Kanuma: Package Insert / Prescribing Info
Package insert / product label
Generic name: sebelipase alfa
Dosage form: injection, solution, concentrate
Drug class: Lysosomal enzymes
J Code (medical billing code): J2840 (1 mg, injection)
Medically reviewed by Drugs.com. Last updated on Jul 22, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
KANUMA (sebelipase alfa) injection, for intravenous use
Initial U.S. Approval: 2015
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
See full prescribing information for complete boxed warning.
- Anaphylaxis has occurred during the early course of enzyme replacement therapy and after extended duration of therapy. (5.1)
- Initiate KANUMA in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation. (5.1)
- If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue KANUMA and immediately initiate appropriate medical treatment, including use of epinephrine. (5.1)
Recent Major Changes
| Boxed Warning | 7/2024 |
| Dosage and Administration (2.1) | 7/2024 |
| Warnings and Precautions (5.1) | 7/2024 |
Indications and Usage for Kanuma
KANUMA® is a hydrolytic lysosomal cholesteryl ester and triacylglycerol-specific enzyme indicated for the treatment of patients with a diagnosis of Lysosomal Acid Lipase (LAL) deficiency. (1)
Kanuma Dosage and Administration
Administration of KANUMA should be supervised by a healthcare provider knowledgeable in the management of hypersensitivity reactions including anaphylaxis. (2.1)
Infants with Rapidly Progressive LAL Deficiency Presenting within the First 6 Months of Life:
- The recommended starting dosage is 1 mg/kg as an intravenous infusion once weekly. (2.2)
- For patients with a suboptimal clinical response, increase the dosage to 3 mg/kg once weekly. (2.2)
- For patients with continued suboptimal clinical response, further increase the dosage to 5 mg/kg once weekly. (2.2)
Pediatric and Adult Patients with LAL Deficiency:
- The recommended dosage is 1 mg/kg as an intravenous infusion once every other week. (2.2)
- For patients with a suboptimal clinical response, increase the dosage to 3 mg/kg once every other week. (2.2)
See Full Prescribing Information for complete Dosage and Administration Information.
Administration Instructions
Dosage Forms and Strengths
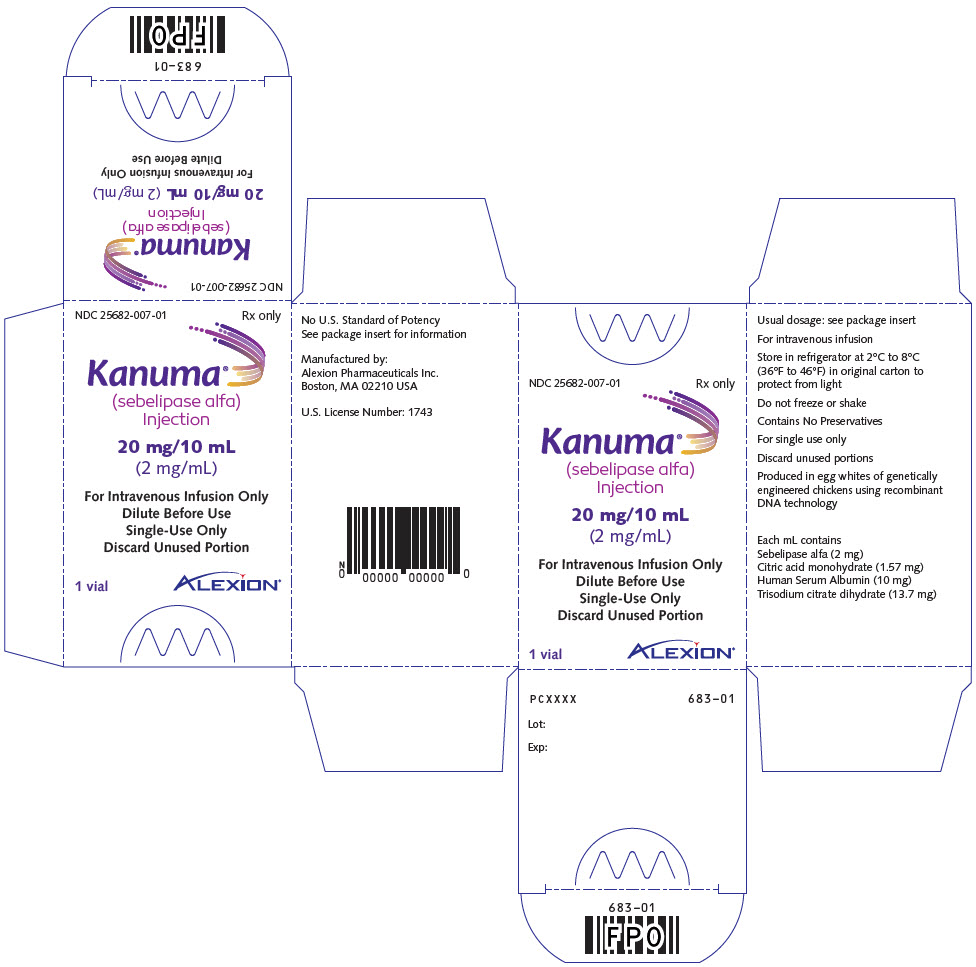
Injection: 20 mg/10 mL (2 mg/mL) solution in single-dose vials. (3)
Contraindications
None. (4)
Warnings and Precautions
- Hypersensitivity to Eggs or Egg Products: Consider the risks and benefits of treatment in patients with known systemic hypersensitivity reactions to eggs or egg products. (5.2)
Adverse Reactions/Side Effects
The most common adverse reactions are:
- Infants with Rapidly Progressive LAL Deficiency Presenting within the First 6 Months of Life (≥30%): diarrhea, vomiting, fever, rhinitis, anemia, cough, nasopharyngitis, and urticaria. (6.1)
- Pediatric and Adult Patients with LAL Deficiency (≥8%): headache, fever, oropharyngeal pain, nasopharyngitis, asthenia, constipation, and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Alexion at 1-844-259-6783 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2024
Full Prescribing Information
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
Patients treated with enzyme replacement therapies have experienced life-threatening hypersensitivity reactions, including anaphylaxis. Anaphylaxis has occurred during the early course of enzyme replacement therapy and after extended duration of therapy.
Initiate KANUMA in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment. If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue KANUMA and immediately initiate appropriate medical treatment, including use of epinephrine. Inform patients of the symptoms of life-threatening hypersensitivity reactions, including anaphylaxis and to seek immediate medical care should symptoms occur [see Warnings and Precautions (5.1)].
1. Indications and Usage for Kanuma
KANUMA® is indicated for the treatment of patients with a diagnosis of Lysosomal Acid Lipase (LAL) deficiency.
2. Kanuma Dosage and Administration
2.1 Recommendations Prior to KANUMA Treatment
Administration of KANUMA should be supervised by a healthcare provider knowledgeable in the management of hypersensitivity reactions including anaphylaxis [see Warnings and Precautions (5.1)].
Initiate KANUMA in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage
Infants with Rapidly Progressive LAL Deficiency Presenting within the First 6 Months of Life:
- The recommended starting dosage is 1 mg/kg administered as an intravenous infusion once weekly.
- For patients with a suboptimal clinical response, increase the dosage to 3 mg/kg once weekly.
- For patients with continued suboptimal clinical response on the 3 mg/kg once weekly dosage, further increase the dosage to 5 mg/kg once weekly.
- A suboptimal clinical response is defined as any of the following: poor growth, deteriorating biochemical markers, or persistent or worsening organomegaly.
Pediatric and Adult Patients with LAL Deficiency:
- The recommended dosage is 1 mg/kg administered as an intravenous infusion once every other week.
- For patients with a suboptimal clinical response, increase the dosage to 3 mg/kg once every other week.
- A suboptimal clinical response is defined as any of the following: poor growth, deteriorating biochemical markers [e.g., alanine aminotransferase (ALT), aspartate aminotransferase (AST)], and/or parameters of lipid metabolism [e.g., low-density lipoprotein cholesterol (LDL-c), triglycerides (TG)].
2.3 Preparation Instructions
KANUMA is for intravenous infusion only. Prepare KANUMA using the following steps.
- Determine the number of vials needed based on the patient's weight and the recommended dose of 1 mg/kg, 3 mg/kg, or 5 mg/kg using the following calculations (a-b):
- Total dose (mg) = Patient's weight (kg) × Recommended dose (mg/kg)
- Total number of vials = Total dose (mg) divided by 20 mg/vial
- Round to the next whole vial and remove the required number of vials from the refrigerator to allow them to reach room temperature.
- Volume (mL) of calculated total dose = Total dose (mg) divided by the 2 mg/mL concentration
- Volume (mL) of 0.9% Sodium Chloride for dilution = Total infusion volume (mL) for patient's weight (see Table 1) - Volume (mL) of calculated total dose
Table 1: Total Infusion Volumes* Weight Range (kg) 1 mg/kg dose 3 mg/kg dose 5 mg/kg dose Total Infusion Volume (mL) Total Infusion Volume (mL) Total Infusion Volume (mL) - *
- The infusion volume should be based on the prescribed dose and should be prepared to a final KANUMA concentration of 0.1 mg/mL to 1.5 mg/mL.
1 to 2.9 4 8 12 3 to 5.9 6 12 20 6 to 10.9 10 25 50 11 to 24.9 25 50 150 25 to 49.9 50 100 250 50 to 99.9 100 250 500 100 to 120.9 250 500 600 - Mix gently by inversion. Do not shake the vials or the prepared infusion.
- The solution should be inspected visually for particulate matter and discoloration prior to administration. The solution should be a clear to slightly opalescent, colorless to slightly colored solution. Thin, translucent particles or fibers may be present in the vials or diluted solution. Do not use if the solution is cloudy or if other particulate matter is observed.
- Vials are for single-use only. Discard any unused product. Do not freeze.
2.4 Administration Instructions
Administer the diluted solution as an intravenous infusion using a low-protein binding infusion set with an in-line, low-protein binding 0.2 micron filter.
Infuse over at least 2 hours. Consider further prolonging the infusion time for patients receiving dosages greater than 1 mg/kg or those who have experienced hypersensitivity reactions [see Warnings and Precautions (5.1)]. A 1-hour infusion may be considered for those patients receiving the 1 mg/kg dose who tolerate the infusion.
2.5 Storage of Diluted Solution
KANUMA contains no preservatives; therefore, product should be used immediately after dilution. If immediate use is not possible, the diluted product may be stored up to 24 hours in the refrigerator at 2°C to 8°C (36°F to 46°F). Do not freeze or shake. Protect from light.
3. Dosage Forms and Strengths
Injection: 20 mg/10 mL (2 mg/mL) clear to slightly opalescent, colorless to slightly colored solution in single-dose vials.
5. Warnings and Precautions
5.1 Hypersensitivity Reactions Including Anaphylaxis
Life-threatening hypersensitivity reactions, including anaphylaxis, have been reported in patients treated with enzyme replacement therapies, including KANUMA. These reactions in KANUMA-treated patients were based on application of Sampson criteria to identify signs/symptoms consistent with anaphylaxis. In clinical trials, 3 (infants) of 106 (3%) patients treated with KANUMA experienced signs and symptoms consistent with anaphylaxis. These patients experienced reactions during infusion with signs and symptoms including chest discomfort, conjunctival injection, dyspnea, generalized and itchy rash, hyperemia, swelling of eyelids, rhinorrhea, severe respiratory distress, tachycardia, tachypnea, and urticaria.
In clinical trials, 21 of 106 (20%) KANUMA-treated patients, including 9 of 14 (64%) infants and 12 of 92 (13%) pediatric patients who were 4 years and older and adults, experienced signs and symptoms either consistent with or that may be related to a hypersensitivity reaction. Signs and symptoms of hypersensitivity reactions, occurring in two or more patients, included abdominal pain, agitation, fever, chills, diarrhea, eczema, edema, hypertension, irritability, laryngeal edema, nausea, pallor, pruritus, rash, and vomiting. The majority of reactions occurred during or within 4 hours of the completion of the infusion. Patients were not routinely pre-medicated prior to infusion of KANUMA in these clinical trials.
Anaphylaxis has occurred during the early course of enzyme replacement therapy and after extended duration of therapy. Administration of KANUMA should be supervised by a healthcare provider knowledgeable in the management of hypersensitivity reactions including anaphylaxis. Initiate KANUMA in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment. Observe patients closely during and after the infusion.
The management of hypersensitivity reactions should be based on the severity of the reaction and may include temporarily interrupting the infusion, lowering the infusion rate, and/or treatment with antihistamines, antipyretics, and/or corticosteroids. If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue KANUMA and immediately initiate appropriate medical treatment, including use of epinephrine. If interrupted, the infusion may be resumed at a slower rate with increases as tolerated. Pre-treatment with antipyretics and/or antihistamines may prevent subsequent reactions in those cases where symptomatic treatment was required.
Inform patients of the symptoms of life-threatening hypersensitivity reactions, including anaphylaxis and to seek immediate medical care should symptoms occur.
Consider the risks and benefits of re-administering KANUMA following a severe reaction. Monitor patients, with appropriate resuscitation measures available, if the decision is made to re-administer the product.
5.2 Hypersensitivity to Eggs or Egg Products
KANUMA is produced in the egg whites of genetically engineered chickens. Patients with a known history of egg allergies were excluded from the clinical trials. Consider the risks and benefits of treatment with KANUMA in patients with known systemic hypersensitivity reactions to eggs or egg products.
6. Adverse Reactions/Side Effects
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
In clinical trials, a total of 106 patients received treatment with KANUMA. The data described below reflect exposure to KANUMA in 75 patients who received KANUMA at dosages up to 3 mg/kg once weekly in clinical trials:
- Nine patients (5 males, 4 females) who had growth failure or other evidence of rapidly progressive LAL deficiency presenting within the first 6 months of life received KANUMA for up to 165 weeks (median 60 weeks) at escalating doses ranging between 0.35 mg/kg and 5 mg/kg once weekly [see Clinical Studies (14.1)].
- 66 pediatric and adult patients with LAL deficiency aged 4 to 58 years (33 males, 33 females) received KANUMA 1 mg/kg every other week for up to 36 weeks.
Table 2 summarizes the most common adverse reactions occurring in >30% of patients with rapidly progressive LAL deficiency presenting within the first 6 months of life receiving KANUMA in Study 1.
| Adverse Reactions | KANUMA N=9 |
|---|---|
| n (%) | |
| Diarrhea | 6 (67) |
| Vomiting | 6 (67) |
| Fever | 5 (56) |
| Rhinitis | 5 (56) |
| Anemia | 4 (44) |
| Cough | 3 (33) |
| Nasopharyngitis | 3 (33) |
| Urticaria | 3 (33) |
Other less common adverse reactions reported in patients with rapidly progressive disease presenting within the first 6 months of life who received KANUMA included hypotonia, decreased oxygen saturation, retching, sneezing, and tachycardia.
For infant patients within Study 1 and Study 3 (n = 19), the following additional adverse reactions were reported in ≥ 30% of infants who received KANUMA since the time of marketing authorization, including patients who received an escalated dose to 5 mg/kg qw: hypersensitivity, respiratory distress, and tachycardia.
Table 3 summarizes the most common adverse reactions that occurred in ≥8% of pediatric and adult patients with LAL deficiency receiving KANUMA in Study 2.
| Adverse Reactions | KANUMA N = 36 | Placebo N = 30 |
|---|---|---|
| n (%) | n (%) | |
| Headache | 10 (28) | 6 (20) |
| Fever | 9 (25) | 7 (23) |
| Oropharyngeal pain | 6 (17) | 1 (3) |
| Nasopharyngitis | 4 (11) | 3 (10) |
| Asthenia | 3 (8) | 1 (3) |
| Constipation | 3 (8) | 1 (3) |
| Nausea | 3 (8) | 2 (7) |
Other less common adverse reactions reported in pediatric and adult patients who received KANUMA included anxiety and chest discomfort.
For pediatric and adult patients (n = 106), the following additional adverse reactions were reported in ≥ 8% of pediatric and adult patients who received KANUMA since the time of marketing authorization, including patients who received an escalated dose to 3 mg/kg qw: hypersensitivity, diarrhea, abdominal pain, and dizziness.
Hyperlipidemia
Increases in circulating LDL-cholesterol (LDL-c) and triglycerides above pre-treatment values were observed in 29 of 36 (81%) and 21 of 36 (58%) patients, respectively, at 2 and 4 weeks following initiation of KANUMA [see Clinical Pharmacology (12.2)]. The maximum mean percentage increase was 18% for LDL-c at Week 2 and 5% for triglycerides at Week 4.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay and may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other sebelipase alfa products may be misleading.
Approximately 8% (9/106) of pediatric and adult patients with LAL deficiency developed antibodies to sebelipase alfa (anti-drug antibodies or ADA) following treatment with KANUMA across all clinical studies. Among the 9 patients who developed ADA, 2 patients were positive for neutralizing antibodies (NAb). Approximately 53% (10/19) of infants with rapidly progressive LAL deficiency developed ADA following treatment with KANUMA across all clinical studies. Among the 10 patients who developed ADA, 9 patients were positive for NAb.
Study 1: Patients with Rapidly Progressive LAL Deficiency Presenting within the First 6 Months of Life
Seven of the 9 infants with rapidly progressive disease had at least one post-treatment ADA assessment, and 4 of these 7 (57%) patients developed ADA during treatment with KANUMA. All of the 4 ADA-positive patients were determined to be positive for neutralizing antibodies that inhibit in vitro enzyme activity and/or cellular uptake of the enzyme. At the time of initial ADA positivity, 3 patients were receiving a dosage of 1 mg/kg once weekly and 1 patient was receiving a dosage of 3 mg/kg once weekly. Three of the 4 ADA-positive patients had ADA titers monitored from the initiation of treatment and developed measurable ADA titers within the first 2 months of exposure. Persistent ADA was observed in all 4 patients.
Hypersensitivity reactions occurred in all 4 of the ADA-positive patients, whereas they occurred in only 1 of the 3 ADA-negative patients. None of the patients discontinued treatment. In 1 patient, decreased growth velocity in a setting of neutralizing antibodies to KANUMA was observed.
Study 2: Pediatric and Adult Patients with LAL Deficiency
Five of 35 (14%) KANUMA-treated pediatric and adult patients who completed the 20-week double-blind period of study treatment developed ADA. All patients were receiving 1 mg/kg once every other week. All 5 ADA-positive patients first developed measurable ADA titers within the first 3 months of exposure. Two of the 5 ADA-positive patients had a measurable ADA titer at only one time point. In the 3 patients with measurable ADA titers at multiple time points, ADA titers decreased to undetectable levels during continued treatment. Two patients developed in vitro neutralizing antibodies during the open-label extension phase after 20 weeks and 52 weeks of treatment with KANUMA, respectively. There is no clear association between the development of ADA and decreased efficacy in pediatric and adult patients treated with KANUMA.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Available data with KANUMA use in pregnant women are insufficient to identify a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. Animal reproductive studies conducted with sebelipase alfa showed no evidence of embryolethality, fetotoxicity, teratogenicity, or abnormal early embryonic development at dosages up to 164 and 526 times the human dosage of 1 mg/kg every other week (based on AUC) in rats and rabbits, respectively.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Animal Data
Sebelipase alfa administered during the period of organogenesis to rats (on gestation days 6, 9, 12, 15 and 17) and rabbits (on gestation days 7, 10, 13, 16 and 19) at intravenous doses up to 60 and 50 mg/kg, respectively, (approximately 164 and 526 times the human AUC of 1387 ng.h/mL at 1 mg/kg dose administered once every other week, respectively) did not cause any adverse effects on embryofetal development. A pre- and post-natal development study in rats showed no evidence of adverse effects on pre- and postnatal development at intravenous doses (administered on gestation days 6, 9, 12, 15, 18, and 20 and days 4, 7, 10, 14, and 17 postpartum) of sebelipase alfa up to 60 mg/kg/day (approximately 164 times the human AUC of 1387 ng.h/mL at 1 mg/kg dose administered once every other week).
8.2 Lactation
Risk Summary
There are no data on the presence of sebelipase alfa in human milk, the effects on the breastfed infant, or the effects on milk production. It is not known if sebelipase alfa is present in animal milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for KANUMA and any potential adverse effects on the breastfed infant from sebelipase alfa or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of KANUMA have been established in pediatric patients aged 1 month and older. Clinical trials with KANUMA were conducted in 56 pediatric patients (range 1 month to <18 years old) [see Clinical Studies (14)].
11. Kanuma Description
Sebelipase alfa is a recombinant human lysosomal acid lipase (rhLAL) that is a lysosomal glycoprotein enzyme produced by recombinant DNA technology in the egg white of eggs laid by genetically engineered chickens. Purified sebelipase alfa is a monomeric glycoprotein containing 6 N-linked glycosylation sites and has a molecular mass of approximately 55 kDa. The amino acid sequence for sebelipase alfa is the same as the amino acid sequence for human LAL. The specific activity of sebelipase alfa is 195 to 345 units/mg. One unit is the amount of enzyme activity that catalyzes the hydrolysis of 1 micromole of the synthetic substrate 4-methylumbelliferyl oleate per minute at 37°C under specified assay conditions.
KANUMA (sebelipase alfa) injection is supplied as a sterile, preservative-free, non-pyrogenic clear to slightly opalescent, colorless to slightly colored aqueous solution in single-dose vials for intravenous infusion. Each vial contains sebelipase alfa 20 mg/10 mL. Each mL of solution contains sebelipase alfa (2 mg), citric acid monohydrate (1.57 mg), Human Serum Albumin (10 mg), and trisodium citrate dihydrate (13.7 mg) at pH 5.9.
12. Kanuma - Clinical Pharmacology
12.1 Mechanism of Action
LAL deficiency is an autosomal recessive lysosomal storage disorder characterized by a genetic defect resulting in a marked decrease or loss in activity of the lysosomal acid lipase (LAL) enzyme. The primary site of action of the LAL enzyme is the lysosome, where the enzyme normally causes the breakdown of lipid particles, including LDL-c and triglycerides. Deficient LAL enzyme activity results in progressive complications due to the lysosomal accumulation of cholesteryl esters and triglycerides in multiple organs, including the liver, spleen, intestine, and the walls of blood vessels. The resulting lipid accumulation in the liver may lead to increased liver fat content and progression of liver disease, including fibrosis and cirrhosis. Lipid accumulation in the intestinal wall leads to malabsorption and growth failure. In parallel, dyslipidemia due to impaired degradation of lysosomal lipid is common with elevated LDL-c and triglycerides and low HDL-cholesterol (HDL-c).
Sebelipase alfa binds to cell surface receptors via glycans expressed on the protein and is subsequently internalized into lysosomes. Sebelipase alfa catalyzes the lysosomal hydrolysis of cholesteryl esters and triglycerides to free cholesterol, glycerol and free fatty acids.
12.2 Pharmacodynamics
In clinical trials, after initiation of dosing with KANUMA, breakdown of accumulated lysosomal lipid led to initial increases in LDL-c and triglycerides within the first 2 to 4 weeks of treatment. In general, following increases in LDL-c and triglycerides, these parameters decreased to below pre-treatment values within 8 weeks of treatment with KANUMA.
In all patients with elevated alanine aminotransferase (ALT) values at baseline (82 of 84 patients in clinical trials), reductions in ALT values were observed, generally within 2 weeks after initiation of treatment with KANUMA. Treatment interruption resulted in increases in LDL-c and ALT values and decreases in HDL-c.
12.3 Pharmacokinetics
The pharmacokinetic profile of sebelipase alfa was nonlinear with a greater than dose-proportional increase in exposure between 1 and 3 mg/kg based on non-compartmental analysis of data from 26 adults. No accumulation was observed following once weekly or once every other week dosing.
Using a population pharmacokinetic model, sebelipase alfa pharmacokinetic parameters were estimated for 65 pediatric and adult patients who received intravenous infusions of KANUMA at 1 mg/kg at Week 22 (Table 4); 24 patients were 4 to 11 years old, 23 were 12 to 17 years old, and 18 were adults. The pharmacokinetic profiles of sebelipase alfa were similar between adolescents and adults. The Tmax and T1/2 were similar across all age groups.
| Parameter | 4-11 years old | 12-17 years old | ≥18 years old |
|---|---|---|---|
| N=24 | N=23 | N=18 | |
| Parameter values were estimated using a population pharmacokinetic model. | |||
| AUC = Area under the plasma concentration time curve. Cmax = Maximum concentration. Tmax = Time to maximum concentration. CL = Clearance. Vc = Central volume of distribution. T1/2 = Half-life. |
|||
| AUC (ng∙hr/mL) | 942 (388) | 1454 (699) | 1861 (599) |
| Cmax (ng/mL) | 490 (205) | 784 (480) | 957 (303) |
| Tmax (hr) | 1.3 (0.6) | 1.1 (0.3) | 1.3 (0.6) |
| CL (L/hr) | 31.1 (7.1) | 37.4 (12.4) | 38.2 (12.5) |
| Vc (L) | 3.6 (3.0) | 5.4 (2.4) | 5.3 (1.6) |
| T1/2 (min) | 5.4 (4.3) | 6.6 (3.7) | 6.6 (3.7) |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to evaluate carcinogenic potential or studies to evaluate mutagenic potential have not been performed with sebelipase alfa. Sebelipase alfa at intravenous doses up to 60 mg/kg administered twice weekly (approximately 164 times the human AUC of 1387 ng.h/mL at 1 mg/kg dose administered once every other week) was found to have no adverse effect on fertility and reproductive performance of male and female rats.
13.2 Animal Toxicology and/or Pharmacology
In a rat disease model of LAL deficiency that exhibits several abnormalities analogous to the human disease, sebelipase alfa administered intravenously at dosages up to 3 mg/kg once weekly showed improvements in survival, body weight gain, organ weight reduction, reduction in serum transaminases (ALT and aspartate aminotransferase [AST]), reduction in serum and hepatic lipids, and improvement in liver histopathology.
14. Clinical Studies
14.1 Infants with Rapidly Progressive LAL Deficiency Presenting within the First 6 Months of Life
A multicenter, open-label, single-arm clinical study of KANUMA was conducted in 9 infants with LAL deficiency who had growth failure or other evidence of rapidly progressive disease prior to 6 months of age. The age range at entry was 1 to 6 months. Patients received KANUMA at 0.35 mg/kg once weekly for the first 2 weeks and then 1 mg/kg once weekly. Due to suboptimal clinical response, doses in all 6 surviving patients were escalated to 3 mg/kg once weekly, between 4 and 88 weeks (median 11 weeks) after starting treatment at 1 mg/kg.
The efficacy of KANUMA was assessed by comparing the survival of the 9 KANUMA-treated patients (followed in the open-label, single-arm trial) at 12 months of age to the survival in an untreated, historical cohort of 21 patients with a similar age at disease presentation and clinical characteristics. Of the 9 KANUMA-treated patients, 6 patients survived beyond 12 months of age, compared to 0 of 21 patients who survived in the historical cohort, all of whom had died by 8 months of age. The median age of the 6 surviving KANUMA-treated patients was 18.1 months (range 12 to 42.2 months).
Following initiation of treatment with KANUMA 1 mg/kg once weekly, weight-for-age (WFA) z-scores improved in 3 of 5 surviving patients with growth failure, and all 6 surviving patients demonstrated improvements in WFA z-scores following dose escalation to 3 mg/kg once weekly.
Continuation Treatment
Across Study 1 and another study, Study 3, in infants with rapidly progressive LAL Deficiency, 9 patients received successive dose escalations up to 5 mg/kg once weekly due to suboptimal clinical response [see Recommended Dosage (2.2)]. The median duration of exposure to 5 mg/kg for the 9 patients whose doses were escalated to 5 mg/kg once weekly was 33 months (range 27 to 39 months) for patients in Study 1 and 15 months (range 5 to 24 months) in Study 3.
Of the 9 patients whose KANUMA dose was escalated to 5 mg/kg once weekly, 6 were alive at their last follow up at 3 years, and 2 were alive at their last follow up at 5 years. Of these 9 patients, 6 experienced normalization of ALT and/or AST which had remained abnormal on the lower KANUMA dose.
14.2 Pediatric and Adult Patients with LAL Deficiency
The safety and efficacy of KANUMA were assessed in 66 pediatric and adult patients with LAL deficiency, aged 4 to 58 years (71% were less than 18 years old), in a multicenter, double-blind, placebo-controlled trial. Patients were randomized to receive KANUMA at a dosage of 1 mg/kg (n=36) or placebo (n=30) once every other week for 20 weeks in the double-blind period. Sixty-two of the 66 (94%) patients had LDL-c of 130 mg/dL or greater at study entry. The majority of patients (58%) had LDL-c above 190 mg/dL at study entry, and 24% of patients with LDL-c above 190 mg/dL remained on lipid lowering medications.
At the completion of the 20-week double-blind period of the trial, a statistically significant improvement in percent change from baseline in LDL-c was observed in the KANUMA-treated group as compared to the placebo group (mean difference and 95% C.I.: -22%, [-33%, -15%]; p<0.0001). LDL-c of less than 130 mg/dL was achieved in 13 of 32 (41%; 95% C.I.: [24%, 58%]) KANUMA-treated patients and in only 2 of 30 (7%; 95% C.I.: [0%, 16%]) placebo-treated patients with baseline LDL-c of 130 mg/dL or greater. A statistically significant improvement in percent change from baseline at 20 weeks was also observed in the KANUMA-treated group compared to the placebo group for other parameters related to LAL deficiency, including decreases in non-HDL-c (mean difference and 95% C.I.: -21%, [-30%, -15%]; p<0.0001) and triglycerides (mean difference and 95% C.I.: -14%, [-28%, -1%]; p=0.0375), and increases in HDL-c (mean difference and 95% C.I.: 20%, [12%, 26%]; p<0.0001). The effect of KANUMA on cardiovascular morbidity and mortality has not been established.
Patients treated with KANUMA had larger reductions from baseline in ALT values and liver fat content (measured by MRI), compared to patients treated with placebo. The significance of these findings as they relate to progression of liver disease in LAL deficiency has not been established.
Open Label Treatment
Pediatric and adult patients who participated in the randomized, placebo-controlled trial were eligible to continue treatment in an open-label extension. Sixty-five of the 66 patients entered the open-label extension and were treated with KANUMA at a dosage of 1 mg/kg once every other week. During the open-label extension, patients treated with KANUMA for up to 36 weeks demonstrated improvements in lipid parameters, including LDL-c and HDL-c levels, and ALT.
Continuation Treatment
Across Study 2 and another study, Study 4, in children and adults with LAL Deficiency, 23 of 97 patients received dose escalations from the protocol-defined starting dose of 1 mg/kg every other week (12 patients in Study 2 and 11 patients in Study 4). The median duration of exposure to the 3 mg/kg every other week regimen was 28 months (range 6 to 33 months) for patients in Study 2 and 12 months (range 3 to 27 months) in Study 4. Before being escalated to 3 mg/kg every other week, patients were on 1 mg/kg every other week for a median of 19 months (range 6 to 33 months), and most dose escalations were initiated in response to an increase in serum transaminase levels, an increase in serum lipids, or a decrease in WFA z-scores in children.
Of the 23 patients whose KANUMA dose was escalated to 3 mg/kg every other week, 20 were children. After the dose escalation, 14 of the 23 patients experienced normalization of one or more of the following biomarkers which had remained abnormally high on the lower KANUMA dose: serum transaminases, triglycerides, and/or LDL-c.
16. How is Kanuma supplied
KANUMA (sebelipase alfa) injection is a preservative-free, clear to slightly opalescent, colorless to slightly colored, nonpyrogenic solution supplied as 20 mg/10 mL (2 mg/mL) in single-dose, glass vials.
NDC 25682-007-01: 20 mg/10 mL vial
17. Patient Counseling Information
Hypersensitivity Reactions Including Anaphylaxis
Advise patients and caregivers that life-threatening hypersensitivity reactions, including anaphylaxis may occur with KANUMA treatment.
Advise patients and caregivers that anaphylaxis has occurred during the early course of enzyme replacement therapy and after extended duration of therapy.
Inform patients and caregivers of the symptoms of life-threatening hypersensitivity reactions, including anaphylaxis and to seek immediate medical care should symptoms occur [see Warnings and Precautions (5.1)].
Hypersensitivity to Eggs or Egg Products
KANUMA is produced in the egg whites of genetically engineered chickens. Patients with a known history of egg allergies were excluded from the clinical trials. Consider the risks and benefits of treatment with KANUMA in patients with known systemic hypersensitivity reactions to eggs or egg products [see Warnings and Precautions (5.2)].
| KANUMA
sebelipase alfa injection, solution, concentrate |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Alexion Pharmaceuticals, Inc. (789359510) |
More about Kanuma (sebelipase alfa)
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: lysosomal enzymes
- Breastfeeding
- En español