Primaxin IV Prescribing Information
Package insert / product label
Generic name: imipenem and cilastatin sodium
Dosage form: injection, powder, for solution
Drug class: Carbapenems
J Code (medical billing code): J0743 (Per 250 mg, injection)
Medically reviewed by Drugs.com. Last updated on Jul 16, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
PRIMAXIN® (imipenem and cilastatin) for Injection, for intravenous use
Initial U.S. Approval: 1985
Indications and Usage for Primaxin IV
PRIMAXIN for intravenous use is a combination of imipenem, a penem antibacterial, and cilastatin, a renal dehydropeptidase inhibitor, indicated for the treatment of the following serious infections caused by designated susceptible bacteria:
- Lower respiratory tract infections. (1.1)
- Urinary tract infections. (1.2)
- Intra-abdominal infections. (1.3)
- Gynecologic infections. (1.4)
- Bacterial septicemia. (1.5)
- Bone and joint infections. (1.6)
- Skin and skin structure infections. (1.7)
- Endocarditis. (1.8)
Limitations of Use:
- PRIMAXIN is not indicated in patients with meningitis because safety and efficacy have not been established (1.9).
- PRIMAXIN is not recommended in pediatric patients with CNS infections because of the risk of seizures (1.9).
- PRIMAXIN is not recommended in pediatric patients weighing less than 30 kg with impaired renal function (1.9).
Usage:
To reduce the development of drug resistant bacteria and maintain the effectiveness of PRIMAXIN and other antibacterial drugs, PRIMAXIN should be used only to treat infections that are proven or strongly suspected to be caused by bacteria (1.10).
Primaxin IV Dosage and Administration
- The dosage of PRIMAXIN in adult patients should be based on suspected or confirmed pathogen susceptibility (2.1).
- For adult patients with normal renal function (creatinine clearance of greater than or equal to 90 mL/min), the recommended dosage regimens are: 500 mg every 6 hours OR 1000 mg every 8 hours OR 1000 mg every 6 hours (2.1).
- See full prescribing information for dosage recommendations in pediatric patients (2.2).
- A reduction in dose must be made for a patient with a creatinine clearance of less than 90 mL/min (2.3).
- Patients with creatinine clearances of less than 15 mL/min should not receive PRIMAXIN unless hemodialysis is instituted within 48 hours (2.4).
- Reconstitute PRIMAXIN vial with appropriate diluent and dilute the reconstituted suspension with an appropriate infusion solution before administering by intravenous infusion (2.5).
Dosage Forms and Strengths
For Injection: PRIMAXIN is a sterile powder mixture for reconstitution in single-dose containers including vials containing:
- 500 mg imipenem (anhydrous equivalent) and 500 mg cilastatin (free acid equivalent) (3)
Contraindications
- Known hypersensitivity to any component of PRIMAXIN (4)
Warnings and Precautions
- Hypersensitivity Reactions: Serious and occasionally fatal hypersensitivity (anaphylactic) reactions have been reported in patients receiving therapy with beta-lactams. If an allergic reaction to PRIMAXIN occurs, discontinue the drug immediately (5.1).
- Seizure Potential: Seizures and other CNS adverse reactions, such as confusional states and myoclonic activity, have been reported during treatment with PRIMAXIN. If focal tremors, myoclonus, or seizures occur, patients should be evaluated neurologically, placed on anticonvulsant therapy if not already instituted, and the dosage of PRIMAXIN re-examined to determine whether it should be decreased, or the antibacterial drug discontinued (5.2).
- Increased Seizure Potential Due to Interaction with Valproic Acid: Co-administration of PRIMAXIN, to patients receiving valproic acid or divalproex sodium results in a reduction in valproic acid concentrations. The valproic acid concentrations may drop below the therapeutic range as a result of this interaction, therefore increasing the risk of breakthrough seizures. The concomitant use of PRIMAXIN and valproic acid/divalproex sodium is generally not recommended (5.3, 7.3).
- Clostridioides difficile-Associated Diarrhea (CDAD): has been reported with use of PRIMAXIN and may range in severity from mild diarrhea to fatal colitis. Evaluate if diarrhea occurs (5.4).
Adverse Reactions/Side Effects
- The most frequently occurring adverse reactions (≥0.2%) in adults were phlebitis, nausea, diarrhea, vomiting, rash, pain injection site, fever, hypotension, seizures, erythema at injection site, dizziness, pruritus, vein induration, urticaria, somnolence (6.1).
- The most frequently occurring adverse reactions (>1%) in pediatric patients greater than or equal to 3 months of age were diarrhea, rash, phlebitis, gastroenteritis, vomiting, IV site irritation, urine discoloration (6.1).
- The most frequently occurring adverse reactions (>1%) in neonates to 3 months of age were convulsions, diarrhea, oliguria/anuria, oral candidiasis, rash, tachycardia (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme LLC at 1-877-888-4231 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch .
Drug Interactions
- Ganciclovir: Generalized seizures have been reported in patients who received ganciclovir. Do not co-administer unless benefit outweighs risk (7.1).
- Probenecid: Concomitant administration of PRIMAXIN and probenecid results in increases in the plasma level and half-life of imipenem. Concomitant administration is not recommended (7.2).
- Valproic acid/divalproex sodium: Concomitant use with PRIMAXIN is generally not recommended. Consider other antibacterial drugs to treat infections in patients whose seizures are well-controlled on valproic acid or divalproex sodium (5.3, 7.3).
Use In Specific Populations
- Renal Impairment: Dosage adjustment is necessary in patients with renal impairment (2.3).
- Adult patients with creatinine clearances of less than or equal to 30 mL/min, whether or not undergoing hemodialysis, had a higher risk of seizure activity than those without impairment of renal function (5.2).
- Therefore, close adherence to the dosing guidelines and regular monitoring of creatinine clearance for these patients is recommended (8.6).
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2022
Related/similar drugs
prednisone, amoxicillin, doxycycline, ciprofloxacin, cephalexin, azithromycin, metronidazole
Full Prescribing Information
1. Indications and Usage for Primaxin IV
1.1 Lower Respiratory Tract Infections
PRIMAXIN for intravenous use is indicated for the treatment of lower respiratory tract infections caused by susceptible strains of Staphylococcus aureus (penicillinase-producing isolates), Acinetobacter species, Enterobacter species, Escherichia coli, Haemophilus influenzae, Haemophilus parainfluenzae, Klebsiella species, Serratia marcescens.
1.2 Urinary Tract Infections (complicated and uncomplicated)
PRIMAXIN is indicated for the treatment of urinary tract infections (complicated and uncomplicated) caused by susceptible strains of Enterococcus faecalis, Staphylococcus aureus (penicillinase-producing isolates), Enterobacter species, Escherichia coli, Klebsiella species, Morganella morganii, Proteus vulgaris, Providencia rettgeri, Pseudomonas aeruginosa.
1.3 Intra-Abdominal Infections
PRIMAXIN is indicated for the treatment of intra-abdominal infections caused by susceptible strains of Enterococcus faecalis, Staphylococcus aureus (penicillinase-producing isolates), Staphylococcus epidermidis, Citrobacter species, Enterobacter species, Escherichia coli, Klebsiella species, Morganella morganii, Proteus species, Pseudomonas aeruginosa, Bifidobacterium species, Clostridium species, Eubacterium species, Peptococcus species, Peptostreptococcus species, Propionibacterium species, Bacteroides species including B. fragilis, Fusobacterium species.
1.4 Gynecologic Infections
PRIMAXIN is indicated for the treatment of gynecologic infections caused by susceptible strains of Enterococcus faecalis, Staphylococcus aureus (penicillinase-producing isolates), Staphylococcus epidermidis, Streptococcus agalactiae (Group B streptococci), Enterobacter species, Escherichia coli, Gardnerella vaginalis, Klebsiella species, Proteus species, Bifidobacterium species, Peptococcus species, Peptostreptococcus species, Propionibacterium species, Bacteroides species including B. fragilis.
1.5 Bacterial Septicemia
PRIMAXIN is indicated for the treatment of bacterial septicemia caused by susceptible strains of Enterococcus faecalis, Staphylococcus aureus (penicillinase-producing isolates), Enterobacter species, Escherichia coli, Klebsiella species, Pseudomonas aeruginosa, Serratia species, Bacteroides species including B. fragilis.
1.6 Bone and Joint Infections
PRIMAXIN is indicated for the treatment of bone and joint infections caused by susceptible strains of Enterococcus faecalis, Staphylococcus aureus (penicillinase-producing isolates), Staphylococcus epidermidis, Enterobacter species, Pseudomonas aeruginosa.
1.7 Skin and Skin Structure Infections
PRIMAXIN is indicated for the treatment of skin and skin structure infections caused by susceptible strains of Enterococcus faecalis, Staphylococcus aureus (penicillinase-producing isolates), Staphylococcus epidermidis, Acinetobacter species, Citrobacter species, Enterobacter species, Escherichia coli, Klebsiella species, Morganella morganii, Proteus vulgaris, Providencia rettgeri, Pseudomonas aeruginosa, Serratia species, Peptococcus species, Peptostreptococcus species, Bacteroides species including B. fragilis, Fusobacterium species.
1.8 Endocarditis
PRIMAXIN is indicated for the treatment of endocarditis caused by susceptible strains of Staphylococcus aureus (penicillinase-producing isolates).
1.9 Limitations of Use
- PRIMAXIN is not indicated in patients with meningitis because safety and efficacy have not been established.
- PRIMAXIN is not recommended in pediatric patients with CNS infections because of the risk of seizures [see Dosage and Administration (2.2), Warnings and Precautions (5.2), and Use in Specific Populations (8.4)].
- PRIMAXIN is not recommended in pediatric patients less than 30 kg with impaired renal function, as no data are available [see Use in Specific Populations (8.4), and Dosage and Administration (2.2)].
- Periodic assessment of organ system functions, including renal, hepatic and hematopoietic, is advisable during prolonged therapy.
1.10 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of PRIMAXIN and other antibacterial drugs, PRIMAXIN should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
2. Primaxin IV Dosage and Administration
2.1 Dosage in Adults
For Intravenous Injection Only
- The dosage of PRIMAXIN in adult patients should be based on suspected or confirmed pathogen susceptibility as shown in Table 1 below. The dosage recommendations for PRIMAXIN represent the quantity of imipenem to be administered. An equivalent amount of cilastatin is also present in the solution.
- These doses should be used for patients with creatinine clearance of greater than or equal to 90 mL/min. A reduction in dose must be made for patients with creatinine clearance less than 90 mL/min as shown in Table 3 [see Dosage and Administration (2.3)].
- Recommend that the maximum total daily dosage not exceed 4 g/day.
- Administer 500 mg by intravenous infusion over 20 to 30 minutes.
- Administer 1000 mg by intravenous infusion over 40 to 60 minutes.
- In patients who develop nausea during the infusion, the rate of infusion may be slowed.
| Suspected or Proven Pathogen Susceptibility | Dosage of PRIMAXIN |
|---|---|
| If the infection is suspected or proven to be due to a susceptible bacterial species | 500 mg every 6 hours OR 1000 mg every 8 hours |
| If the infection is suspected or proven to be due to bacterial species with intermediate susceptibility [see Microbiology (12.4)] | 1000 mg every 6 hours |
2.2 Dosage in Pediatric Patients
PRIMAXIN is not recommended in pediatric patients with CNS infections because of the risk of seizures [see Use in Specific Populations (8.4)].
PRIMAXIN is not recommended in pediatric patients <30 kg with renal impairment, as no data are available [see Use in Specific Populations (8.4)].
Based on studies in adults, the maximum total daily dose in pediatric patients should not exceed 4 g/day [see Dosage and Administration (2.1)].
The recommended dosage for pediatric patients with non-CNS infections is shown in Table 2 below:
| Age | Dose (mg/kg) *,† | Frequency (hours) |
|---|---|---|
| Greater than or equal to 3 Months of Age | ||
| 15-25 mg/kg | Every 6 hours | |
| Less than or equal to 3 months of age (Greater than or equal to 1,500 g body weight) | ||
| 4 weeks to 3 months of age | 25 mg/kg | Every 6 hours |
| 1 to 4 weeks of age | 25 mg/kg | Every 8 hours |
| Less than 1 week of age | 25 mg/kg | Every 12 hours |
2.3 Dosage in Adult Patients with Renal Impairment
Patients with creatinine clearance less than 90 mL/min require dosage reduction of PRIMAXIN as indicated in Table 3. The serum creatinine should represent a steady state of renal function. Use the Cockcroft-Gault method described below to calculate the creatinine clearance:
| Males: | (weight in kg) × (140-age in years) |
| (72) × serum creatinine (mg/100 mL) | |
| Females: | (0.85) × (value calculated for males) |
| Creatinine clearance (mL/min) | ||||
|---|---|---|---|---|
| Greater than or equal to 90 | Less than 90 to greater than or equal to 60 | Less than 60 to greater than or equal to 30 | Less than 30 to greater than or equal to 15 | |
|
||||
| Dosage of PRIMAXIN*,†
If the infection is suspected or proven to be due to a susceptible bacterial species: | 500 mg every 6 hours | 400 mg every 6 hours | 300 mg every 6 hours | 200 mg every 6 hours |
| OR | ||||
| 1000 mg every 8 hours | 500 mg every 6 hours | 500 mg every 8 hours | 500 mg every 12 hours | |
| Dosage of PRIMAXIN*,†
If the infection is suspected or proven to be due to bacterial species with intermediate susceptibility [see Microbiology (12.4)]: | 1000 mg every 6 hours | 750 mg every 8 hours | 500 mg every 6 hours | 500 mg every 12 hours |
In patients with creatinine clearances of less than 30 to greater than or equal to 15 mL/min, there may be an increased risk of seizures [see Warnings and Precautions (5.2) and Use in Specific Populations (8.6)]. Patients with creatinine clearance less than 15 mL/min should not receive PRIMAXIN unless hemodialysis is instituted within 48 hours. There is inadequate information to recommend usage of PRIMAXIN for patients undergoing peritoneal dialysis.
2.4 Dosage in Hemodialysis Patients
When treating patients with creatinine clearances of less than 15 mL/min who are undergoing hemodialysis, use the dosage recommendations for patients with creatinine clearances of less than 30 to greater than or equal to 15 mL/min in Table 3 above [see Dosage and Administration (2.3)]. Both imipenem and cilastatin are cleared from the circulation during hemodialysis. The patient should receive PRIMAXIN after hemodialysis and at intervals timed from the end of that hemodialysis session. Dialysis patients, especially those with background CNS disease, should be carefully monitored; for patients on hemodialysis, PRIMAXIN is recommended only when the benefit outweighs the potential risk of seizures [see Warnings and Precautions (5.2)].
2.5 Reconstitution and Preparation of PRIMAXIN Solution for Intravenous Administration
PRIMAXIN Vials
- Do not use diluents containing benzyl alcohol to reconstitute PRIMAXIN for administration to neonates because it has been associated with toxicity in neonates. While toxicity has not been demonstrated in pediatric patients greater than three months of age, small pediatric patients in this age range may also be at risk for benzyl alcohol toxicity.
- Contents of the vials must be reconstituted by adding approximately 10 mL of the appropriate diluent to the vial. List of appropriate diluents are as follows:
- 0.9% Sodium Chloride Injection
- 5% Dextrose Injection
- 5% Dextrose and 0.9% Sodium Chloride Injection
- 5% Dextrose Injection with 0.225% or 0.45% saline solution
- Reconstituted Solutions of PRIMAXIN range from colorless to yellow. Variations of color within this range do not affect the potency of the product.
- The reconstituted suspension must not be administered by direct Intravenous Infusion
- After reconstitution, shake vial well and transfer the resulting suspension to 100 mL of an appropriate infusion solution before administering by intravenous infusion.
- Repeat transfer of the resulting suspension with an additional 10 mL of infusion solution to ensure complete transfer of vial contents to the infusion solution. Agitate the resulting mixture until clear.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- Discard unused portion of the infusion solution where applicable.
2.6 Storage of Reconstituted Solutions
Vials (After Reconstitution)
- PRIMAXIN, as supplied in single dose vials and reconstituted with the appropriate diluents [see Dosage and Administration (2.5)], maintains satisfactory potency for 4 hours at room temperature or for 24 hours under refrigeration (5°C). Do not freeze solutions of PRIMAXIN.
3. Dosage Forms and Strengths
For Injection PRIMAXIN is a sterile powder mixture for reconstitution in single-dose containers including vials containing:
- 500 mg imipenem (anhydrous equivalent) and 500 mg cilastatin (free acid equivalent)
4. Contraindications
PRIMAXIN is contraindicated in patients who have shown hypersensitivity to any component of this product.
5. Warnings and Precautions
5.1 Hypersensitivity Reactions
Serious and occasionally fatal hypersensitivity (anaphylactic) reactions have been reported in patients receiving therapy with beta-lactams. These reactions are more likely to occur in individuals with a history of sensitivity to multiple allergens. There have been reports of individuals with a history of penicillin hypersensitivity who have experienced severe hypersensitivity reactions when treated with another beta-lactam. Before initiating therapy with PRIMAXIN, careful inquiry should be made concerning previous hypersensitivity reactions to penicillins, cephalosporins, other beta-lactams and other allergens. If an allergic reaction to PRIMAXIN occurs, discontinue the drug immediately. Serious anaphylactic reactions require immediate emergency treatment as clinically indicated.
5.2 Seizure Potential
Seizures and other CNS adverse experiences, such as confusional states and myoclonic activity, have been reported during treatment with PRIMAXIN, especially when recommended dosages were exceeded [see Adverse Reactions (6.1, 6.2)]. These experiences have occurred most commonly in patients with CNS disorders (e.g., brain lesions or history of seizures) and/or compromised renal function [see Use in Specific Populations (8.6)]. However, there have been reports of CNS adverse experiences in patients who had no recognized or documented underlying CNS disorder or compromised renal function.
Anticonvulsant therapy should be continued in patients with known seizure disorders. If focal tremors, myoclonus, or seizures occur, patients should be evaluated neurologically, placed on anticonvulsant therapy if not already instituted, and the dosage of PRIMAXIN re-examined to determine whether it should be decreased, or the antibacterial drug discontinued.
5.3 Increased Seizure Potential Due to Interaction with Valproic Acid
Case reports in the literature have shown that co-administration of carbapenems, including PRIMAXIN, to patients receiving valproic acid or divalproex sodium results in a reduction in valproic acid concentrations. The valproic acid concentrations may drop below the therapeutic range as a result of this interaction, therefore increasing the risk of breakthrough seizures. Increasing the dose of valproic acid or divalproex sodium may not be sufficient to overcome this interaction. The concomitant use of PRIMAXIN and valproic acid/divalproex sodium is generally not recommended. Antibacterials other than carbapenems should be considered to treat infections in patients whose seizures are well controlled on valproic acid or divalproex sodium. If administration of PRIMAXIN is necessary, supplemental anti-convulsant therapy should be considered [see Drug Interactions (7.3)]. Close adherence to the recommended dosage and dosage schedules is urged, especially in patients with known factors that predispose to convulsive activity.
5.4 Clostridioides difficile-Associated Diarrhea (CDAD)
Clostridioides difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including PRIMAXIN, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD.
Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial drug use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibacterial drug use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibacterial drug treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
5.5 Development of Drug-Resistant Bacteria
As with other antibacterial drugs, prolonged use of PRIMAXIN may result in overgrowth of nonsusceptible organisms. Repeated evaluation of the patient's condition is essential. If superinfection occurs during therapy, appropriate measures should be taken.
Prescribing PRIMAXIN in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described in greater detail in the Warnings and Precautions section.
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Seizure Potential [see Warnings and Precautions (5.2)]
- Increased Seizure Potential Due to Interaction with Valproic Acid [see Warnings and Precautions (5.3)]
- Clostridioides difficile-Associated Diarrhea (CDAD) [see Warnings and Precautions (5.4)]
- Development of Drug-Resistant Bacteria [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult Patients
During clinical investigations 1,723 patients were treated with PRIMAXIN. Table 4 shows the incidence of adverse reactions reported during the clinical investigations of adult patients treated with PRIMAXIN.
| Body System | Adverse Reactions | Frequency (%) |
|---|---|---|
|
||
| Local Administration site | Phlebitis/thrombophlebitis | 3.1% |
| Pain at the injection site | 0.7% | |
| Erythema at the injection site | 0.4% | |
| Vein induration | 0.2% | |
| Gastrointestinal | Nausea | 2.0% |
| Diarrhea | 1.8% | |
| Vomiting | 1.5% | |
| Skin | Rash | 0.9% |
| Pruritus | 0.3% | |
| Urticaria | 0.2% | |
| Vascular | Hypotension | 0.4% |
| Body as a Whole | Fever | 0.5% |
| Nervous system | Seizures | 0.4% |
| Dizziness | 0.3% | |
| Somnolence | 0.2% | |
Additional adverse reactions reported in less than 0.2% of the patients or reported since the drug was marketed are listed within each body system in order of decreasing severity [see Table 5].
| Body System | Adverse Reactions |
|---|---|
| Gastrointestinal | Pseudomembranous Colitis (the onset of Pseudomembranous colitis symptoms), Hemorrhagic Colitis |
| Gastroenteritis | |
| Abdominal Pain | |
| Glossitis | |
| Tongue Papillar | |
| Hypertrophy | |
| Heartburn | |
| Pharyngeal Pain | |
| Increased Salivation | |
| CNS | Encephalopathy |
| Confusion | |
| Myoclonus | |
| Paresthesia | |
| Vertigo | |
| Headache | |
| Special Senses | Hearing Loss |
| Tinnitus | |
| Respiratory | Chest Discomfort |
| Dyspnea | |
| Hyperventilation | |
| Thoracic Spine Pain | |
| Cardiovascular | Palpitations |
| Tachycardia | |
| Skin | Erythema Multiforme |
| Angioneurotic Edema | |
| Flushing | |
| Cyanosis | |
| Hyperhidrosis | |
| Skin Texture Changes | |
| Candidiasis | |
| Pruritus Vulvae | |
| Local Administration site | Infused vein infection |
| Body as a Whole | Polyarthralgia |
| Asthenia/Weakness | |
| Renal | Oliguria/Anuria |
| Polyuria |
Adverse Laboratory Changes
The following adverse laboratory changes were reported during clinical trials:
Hepatic: Increased alanine aminotransferase (ALT or SGPT), aspartate aminotransferase (AST or SGOT), alkaline phosphatase, bilirubin, and lactate dehydrogenase (LDH)
Hemic: Increased eosinophils, positive Coombs test, increased WBC, increased platelets, decreased hemoglobin and hematocrit, increased monocytes, abnormal prothrombin time, increased lymphocytes, increased basophils
Electrolytes: Decreased serum sodium, increased potassium, increased chloride
Renal: Increased BUN, creatinine
Urinalysis: Presence of urine protein, urine red blood cells, urine white blood cells, urine casts, urine bilirubin, and urine urobilinogen.
Pediatric Patients
| Body System | Adverse Reactions | Frequency (%) |
|---|---|---|
|
||
| Local Administration Site | Phlebitis | 2.2% |
| Intravenous Site Irritation | 1.1% | |
| Gastrointestinal | Diarrhea | 3.9% |
| Gastroenteritis | 1.1% | |
| Vomiting | 1.1% | |
| Skin | Rash | 2.2% |
| Renal | Urine Discoloration | 1.1% |
| Body System | Adverse Reactions | Frequency (%) |
|---|---|---|
|
||
| Gastrointestinal | Diarrhea | 3% |
| CNS | Convulsions | 5.9% |
| Cardiovascular | Tachycardia | 1.5% |
| Skin | Rash | 1.5% |
| Body as a Whole | Oral Candidiasis | 1.5% |
| Renal | Oliguria/Anuria | 2.2% |
Adverse Laboratory Changes
The following adverse laboratory changes were reported in studies of 178 pediatric patients 3 months of age: increased AST (SGOT), decreased hemoglobin/hematocrit, increased platelets, increased eosinophils, increased ALT (SGPT), increased urine protein, decreased neutrophils.
The following adverse laboratory changes were reported in studies of 135 patients (neonates to 3 months of age): increased eosinophils, increased AST (SGPT), increased serum creatinine, increased/decreased platelet count, increased/decreased bilirubin, increased ALT (SGPT), increased alkaline phosphatase, increased/decreased hematocrit.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of PRIMAXIN. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
| Body System | Adverse Reactions |
|---|---|
| Gastrointestinal | Hepatitis (including fulminant hepatitis) |
| Hepatic failure | |
| Jaundice | |
| Staining of the teeth and/or tongue | |
| Hematologic | Pancytopenia |
| Bone marrow depression | |
| Thrombocytopenia | |
| Neutropenia | |
| Leukopenia | |
| Hemolytic anemia | |
| CNS | Tremor |
| Psychic disturbances including hallucinations | |
| Dyskinesia | |
| Agitation | |
| Special Senses | Taste perversion |
| Skin | Stevens-Johnson syndrome |
| Toxic epidermal necrolysis | |
| Body as a whole | Drug fever |
| Renal | Acute renal failure |
| Urine discoloration |
7. Drug Interactions
7.1 Ganciclovir
Generalized seizures have been reported in patients who received ganciclovir and PRIMAXIN. These drugs should not be used concomitantly with PRIMAXIN unless the potential benefits outweigh the risks.
7.2 Probenecid
Concomitant administration of PRIMAXIN and probenecid results in increases in the plasma level and half-life of imipenem. Therefore, it is not recommended that probenecid be given concomitantly with PRIMAXIN.
7.3 Valproic Acid
Case reports in the literature have shown that co-administration of carbapenems, including PRIMAXIN, to patients receiving valproic acid or divalproex sodium results in a reduction in valproic acid concentrations. The valproic acid concentrations may drop below the therapeutic range as a result of this interaction, therefore increasing the risk of breakthrough seizures. Although the mechanism of this interaction is unknown, data from in vitro and animal studies suggest that carbapenems may inhibit the hydrolysis of valproic acid's glucuronide metabolite (VPA-g) back to valproic acid, thus decreasing the serum concentrations of valproic acid [see Warnings and Precautions (5.3)]. The concomitant use of PRIMAXIN and valproic acid/divalproex sodium is generally not recommended. Antibacterials other than carbapenems should be considered to treat infections in patients whose seizures are well-controlled on valproic acid or divalproex sodium.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Available data from a small number of postmarketing cases with PRIMAXIN use in pregnancy are not sufficient to identify any drug-associated risks for major birth defects, miscarriage, or adverse maternal or fetal outcomes.
Developmental toxicity studies with imipenem and cilastatin sodium (alone or in combination) administered to mice, rats, rabbits, and monkeys at doses 0.4 to 2.9 times the recommended human dose (RHD), (based on body surface area), showed no drug-induced fetal malformations.
Embryofetal development studies with imipenem/cilastatin administered to cynomolgus monkeys at doses similar to the RHD (based on body surface area) showed an increase in embryonic loss (see Data).
The background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies within the general population.
Data
Animal Data
Reproductive toxicity studies with imipenem and cilastatin (alone or in combination) administered to mice, rats, and rabbits showed no evidence of effects on embryofetal (mice, rats and rabbits) or pre/postnatal (rats) development.
Imipenem was administered intravenously to rats (gestation days (GD) 7 to 17) and rabbits (GD 6 to 18) at doses up to 900 and 60 mg/kg/day, respectively, approximately 2.9 and 0.4 times the RHD (based on body surface area).
Cilastatin was administered subcutaneously to rats (GD 6 to 17) and intravenously to rabbits (GD 6 to 18) at doses up to 1000 and 300 mg/kg/day, respectively, approximately 3.2 and 1.9 times the RHD (based on body surface area).
Imipenem/cilastatin was administered intravenously to mice at doses up to 320 mg/kg/day (GD 6 to 15). In two separate studies, imipenem/cilastatin was administered to rats (GD 6 to 17 and GD 15 to day 21 postpartum) both intravenously at doses up to 80 mg/kg/day and subcutaneously at 320 mg/kg/day. The higher dose is approximately equal to the RHD (based on body surface area).
Imipenem/cilastatin administered intravenously to pregnant cynomolgus monkeys during organogenesis at 100 mg/kg/day, approximately 0.6 times the RHD (based on body surface area), at an infusion rate mimicking human clinical use, was not associated with fetal malformations, but there was an increase in embryonic loss relative to controls. Imipenem/cilastatin administered to pregnant cynomolgus monkeys during organogenesis at 40 mg/kg/day by bolus intravenous injection caused significant maternal toxicity including death and embryofetal loss.
8.2 Lactation
Risk Summary
There are insufficient data on the presence of imipenem/cilastatin in human milk, and no data on the effects on the breastfed child, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PRIMAXIN and any potential adverse effects on the breastfed child from PRIMAXIN or from the underlying maternal condition.
8.4 Pediatric Use
Use of PRIMAXIN in pediatric patients is supported by evidence from adequate and well-controlled trials of PRIMAXIN in adults and clinical studies in pediatric patients [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
PRIMAXIN is not recommended in pediatric patients with CNS infections because of the risk of seizures.
PRIMAXIN is not recommended in pediatric patients less than 30 kg with renal impairment, as no data are available.
8.5 Geriatric Use
Of the approximately 3600 subjects ≥18 years of age in clinical studies of PRIMAXIN, including postmarketing studies, approximately 2800 received PRIMAXIN. Of the subjects who received PRIMAXIN, data are available on approximately 800 subjects who were 65 and over, including approximately 300 subjects who were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
No dosage adjustment is required based on age [see Clinical Pharmacology (12.3)]. Dosage adjustment in the case of renal impairment is necessary [see Dosage and Administration (2.3)].
8.6 Renal Impairment
Dosage adjustment is necessary in patients with renal impairment [see Dosage and Administration (2.3)]. Adult patients with creatinine clearances of less than or equal to 30 mL/min, whether or not undergoing hemodialysis, had a higher risk of seizure activity than those without impairment of renal function [see Warnings and Precautions (5.2)]. Therefore, close adherence to the dosing guidelines and regular monitoring of creatinine clearance for these patients is recommended.
10. Overdosage
In the case of overdosage, discontinue PRIMAXIN, treat symptomatically, and institute supportive measures as required. PRIMAXIN is hemodialyzable.
11. Primaxin IV Description
PRIMAXIN (imipenem and cilastatin) for Injection is a sterile formulation of imipenem, a penem antibacterial, and cilastatin, a renal dehydropeptidase inhibitor with sodium bicarbonate added as a buffer. PRIMAXIN is an antibacterial drug for intravenous administration.
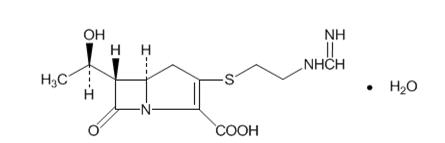
Imipenem (N-formimidoylthienamycin monohydrate) is a crystalline derivative of thienamycin, which is produced by Streptomyces cattleya. Its chemical name is (5R,6S)-3-[[2-(formimidoylamino)ethyl]thio]-6-[(R)-1-hydroxyethyl]-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid monohydrate. It is an off-white, nonhygroscopic crystalline compound with a molecular weight of 317.37. It is sparingly soluble in water and slightly soluble in methanol. Its empirical formula is C12H17N3O4S∙H2O, and its structural formula is:
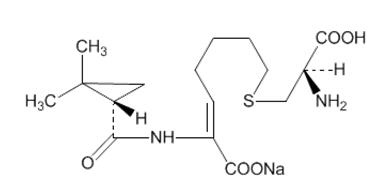
Cilastatin sodium is the sodium salt of a derivatized heptenoic acid. Its chemical name is sodium (Z)-7[[(R)-2-amino-2-carboxyethyl]thio]-2-[(S)-2,2-dimethylcyclopropanecarboxamido]-2-heptenoate. It is an off-white to yellowish-white, hygroscopic, amorphous compound with a molecular weight of 380.43. It is very soluble in water and in methanol. Its empirical formula is C16H25N2O5SNa, and its structural formula is:
Each single-dose vial of PRIMAXIN contains 500 mg imipenem (equivalent to 530 mg imipenem monohydrate), 500 mg cilastatin (equivalent to 531 mg cilastatin sodium), and 20 mg sodium bicarbonate (used as a buffer). PRIMAXIN is buffered to provide solutions in the pH range of 6.5 to 8.5. There is no significant change in pH when solutions are prepared and used as directed [see How Supplied/Storage and Handling (16.1)]. PRIMAXIN 500 contains 37.5 mg of sodium (1.6 mEq). Solutions of PRIMAXIN range from colorless to yellow. Variations of color within this range do not affect the potency of the product.
12. Primaxin IV - Clinical Pharmacology
12.1 Mechanism of Action
PRIMAXIN is a combination of imipenem and cilastatin. Imipenem is a penem antibacterial drug [see Microbiology (12.4)]. Cilastatin is a renal dehydropeptidase inhibitor that limits the renal metabolism of imipenem.
12.3 Pharmacokinetics
Intravenous infusion of PRIMAXIN over 20 minutes results in peak plasma levels of imipenem antimicrobial activity that range from 21 to 58 mcg/mL for the 500 mg dose, and from 41 to 83 mcg/mL for the 1000 mg dose. At these doses, plasma levels of imipenem antimicrobial activity decline to below 1 mcg/mL or less in 4 to 6 hours. Peak plasma levels of cilastatin following a 20-minute intravenous infusion of PRIMAXIN range from 31 to 49 mcg/mL for the 500 mg dose, and from 56 to 88 mcg/mL for the 1000 mg dose.
Distribution
The binding of imipenem to human serum proteins is approximately 20% and that of cilastatin is approximately 40%.
Imipenem has been shown to penetrate into human tissues, including vitreous humor, aqueous humor, lung, peritoneal fluid, CSF, bone, interstitial fluid, skin, and fascia. As there are no adequate and well-controlled studies of imipenem treatment in these additional body sites, the clinical significance of these tissue concentration data is unknown.
After a 1 gram dose of PRIMAXIN, the following average levels of imipenem were measured (usually at 1 hour post dose except where indicated) in the tissues and fluids listed in Table 9:
| Tissue or Fluid | N | Imipenem Level mcg/mL or mcg/g | Range |
|---|---|---|---|
| Vitreous Humor | 3 | 3.4 (3.5 hours post dose) | 2.88–3.6 |
| Aqueous Humor | 5 | 2.99 (2 hours post dose) | 2.4–3.9 |
| Lung Tissue | 8 | 5.6 (median) | 3.5–15.5 |
| Sputum | 1 | 2.1 | — |
| Pleural | 1 | 22.0 | — |
| Peritoneal | 12 | 23.9 S.D.±5.3 (2 hours post dose) | — |
| Bile | 2 | 5.3 (2.25 hours post dose) | 4.6–6.0 |
| CSF (uninflamed) | 5 | 1.0 (4 hours post dose) | 0.26–2.0 |
| CSF (inflamed) | 7 | 2.6 (2 hours post dose) | 0.5–5.5 |
| Fallopian Tubes | 1 | 13.6 | — |
| Endometrium | 1 | 11.1 | — |
| Myometrium | 1 | 5.0 | — |
| Bone | 10 | 2.6 | 0.4–5.4 |
| Interstitial Fluid | 12 | 16.4 | 10.0–22.6 |
| Skin | 12 | 4.4 | NA |
| Fascia | 12 | 4.4 | NA |
Metabolism
Imipenem, when administered alone, is metabolized in the kidneys by dehydropeptidase I, resulting in relatively low levels in urine. Cilastatin, an inhibitor of this enzyme, effectively prevents renal metabolism of imipenem so that when imipenem and cilastatin sodium are given concomitantly, adequate antibacterial levels of imipenem are achieved in the urine.
Elimination
The plasma half-life of each component is approximately 1 hour. Approximately 70% of the administered imipenem is recovered in the urine within 10 hours after which no further urinary excretion is detectable. Urine concentrations of imipenem in excess of 10 mcg/mL can be maintained for up to 8 hours with PRIMAXIN at the 500 mg dose. Approximately 70% of the cilastatin sodium dose is recovered in the urine within 10 hours of administration of PRIMAXIN. PRIMAXIN is hemodialyzable [see Overdosage (10)].
No accumulation of imipenem/cilastatin in plasma or urine is observed with regimens administered as frequently as every 6 hours in patients with normal renal function.
Specific Populations
Geriatric Patients
In healthy elderly volunteers (65 to 75 years of age with normal renal function for their age), the pharmacokinetics of a single dose of imipenem 500 mg and cilastatin 500 mg administered intravenously over 20 minutes are consistent with those expected in subjects with slight renal impairment for which no dosage alteration is considered necessary. The mean plasma half-lives of imipenem and cilastatin are 91±7 minutes and 69±15 minutes, respectively. Multiple dosing has no effect on the pharmacokinetics of either imipenem or cilastatin, and no accumulation of imipenem/cilastatin is observed.
Pediatric Patients
Doses of 25 mg/kg/dose in patients 3 months to <3 years of age, and 15 mg/kg/dose in patients 3-12 years of age were associated with mean trough plasma concentrations of imipenem of 1.1±0.4 mcg/mL and 0.6±0.2 mcg/mL following multiple 60-minute infusions, respectively; trough urinary concentrations of imipenem were in excess of 10 mcg/mL for both doses. These doses have provided adequate plasma and urine concentrations for the treatment of non-CNS infections.
In a dose-ranging study of smaller premature infants (670-1,890 g) in the first week of life, a dose of 20 mg/kg q12h by 15-30 minutes infusion was associated with mean peak and trough plasma imipenem concentrations of 43 mcg/mL and 1.7 mcg/mL after multiple doses, respectively. However, moderate accumulation of cilastatin in neonates may occur following multiple doses of PRIMAXIN. The safety of this accumulation is unknown.
12.4 Microbiology
Mechanism of Action
PRIMAXIN is a combination of imipenem and cilastatin. The bactericidal activity of imipenem results from the inhibition of cell wall synthesis. Its greatest affinity is for penicillin binding proteins (PBPs) 1A, 1B, 2, 4, 5 and 6 of Escherichia coli, and 1A, 1B, 2, 4 and 5 of Pseudomonas aeruginosa. The lethal effect is related to binding to PBP 2 and PBP 1B.
Imipenem has a high degree of stability in the presence of beta-lactamases, both penicillinases and cephalosporinases produced by Gram-negative and Gram-positive bacteria. It is a potent inhibitor of beta-lactamases from certain Gram-negative bacteria which are inherently resistant to most beta-lactam antibacterials, e.g., Pseudomonas aeruginosa, Serratia spp., and Enterobacter spp.
Resistance
Imipenem is inactive in vitro against Enterococcus faecium, Stenotrophomonas maltophilia and some isolates of Burkholderia cepacia. Methicillin-resistant staphylococci should be reported as resistant to imipenem.
Interaction with Other Antimicrobials
In vitro tests show imipenem to act synergistically with aminoglycoside antibacterials against some isolates of Pseudomonas aeruginosa.
Antimicrobial Activity
Imipenem has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1)].
Aerobic bacteria
- Gram-positive bacteria
- Enterococcus faecalis
- Staphylococcus aureus
- Staphylococcus epidermidis
- Streptococcus agalactiae (Group B streptococci)
- Streptococcus pneumoniae
- Streptococcus pyogenes
- Gram-negative bacteria
- Acinetobacter spp.
- Citrobacter spp.
- Enterobacter spp.
- Escherichia coli
- Gardnerella vaginalis
- Haemophilus influenzae
- Haemophilus parainfluenzae
- Klebsiella spp.
- Morganella morganii
- Proteus vulgaris
- Providencia rettgeri
- Pseudomonas aeruginosa
- Serratia spp., including S. marcescens
Anaerobic bacteria
- Gram-positive bacteria
- Bifidobacterium spp.
- Clostridium spp.
- Eubacterium spp.
- Peptococcus spp.
- Peptostreptococcus spp.
- Propionibacterium spp.
- Gram-negative bacteria
- Bacteroides spp., including B. fragilis
- Fusobacterium spp.
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for imipenem against isolates of similar genus or organism group. However, the efficacy of imipenem in treating clinical infections due to these bacteria has not been established in adequate and well-controlled clinical trials.
Aerobic bacteria
- Gram-positive bacteria
- Bacillus spp.
- Listeria monocytogenes
- Nocardia spp.
- Staphylococcus saprophyticus
- Group C streptococci
- Group G streptococci
- Viridans group streptococci
- Gram-negative bacteria
- Aeromonas hydrophila
- Alcaligenes spp.
- Capnocytophaga spp.
- Haemophilus ducreyi
- Neisseria gonorrhoeae
- Pasteurella spp.
- Providencia stuartii
Anaerobic bacteria
- Prevotella bivia
- Prevotella disiens
- Prevotella melaninogenica
- Veillonella spp.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long term studies in animals have not been performed to evaluate carcinogenic potential of imipenem/cilastatin. A variety of bacterial and mammalian tests were performed to evaluate genetic toxicity. The tests used were: V79 mammalian cell mutagenesis assay (cilastatin sodium alone and imipenem alone), Ames test (cilastatin sodium alone and imipenem alone), unscheduled DNA synthesis assay (imipenem/cilastatin sodium) and in vivo mouse cytogenetics test (imipenem/cilastatin sodium). None of these tests showed any evidence of genetic alterations.
Impairment of fertility or reproductive performance was not observed in male and female rats given imipenem/cilastatin at intravenous doses up to 80 mg/kg/day and at a subcutaneous dose of 320 mg/kg/day. In rats, a dose of 320 mg/kg/day was approximately equal to the highest recommended human dose based on body surface area.
16. How is Primaxin IV supplied
16.1 How Supplied
PRIMAXIN is supplied as a sterile powder mixture in single dose containers including vials containing imipenem (anhydrous equivalent) and cilastatin (free acid equivalent) as follows:
| Each PRIMAXIN Package Contains: | National Drug Code (NDC) Number |
|---|---|
| A tray of 25 vials, each vial containing 500 mg imipenem equivalent, 500 mg cilastatin equivalent, and 20 mg sodium bicarbonate as a buffer. | (NDC 0006-3516-59) |
17. Patient Counseling Information
- Advise patients that allergic reactions, including serious allergic reactions, could occur and that serious reactions require immediate treatment. They should report any previous hypersensitivity reactions to PRIMAXIN, other carbapenems, beta-lactams or other allergens.
- Counsel patients that antibacterial drugs including PRIMAXIN should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When PRIMAXIN is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by PRIMAXIN or other antibacterial drugs in the future.
- Counsel patients to inform their physician:
- if they have central nervous system disorders such as stroke or history of seizures. Seizures have been reported during treatment with PRIMAXIN and with closely related antibacterial drugs.
- if they are taking valproic acid or sodium valproate. Valproic acid concentrations in the blood may drop below the therapeutic range upon co-administration with PRIMAXIN. If treatment with PRIMAXIN is necessary and continued, alternative or supplemental anti-convulsant medication to prevent and/or treat seizures may be needed.
- Advise patients that diarrhea is a common problem caused by antibacterial drugs and usually resolves when the drug is discontinued. Sometimes, frequent watery or bloody diarrhea may occur and may be a sign of a more serious intestinal infection. If severe watery or bloody diarrhea develops, patients should contact their healthcare provider.
PRINCIPAL DISPLAY PANEL 250 mg Vial Label
NDC 0006-3514-58
250
PRIMAXIN® I.V.
(IMIPENEM AND CILASTATIN FOR INJECTION)
IMIPENEM 250 mg (Anhydrous Equivalent)
CILASTATIN EQUIVALENT 250 mg
CAUTION: SINGLE DOSE VIAL / FOR I.V. USE ONLY
NOT FOR DIRECT INFUSION
Rx only
250 mg
Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC., Whitehouse Station, NJ 08889, USA
3514

PRINCIPAL DISPLAY PANEL 500 mg Vial Label
NDC 0006-3516-59
500
PRIMAXIN® I.V.
(IMIPENEM AND CILASTATIN FOR INJECTION)
Each vial contains 500 mg imipenem (equivalent to 530 mg
imipenem monohydrate), 500 mg cilastatin (equivalent to 531 mg
cilastatin sodium), and 20 mg sodium bicarbonate (used as a buffer).
CAUTION: SINGLE DOSE VIAL / FOR I.V. USE ONLY
NOT FOR DIRECT INFUSION
Rx only
500 mg

PRINCIPAL DISPLAY PANEL - 500 mg Vial Package
1 PACKAGE (25 VIALS)
NDC 0006-3516-59
500
PRIMAXIN® I.V.
(IMIPENEM AND CILASTATIN FOR INJECTION)
Not to be divided.
Each vial contains 500 mg imipenem (equivalent to 530 mg imipenem monohydrate), 500 mg cilastatin
(equivalent to 531 mg cilastatin sodium), and 20 mg sodium bicarbonate (used as a buffer).
CAUTION: SINGLE DOSE VIAL / FOR I.V. USE ONLY / NOT FOR DIRECT INFUSION
For the Preparation of Intravenous Solutions and USUAL ADULT DOSAGE: See
accompanying circular. Color changes in solution from colorless to
yellow do not affect potency. Store dry material below 25ºC.
Rx only
Merck Sharp & Dohme LLC
Rahway, NJ 07065, USA
Copyright © 2022 Merck & Co., Inc.
Rahway, NJ, USA, and its affiliates.
All rights reserved.

PRINCIPAL DISPLAY PANEL - Single-Dose ADD-Vantage Vial 250 mg
NDC 0006-3551-58
Single-dose ADD-Vantage® vial
250
PRIMAXIN® I.V.
(IMIPENEM AND CILASTATIN FOR INJECTION)
IMIPENEM 250 mg (Anhydrous Equivalent)
CILASTATIN EQUIVALENT 250 mg
CAUTION: SINGLE DOSE VIAL / FOR I.V. USE ONLY
NOT FOR DIRECT INFUSION
PRIMAXIN is a registered trademark of Merck Sharp & Dohme Corp.
ADD-Vantage is a registered trademark of ABBOTT LABORATORIES, Inc.
9960800
For the Preparation of Intravenous Solutions and USUAL ADULT DOSAGE: See accompanying circular.
Store dry material below 25°C.
After constitution as directed, the solution maintains satisfactory potency for 4 hours at room temperature.
Consult accompanying INSTRUCTIONS FOR USE.
Use only with ADD-Vantage® Diluent Containers.
Rx only
250 mg | No. 3551
Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC.
Whitehouse Station, NJ 08889, USA

PRINCIPAL DISPLAY PANEL - Single-Dose ADD-Vantage Vial 500 mg
NDC 0006-3552-59
Single-dose ADD-Vantage® vial
500
PRIMAXIN® I.V.
(IMIPENEM AND CILASTATIN FOR INJECTION)
IMIPENEM 500 mg (Anhydrous Equivalent)
CILASTATIN EQUIVALENT 500 mg
CAUTION: SINGLE DOSE VIAL / FOR I.V. USE ONLY
NOT FOR DIRECT INFUSION
PRIMAXIN is a registered trademark of Merck Sharp & Dohme Corp.
ADD-Vantage is a registered trademark of ABBOTT LABORATORIES, Inc.
9940700
For the Preparation of Intravenous Solutions and USUAL ADULT DOSAGE: See accompanying circular.
Store dry material below 25°C.
After constitution as directed, the solution maintains satisfactory potency for 4 hours at room temperature.
Consult accompanying INSTRUCTIONS FOR USE.
Use only with ADD-Vantage® Diluent Containers.
Rx only
500 mg | No. 3552
Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC.
Whitehouse Station, NJ 08889, USA

| PRIMAXIN
IV
imipenem and cilastatin sodium injection, powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| PRIMAXIN
IV
imipenem and cilastatin sodium injection, powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| PRIMAXIN
IV
imipenem and cilastatin sodium injection, powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| PRIMAXIN
IV
imipenem and cilastatin sodium injection, powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Merck Sharp & Dohme LLC (118446553) |
More about Primaxin IV (cilastatin / imipenem)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- Drug class: carbapenems
- En español
Patient resources
Professional resources
Related treatment guides
Copyright © 1985-2022 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates.
All rights reserved.