.9% Sodium Chloride Injection, USP. Withdraw 50 mL from each of 2 single-use 1,000-mg vials of ARZERRA and add to the bag. Mix diluted solution by gentle inversion.
- Store diluted solution between 2° to 8°C (36° to 46°F).
- No incompatibilities between ARZERRA and polyvinylchloride or polyolefin bags and administration sets have been observed.
Administration Instructions:
- Do not mix ARZERRA with, or administer as an infusion with other medicinal products.
- Administer using an infusion pump and an administration set.
- Flush the intravenous line with 0.9% Sodium Chloride Injection, USP before and after each dose.
- Start infusion within 12 hours of preparation.
- Discard prepared solution after 24 hours.
3. Dosage Forms and Strengths
- 100 mg/5 mL single‑use vial for intravenous infusion.
- 1,000 mg/50 mL single‑use vial for intravenous infusion.
5. Warnings and Precautions
5.1 Infusion Reactions
ARZERRA can cause serious, including fatal, infusion reactions manifesting as bronchospasm, dyspnea, laryngeal edema, pulmonary edema, flushing, hypertension, hypotension, syncope, cardiac events (e.g., myocardial ischemia/infarction, acute coronary syndrome, arrhythmia, bradycardia), back pain, abdominal pain, pyrexia, rash, urticaria, angioedema, cytokine release syndrome, and anaphylactoid/anaphylactic reactions. Infusion reactions occur more frequently with the first 2 infusions. These reactions may result in temporary interruption or withdrawal of treatment [see Adverse Reactions (6.1)].
Pre-medicate with acetaminophen, an antihistamine, and a corticosteroid [see Dosage and Administration (2.1, 2.4)]. Infusion reactions may occur despite premedication. Interrupt infusion with ARZERRA for infusion reactions of any severity. Institute medical management for severe infusion reactions including angina or other signs and symptoms of myocardial ischemia [see Dosage and Administration (2.3)]. If an anaphylactic reaction occurs, immediately and permanently discontinue ARZERRA and initiate appropriate medical treatment.
5.2 Hepatitis B Virus Reactivation
Hepatitis B virus (HBV) reactivation, in some cases resulting in fulminant hepatitis, hepatic failure, and death, has occurred in patients treated with ARZERRA. Cases have been reported in patients who are hepatitis B surface antigen (HBsAg) positive and also in patients who are HBsAg negative but are hepatitis B core antibody (anti-HBc) positive. Reactivation also has occurred in patients who appear to have resolved hepatitis B infection (i.e., HBsAg negative, anti-HBc positive, and hepatitis B surface antibody [anti-HBs] positive).
HBV reactivation is defined as an abrupt increase in HBV replication manifesting as a rapid increase in serum HBV DNA level or detection of HBsAg in a person who was previously HBsAg negative and anti-HBc positive. Reactivation of HBV replication is often followed by hepatitis, i.e., increase in transaminase levels and, in severe cases, increase in bilirubin levels, liver failure, and death.
Screen all patients for HBV infection by measuring HBsAg and anti-HBc before initiating treatment with ARZERRA. For patients who show evidence of hepatitis B infection (HBsAg positive [regardless of antibody status] or HBsAg negative but anti-HBc positive), consult physicians with expertise in managing hepatitis B regarding monitoring and consideration for HBV antiviral therapy.
Monitor patients with evidence of current or prior HBV infection for clinical and laboratory signs of hepatitis or HBV reactivation during and for several months following treatment with ARZERRA. HBV reactivation has been reported for at least 12 months following completion of therapy.
In patients who develop reactivation of HBV while receiving ARZERRA, immediately discontinue ARZERRA and any concomitant chemotherapy, and institute appropriate treatment. Resumption of ARZERRA in patients whose HBV reactivation resolves should be discussed with physicians with expertise in managing hepatitis B. Insufficient data exist regarding the safety of resuming ARZERRA in patients who develop HBV reactivation.
5.3 Hepatitis B Virus Infection
Fatal infection due to hepatitis B in patients who have not been previously infected has been observed with ARZERRA. Monitor patients for clinical and laboratory signs of hepatitis.
5.4 Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) resulting in death has occurred with ARZERRA. Consider PML in any patient with new onset of or changes in pre-existing neurological signs or symptoms. If PML is suspected, discontinue ARZERRA and initiate evaluation for PML including neurology consultation.
5.5 Tumor Lysis Syndrome
Tumor lysis syndrome (TLS), including the need for hospitalization, has occurred in patients treated with ARZERRA. Patients with high tumor burden and/or high circulating lymphocyte counts (>25 x 109/L) are at greater risk for developing TLS. Consider tumor lysis prophylaxis with anti-hyperuricemics and hydration beginning 12 to 24 hours prior to infusion of ARZERRA. For treatment of TLS, administer aggressive intravenous hydration and anti-hyperuricemic agents, correct electrolyte abnormalities, and monitor renal function.
5.6 Cytopenias
Severe cytopenias, including neutropenia, thrombocytopenia, and anemia, can occur with ARZERRA. Pancytopenia, agranulocytosis, and fatal neutropenic sepsis have occurred in patients who received ARZERRA in combination with chlorambucil. Grade 3 or 4 late-onset neutropenia (onset at least 42 days after last treatment dose) and/or prolonged neutropenia (not resolved between 24 and 42 days after last treatment dose) were reported in patients who received ARZERRA [see Adverse Reactions (6.1)]. Monitor complete blood counts at regular intervals during and after conclusion of therapy, and increase the frequency of monitoring in patients who develop Grade 3 or 4 cytopenias.
5.7 Immunizations
The safety of immunization with live viral vaccines during or following administration of ARZERRA has not been studied. Do not administer live viral vaccines to patients who have recently received ARZERRA. The ability to generate an immune response to any vaccine following administration of ARZERRA has not been studied.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are discussed in greater detail in other sections of the labeling:
- Infusion Reactions [see Warnings and Precautions (5.1)]
- Hepatitis B Virus Reactivation [see Warnings and Precautions (5.2)]
- Hepatitis B Virus Infection [see Warnings and Precautions (5.3)]
- Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.4)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.5)]
- Cytopenias [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Previously Untreated CLL: The safety of ARZERRA was evaluated in an open-label, parallel-arm, randomized trial (Study 1) in 444 patients with previously untreated CLL. Patients were randomized to receive either ARZERRA as an intravenous infusion every 28 days in combination with chlorambucil (n = 217) or chlorambucil as a single agent (n = 227). In both arms, patients received chlorambucil 10 mg/m2 orally on Days 1 to 7 every 28 days. The infusion schedule for ARZERRA was 300 mg administered on Cycle 1 Day 1, 1,000 mg administered on Cycle 1 Day 8, and 1,000 mg administered on Day 1 of subsequent 28-day cycles. The median number of cycles of ARZERRA completed was 6.
The most common adverse reactions (≥10%) were infusion reactions and neutropenia (Table 4).
The data described in Table 4 include relevant adverse reactions occurring up to 60 days after the last dose of study medication; Table 5 includes relevant hematologic laboratory abnormalities.
| a Includes events which occurred on the day of an infusion or within 24 hours of the end of an infusion and resulted in an interruption or discontinuation of treatment. Infusion reactions may include, but are not limited to, chills, dyspnea, flushing, hypotension, nausea, pain, pruritus, pyrexia, rash, and urticaria. b Includes oral herpes, herpes, herpes virus infection, genital herpes, and herpes simplex. |
||||
|
Adverse Reactions |
ARZERRA plus Chlorambucil (N = 217) |
Chlorambucil (N = 227) |
||
|
All Grades % |
Grade ≥3 % |
All Grades % |
Grade ≥3 % |
|
|
Infusion reactionsa |
67 |
10 |
0 |
0 |
|
Neutropenia |
27 |
26 |
18 |
14 |
|
Asthenia |
8 |
<1 |
5 |
0 |
|
Headache |
7 |
<1 |
3 |
0 |
|
Leukopenia |
6 |
3 |
2 |
<1 |
|
Herpes simplexb |
6 |
0 |
4 |
<1 |
|
Lower respiratory tract infection |
5 |
1 |
3 |
<1 |
|
Arthralgia |
5 |
<1 |
3 |
0 |
|
Upper abdominal pain |
5 |
0 |
3 |
0 |
|
Investigations |
ARZERRA plus Chlorambucil (N = 217) |
Chlorambucil (N = 227) |
||
|
All Grades % |
Grade ≥3 % |
All Grades % |
Grade ≥3 % |
|
|
Leukopenia |
67 |
23 |
28 |
4 |
|
Neutropenia |
66 |
29 |
56 |
24 |
|
Lymphopenia |
52 |
29 |
20 |
7 |
Infusion Reactions: Overall, 67% of patients who received ARZERRA in combination with chlorambucil experienced one or more symptoms of infusion reactions (10% were Grade 3 or greater; none were fatal). Infusion reactions occurred most frequently during Cycle 1 (56% on Day 1 [6% were Grade 3 or greater] and 23% on Day 8 [3% were Grade 3 or greater]) and decreased with subsequent infusions. Infusion reactions led to discontinuation of treatment in 3% of patients. Serious adverse events of infusion reactions occurred in 2% of patients.
Neutropenia: Overall, 3% of patients had neutropenia as a serious adverse event, reported up to 60 days after the last dose. One patient died with neutropenic sepsis and agranulocytosis. Prolonged neutropenia occurred in 6% of patients receiving ARZERRA in combination with chlorambucil compared with 4% of patients receiving chlorambucil. Late-onset neutropenia occurred in 6% of patients receiving ARZERRA in combination with chlorambucil compared with 1% of patients receiving chlorambucil alone.
Relapsed CLL: The safety of ARZERRA in combination with fludarabine and cyclophosphamide compared with fludarabine and cyclophosphamide was evaluated in a randomized, open-label, parallel-arm, multicenter trial (Study 2) in 359 patients with relapsed CLL. Patients were randomized to receive ARZERRA as an intravenous infusion (Cycle 1: 300 mg on Day 1 and 1,000 mg on Day 8; followed by 1,000 mg on Day 1 of subsequent 28-day cycles for a maximum of 6 cycles). Standard fludarabine and cyclophosphamide therapy was administered as a 3-day course starting on the first day of each cycle, with initial dosages of 25 mg/m2 for fludarabine and 250 mg/m2 for cyclophosphamide. Table 6 includes adverse reactions occurring up to 60 days after the last dose of study medication.
The most common adverse reactions (≥10%) were infusion reactions, neutropenia, leukopenia and febrile neutropenia (Table 6).
| a Includes events which occurred on the day of an infusion or within 24 hours of the end of an infusion and resulted in an interruption or discontinuation of treatment. Infusion reactions may include, but are not limited to, chills, dyspnea, flushing, hypotension, nausea, pain, pruritus, pyrexia, rash, and urticaria. | ||||
|
Adverse Reactions |
ARZERRA plus Fludarabine and Cyclophosphamide (N = 181) |
Fludarabine and Cyclophosphamide Arm (N = 178) |
||
|
All Grades % |
Grade >3 % |
All Grades % |
Grade >3 % |
|
|
Infusion reactionsa |
60 |
9 |
28 |
3 |
|
Neutropenia |
55 |
49 |
39 |
36 |
|
Leukopenia |
15 |
12 |
6 |
3 |
|
Febrile neutropenia |
10 |
10 |
8 |
8 |
|
Bronchitis |
6 |
1 |
4 |
<1 |
Adverse reactions associated with decreased platelet counts (including but not limited to thrombocytopenia, platelet count decreased and pancytopenia) and decreased hemoglobin (including but not limited to anemia, hemoglobin decreased and pancytopenia) occurred less frequently in the ARZERRA plus fludarabine and cyclophosphamide arm than in the fludarabine and cyclophosphamide arm up to 60 days after the last dose of study treatment: 30% (all grades) and 15% (Grade ≥3) vs 38% (all grades) and 28% (Grade ≥3), respectively for decreased platelet counts; and 23% (all grades) and 10% (Grade ≥3) vs 33% (all grades) and 16% (Grade ≥3), respectively for decreased hemoglobin.
Infusion Reactions: On Day 1 of infusion, infusion reactions occurred in 49% (7% were >Grade 3) of patients treated with ARZERRA plus fludarabine and cyclophosphamide, compared to 16% (1% were >Grade 3) of patients treated with fludarabine and cyclophosphamide and decreased with subsequent infusions. Infusion reactions led to discontinuation of treatment in 3% of patients in the ARZERRA plus fludarabine and cyclophosphamide. Serious adverse events of infusion reactions occurred in 2% of patients in the ARZERRA plus fludarabine and cyclophosphamide compared to <1% of patients treated with fludarabine and cyclophosphamide.
Neutropenia: The proportion of patients that had Grade 3 or greater neutropenia reported up to 60 days after the last dose of study medication was higher in patients treated with ARZERRA plus fludarabine and cyclophosphamide (51%) compared to the fludarabine and cyclophosphamide arm (37%). Grade 3 or greater neutropenic sepsis occurred in 2 patients (1%) treated with ARZERRA plus fludarabine and cyclophosphamide vs. 3 patients (2%) in the fludarabine and cyclophosphamide arm. Prolonged neutropenia occurred in 18 patients (10%) treated with ARZERRA plus fludarabine and cyclophosphamide vs. 20 patients (11%) in the fludarabine and cyclophosphamide arm. Late-onset neutropenia occurred in 13 patients (7%) treated with ARZERRA plus fludarabine and cyclophosphamide vs. 5 patients (3%) in the fludarabine and cyclophosphamide arm.
During the period between the first dose and 60 days after last dose there were five (3%) patients who died in the ARZERRA plus fludarabine and cyclophosphamide arm and ten (6%) patients who died in the fludarabine and cyclophosphamide arm.
Extended Treatment in CLL: The safety of ARZERRA was evaluated in an open-label, parallel-arm, randomized trial (Study 3) in 474 patients who had responded to therapy for their recurrent or progressive disease. Patients were randomized to receive ARZERRA as an intravenous infusion every 8 weeks or observation. The infusion schedule for ARZERRA was 300 mg on Day 1 followed 1 week later by 1,000 mg on Day 8 followed 7 weeks later by 1,000 mg and every 8 weeks thereafter for up to a maximum of 2 years. The data described in Table 7 include relevant adverse reactions occurring up to 60 days after the last dose of study medication (last visit for observation arm). The most common adverse reactions (≥10%) were infusion reactions, neutropenia, and upper respiratory tract infection (Table 7).
| a Includes events which occurred on the day of an infusion or within 24 hours of the end of an infusion and resulted in an interruption or discontinuation of treatment. Infusion reactions may include, but are not limited to, chills, dyspnea, flushing, hypotension, nausea, pain, pruritus, pyrexia, rash, and urticaria. | ||||
| ARZERRA (N = 237) | Observation Arm (N = 237) |
|||
| Adverse Reactions | All Grades % | Grade ≥3 % | All Grades % | Grade ≥3 % |
| Infusion reactionsa | 46 | 4 | - | - |
| Neutropenia | 24 | 22 | 9 | 8 |
| Upper respiratory tract infection | 19 | 1 | 9 | 0 |
| Bronchitis | 9 | <1 | 7 | <1 |
| Pneumonia | 8 | 5 | 5 | 3 |
| Influenza | 6 | 0 | 3 | 0 |
| Herpes zoster | 5 | <1 | 3 | <1 |
| Insomnia | 5 | <1 | 2 | 0 |
| Back pain | 5 | 0 | 3 | 0 |
| Hypogammaglobulinemia | 5 | <1 | <1 | <1 |
Infusion Reactions: Infusion reactions occurred in 25% of patients on the day of Infusion 1 (300 mg) and decreased with subsequent infusions (between 2% to 10%).
Infections: A total of 154 patients (65%) treated with ARZERRA compared with 120 patients (51%) in the observation arm experienced bacterial, viral, or fungal infections. The incidence of serious infections, however, was similar for patients treated with ARZERRA (20%) and the observation arm (18%). The proportions of fatal infections in patients treated with ARZERRA and in the observation arm were 2% and 3% respectively.
Neutropenia: The proportion of patients that had Grade 3 or greater neutropenia reported up to 60 days after the last dose of study medication was higher in patients treated with ARZERRA (22%) compared with the observation arm (8%). There were no cases of neutropenic sepsis reported with ARZERRA. Prolonged neutropenia occurred in 13 patients (5%) treated with ARZERRA and in 5 patients (2%) in the observation arm. Late-onset neutropenia occurred in 2 patients (<1%) treated with ARZERRA and 1 patient (<1%) in the observation arm.
During the period between the first dose and 60 days after last dose there were two (1%) patients in the ofatumumab group who died due to adverse events and five (2%) patients in the observation group.
Refractory CLL: The safety of monotherapy with ARZERRA was evaluated in 181 patients with relapsed or refractory CLL in 2 open-label, non-randomized, single-arm studies. In these studies, ARZERRA was administered at 2,000 mg beginning with the second dose for 11 doses (Study 4 [n = 154]) or 3 doses (Study 5 [n = 27]).
The data described in Table 8 and other sections below are derived from 154 patients in Study 4. All patients received 2,000 mg weekly from the second dose onward. Ninety percent (90%) of patients received at least 8 infusions of ARZERRA and 55% received all 12 infusions. The median age was 63 years (range: 41 to 86 years), 72% were male, and 97% were white.
In refractory CLL, the most common adverse reactions (≥10%) were neutropenia, pneumonia, pyrexia, cough, diarrhea, anemia, fatigue, dyspnea, rash, nausea, bronchitis, and upper respiratory tract infections (Table 8). The most common serious adverse reactions were infections (including pneumonia and sepsis), neutropenia, and pyrexia. Infections were the most common adverse reactions leading to drug discontinuation.
| a Includes pneumonia, lung infection, lobar pneumonia, and bronchopneumonia. b Includes rash, rash macular, and rash vesicular. c Includes sepsis, neutropenic sepsis, bacteremia, and septic shock. |
||||
|
Adverse Reaction |
Total Population (N = 154) |
Fludarabine‑ and Alemtuzumab‑refractory (N = 59) |
||
|
All Grades % |
Grade ≥3 % |
All Grades % |
Grade ≥3 % |
|
|
Pneumoniaa |
23 |
14 |
25 |
15 |
|
Pyrexia |
20 |
3 |
25 |
5 |
|
Cough |
19 |
0 |
19 |
0 |
|
Diarrhea |
18 |
0 |
19 |
0 |
|
Anemia |
16 |
5 |
17 |
8 |
|
Fatigue |
15 |
0 |
15 |
0 |
|
Dyspnea |
14 |
2 |
19 |
5 |
|
Rashb |
14 |
<1 |
17 |
2 |
|
Bronchitis |
11 |
<1 |
19 |
2 |
|
Nausea |
11 |
0 |
12 |
0 |
|
Upper respiratory tract infection |
11 |
0 |
3 |
0 |
|
Edema peripheral |
9 |
<1 |
8 |
2 |
|
Back pain |
8 |
1 |
12 |
2 |
|
Chills |
8 |
0 |
10 |
0 |
|
Nasopharyngitis |
8 |
0 |
8 |
0 |
|
Sepsisc |
8 |
8 |
10 |
10 |
|
Urticaria |
8 |
0 |
5 |
0 |
|
Insomnia |
7 |
0 |
10 |
0 |
|
Headache |
6 |
0 |
7 |
0 |
|
Herpes zoster |
6 |
1 |
7 |
2 |
|
Hyperhidrosis |
5 |
0 |
5 |
0 |
|
Hypertension |
5 |
0 |
8 |
0 |
|
Hypotension |
5 |
0 |
3 |
0 |
|
Muscle spasms |
5 |
0 |
3 |
0 |
|
Sinusitis |
5 |
2 |
3 |
2 |
|
Tachycardia |
5 |
<1 |
7 |
2 |
Infusion Reactions: Infusion reactions occurred in 44% of patients on the day of the first infusion (300 mg), 29% on the day of the second infusion (2,000 mg), and less frequently during subsequent infusions.
Infections: A total of 108 patients (70%) experienced bacterial, viral, or fungal infections. A total of 45 patients (29%) experienced Grade 3 or greater infections, of which 19 (12%) were fatal. The proportion of fatal infections in the fludarabine‑ and alemtuzumab‑refractory group was 17%.
Neutropenia: Of 108 patients with normal neutrophil counts at baseline, 45 (42%) developed Grade 3 or greater neutropenia. Nineteen (18%) developed Grade 4 neutropenia. Some patients experienced new onset Grade 4 neutropenia >2 weeks in duration.
6.2 Immunogenicity
There is a potential for immunogenicity with therapeutic proteins such as ofatumumab. Serum samples from more than 926 patients with CLL were tested during and after treatment for antibodies to ARZERRA. Formation of anti-ofatumumab antibodies was observed in less than 1% of patients with CLL after treatment with ofatumumab.
Immunogenicity assay results are highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to ARZERRA with the incidence of antibodies to other products may be misleading.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ARZERRA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Infusion-related Cardiac Events
Cardiac arrest
Mucocutaneous Reactions
Stevens-Johnson syndrome, porphyria cutanea tarda
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
ARZERRA may cause fetal B-cell depletion based on findings from animal studies and the drug’s mechanism of action [see Clinical Pharmacology (12.1)]. There are no data on ARZERRA use in pregnant women to inform a drug-associated risk. However, there are clinical considerations [see Clinical Considerations]. No teratogenicity was observed in animal reproduction studies with administration of ARZERRA to pregnant monkeys during organogenesis at doses 0.3 and 2.4 times the maximum recommended human dose (MRHD) of 2000 mg based on monkey geometric mean AUCinf of 213 mg.h/mL and 1646 mg.h/mL, respectively. However, ofatumumab caused depletion of maternal circulating B-cells, depletion of peripheral and splenic fetal B-cells, and decreased fetal spleen weights [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population the estimated background risk of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies, respectively.
Clinical Considerations
Fetal/neonatal adverse reactions
ARZERRA may cause fetal B-cell depletion [see Data]. Avoid administering live vaccines to neonates and infants exposed to ARZERRA in utero until B-cell recovery occurs [see Warnings and Precautions (5.7) and Clinical Pharmacology (12.2)].
Data
Animal Data
In an embryo-fetal development study, pregnant cynomolgus monkeys received 20 or 100 mg/kg/day of ofatumumab intravenously (30 minute infusion) once weekly during the period of organogenesis [Gestation Days (GD) 20 to 50] with systemic exposure throughout pregnancy due to the long half-life as drug was detected in maternal serum on GD 100 (early fetal period of development). At the end of organogenesis on GD 48, the exposure in pregnant monkeys receiving ofatumumab 20 and 100 mg/kg/day was approximately 0.3 and 2.4 times the human exposure after the 8th infusion of the MRHD of 2,000 mg based on monkey geometric mean AUCinf of 213 mg.h/mL and 1,646 mg.h/mL, respectively. Ofatumumab crossed the placenta, as it was detected in fetal cord blood on GD 100 in both dose groups. There was no maternal toxicity and no effect on pregnancy success. As expected, both dose levels of ofatumumab depleted circulating B cells in the mothers; recovery of B lymphocytes in dosed animals was not observed during the dosing-free period. Following Caesarean section at GD 100, fetuses from treated mothers exhibited decreases in mean peripheral and splenic B-cell counts (decreased to approximately 12% and 15% of control values at both dose levels, respectively and spleen weights (decreased by approximately 15% in low-dose group and by approximately 30% in high-dose group, compared with control values). There was no effect on prenatal survival and no evidence of teratogenicity in this monkey study.
The kinetics of B-lymphocyte recovery and the potential long-term effects of perinatal B-cell depletion in offspring from ofatumumab-treated maternal monkeys have not been studied.
8.2 Lactation
Risk Summary
There is no information regarding the presence of ARZERRA in human milk, the effects on the breastfed infant, or the effects on milk production. Human IgG is known to be present in human milk. Published data suggest that antibodies in breast milk do not enter the neonatal and infant circulations in substantial amounts.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ARZERRA and any potential adverse effects on the breastfed infant from ARZERRA or from the underlying maternal condition.
8.5 Geriatric Use
In patients with previously untreated CLL (Study 1), 68% (148/217) receiving ARZERRA plus chlorambucil were 65 years and older. Patients age 65 years and older experienced a higher incidence of the following Grade 3 or greater adverse reactions compared with patients younger than 65 years of age: neutropenia (30% versus 17%) and pneumonia (5% versus 1%) [see Adverse Reactions (6.1)]. In patients 65 years and older, 29% experienced serious adverse events compared with 13% of patients younger than 65 years. No clinically meaningful differences in the effectiveness of ARZERRA plus chlorambucil were observed between older and younger patients [see Clinical Studies (14.1)].
In patients with relapsed CLL (Study 2), 33% (60/181) of patients receiving ARZERRA plus fludarabine and cyclophosphamide were 65 years and older. No clinically meaningful differences in the safety or effectiveness of ARZERRA were observed between patients age 65 years and older, and those younger than 65 years [see Adverse Reactions (6.1), Clinical Studies (14.2)].
With extended treatment in patients with CLL (Study 3), 49% (117/237) receiving ARZERRA were 65 years and older. No clinically meaningful differences in the safety or effectiveness of ARZERRA were observed between patients age 65 years and older and those younger than 65 years of age [see Adverse Reactions (6.1), Clinical Studies (14.3)].
In refractory CLL clinical studies there were insufficient numbers of patients aged 65 years and older to determine whether they respond differently from younger patients [see Clinical Pharmacology (12.3)].
11. Arzerra Description
ARZERRA (ofatumumab) is an IgG1κ human monoclonal antibody with a molecular weight of approximately 149 kDa. The antibody was generated via transgenic mouse and hybridoma technology and is produced in a recombinant murine cell line (NS0) using standard mammalian cell cultivation and purification technologies.
ARZERRA is a sterile, clear to opalescent, colorless, preservative-free liquid concentrate for intravenous administration. ARZERRA is supplied at a concentration of 20 mg/mL in single-use vials. Each single-use vial contains either 100 mg ofatumumab in 5 mL of solution or 1,000 mg ofatumumab in 50 mL of solution.
Inactive ingredients include: 10 mg/mL arginine, diluted hydrochloric acid, 0.019 mg/mL edetate disodium, 0.2 mg/mL polysorbate 80, 6.8 mg/mL sodium acetate, 2.98 mg/mL sodium chloride, and Water for Injection, USP. The pH is 5.5.
12. Arzerra - Clinical Pharmacology
12.1 Mechanism of Action
Ofatumumab binds specifically to both the small and large extracellular loops of the CD20 molecule. The CD20 molecule is expressed on normal B lymphocytes (pre–B- to mature B-lymphocyte) and on B-cell CLL. The CD20 molecule is not shed from the cell surface and is not internalized following antibody binding.
The Fab domain of ofatumumab binds to the CD20 molecule and the Fc domain mediates immune effector functions to result in B-cell lysis in vitro. Data suggest that possible mechanisms of cell lysis include complement-dependent cytotoxicity and antibody-dependent, cell-mediated cytotoxicity.
12.2 Pharmacodynamics
B-cell Depletion: In patients with previously untreated CLL, at 6 months after the last dose, the median reductions in CD19-positive B cells were >99% (n = 155) for ARZERRA in combination with chlorambucil and 94% (n = 121) for chlorambucil alone.
In patients with relapsed CLL, the proportion of responders in the ofatumumab in combination with fludarabine and cyclophosphamide (O+FC) arm who showed complete or near complete B cell depletion was 39% (n = 59) and 82% (n = 126) , respectively. The proportion of responders in the fludarabine and cyclophosphamide (FC) arm with complete or near complete B cell depletion was 4% (n = 5) and 23% (n = 28), respectively.
In patients treated with extended treatment for CLL after response to therapy for their recurrent or progressive disease, the median decreases in B-cell counts were 61% (n = 168) after the first infusion and 80% (n = 114) prior to the sixth infusion; in the observation arm, the median changes in B-cell counts at the same time points were increases of 32% (n = 148) and 1,328% (n = 95).
In patients with CLL refractory to fludarabine and alemtuzumab, the median decrease in circulating CD19-positive B cells was 91% (n = 50) with the 8th infusion and 85% (n = 32) with the 12th infusion. The time to recovery of lymphocytes, including CD19-positive B cells, to normal levels has not been determined.
Although the depletion of B-cells in the peripheral blood is a measurable pharmacodynamic effect, it is not directly correlated with the depletion of B cells in solid organs or in malignant deposits. B-cell depletion has not been shown to be directly correlated to clinical response.
Cardiac Electrophysiology: The effect of multiple doses of ARZERRA on the QTc interval was evaluated in a pooled analysis of 3 open-label studies in patients with CLL (N = 85). Patients received ARZERRA 300 mg on Day 1 followed by either 1,000 mg or 2,000 mg for subsequent doses. No large changes in the mean QTc interval (i.e., >20 milliseconds) were detected in the pooled analysis.
12.3 Pharmacokinetics
Ofatumumab is eliminated through both a target-independent route and a B cell-mediated route. Ofatumumab exhibited dose-dependent clearance in the dose range of 100 to 2,000 mg. Due to the depletion of B cells, the clearance of ofatumumab decreased substantially after subsequent infusions compared with the first infusion.
Pharmacokinetic data were obtained after repeated administration (4, 5, 6, 8, or 12 infusions) of 1,000 mg or 2,000 mg doses in 774 patients with CLL (Studies 1, 2, 3, 4 and 5). The geometric mean (% CV) values for clearance, volume of distribution at steady state (Vss), and half-life for ofatumumab in these patients were 9.3 mL/hour (91%), 6.1 L (52%), and 17.6 days (83%). The pharmacokinetic profile was similar across doses in patients with CLL.
Specific Populations: The following population characteristics do not have a clinically meaningful effect on the pharmacokinetics of ofatumumab: body size, gender, age, and renal impairment (evaluated in patients with a calculated creatinine clearance ≥30 mL/min).
No formal studies of ARZERRA in patients with hepatic impairment have been conducted. The effect of a calculated CrCL < 30 mL/min on the pharmacokinetics of ARZERRA has not been evaluated.
Drug Interactions: Coadministration of ARZERRA did not result in clinically relevant effects on the pharmacokinetics of fludarabine, cyclophosphamide, chlorambucil, or its active metabolite, phenylacetic acid mustard.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or mutagenicity studies of ofatumumab have been conducted. In a repeat-dose toxicity study, no tumorigenic or unexpected mitogenic responses were noted in cynomolgus monkeys treated for 7 months with up to 3.5 times the maximum human dose (2,000 mg) of ofatumumab. Effects on male and female fertility have not been evaluated in animal studies.
14. Clinical Studies
14.1 Previously Untreated CLL
The efficacy of ARZERRA was evaluated in a randomized, open-label, parallel-arm study; 447 patients previously untreated for CLL were randomized to receive either ARZERRA as monthly intravenous infusions (Cycle 1: 300 mg on Day 1 and 1,000 mg on Day 8; subsequent cycles: 1,000 mg on Day 1 every 28 days) in combination with chlorambucil (10 mg/m2 orally on Days 1 to 7 every 28 days) or chlorambucil alone (10 mg/m2 orally on Days 1 to 7 every 28 days). Patients received treatment for a minimum of 3 cycles. Treatment was continued for 3 cycles beyond maximal response (2 consecutive response assessments of stable disease, partial response, or complete response) for up to 12 cycles. Approximately 60% of patients received 3 to 6 cycles of ARZERRA and 30% received 7 to 12 cycles.
This trial enrolled patients for whom fludarabine-based therapy was considered to be inappropriate by the investigator for reasons that included advanced age or presence of co-morbidities. In the overall trial population, the median age was 69 years (range: 35 to 92 years) and 69% of patients in both arms were at least 65 years of age. In the overall trial population, 72% of patients had 2 or more co-morbidities and 48% of patients had a creatinine clearance of less than 70 mL/min. Sixty-three percent (63%) of patients were male and 89% were white. Elevated beta-2 microglobulin (β2m) >3,500 mcg/L was present in 72% of patients at baseline.
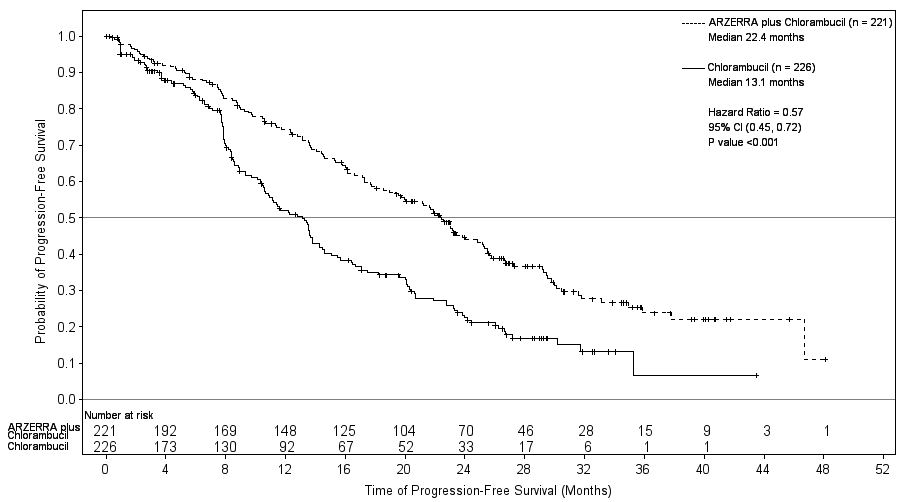
Efficacy was evaluated by progression-free-survival (PFS) as assessed by a blinded Independent Review Committee (IRC) using the International Workshop for Chronic Lymphocytic Leukemia (IWCLL) updated National Cancer Institute-sponsored Working Group (NCI-WG) guidelines (2008). ARZERRA plus chlorambucil resulted in statistically significant improvement in IRC-assessed median PFS compared with chlorambucil alone (22.4 months versus 13.1 months; hazard ratio: 0.57 [0.45, 0.72]) (Table 9; Figure 1).
Secondary efficacy endpoints, including overall response (OR), complete response (CR), and duration of response, were also assessed by the IRC using the 2008 IWCLL Guidelines (Table 9).
|
IRC = Independent Review Committee; ITT = Intention to treat; CI = Confidence interval. a Intention-to-treat population includes all 447 randomized patients. b Pike Estimator. |
||
|
Primary and Key Secondary Endpoints |
ARZERRA plus Chlorambucil (N = 221) |
Chlorambucil (N = 226) |
|
Progression-free survival (PFS)
|
22.4 (19.0, 25.2) |
13.1 (10.6, 13.8) |
|
Hazard ratiob (95% CI)
|
0.57 (0.45, 0.72) P <0.001 |
|
|
82.4 (76.7, 87.1) |
68.6 (62.1, 74.6) |
|
P = 0.001 |
|
|
12 |
1 |
|
22.1 (19.1, 24.6) |
13.2 (10.8, 16.4) |
Figure 1. Kaplan-Meier Estimates of IRC-assessed Progression-free Survival
14.2 Relapsed CLL
Study 2 (randomized, open-label, parallel-arm, multicenter trial) evaluated the efficacy of ARZERRA in combination with fludarabine and cyclophosphamide compared with fludarabine and cyclophosphamide in 365 patients with relapsed CLL. Baseline disease characteristics and prognostic markers were balanced between treatment arms. Patient median age was 61 years (range: 32 to 90 years), 60% were male and 55% and 28% of patients were Binet stage B and C, respectively. Eighty one percent (81%) of patients received 1-2 prior treatments and 21% of patients had received prior rituximab.
Patients received ARZERRA as intravenous infusions (Cycle 1: 300 mg on Day 1 and 1,000 mg on Day 8; followed by 1,000 mg on Day 1 of subsequent cycles). Approximately 90% of patients received 3-6 cycles of ARZERRA and 66% completed all 6 cycles.
Efficacy was evaluated by progression-free survival (PFS), as assessed by an independent review committee (IRC). PFS was prolonged in the ARZERRA plus fludarabine and cyclophosphamide (O+FC) arm compared to the fludarabine and cyclophosphamide (FC) arm (Table 10) resulting in a 10 months improvement in median PFS (mPFS) (see Figure 2).
|
a Confidence intervals were obtained using the Brookmeyer-Crowley method. b Hazard ratios were obtained using the Pike estimator. A hazard ratio<1 indicates a lower risk with O+FC compared with FC. The hazard ratio and p-value from the stratified log-rank test are adjusted for Binet stage and number of prior therapies. c Cochran-Mantel-Haenzel test, adjusting for stratification factors: Binet stage and number of prior therapies. |
||
|
Primary and Key Secondary Endpoints |
ARZERRA plus Fludarabine and Cyclophosphamide (N = 183) |
Fludarabine and Cyclophosphamide (N = 182) |
|
Progression-free survival (PFS)
|
28.9 (22.8, 35.9) |
18.8 (14.4, 25.8) |
|
Hazard ratiob (95% CI)
|
0.67 (0.51, 0.88) P = 0.0032 |
|
|
Overall response, % (95% CI) |
84 (77, 89) |
68 (60, 74) |
|
P = 0.0003 |
|
|
Complete response |
27% |
7% |
|
Complete response with incomplete bone marrow recovery |
2% |
1% |
Figure 2. Kaplan-Meier Estimates of IRC-assessed Progression-free Survival
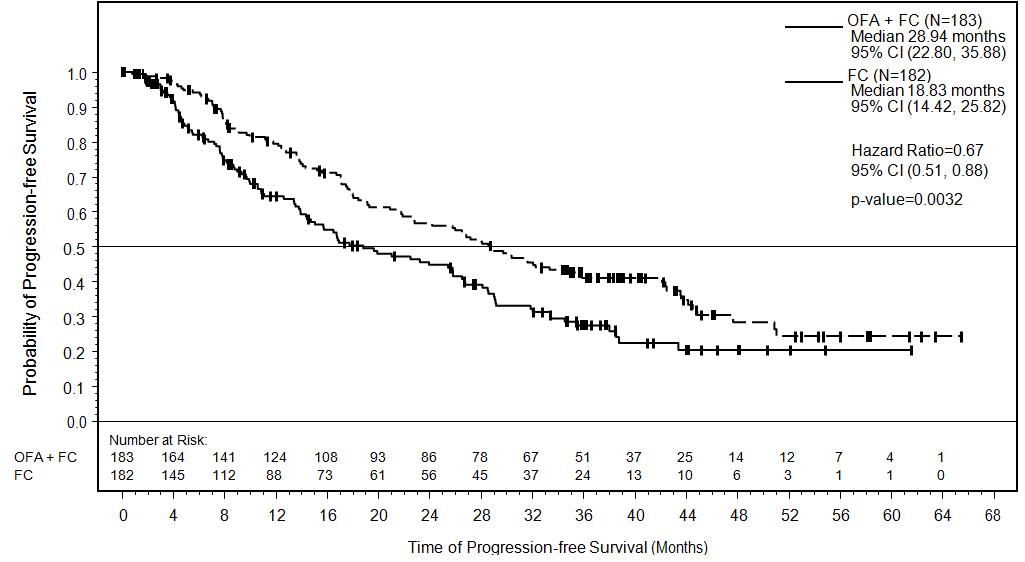
With a median follow-up of approximately 34 months, overall survival results showed a HR of 0.78 [95% CI: 0.56-1.09]. The median overall survival was 56.4 months [95% CI: 44.2, NC] in the O+FC arm and was 45.8 months [95% CI: 37.3, NC] in the FC arm.
14.3 Extended Treatment in CLL
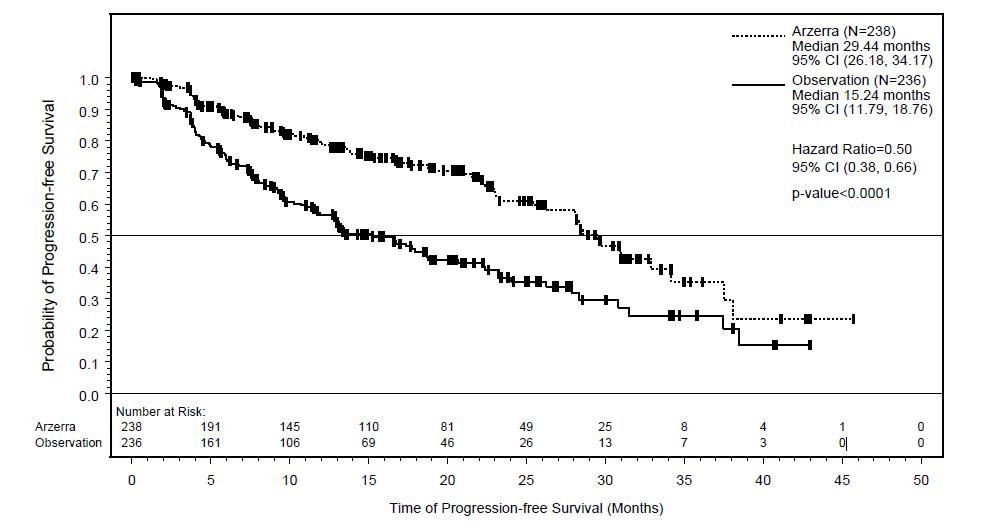
The efficacy of ARZERRA as extended treatment in CLL was evaluated in a randomized, parallel arm, open-label trial. In this trial, 474 patients who were in complete or partial response after at least two lines of prior therapy, were randomized to receive ARZERRA as intravenous infusions (300 mg on Day 1 followed 1 week later by 1,000 mg on Day 8 followed 7 weeks later by 1,000 mg and every 8 weeks thereafter for up to a maximum of 2 years) or observation.
In the overall trial population, the median age was 65 years (range: 33 to 87 years), 68% were male, and 96% were white. Most patients were in partial remission (81%), had two prior treatments (70%), and had received chemoimmunotherapy (80%) as prior therapy. The main efficacy outcome was progression-free survival (PFS) as assessed by the investigator.
At the time of the efficacy analysis, the median follow-up was 19.4 months with the ARZERRA arm and 18.7 months with the observation arm. The event rate (progressed or died) was 33% in the ARZERRA arm and 51% in the observation arm. The investigator-assessed median PFS was 29.4 months in the ARZERRA arm and 15.2 months in the observation arm (hazard ratio: 0.50 with 95% confidence interval [0.38, 0.66]; P <0.0001).
Figure 3. Kaplan-Meier Estimates of Investigator-assessed Progression-free Survival
14.4 Refractory CLL
Study 4 was a single-arm, multicenter trial in 154 patients with relapsed or refractory CLL. ARZERRA was administered by intravenous infusion according to the following schedule: 300 mg (Week 0), 2,000 mg weekly for 7 infusions (Weeks 1 through 7), and 2,000 mg every 4 weeks for 4 infusions (Weeks 12 through 24). Patients with CLL refractory to fludarabine and alemtuzumab (n = 59) comprised the efficacy population. Drug refractoriness was defined as failure to achieve at least a partial response to, or disease progression within 6 months of, the last dose of fludarabine or alemtuzumab. The main efficacy outcome was durable objective tumor response rate. Objective tumor responses were determined using the 1996 NCI-WG Guidelines for CLL.
In patients with CLL refractory to fludarabine and alemtuzumab, the median age was 64 years (range: 41 to 86 years), 75% were male, and 95% were white. The median number of prior therapies was 5; 93% received prior alkylating agents, 59% received prior rituximab, and all received prior fludarabine and alemtuzumab. Eighty-eight percent of patients received at least 8 infusions of ARZERRA and 54% received 12 infusions.
The investigator-determined overall response rate in patients with CLL refractory to fludarabine and alemtuzumab was 42% (99% CI: 26, 60) with a median duration of response of 6.5 months (95% CI: 5.8, 8.3). There were no complete responses. Anti-tumor activity was also observed in additional patients in Study 4 and in a multicenter, open-label, dose-escalation study (Study 5) conducted in patients with relapsed or refractory CLL.
16. How is Arzerra supplied
ARZERRA (ofatumumab) is a sterile, clear to opalescent, colorless, preservative-free liquid concentrate (20 mg/mL) for dilution and intravenous administration provided in single-use glass vials with a rubber stopper (not made with natural rubber latex) and an aluminum overseal. Each vial contains either 100 mg ofatumumab in 5 mL of solution or 1,000 mg ofatumumab in 50 mL of solution.
ARZERRA is available as follows:
|
Carton Contents |
NDC |
|
3 single-use 100 mg/5 mL vials |
Vial: NDC 0078-0669-61 Carton of 3 vials: NDC 0078-0669-13 |
|
1 single-use 1,000 mg/50 mL vial |
Vial and Carton: NDC 0078-0690-61 |
17. Patient Counseling Information
Advise patients to contact a healthcare professional for any of the following:
- Signs and symptoms of infusion reactions including fever, chills, rash, or breathing problems within 24 hours of infusion [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]
- Symptoms of hepatitis including worsening fatigue or yellow discoloration of skin or eyes [see Warnings and Precautions (5.2, 5.3)]
- New neurological symptoms such as confusion, dizziness or loss of balance, difficulty talking or walking, or vision problems [see Warnings and Precautions (5.4)]
- Bleeding, easy bruising, petechiae, pallor, worsening weakness, or fatigue [see Warnings and Precautions (5.6)]
- Signs of infections including fever and cough [see Warnings and Precautions (5.6), Adverse Reactions (6.1)]
- Pregnancy- Advise pregnant women of potential fetal B-cell depletion [see Use in Specific Populations (8.1, 8.2)]
Advise patients of the need for:
- Monitoring and possible need for treatment if they have a history of hepatitis B infection (based on the blood test) [see Warnings and Precautions (5.2)].
- Periodic monitoring for blood counts [see Warnings and Precautions (5.6)]
- Avoiding vaccination with live viral vaccines [see Warnings and Precautions (5.7)]
Manufactured by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
US License 1244
At
Glaxo Group Ltd
Brentford, Middlesex, UK
© Novartis
T2016-73
August 2016

| ARZERRA
ofatumumab injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ARZERRA
ofatumumab injection, solution |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |
Frequently asked questions
More about Arzerra (ofatumumab)
- Check interactions
- Compare alternatives
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- FDA approval history
- Drug class: CD20 monoclonal antibodies
- Breastfeeding
- En español