Tasmar Prescribing Information
Package insert / product label
Generic name: tolcapone
Dosage form: tablet, film coated
Drug class: Dopaminergic antiparkinsonism agents
Medically reviewed by Drugs.com. Last updated on Jan 11, 2024.
On This Page
- Description
- Clinical Pharmacology
- Drug Interactions
- Clinical Studies
- Indications and Usage
- Contraindications
- Warnings
- Precautions
- Patient Counseling Information
- Adverse Reactions/Side Effects
- Drug Abuse and Dependence
- Overdosage
- Dosage and Administration
- How Supplied/Storage and Handling
- Storage and Handling
Before prescribing TASMAR®, the physician should be thoroughly familiar with the details of this prescribing information.
TASMAR SHOULD NOT BE USED BY PATIENTS UNTIL THERE HAS BEEN A COMPLETE DISCUSSION OF THE RISKS AND THE PATIENT HAS PROVIDED WRITTEN ACKNOWLEDGEMENT THAT THE RISKS HAVE BEEN EXPLAINED (SEE PATIENT ACKNOWLEDGEMENT OF RISKS SECTION).
WARNING
Because of the risk of potentially fatal, acute fulminant liver failure, TASMAR (tolcapone) should ordinarily be used in patients with Parkinson's disease on l-dopa/carbidopa who are experiencing symptom fluctuations and are not responding satisfactorily to or are not appropriate candidates for other adjunctive therapies (see INDICATIONS and DOSAGE AND ADMINISTRATION sections).
Because of the risk of liver injury and because TASMAR, when it is effective, provides an observable symptomatic benefit, the patient who fails to show substantial clinical benefit within 3 weeks of initiation of treatment, should be withdrawn from TASMAR.
TASMAR therapy should not be initiated if the patient exhibits clinical evidence of liver disease or two SGPT/ALT or SGOT/AST values greater than the upper limit of normal. Patients with severe dyskinesia or dystonia should be treated with caution (see PRECAUTIONS: Rhabdomyolysis).
PATIENTS WHO DEVELOP EVIDENCE OF HEPATOCELLULAR INJURY WHILE ON TASMAR AND ARE WITHDRAWN FROM THE DRUG FOR ANY REASON MAY BE AT INCREASED RISK FOR LIVER INJURY IF TASMAR IS REINTRODUCED. ACCORDINGLY, SUCH PATIENTS SHOULD NOT ORDINARILY BE CONSIDERED FOR RETREATMENT.
Cases of severe hepatocellular injury, including fulminant liver failure resulting in death, have been reported in postmarketing use. As of May 2005, three cases of fatal fulminant hepatic failure have been reported from more than 40,000 patient years of worldwide use. This incidence may be 10- to 100-fold higher than the background incidence in the general population. Underreporting of cases may lead to significant underestimation of the increased risk associated with the use of TASMAR. All three cases were reported within the first six months of initiation of treatment with TASMAR. Analysis of the laboratory monitoring data in over 3,400 TASMAR-treated patients participating in clinical trials indicated that increases in SGPT/ALT or SGOT/AST, when present, generally occurred within the first 6 months of treatment with TASMAR.
A prescriber who elects to use TASMAR in face of the increased risk of liver injury is strongly advised to monitor patients for evidence of emergent liver injury. Patients should be advised of the need for self-monitoring for both the classical signs of liver disease (e.g., clay colored stools, jaundice) and the nonspecific ones (e.g., fatigue, loss of appetite, lethargy).
Although a program of periodic laboratory monitoring for evidence of hepatocellular injury is recommended, it is not clear that periodic monitoring of liver enzymes will prevent the occurrence of fulminant liver failure. However, it is generally believed that early detection of drug-induced hepatic injury along with immediate withdrawal of the suspect drug enhances the likelihood for recovery. Accordingly, the following liver monitoring program is recommended.
Before starting treatment with TASMAR, the physician should conduct appropriate tests to exclude the presence of liver disease. In patients determined to be appropriate candidates for treatment with TASMAR, serum glutamic-pyruvic transaminase (SGPT/ALT) and serum glutamic-oxaloacetic transaminase (SGOT/AST) levels should be determined at baseline and periodically (i.e., every 2 to 4 weeks) for the first 6 months of therapy. After the first six months, periodic monitoring is recommended at intervals deemed clinically relevant. Although more frequent monitoring increases the chances of early detection, the precise schedule for monitoring is a matter of clinical judgment. If the dose is increased to 200 mg tid (see DOSAGE AND ADMINISTRATION section), liver enzyme monitoring should take place before increasing the dose and then be conducted every 2 to 4 weeks for the following 6 months of therapy. After six months, periodic monitoring is recommended at intervals deemed clinically relevant.
TASMAR should be discontinued if SGPT/ALT or SGOT/AST levels exceed 2 times the upper limit of normal or if clinical signs and symptoms suggest the onset of hepatic dysfunction (persistent nausea, fatigue, lethargy, anorexia, jaundice, dark urine, pruritus, and right upper quadrant tenderness).
Tasmar Description
TASMAR is available as tablets containing 100 mg tolcapone.
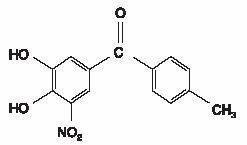
Tolcapone, an inhibitor of catechol-O-methyltransferase (COMT), is used in the treatment of Parkinson's disease as an adjunct to levodopa/carbidopa therapy. It is a yellow, odorless, non-hygroscopic, crystalline compound with a relative molecular mass of 273.25. The chemical name of tolcapone is 3,4-dihydroxy-4'-methyl-5-nitrobenzophenone. Its empirical formula is C14H11NO5 and its structural formula is:
Inactive ingredients: Core: microcrystalline cellulose, lactose monohydrate, anhydrous dibasic calcium phosphate, sodium starch glycolate, povidone, talc, magnesium stearate, and purified water. Film Coating: hypromellose, titanium dioxide, talc, ethylcellulose, triacetin, sodium lauryl sulfate, yellow iron oxide, red iron oxide, and purified water.
Tasmar - Clinical Pharmacology
Mechanism of Action:
Tolcapone is a selective and reversible inhibitor of catechol-O-methyltransferase (COMT).
In mammals, COMT is distributed throughout various organs. The highest activities are in the liver and kidney. COMT also occurs in the heart, lung, smooth and skeletal muscles, intestinal tract, reproductive organs, various glands, adipose tissue, skin, blood cells and neuronal tissues, especially in glial cells. COMT catalyzes the transfer of the methyl group of S-adenosyl-L-methionine to the phenolic group of substrates that contain a catechol structure. Physiological substrates of COMT include dopa, catecholamines (dopamine, norepinephrine, epinephrine) and their hydroxylated metabolites. The function of COMT is the elimination of biologically active catechols and some other hydroxylated metabolites. In the presence of a decarboxylase inhibitor, COMT becomes the major metabolizing enzyme for levodopa catalyzing the metabolism to 3-methoxy-4-hydroxy-L-phenylalanine (3-OMD) in the brain and periphery.
The precise mechanism of action of tolcapone is unknown, but it is believed to be related to its ability to inhibit COMT and alter the plasma pharmacokinetics of levodopa. When tolcapone is given in conjunction with levodopa and an aromatic amino acid decarboxylase inhibitor, such as carbidopa, plasma levels of levodopa are more sustained than after administration of levodopa and an aromatic amino acid decarboxylase inhibitor alone. It is believed that these sustained plasma levels of levodopa result in more constant dopaminergic stimulation in the brain, leading to greater effects on the signs and symptoms of Parkinson's disease in patients as well as increased levodopa adverse effects, sometimes requiring a decrease in the dose of levodopa. Tolcapone enters the CNS to a minimal extent, but has been shown to inhibit central COMT activity in animals.
Pharmacodynamics:
COMT Activity in Erythrocytes:
Studies in healthy volunteers have shown that tolcapone reversibly inhibits human erythrocyte catechol-O-methyltransferase (COMT) activity after oral administration. The inhibition is closely related to plasma tolcapone concentrations. With a 200-mg single dose of tolcapone, maximum inhibition of erythrocyte COMT activity is on average greater than 80%. During multiple dosing with tolcapone (200 mg tid), erythrocyte COMT inhibition at trough tolcapone blood concentrations is 30% to 45%.
Effect on the Pharmacokinetics of Levodopa and its Metabolites:
When tolcapone is administered together with levodopa/carbidopa, it increases the relative bioavailability (AUC) of levodopa by approximately twofold. This is due to a decrease in levodopa clearance resulting in a prolongation of the terminal elimination half-life of levodopa (from approximately 2 hours to 3.5 hours). In general, the average peak levodopa plasma concentration (Cmax) and the time of its occurrence (Tmax) are unaffected. The onset of effect occurs after the first administration and is maintained during long-term treatment. Studies in healthy volunteers and Parkinson's disease patients have confirmed that the maximal effect occurs with 100 mg to 200 mg tolcapone. Plasma levels of 3-OMD are markedly and dose-dependently decreased by tolcapone when given with levodopa/carbidopa.
Population pharmacokinetic analyses in patients with Parkinson's disease have shown the same effects of tolcapone on levodopa plasma concentrations that occur in healthy volunteers.
Pharmacokinetics of Tolcapone:
Tolcapone pharmacokinetics are linear over the dose range of 50 mg to 400 mg, independent of levodopa/carbidopa co-administration. The elimination half-life of tolcapone is 2 to 3 hours and there is no significant accumulation. With tid dosing of 100 mg or 200 mg, Cmax is approximately 3 mcg/mL and 6 mcg/mL, respectively.
Absorption:
Tolcapone is rapidly absorbed, with a Tmax of approximately 2 hours. The absolute bioavailability following oral administration is about 65%. Food given within 1 hour before and 2 hours after dosing of tolcapone decreases the relative bioavailability by 10% to 20% (see DOSAGE AND ADMINISTRATION).
Distribution:
The steady-state volume of distribution of tolcapone is small (9 L). Tolcapone does not distribute widely into tissues due to its high plasma protein binding. The plasma protein binding of tolcapone is >99.9% over the concentration range of 0.32 to 210 mcg/mL. In vitro experiments have shown that tolcapone binds mainly to serum albumin.
Metabolism and Elimination:
Tolcapone is almost completely metabolized prior to excretion, with only a very small amount (0.5% of dose) found unchanged in urine. The main metabolic pathway of tolcapone is glucuronidation; the glucuronide conjugate is inactive. In addition, the compound is methylated by COMT to 3-O-methyl-tolcapone. Tolcapone is metabolized to a primary alcohol (hydroxylation of the methyl group), which is subsequently oxidized to the carboxylic acid. In vitro experiments suggest that the oxidation may be catalyzed by cytochrome P450 3A4 and P450 2A6. The reduction to an amine and subsequent N-acetylation occur to a minor extent. After oral administration of a 14C-labeled dose of tolcapone, 60% of labeled material is excreted in urine and 40% in feces. Tolcapone is a low-extraction-ratio drug (extraction ratio = 0.15) with a moderate systemic clearance of about 7 L/h.
Special Populations:
Tolcapone pharmacokinetics are independent of sex, age, body weight, and race (Japanese, Black and Caucasian). Polymorphic metabolism is unlikely based on the metabolic pathways involved.
Hepatic Impairment:
A study in patients with hepatic impairment has shown that moderate non-cirrhotic liver disease had no impact on the pharmacokinetics of tolcapone. In patients with moderate cirrhotic liver disease (Child-Pugh Class B), however, clearance and volume of distribution of unbound tolcapone was reduced by almost 50%. This reduction may increase the average concentration of unbound drug by twofold (see DOSAGE AND ADMINISTRATION). TASMAR therapy should not be initiated if the patient exhibits clinical evidence of active liver disease or two SGPT/ALT or SGOT/AST values greater than the upper limit of normal (see BOXED WARNING).
Renal Impairment:
The pharmacokinetics of tolcapone have not been investigated in a specific renal impairment study. However, the relationship of renal function and tolcapone pharmacokinetics has been investigated using population pharmacokinetics during clinical trials. The data of more than 400 patients have confirmed that over a wide range of creatinine clearance values (30 mL/min to 130 mL/min) the pharmacokinetics of tolcapone are unaffected by renal function. This could be explained by the fact that only a negligible amount of unchanged tolcapone (0.5%) is excreted in the urine. The glucuronide conjugate of tolcapone is mainly excreted in the urine but is also excreted in the bile. Accumulation of this stable and inactive metabolite should not present a risk in renally impaired patients with creatinine clearance above 25 mL/min (see DOSAGE AND ADMINISTRATION). Given the very high protein binding of tolcapone, no significant removal of the drug by hemodialysis would be expected.
Clinical Studies:
The effectiveness of TASMAR as an adjunct to levodopa in the treatment of Parkinson's disease was established in three multicenter randomized controlled trials of 13 to 26 weeks' duration, supported by four 6-week trials whose results were consistent with those of the longer trials. In two of the longer trials, tolcapone was evaluated in patients whose Parkinson's disease was characterized by deterioration in their response to levodopa at the end of a dosing interval (so-called fluctuating patients with wearing-off phenomena). In the remaining trial, tolcapone was evaluated in patients whose response to levodopa was relatively stable (so-called non-fluctuators).
Fluctuating Patients:
In two 3-month trials, patients with documented episodes of wearing-off phenomena, despite optimum levodopa therapy, were randomized to receive placebo, tolcapone 100 mg tid or 200 mg tid. The formal double-blind portion of the trial was 3 months long, and the primary outcome was a comparison between treatments in the change from baseline in the amount of time spent "On" (a period of relatively good functioning) and "Off" (a period of relatively poor functioning). Patients recorded periodically, throughout the duration of the trial, the time spent in each of these states.
In addition to the primary outcome, patients were also assessed using sub-parts of the Unified Parkinson's Disease Rating Scale (UPDRS), a frequently used multi-item rating scale intended to evaluate mentation (Part I), activities of daily living (Part II), motor function (Part III), complications of therapy (Part IV), and disease staging (Parts V and VI); an Investigator's Global Assessment of Change (IGA), a subjective scale designed to assess global functioning in 5 areas of Parkinson's disease; the Sickness Impact Profile (SIP), a multi-item scale in 12 domains designed to assess the patient's functioning in multiple areas; and the change in daily levodopa/carbidopa dose.
In one of the studies, 202 patients were randomized in 11 centers in the United States and Canada. In this trial, all patients were receiving concomitant levodopa and carbidopa. In the second trial, 177 patients were randomized in 24 centers in Europe. In this trial, all patients were receiving concomitant levodopa and benserazide.
The following tables display the results of these 2 trials:
|
Primary Measure |
|||
|
Baseline
|
Change from Baseline at Month 3
|
p-value* |
|
|
Hours of Wake Time "Off" † | |||
|
Placebo |
6.2 |
-1.2 |
— |
|
100 mg tid |
6.4 |
-2.0 |
0.169 |
|
200 mg tid |
5.9 |
-3.0 |
<0.001 |
|
Hours of Wake Time "On" † | |||
|
Placebo |
8.7 |
1.4 |
— |
|
100 mg tid |
8.1 |
2.0 |
0.267 |
|
200 mg tid |
9.1 |
2.9 |
0.008 |
|
Secondary Measures |
|||
|
Baseline |
Change from Baseline | ||
|
at Month 3 |
p-value* |
||
|
Levodopa Total Daily Dose (mg) | |||
|
Placebo |
948 |
16 |
— |
|
100 mg tid |
788 |
-166 |
<0.001 |
|
200 mg tid |
865 |
-207 |
<0.001 |
|
Global (overall) % Improved | |||
|
Placebo |
— |
42 |
— |
|
100 mg tid |
— |
71 |
<0.001 |
|
200 mg tid |
— |
91 |
<0.001 |
|
UPDRS Motor | |||
|
Placebo |
19.5 |
-0.4 |
— |
|
100 mg tid |
17.6 |
-1.9 |
0.217 |
|
200 mg tid |
20.6 |
-2.0 |
0.210 |
|
UPDRS ADL | |||
|
Placebo |
7.5 |
-0.3 |
— |
|
100 mg tid |
7.7 |
-0.8 |
0.487 |
|
200 mg tid |
8.3 |
0.2 |
0.412 |
|
SIP (total) | |||
|
Placebo |
14.7 |
-2.2 |
— |
|
100 mg tid |
14.9 |
-0.4 |
0.210 |
|
200 mg tid |
17.6 |
-0.3 |
0.216 |
|
Primary Measure |
|||
|
Baseline
|
Change from Baseline at Month 3
|
p-value* |
|
|
Hours of Wake Time "Off" † | |||
|
Placebo |
6.1 |
-0.7 |
— |
|
100 mg tid |
6.5 |
-2.0 |
0.008 |
|
200 mg tid |
6.0 |
-1.6 |
0.081 |
|
Hours of Wake Time "On" † | |||
|
Placebo |
8.5 |
-0.1 |
— |
|
100 mg tid |
8.1 |
1.7 |
0.003 |
|
200 mg tid |
8.4 |
1.7 |
0.003 |
|
Secondary Measures |
|||
|
Baseline |
Change from Baseline | ||
|
at Month 3 |
p-value* |
||
|
Levodopa Total Daily Dose (mg) | |||
|
Placebo |
660 |
-29 |
— |
|
100 mg tid |
667 |
-109 |
0.025 |
|
200 mg tid |
675 |
-122 |
0.010 |
|
Global (overall) % Improved | |||
|
Placebo |
— |
37 |
— |
|
100 mg tid |
— |
70 |
0.003 |
|
200 mg tid |
— |
78 |
<0.001 |
|
UPDRS Motor | |||
|
Placebo |
24.0 |
-2.1 |
— |
|
100 mg tid |
22.4 |
-4.2 |
0.163 |
|
200 mg tid |
22.4 |
-6.5 |
0.004 |
|
UPDRS ADL | |||
|
Placebo |
7.9 |
-0.5 |
— |
|
100 mg tid |
7.5 |
-0.9 |
0.408 |
|
200 mg tid |
7.7 |
-1.3 |
0.097 |
|
SIP (total) | |||
|
Placebo |
21.6 |
-0.9 |
— |
|
100 mg tid |
16.6 |
-1.9 |
0.419 |
|
200 mg tid |
18.4 |
-4.2 |
0.011 |
Effects on “Off” time and levodopa dose did not differ by age or sex.
Non-fluctuating Patients:
In this study, 298 patients with idiopathic Parkinson's disease on stable doses of levodopa/carbidopa who were not experiencing wearing-off phenomena were randomized to placebo, tolcapone 100 mg tid, or tolcapone 200 mg tid for 6 months at 20 centers in the United States and Canada. The primary measure of effectiveness was the Activities of Daily Living portion (Subscale II) of the UPDRS. In addition, the change in daily levodopa dose, other subscales of the UPDRS, and the SIP were assessed as secondary measures. The results are displayed in the following table:
|
|||
|
Primary Measure |
|||
|
Baseline |
Change from Baseline | ||
|
at Month 6 |
p-value* |
||
|
UPDRS ADL | |||
|
Placebo |
8.5 |
0.1 |
— |
|
100 mg tid |
7.5 |
-1.4 |
<0.001 |
|
200 mg tid |
7.9 |
-1.6 |
<0.001 |
|
Secondary Measures |
|||
|
Baseline |
Change from Baseline | ||
|
at Month 6 |
p-value* |
||
|
Levodopa Total Daily Dose (mg) | |||
|
Placebo |
364 |
47 |
— |
|
100 mg tid |
370 |
-21 |
<0.001 |
|
200 mg tid |
381 |
-32 |
<0.001 |
|
UPDRS Motor | |||
|
Placebo |
19.7 |
0.1 |
— |
|
100 mg tid |
17.3 |
-2.0 |
0.018 |
|
200 mg tid |
16.0 |
-2.3 |
0.008 |
|
SIP (total) | |||
|
Placebo |
6.9 |
0.4 |
— |
|
100 mg tid |
7.3 |
-0.9 |
0.044 |
|
200 mg tid |
7.3 |
-0.7 |
0.078 |
|
Percent of Patients who | |||
|
Developed Fluctuations | |||
|
Placebo |
— |
26 |
— |
|
100 mg tid |
— |
19 |
0.297 |
|
200 mg tid |
— |
14 |
0.047 |
Effects on Activities of Daily Living did not differ by age or sex.
Related/similar drugs
ropinirole, benztropine, pramipexole, carbidopa / levodopa, Exelon, Gocovri
Indications and Usage for Tasmar
TASMAR is indicated as an adjunct to levodopa and carbidopa for the treatment of the signs and symptoms of idiopathic Parkinson's disease. Because of the risk of potentially fatal, acute fulminant liver failure, TASMAR (tolcapone) should ordinarily be used in patients with Parkinson's disease on l-dopa/carbidopa who are experiencing symptom fluctuations and are not responding satisfactorily to or are not appropriate candidates for other adjunctive therapies. Because of the risk of liver injury and because TASMAR, when it is effective, provides an observable symptomatic benefit, the patient who fails to show substantial clinical benefit within 3 weeks of initiation of treatment, should be withdrawn from TASMAR.
The effectiveness of TASMAR was demonstrated in randomized controlled trials in patients receiving concomitant levodopa therapy with carbidopa or another aromatic amino acid decarboxylase inhibitor who experienced end of dose wearing-off phenomena as well as in patients who did not experience such phenomena (see CLINICAL PHARMACOLOGY: Clinical Studies).
Contraindications
TASMAR tablets are contraindicated in patients with liver disease, in patients who were withdrawn from TASMAR because of evidence of TASMAR-induced hepatocellular injury or who have demonstrated hypersensitivity to the drug or its ingredients.
TASMAR is also contraindicated in patients with a history of nontraumatic rhabdomyolysis or hyperpyrexia and confusion possibly related to medication (see PRECAUTIONS: Events Reported With Dopaminergic Therapy).
Warnings
(see BOXED WARNING) Because of the risk of potentially fatal, acute fulminant liver failure, TASMAR (tolcapone) should ordinarily be used in patients with Parkinson's disease on l-dopa/carbidopa who are experiencing symptom fluctuations and are not responding satisfactorily to or are not appropriate candidates for other adjunctive therapies (see INDICATIONS and DOSAGE AND ADMINISTRATION sections).
Because of the risk of liver injury and because TASMAR, when it is effective, provides an observable symptomatic benefit, the patient who fails to show substantial clinical benefit within 3 weeks of initiation of treatment, should be withdrawn from TASMAR.
TASMAR therapy should not be initiated if the patient exhibits clinical evidence of liver disease or two SGPT/ALT or SGOT/AST values greater than the upper limit of normal. Patients with severe dyskinesia or dystonia should be treated with caution (see PRECAUTIONS: Rhabdomyolysis).
Patients who develop evidence of hepatocellular injury while on TASMAR and are withdrawn from the drug for any reason may be at increased risk for liver injury if TASMAR is reintroduced. Accordingly, such patients should not ordinarily be considered for retreatment.
In controlled Phase 3 trials, increases to more than 3 times the upper limit of normal in ALT or AST occurred in approximately 1% of patients at 100 mg tid and 3% of patients at 200 mg tid. Females were more likely than males to have an increase in liver enzymes (approximately 5% vs 2%). Approximately one third of patients with elevated enzymes had diarrhea. Increases to more than 8 times the upper limit of normal in liver enzymes occurred in 0.3% at 100 mg tid and 0.7% at 200 mg tid. Elevated enzymes led to discontinuation in 0.3% and 1.7% of patients treated with 100 mg tid and 200 mg tid, respectively. Elevations usually occurred within 6 weeks to 6 months of starting treatment. In about half the cases with elevated liver enzymes, enzyme levels returned to baseline values within 1 to 3 months while patients continued TASMAR treatment. When treatment was discontinued, enzymes generally declined within 2 to 3 weeks but in some cases took as long as 1 to 2 months to return to normal.
Monoamine oxidase (MAO) and COMT are the two major enzyme systems involved in the metabolism of catecholamines. It is theoretically possible, therefore, that the combination of TASMAR and a non-selective MAO inhibitor (e.g., phenelzine and tranylcypromine) would result in inhibition of the majority of the pathways responsible for normal catecholamine metabolism. For this reason, patients should ordinarily not be treated concomitantly with TASMAR and a non-selective MAO inhibitor.
Tolcapone can be taken concomitantly with a selective MAO-B inhibitor (e.g., selegiline).
Falling Asleep During Activities of Daily Living and Somnolence
Tolcapone (TASMAR) increases plasma levels of levodopa in patients taking concomitant carbidopa levodopa products (see DOSAGE AND ADMINISTRATION). Patients taking carbidopa levodopa products alone or with other dopaminergic medications have reported suddenly falling asleep without prior warning of sleepiness while engaged in activities of daily living (includes the operation of motor vehicles). Some of these episodes resulted in automobile accidents. Although many of these patients reported somnolence while on TASMAR, some did perceive that they had no warning signs, such as excessive drowsiness, and believed that they were alert immediately prior to the event. Some patients reported these events one year after the initiation of treatment.
The risk for somnolence was increased with TASMAR treatment (TASMAR 100 mg-18%, 200 mg-14%, vs placebo-13%) compared to placebo treatment. In clinical trials, discontinuation due to somnolence occurred in 1% of patients treated with 200 mg TASMAR and 0% of patients treated with 100 mg TASMAR or placebo. Falling asleep while engaged in activities of daily living usually occurs in patients experiencing pre-existing somnolence, although some patients may not give such a history. For this reason, prescribers should continually reassess patients for drowsiness or sleepiness especially since some of the events occur well after the start of treatment. Prescribers should be aware that patients may not acknowledge drowsiness or sleepiness until directly questioned about drowsiness or sleepiness during specific activities. Patients who have already experienced somnolence or an episode of sudden sleep onset should not participate in these activities during treatment with TASMAR.
Before initiating treatment with TASMAR, advise patients about the potential to develop drowsiness and ask specifically about factors that may increase the risk for somnolence with TASMAR such as the use of concomitant sedating medications and the presence of sleep disorders. Consider discontinuing TASMAR in patients who report significant daytime sleepiness or episodes of falling asleep during activities that require active participation (e.g., conversations, eating, etc.). If treatment with TASMAR continues, patients should be advised not to drive and to avoid other potentially dangerous activities that might result in harm if patients become somnolent. There is insufficient information to establish that dose reduction will eliminate episodes of falling asleep while engaged in activities of daily living.
Precautions
Hypotension/Syncope:
Dopaminergic therapy in Parkinson's disease patients has been associated with orthostatic hypotension. Tolcapone enhances levodopa bioavailability and, therefore, may increase the occurrence of orthostatic hypotension. In TASMAR clinical trials, orthostatic hypotension was documented at least once in 8%, 14% and 13% of the patients treated with placebo, 100 mg and 200 mg TASMAR tid, respectively. A total of 2%, 5% and 4% of the patients treated with placebo, 100 mg and 200 mg TASMAR tid, respectively, reported orthostatic symptoms at some time during their treatment and also had at least one episode of orthostatic hypotension documented (however, the episode of orthostatic symptoms itself was invariably not accompanied by vital sign measurements). Patients with orthostasis at baseline were more likely than patients without symptoms to have orthostatic hypotension during the study, irrespective of treatment group. In addition, the effect was greater in tolcapone-treated patients than in placebo-treated patients. Baseline treatment with dopamine agonists or selegiline did not appear to increase the likelihood of experiencing orthostatic hypotension when treated with TASMAR. Approximately 0.7% of the patients treated with TASMAR (5% of patients who were documented to have had at least one episode of orthostatic hypotension) eventually withdrew from treatment due to adverse events presumably related to hypotension.
In controlled Phase 3 trials, approximately 5%, 4% and 3% of tolcapone 200 mg tid, 100 mg tid and placebo patients, respectively, reported at least one episode of syncope. Reports of syncope were generally more frequent in patients in all three treatment groups who had an episode of documented hypotension (although the episodes of syncope, obtained by history, were themselves not documented with vital sign measurement) compared to patients who did not have any episodes of documented hypotension.
Diarrhea:
In clinical trials, diarrhea developed in approximately 8%, 16% and 18% of patients treated with placebo, 100 mg and 200 mg TASMAR tid, respectively. While diarrhea was generally regarded as mild to moderate in severity, approximately 3% to 4% of patients on tolcapone had diarrhea which was regarded as severe. Diarrhea was the adverse event which most commonly led to discontinuation, with approximately 1%, 5% and 6% of patients treated with placebo, 100 mg and 200 mg TASMAR tid, respectively, withdrawing from the trials prematurely. Discontinuing TASMAR for diarrhea was related to the severity of the symptom. Diarrhea resulted in withdrawal in approximately 8%, 40% and 70% of patients with mild, moderate and severe diarrhea, respectively. Although diarrhea generally resolved after discontinuation of TASMAR, it led to hospitalization in 0.3%, 0.7% and 1.7% of patients in the placebo, 100 mg and 200 mg TASMAR tid groups.
Typically, diarrhea presents 6 to 12 weeks after tolcapone is started, but it may appear as early as 2 weeks and as late as many months after the initiation of treatment. Clinical trial data suggested that diarrhea associated with tolcapone use may sometimes be associated with anorexia (decreased appetite).
No consistent description of tolcapone-induced diarrhea has been derived from clinical trial data, and the mechanism of action is currently unknown.
It is recommended that all cases of persistent diarrhea should be followed up with an appropriate work-up (including occult blood samples).
Hallucinations/Psychotic-Like Behavior:
In clinical trials, hallucinations developed in approximately 5% of patients treated with placebo, compared to 8% and 10% of patients treated with 100 mg or 200 mg three times per day, respectively. Hallucinations led to drug discontinuation and premature withdrawal from clinical trials in 0.3% of patients treated with placebo, compared to 1.4% and 1.0% of patients treated with TASMAR 100 mg or 200 mg TASMAR three times per day, respectively. Hallucinations led to hospitalization in 0.0% of patients in the placebo group, compared to 1.7% and 0.0% of patients treated with 100 mg or 200 mg TASMAR three times per day, respectively.
In general, hallucinations present shortly after the initiation of therapy with tolcapone (typically within the first 2 weeks). Clinical trial data suggest that hallucinations associated with tolcapone use may be responsive to levodopa dose reduction. Patients whose hallucinations resolved had a mean levodopa dose reduction of 175 mg to 200 mg (20% to 25%) after the onset of the hallucinations. Hallucinations were commonly accompanied by confusion and to a lesser extent sleep disorder (insomnia) and excessive dreaming. The incidence of hallucination may be increased in elderly patients over 75 years treated with TASMAR (see Geriatric Use).
Postmarketing reports indicate that patients may experience new or worsening mental status and behavioral changes, which may be severe, including psychotic-like behavior during TASMAR treatment or after starting or increasing the dose of TASMAR. Other drugs prescribed to improve the symptoms of Parkinson’s disease may have similar effects on thinking and behavior. This abnormal thinking and behavior may present with one or more symptoms, including paranoid ideation, delusions, hallucinations, confusion, psychotic-like behavior, disorientation, aggressive behavior, agitation, and delirium.
Ordinarily, patients with a major psychotic disorder should not be treated with TASMAR because of the risk of exacerbating psychosis. In addition, certain medications used to treat psychosis may exacerbate the symptoms of Parkinson's disease and may decrease the effectiveness of TASMAR.
Dyskinesia:
TASMAR may potentiate the dopaminergic side effects of levodopa and may cause and/or exacerbate preexisting dyskinesia. Although decreasing the dose of levodopa may ameliorate this side effect, many patients in controlled trials continued to experience frequent dyskinesias despite a reduction in their dose of levodopa. Dyskinesia was the most common adverse reaction observed in controlled trials and developed in approximately 20% of patients treated with placebo, compared to 42% and 51% of patients treated with TASMAR 100 mg or 200 mg three times daily, respectively. The rates of withdrawal for dyskinesia were 0.0% in the placebo group, compared to 0.3% and 1.0% in the groups receiving TASMAR 100 mg or 200 mg three times a day, respectively.
Impulse Control/Compulsive Behaviors:
Reports suggest that patients may experience an intense urge to gamble, increased sexual urges, intense urges to spend money, binge eating, and/or other intense urges, and the inability to control these urges. These reports are associated with patients taking TASMAR in conjunction with carbidopa/levodopa, as well as other medications that increase central dopaminergic tone and that are used to treat patients with Parkinson’s disease. In some cases, although not all, these urges were reported to have stopped when the dose was reduced or the medication was discontinued. Because patients may not recognize these behaviors as abnormal, it is important for prescribers to specifically ask patients or their caregivers about the development of new or increased gambling urges, sexual urges, uncontrolled spending or other urges while being treated with TASMAR. Physicians should consider dose reduction or stopping the medication if a patient develops such urges while taking TASMAR (see Information for Patients).
Rhabdomyolysis:
Cases of severe rhabdomyolysis, with one case of multi-organ system failure rapidly progressing to death, have been reported. The complicated nature of these cases makes it impossible to determine what role, if any, TASMAR played in their pathogenesis. Severe prolonged motor activity including dyskinesia may account for rhabdomyolysis. Some cases, however, included fever, alteration of consciousness and muscular rigidity. It is possible, therefore, that the rhabdomyolysis may be a result of the syndrome described in Hyperpyrexia and Confusion (see PRECAUTIONS: Events Reported With Dopaminergic Therapy).
Renal Impairment:
No dosage adjustment is needed in patients with mild to moderate renal impairment, however, patients with severe renal impairment should be treated with caution (see CLINICAL PHARMACOLOGY: Pharmacokinetics of Tolcapone and DOSAGE AND ADMINISTRATION).
Renal Toxicity:
When rats were dosed daily for 1 or 2 years (exposures 6 times the human exposure or greater) there was a high incidence of proximal tubule cell damage consisting of degeneration, single cell necrosis, hyperplasia, karyocytomegaly and atypical nuclei. These effects were not associated with changes in clinical chemistry parameters, and there is no established method for monitoring for the possible occurrence of these lesions in humans. Although it has been speculated that these toxicities may occur as the result of a species-specific mechanism, experiments that would confirm the theory have not been conducted.
Hepatic Impairment:
Because of the risk of liver injury, TASMAR therapy should not be initiated in any patient with liver disease. For similar reasons, treatment should not be initiated in patients who have two SGPT/ALT or SGOT/AST values greater than the upper limit of normal (see BOXED WARNING) or any other evidence of hepatocellular dysfunction.
Hematuria:
The rates of hematuria in placebo-controlled trials were approximately 2%, 4% and 5% in placebo, 100 mg and 200 mg TASMAR tid, respectively. The etiology of the increase with TASMAR has not always been explained (for example, by urinary tract infection or warfarin therapy). In placebo-controlled trials in the United States (N=593) rates of microscopically confirmed hematuria were approximately 3%, 2% and 2% in placebo, 100 mg and 200 mg TASMAR tid, respectively.
Events Reported With Dopaminergic Therapy:
The events listed below are known to be associated with the use of drugs that increase dopaminergic activity, although they are most often associated with the use of direct dopamine agonists. While cases of Hyperpyrexia and Confusion have been reported in association with tolcapone withdrawal (see paragraph below), the expected incidence of fibrotic complications is so low that even if tolcapone caused these complications at rates similar to those attributable to other dopaminergic therapies, it is unlikely that even a single example would have been detected in a cohort of the size exposed to tolcapone.
Hyperpyrexia and Confusion:
In clinical trials, four cases of a symptom complex resembling the neuroleptic malignant syndrome (characterized by elevated temperature, muscular rigidity, and altered consciousness), similar to that reported in association with the rapid dose reduction or withdrawal of other dopaminergic drugs, have been reported in association with the abrupt withdrawal or lowering of the dose of tolcapone. In three of these cases, CPK was elevated as well. One patient died, and the other three patients recovered over periods of approximately 2, 4 and 6 weeks. Rare cases of this symptom complex have been reported during marketed use. It is difficult to determine if TASMAR played a role in the pathogenesis of these events because these patients received several concomitant medications affecting the central nervous system such as monoaminergic (i.e., MAO-I, tricyclic and selective serotonin reuptake inhibitors) and anticholinergic agents.
Fibrotic Complications:
Cases of retroperitoneal fibrosis, pulmonary infiltrates, pleural effusion, and pleural thickening have been reported in some patients treated with ergot derived dopaminergic agents. While these complications may resolve when the drug is discontinued, complete resolution does not always occur. Although these adverse events are believed to be related to the ergoline structure of these compounds, whether other, nonergot derived drugs (e.g., tolcapone) that increase dopaminergic activity can cause them is unknown.
Three cases of pleural effusion, one with pulmonary fibrosis, occurred during clinical trials. These patients were also on concomitant dopamine agonists (pergolide or bromocriptine) and had a prior history of cardiac disease or pulmonary pathology (nonmalignant lung lesion).
Melanoma:
Epidemiological studies have shown that patients with Parkinson's disease have a higher risk (2- to approximately 6-fold higher) of developing melanoma than the general population. Whether the increased risk observed was due to Parkinson's disease or other factors, such as drugs used to treat Parkinson's disease, is unclear.
For the reasons stated above, patients and providers are advised to monitor for melanomas frequently and on a regular basis when using TASMAR for any indication. Ideally, periodic skin examination should be performed by appropriately qualified individuals (e.g., dermatologists).
Patient Counseling Information
Patients should be instructed to take TASMAR only as prescribed.
TASMAR should not be used by patients until there has been a complete discussion of the risks and the patient has provided written acknowledgement that the risks have been explained (see PATIENT ACKNOWLEDGEMENT OF RISKSsection).
Inform patients about clinical signs and symptoms that suggest the onset of hepatic injury (persistent nausea, fatigue, lethargy, anorexia, jaundice, dark urine, pruritus, and right upper quadrant tenderness) (see WARNINGS). If symptoms of hepatic failure occur, patients should be advised to contact their physician immediately.
Inform patients of the need to have regular blood tests to monitor liver enzymes.
Advise patients that sleepiness or drowsiness may occur and that they should not drive a car or operate other complex machinery until they have gained sufficient experience on TASMAR to gauge whether or not it adversely affects their mental and/or motor performance. Advise patients to exercise caution while driving, operating machines, or working at heights during treatment with TASMAR. Because of the possible additive sedative effects, caution should also be used when patients are taking other CNS depressants in combination with TASMAR. Inform patients that nausea may occur, especially at the initiation of treatment with TASMAR.
Inform patients that hallucinations and other psychotic-like behavior may occur.
Advise patients about the possibility of developing or worsening of existing dyskinesia and/or dystonia after starting TASMAR.
Advise patients that they may develop postural (orthostatic) hypotension with or without symptoms such as dizziness, nausea, syncope, and sometimes sweating. Advise patients to rise slowly, especially after long periods of sitting or lying down. Hypotension may be more likely when patients first start treatment with TASMAR.
Instruct patients and caregivers to report intense urges to gamble, increased sexual urges, increase in spending money, binge eating, and other intense urges as well as the inability to control these urges to the prescriber while taking TASMAR.
Although TASMAR has not been shown to be teratogenic in animals, it is always given in conjunction with levodopa/carbidopa, which is known to cause visceral and skeletal malformations in the rabbit. Accordingly, patients should be advised to notify their physicians if they become pregnant or intend to become pregnant during therapy (see PRECAUTIONS: Pregnancy).
Tolcapone is excreted into maternal milk in rats. Because of the possibility that tolcapone may be excreted into human milk, advise patients to notify their physicians if they intend to breastfeed or are breastfeeding an infant.
Laboratory Tests:
Although a program of frequent laboratory monitoring for evidence of hepatocellular injury is deemed essential, it is not clear that periodic monitoring of liver enzymes will prevent the occurrence of fulminant liver failure. However, it is generally believed that early detection of drug-induced hepatic injury along with immediate withdrawal of the suspect drug enhances the likelihood for recovery. Accordingly, the following liver monitoring program is recommended.
Before starting treatment with TASMAR, the physician should conduct appropriate tests to exclude the presence of liver disease. In patients determined to be appropriate candidates for treatment with TASMAR, serum glutamic-pyruvic transaminase (SGPT/ALT) and serum glutamic-oxaloacetic transaminase (SGOT/AST) levels should be determined at baseline and periodically (i.e. every 2 to 4 weeks) for the first 6 months of therapy. After the first six months, periodic monitoring is recommended at intervals deemed clinically relevant. Although more frequent monitoring increases the chances of early detection, the precise schedule for monitoring is a matter of clinical judgment.
If the dose is increased to 200 mg tid (see DOSAGE AND ADMINISTRATION), liver enzyme monitoring should take place before increasing the dose and then be conducted every 2 to 4 weeks for the following 6 months of therapy. After six months, periodic monitoring is recommended at intervals deemed clinically relevant.
Discontinue TASMAR if SGPT/ALT or SGOT/AST levels exceed 2 times the upper limit of normal or if clinical signs and symptoms suggest the onset of hepatic dysfunction (e.g., persistent nausea, fatigue, lethargy, anorexia, jaundice, dark urine, pruritus, and right upper quadrant tenderness).
Special Populations:
TASMAR therapy should not be initiated if the patient exhibits clinical evidence of active liver disease or two SGPT/ALT or SGOT/AST values greater than the upper limit of normal. Patients with severe dyskinesia or dystonia should be treated with caution (see PRECAUTIONS: Rhabdomyolysis). Patients with severe renal impairment should be treated with caution (see INDICATIONS, DOSAGE AND ADMINISTRATION, BOXED WARNING and WARNINGS).
Drug Interactions
Protein Binding:
Although tolcapone is highly protein bound, in vitro studies have shown that tolcapone at a concentration of 50 mcg/mL did not displace other highly protein-bound drugs from their binding sites at therapeutic concentrations. The experiments included warfarin (0.5 to 7.2 mcg/mL), phenytoin (4.0 to 38.7 mcg/mL), tolbutamide (24.5 to 96.1 mcg/mL) and digitoxin (9.0 to 27.0 mcg/mL).
Drugs Metabolized by Catechol-O-Methyltransferase (COMT):
Tolcapone may influence the pharmacokinetics of drugs metabolized by COMT. However, no effects were seen on the pharmacokinetics of the COMT substrate carbidopa. The effect of tolcapone on the pharmacokinetics of other drugs of this class such as α-methyldopa, dobutamine, apomorphine, and isoproterenol has not been evaluated. A dose reduction of such compounds should be considered when they are co-administered with tolcapone.
Effect of Tolcapone on the Metabolism of Other Drugs:
In vitro experiments have been performed to assess the potential of tolcapone to interact with isoenzymes of cytochrome P450 (CYP). No relevant interactions with substrates for CYP 2A6 (warfarin), CYP 1A2 (caffeine), CYP 3A4 (midazolam, terfenadine, cyclosporine), CYP 2C19 (S-mephenytoin) and CYP 2D6 (desipramine) were observed in vitro. The absence of an interaction with desipramine, a drug metabolized by cytochrome P450 2D6, was also confirmed in an in vivo study where tolcapone did not change the pharmacokinetics of desipramine.
Due to its affinity to cytochrome P450 2C9 in vitro, tolcapone may interfere with drugs, whose clearance is dependent on this metabolic pathway, such as tolbutamide and warfarin. However, in an in vivo interaction study, tolcapone did not change the pharmacokinetics of tolbutamide. Therefore, clinically relevant interactions involving cytochrome P450 2C9 appear unlikely. Similarly, tolcapone did not affect the pharmacokinetics of desipramine, a drug metabolized by cytochrome P450 2D6, indicating that interactions with drugs metabolized by that enzyme are unlikely. Since clinical information is limited regarding the combination of warfarin and tolcapone, coagulation parameters should be monitored when these two drugs are co-administered.
Drugs That Increase Catecholamines:
Tolcapone did not influence the effect of ephedrine, an indirect sympathomimetic, on hemodynamic parameters or plasma catecholamine levels, either at rest or during exercise. Since tolcapone did not alter the tolerability of ephedrine, these drugs can be co-administered.
When TASMAR was given together with levodopa/carbidopa and desipramine, there was no significant change in blood pressure, pulse rate and plasma concentrations of desipramine. Overall, the frequency of adverse events increased slightly. These adverse events were predictable based on the known adverse reactions to each of the three drugs individually. Therefore, caution should be exercised when desipramine is administered to Parkinson's disease patients being treated with TASMAR and levodopa/carbidopa.
In clinical trials, patients receiving TASMAR/levodopa preparations reported a similar adverse event profile independent of whether or not they were also concomitantly administered selegiline (a selective MAO-B inhibitor).
Carcinogenesis, Mutagenesis and Impairment of Fertility:
Carcinogenesis:
Carcinogenicity studies in which tolcapone was administered in the diet were conducted in mice and rats. Mice were treated for 80 (female) or 95 (male) weeks with doses of 100, 300 and 800 mg/kg/day, equivalent to 0.8, 1.6 and 4 times human exposure (AUC = 80 mcg∙hr/mL) at the recommended daily clinical dose of 600 mg. Rats were treated for 104 weeks with doses of 50, 250 and 450 mg/kg/day. Tolcapone exposures were 1, 6.3 and 13 times the human exposure in male rats and 1.7, 11.8 and 26.4 times the human exposure in female rats. There was an increased incidence of uterine adenocarcinomas in female rats at exposure equivalent to 26.4 times the human exposure. There was evidence of renal tubular injury and renal tubular tumor formation in rats. A low incidence of renal tubular cell adenomas occurred in middle- and high-dose female rats; tubular cell carcinomas occurred in middle- and high-dose male and high-dose female rats, with a statistically significant increase in high-dose males. Exposures were equivalent to 6.3 (males) or 11.8 (females) times the human exposure or greater; no renal tumors were observed at exposures of 1 (males) or 1.7 (females) times the human exposure. Minimal-to-marked damage to the renal tubules, consisting of proximal tubule cell degeneration, single cell necrosis, hyperplasia and karyocytomegaly, occurred at the doses associated with renal tumors. Renal tubule damage, characterized by proximal tubule cell degeneration and the presence of atypical nuclei, as well as one adenocarcinoma in a high-dose male, were observed in a 1-year study in rats receiving doses of tolcapone of 150 and 450 mg/kg/day. These histopathological changes suggest the possibility that renal tumor formation might be secondary to chronic cell damage and sustained repair, but this relationship has not been established, and the relevance of these findings to humans is not known. There was no evidence of carcinogenic effects in the long-term mouse study. The carcinogenic potential of tolcapone in combination with levodopa/carbidopa has not been examined.
Mutagenesis:
Tolcapone was clastogenic in the in vitro mouse lymphoma/thymidine kinase assay in the presence of metabolic activation. Tolcapone was not mutagenic in the Ames test, the in vitro V79/HPRT gene mutation assay, or the unscheduled DNA synthesis assay. It was not clastogenic in an in vitro chromosomal aberration assay in cultured human lymphocytes, or in an in vivo micronucleus assay in mice.
Pregnancy:
Tolcapone, when administered alone during organogenesis, was not teratogenic at doses of up to 300 mg/kg/day in rats or up to 400 mg/kg/day in rabbits (5.7 times and 15 times the recommended daily clinical dose of 600 mg, on a mg/m2 basis, respectively). In rabbits, however, an increased rate of abortion occurred at a dose of 100 mg/kg/day (3.7 times the daily clinical dose on a mg/m2 basis) or greater. Evidence of maternal toxicity (decreased weight gain, death) was observed at 300 mg/kg in rats and 400 mg/kg in rabbits. When tolcapone was administered to female rats during the last part of gestation and throughout lactation, decreased litter size and impaired growth and learning performance in female pups were observed at a dose of 250/150 mg/kg/day (dose reduced from 250 to 150 mg/kg/day during late gestation due to high rate of maternal mortality; equivalent to 4.8/2.9 times the clinical dose on a mg/m2 basis).
Tolcapone is always given concomitantly with levodopa/carbidopa, which is known to cause visceral and skeletal malformations in rabbits. The combination of tolcapone (100 mg/kg/day) with levodopa/carbidopa (80/20 mg/kg/day) produced an increased incidence of fetal malformations (primarily external and skeletal digit defects) compared to levodopa/carbidopa alone when pregnant rabbits were treated throughout organogenesis. Plasma exposures to tolcapone (based on AUC) were 0.5 times the expected human exposure, and plasma exposures to levodopa were 6 times higher than those in humans under therapeutic conditions. In a combination embryo-fetal development study in rats, fetal body weights were reduced by the combination of tolcapone (10, 30 and 50 mg/kg/day) and levodopa/carbidopa (120/30 mg/kg/day) and by levodopa/carbidopa alone. Tolcapone exposures were 0.5 times expected human exposure or greater: levodopa exposures were 21 times the expected human exposure or greater. The high dose of 50 mg/kg/day of tolcapone given alone was not associated with reduced fetal body weight (plasma exposures of 1.4 times the expected human exposure).
There is no experience from clinical studies regarding the use of TASMAR in pregnant women. Therefore, TASMAR should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Women:
In animal studies, tolcapone was excreted into maternal rat milk.
It is not known whether tolcapone is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when tolcapone is administered to a nursing woman.
Geriatric Use:
Parkinson’s disease is primarily an affliction of the elderly. Consequently, the mean age of patients in tolcapone clinical trials was 60 to 65 years. To investigate safety as it relates to advancing age, three subgroups were identified: less than 65 years, 65 to 75 years, and greater than 75 years. There were generally no consistent age-related trends in safety parameters. However, patients greater than 75 years of age may be more likely to develop hallucinations than patients less than 75 years of age, while patients over 75 may be less likely to develop dystonia (see PRECAUTIONS: Hallucinations/Psychotic-Like Behavior). In tolcapone clinical trials, measures of therapeutic efficacy (effects on “Off” time, levodopa dose, and effects on Activities of Daily Living) were not affected by age (see CLINICAL PHARMACOLOGY: Clinical Studies). Tolcapone pharmacokinetics have not been found to be affected by age (see CLINICAL PHARMACOLOGY: Special Populations).
Adverse Reactions/Side Effects
Cases of severe hepatocellular injury, including fulminant liver failure resulting in death, have been reported in postmarketing use. As of May 2005, three cases of fatal fulminant hepatic failure have been reported from more than 40,000 patient years of worldwide use. This incidence may be 10- to 100-fold higher than the background incidence in the general population. All three cases were reported within the first six months of initiation of treatment with TASMAR. Analysis of the laboratory monitoring data in over 3,400 TASMAR-treated patients participating in clinical trials indicated that increases in SGPT/ALT or SGOT/AST, when present, generally occurred within the first 6 months of treatment with TASMAR.
The imprecision of the estimated increase is due to uncertainties about the base rate and the actual number of cases occurring in association with TASMAR. The incidence of idiopathic potentially fatal fulminant hepatic failure (i.e., not due to viral hepatitis or alcohol) is low. One estimate, based upon transplant registry data, is approximately 3/1,000,000 patients per year in the United States. Whether this estimate is an appropriate basis for estimating the increased risk of liver failure among TASMAR users is uncertain. TASMAR users, for example, differ in age and general health status from candidates for liver transplantation. Similarly, underreporting of cases may lead to significant underestimation of the increased risk associated with the use of TASMAR.
During the premarketing development of tolcapone, two distinct patient populations were studied, patients with end-of-dose wearing-off phenomena and patients with stable responses to levodopa therapy. All patients received concomitant treatment with levodopa preparations, however, and were similar in other clinical aspects. Adverse reactions are shown for these two populations combined.
The most commonly observed adverse reactions in the double-blind, placebo-controlled trials (N=892), with a difference in incidence (TASMAR minus Placebo) of at least 5% or greater in the 100 mg or 200 mg TASMAR-treated groups compared to placebo, were dyskinesia, nausea, diarrhea, anorexia, sleep disorder, vomiting, urine discoloration, somnolence, hallucination, dystonia, and sweating.
Approximately 16% of the 592 patients who participated in the double-blind, placebo-controlled trials discontinued treatment due to adverse reactions compared to 10% of the 298 patients who received placebo. Diarrhea was by far the most frequent cause of discontinuation (approximately 6% in tolcapone patients vs. 1% on placebo).
Adverse Reaction Incidence in Controlled Clinical Studies:
Table 4 lists treatment emergent adverse reactions that occurred in at least 1% of patients treated with tolcapone participating in the double-blind, placebo-controlled studies and were numerically more common in at least one of the tolcapone groups. In these studies, either tolcapone or placebo was added to levodopa/carbidopa (or benserazide).
The prescriber should be aware that these figures cannot be used to predict the incidence of adverse reactions in the course of usual medical practice where patient characteristics and other factors differ from those that prevailed in the clinical studies. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatments, uses, and investigators. However, the cited figures do provide the prescriber with some basis for estimating the relative contribution of drug and nondrug factors to the adverse reactions incidence rate in the population studied.
| Placebo | Tolcapone tid | ||
|---|---|---|---|
| 100 mg | 200 mg | ||
| N = 298 | N = 296 | N = 298 | |
| Adverse Reactions | (%) | (%) | (%) |
|
Dyskinesia |
20 |
42 |
51 |
|
Nausea |
18 |
30 |
35 |
|
Sleep Disorder |
18 |
24 |
25 |
|
Dystonia |
17 |
19 |
22 |
|
Dreaming Excessive |
17 |
21 |
16 |
|
Anorexia |
13 |
19 |
23 |
|
Cramps Muscle |
17 |
17 |
18 |
|
Orthostatic Complaints |
14 |
17 |
17 |
|
Somnolence |
13 |
18 |
14 |
|
Diarrhea |
8 |
16 |
18 |
|
Confusion |
9 |
11 |
10 |
|
Dizziness |
10 |
13 |
6 |
|
Headache |
7 |
10 |
11 |
|
Hallucination |
5 |
8 |
10 |
|
Vomiting |
4 |
8 |
10 |
|
Constipation |
5 |
6 |
8 |
|
Fatigue |
6 |
7 |
3 |
|
Upper Respiratory Tract Infection |
3 |
5 |
7 |
|
Falling |
4 |
4 |
6 |
|
Sweating Increased |
2 |
4 |
7 |
|
Urinary Tract Infection |
4 |
5 |
5 |
|
Xerostomia |
2 |
5 |
6 |
|
Abdominal Pain |
3 |
5 |
6 |
|
Syncope |
3 |
4 |
5 |
|
Urine Discoloration |
1 |
2 |
7 |
|
Dyspepsia |
2 |
4 |
3 |
|
Influenza |
2 |
3 |
4 |
|
Dyspnea |
2 |
3 |
3 |
|
Balance Loss |
2 |
3 |
2 |
|
Flatulence |
2 |
2 |
4 |
|
Hyperkinesia |
1 |
3 |
2 |
|
Chest Pain |
1 |
3 |
1 |
|
Hypotension |
1 |
2 |
2 |
|
Paresthesia |
2 |
3 |
1 |
|
Stiffness |
1 |
2 |
2 |
|
Arthritis |
1 |
2 |
1 |
|
Chest Discomfort |
1 |
1 |
2 |
|
Hypokinesia |
1 |
1 |
3 |
|
Micturition Disorder |
1 |
2 |
1 |
|
Pain Neck |
1 |
2 |
2 |
|
Burning |
0 |
2 |
1 |
|
Sinus Congestion |
0 |
2 |
1 |
|
Agitation |
0 |
1 |
1 |
|
Bleeding Dermal |
0 |
1 |
1 |
|
Irritability |
0 |
1 |
1 |
|
Mental Deficiency |
0 |
1 |
1 |
|
Hyperactivity |
0 |
1 |
1 |
|
Malaise |
0 |
1 |
0 |
|
Panic Reaction |
0 |
1 |
0 |
|
Tumor Skin |
0 |
1 |
0 |
|
Cataract |
0 |
1 |
0 |
|
Euphoria |
0 |
1 |
0 |
|
Fever |
0 |
0 |
1 |
|
Alopecia |
0 |
1 |
0 |
|
Eye Inflamed |
0 |
1 |
0 |
|
Hypertonia |
0 |
0 |
1 |
|
Tumor Uterus |
0 |
1 |
0 |
Effects of Gender on Adverse Reactions:
Female patients may be more likely to develop somnolence than males.
Other Adverse Events Observed During All Trials in Patients With Parkinson's Disease:
During these trials, all adverse events were recorded by the clinical investigators using terminology of their own choosing. To provide a meaningful estimate of the proportion of individuals having adverse events, similar types of adverse events were grouped into a smaller number of standardized categories using COSTART dictionary terminology. These categories are used in the listing below.
All reported events that occurred at least twice (or once for serious or potentially serious events), except those already listed above, trivial events and terms too vague to be meaningful are included, without regard to determination of a causal relationship to TASMAR.
Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are defined as those occurring in at least 1/100 patients; infrequent adverse events are defined as those occurring in between 1/100 and 1/1000 patients; and rare adverse events are defined as those occurring in fewer than 1/1000 patients.
Nervous System — frequent: depression, hypesthesia, tremor, speech disorder, vertigo, emotional lability; infrequent: neuralgia, amnesia, extrapyramidal syndrome, hostility, libido increased, manic reaction, nervousness, paranoid reaction, cerebral ischemia, cerebrovascular accident, delusions, libido decreased, neuropathy, apathy, choreoathetosis, myoclonus, psychosis, thinking abnormal, twitching; rare: antisocial reaction, delirium, encephalopathy, hemiplegia, meningitis.
Digestive System — frequent: tooth disorder; infrequent: dysphagia, gastrointestinal hemorrhage, gastroenteritis, mouth ulceration, increased salivation, abnormal stools, esophagitis, cholelithiasis, colitis, tongue disorder, rectal disorder; rare: cholecystitis, duodenal ulcer, gastrointestinal carcinoma, stomach atony.
Body as a Whole — frequent: flank pain, accidental injury, abdominal pain, infection; infrequent: hernia, pain, allergic reaction, cellulitis, infection fungal, viral infection, carcinoma, chills, infection bacterial, neoplasm, abscess, face edema; rare: death.
Cardiovascular System — frequent: palpitation; infrequent: hypertension, vasodilation, angina pectoris, heart failure, atrial fibrillation, tachycardia, migraine, aortic stenosis, arrhythmia, arteriospasm, bradycardia, cerebral hemorrhage, coronary artery disorder, heart arrest, myocardial infarct, myocardial ischemia, pulmonary embolus; rare: arteriosclerosis, cardiovascular disorder, pericardial effusion, thrombosis.
Musculoskeletal System — frequent: myalgia; infrequent: tenosynovitis, arthrosis, joint disorder.
Urogenital System — frequent: urinary incontinence, impotence; infrequent: prostatic disorder, dysuria, nocturia, polyuria, urinary retention, urinary tract disorder, hematuria, kidney calculus, prostatic carcinoma, breast neoplasm, oliguria, uterine atony, uterine disorder, vaginitis; rare: bladder calculus, ovarian carcinoma, uterine hemorrhage.
Respiratory System — frequent: bronchitis, pharyngitis; infrequent: cough increased, rhinitis, asthma, epistaxis, hyperventilation, laryngitis, hiccup; rare: apnea, hypoxia, lung edema.
Skin and Appendages — frequent: rash; infrequent: herpes zoster, pruritus, seborrhea, skin discoloration, eczema, erythema multiforme, skin disorder, furunculosis, herpes simplex, urticaria.
Special Senses — frequent: tinnitus; infrequent: diplopia, ear pain, eye hemorrhage, eye pain, lacrimation disorder, otitis media, parosmia; rare: glaucoma.
Metabolic and Nutritional — infrequent: edema, hypercholesteremia, thirst, dehydration.
Hemic and Lymphatic System — infrequent: anemia; rare: leukemia, thrombocytopenia.
Endocrine System — infrequent: diabetes mellitus.
Unclassified — infrequent: surgical procedure.
To report SUSPECTED ADVERSE REACTIONS, contact Bausch Health US, LLC at 1-800-321-4576 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Abuse and Dependence
Tolcapone is not a controlled substance.
Studies conducted in rats and monkeys did not reveal any potential for physical or psychological dependence. Although clinical trials have not revealed any evidence of the potential for abuse, tolerance or physical dependence, systematic studies in humans designed to evaluate these effects have not been performed.
Overdosage
The highest dose of tolcapone administered to humans was 800 mg tid, with and without levodopa/carbidopa co-administration. This was in a 1-week study in elderly, healthy volunteers. The peak plasma concentrations of tolcapone at this dose were on average 30 mcg/mL (compared to 3 mcg/mL and 6 mcg/mL with 100 mg and 200 mg tolcapone, respectively). Nausea, vomiting and dizziness were observed, particularly in combination with levodopa/carbidopa.
The threshold for the lethal plasma concentration for tolcapone based on animal data is >100 mcg/mL. Respiratory difficulties were observed in rats at high oral (gavage) and intravenous doses and in dogs with rapidly injected intravenous doses.
Tasmar Dosage and Administration
Because of the risk of potentially fatal, acute fulminant liver failure, TASMAR (tolcapone) should ordinarily be used in patients with Parkinson's disease on l-dopa/carbidopa who are experiencing symptom fluctuations and are not responding satisfactorily to or are not appropriate candidates for other adjunctive therapies (see INDICATIONS and DOSAGE AND ADMINISTRATION sections).
BECAUSE OF THE RISK OF LIVER INJURY AND BECAUSE TASMAR WHEN IT IS EFFECTIVE PROVIDES AN OBSERVABLE SYMPTOMATIC BENEFIT, THE PATIENT WHO FAILS TO SHOW SUBSTANTIAL CLINICAL BENEFIT WITHIN 3 WEEKS OF INITIATION OF TREATMENT, SHOULD BE WITHDRAWN FROM TASMAR.
TASMAR therapy should not be initiated if the patient exhibits clinical evidence of liver disease or two SGPT/ALT or SGOT/AST values greater than the upper limit of normal. Patients with severe dyskinesia or dystonia should be treated with caution (see PRECAUTIONS: Rhabdomyolysis).
Patients who develop evidence of hepatocellular injury while on TASMAR and are withdrawn from the drug for any reason may be at increased risk for liver injury if TASMAR is reintroduced. These patients should not ordinarily be considered for retreatment with TASMAR.
Only prescribe TASMAR for patients taking concomitant carbidopa levodopa therapy. The initial dose of TASMAR is always 100 mg three times per day. The recommended daily dose of TASMAR is also 100 mg tid. In clinical trials, elevations in ALT occurred more frequently at the dose of 200 mg tid. While it is unknown whether the risk of acute fulminant liver failure is increased at the 200-mg dose, it would be prudent to use 200 mg only if the anticipated incremental clinical benefit is justified (see BOXED WARNING, WARNINGS, andPRECAUTIONS: Laboratory Tests). If a patient fails to show the expected incremental benefit on the 200-mg dose after a total of 3 weeks of treatment (regardless of dose), TASMAR should be discontinued.
In clinical trials, the first dose of the day of TASMAR was always taken together with the first dose of the day of levodopa/carbidopa, and the subsequent doses of TASMAR were given approximately 6 and 12 hours later.
In clinical trials, the majority of patients required a decrease in their daily levodopa dose if their daily dose of levodopa was >600 mg or if patients had moderate or severe dyskinesias before beginning treatment.
To optimize an individual patient's response, reductions in daily levodopa dose may be necessary. In clinical trials, the average reduction in daily levodopa dose was about 30% in those patients requiring a levodopa dose reduction. (Greater than 70% of patients with levodopa doses above 600 mg daily required such a reduction.)
TASMAR can be combined with both the immediate and sustained release formulations of levodopa/carbidopa.
TASMAR may be taken with or without food (see CLINICAL PHARMACOLOGY).
Patients With Impaired Hepatic Function:
TASMAR therapy should not be initiated in any patient with liver disease or two SGPT/ALT or SGOT/AST values greater than the upper limit of normal. (see BOXED WARNING, WARNINGS, andCLINICAL PHARMACOLOGY.)
Patients With Impaired Renal Function:
No dose adjustment of TASMAR is recommended for patients with mild to moderate renal impairment. However, patients with severe renal impairment should be treated with caution. The safety of tolcapone has not been examined in subjects who had creatinine clearance less than 25 mL/min (see CLINICAL PHARMACOLOGY).
Withdrawing Patients From TASMAR:
As with any dopaminergic drug, withdrawal or abrupt reduction in the TASMAR dose may lead to emergence of signs and symptoms of Parkinson's disease or Hyperpyrexia and Confusion, a syndrome complex resembling the neuroleptic malignant syndrome (see PRECAUTIONS: Events Reported With Dopaminergic Therapy). If a decision is made to discontinue treatment with TASMAR, then it is recommended to closely monitor the patient and adjust other dopaminergic treatments as needed. This syndrome should be considered in the differential diagnosis for any patient who develops a high fever or severe rigidity. Tapering TASMAR has not been systematically evaluated. As the duration of COMT inhibition with TASMAR is generally 5 to 6 hours on average, decreasing the frequency of dosage to twice or once a day may not in itself prevent withdrawal effects.
How is Tasmar supplied
TASMAR is supplied as film-coated tablets containing 100 mg tolcapone. The 100 mg beige to yellowish beige tablet is hexagonal and biconvex. Debossed on one side of the 100 mg tablet is TASMAR and the tablet strength (100), on the other side is a V.
TASMAR 100 mg Tablets: bottles of 90 (NDC 0187-0938-01).
Distributed by:
Bausch Health US, LLC
Bridgewater, NJ 08807 USA
Manufactured by:
Bausch Health Companies Inc.
Steinbach, MB R5G 1Z7, Canada
TASMAR is a trademark of Bausch Health Companies Inc. or its affiliates.
© 2020 Bausch Health Companies Inc. or its affiliates
|
PATIENT ACKNOWLEDGEMENT OF RISKS |
|
ASSOCIATED WITH TASMAR TREATMENT |
|
The following is important information that patients should know about TASMAR. |
|
|
The above points of information, possibly along with other information, have been explained to me and I have been able to ask my physician questions and discuss risks and benefits associated with TASMAR treatment. |
|
Patient or Patient Caregiver Signature:___________________________________________________ |
|
Date: ________________________________ |
|
NOTE TO PHYSICIAN: It is strongly recommended that you retain a signed copy of this form with the patient's medical records. |
|
SUPPLY OF PATIENT ACKNOWLEDGEMENT FORMS: |
|
A supply of Patient Acknowledgement forms is available, free of charge, from your local Bausch Health representative, by calling 1-800-321-4576. Permission to use the above Patient Acknowledgement form by photocopy reproduction is also hereby granted by Bausch Health US, LLC. |
Rev. 10/2020
20003004
9467003

| TASMAR
tolcapone tablet, film coated |
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
| Labeler - Bausch Health US LLC (831922468) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bausch Health Companies Inc. | 253292734 | MANUFACTURE(0187-0938) , PACK(0187-0938) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Carton Service Incorporated | 928861723 | PACK(0187-0938) | |
More about Tasmar (tolcapone)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- Drug class: dopaminergic antiparkinsonism agents
- Breastfeeding
- En español