Aldurazyme Prescribing Information
Package insert / product label
Generic name: laronidase
Dosage form: injection, solution, concentrate
Drug class: Lysosomal enzymes
J Code (medical billing code): J1931 (0.1 mg, injection)
Medically reviewed by Drugs.com. Last updated on Jan 2, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
ALDURAZYME (laronidase) injection, for intravenous use
Initial U.S. Approval: 2003
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS, and ACUTE RESPIRATORY COMPLICATIONS ASSOCIATED WITH ADMINISTRATION
See full prescribing information for complete boxed warning.
- Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available. If a severe hypersensitivity reaction occurs, discontinue ALDURAZYME immediately and initiate appropriate medical treatment. (5.1)
- Patients with compromised respiratory function or acute respiratory disease may be at risk of serious acute exacerbation of their respiratory compromise due to infusion reactions and require additional monitoring. Appropriate respiratory support should be available during infusion. (5.2)
Recent Major Changes
Indications and Usage for Aldurazyme
ALDURAZYME is a hydrolytic lysosomal glycosaminoglycan (GAG)-specific enzyme indicated for the treatment of adult and pediatric patients with Hurler and Hurler-Scheie forms of Mucopolysaccharidosis I (MPS I) and for the treatment of patients with the Scheie form of MPS I who have moderate to severe symptoms. (1)
Limitations of Use:
Aldurazyme Dosage and Administration
- For pretreatment recommendations, see Full Prescribing Information. (2.1)
- The recommended dosage is 0.58 mg/kg administered once weekly as an intravenous infusion. (2.2)
- For dosage and administration modifications due to hypersensitivity reactions or infusion-associated reactions (IARs), see Full Prescribing Information. (2.3)
- For instructions on preparation, storage, and administration, see Full Prescribing Information. (2.4, 2.5, 2.6)
Dosage Forms and Strengths
Injection: 2.9 mg/5 mL (0.58 mg/mL) of laronidase in a single-dose vial. (3)
Contraindications
None. (4)
Warnings and Precautions
- Risk of Acute Respiratory Complications: Patients with acute febrile or respiratory illness at the time of ALDURAZYME infusion may be at greater risk for infusion reactions. Consider delaying ALDURAZYME infusion. Sleep apnea is common in MPS I patients. Consider evaluating airway patency prior to initiation of treatment with ALDURAZYME. Appropriate respiratory support should be available during infusion. (5.2)
- Risk of Acute Cardiorespiratory Failure: Patients susceptible to fluid overload may be at increased risk for serious exacerbation of their cardiac or respiratory status during infusions. Consider a decreased total infusion volume and infusion rate when administering ALDURAZYME to these patients. Appropriate medical monitoring and support measures should be available during infusion. (2.2, 5.3)
- Infusion Reactions: If severe IARs occur, discontinue ALDURAZYME and initiate appropriate medical treatment. (5.4)
Adverse Reactions/Side Effects
Most common adverse reactions (≥10%) in patients:
- 6 months of age and older are: infusion reactions (pyrexia, chills, blood pressure increased, tachycardia, and oxygen saturation decreased). (6.1)
- 6 years and older are: rash, upper respiratory tract infection, injection site reaction, hyperreflexia, paresthesia, flushing, and poor venous access. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genzyme at 1-800-745-4447 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2023
Full Prescribing Information
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS, and ACUTE RESPIRATORY COMPLICATIONS ASSOCIATED WITH ADMINISTRATION
Hypersensitivity Reactions Including Anaphylaxis
Patients treated with ALDURAZYME have experienced life-threatening hypersensitivity reactions, including anaphylaxis. Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available during ALDURAZYME administration. If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue ALDURAZYME immediately and initiate appropriate medical treatment. In patients with severe hypersensitivity reactions, a desensitization procedure to ALDURAZYME may be considered [see Warnings and Precautions (5.1)].
Acute Respiratory Complications Associated with Administration
Patients with compromised respiratory function or acute respiratory disease may be at risk of serious acute exacerbation of their respiratory compromise due to infusion reactions and require additional monitoring [see Warnings and Precautions (5.2)].
1. Indications and Usage for Aldurazyme
ALDURAZYME® is indicated for the treatment of:
- adult and pediatric patients with Hurler and Hurler-Scheie forms of Mucopolysaccharidosis I (MPS I) and
- patients with the Scheie form of MPS I who have moderate to severe symptoms.
2. Aldurazyme Dosage and Administration
2.1 Recommendations Prior to ALDURAZYME Treatment
Premedication
Prior to ALDURAZYME administration, consider premedicating with antihistamines, with or without antipyretics, 60 minutes before the start of infusion [see Warnings and Precautions (5.1, 5.4)].
2.2 Recommended Dosage and Administration
The recommended dosage of ALDURAZYME is 0.58 mg/kg (actual body weight) administered once weekly as an intravenous infusion.
ALDURAZYME injection must be diluted with 0.9% Sodium Chloride Injection to a final volume of 50 mL, 100 mL or 250 mL as determined by the patient's body weight and cardiopulmonary condition:
Patients with a body weight equal to or greater than 2 kg and less than 4 kg should receive a total volume of 50 mL; patients with a body weight equal to or greater than 4 kg and up to 20 kg should receive a total volume of 100 mL; and those patients with a body weight greater than 20 kg should receive a total volume of 250 mL [see Dosage and Administration (2.6)]. For patients with underlying cardiac or respiratory compromise and weighing up to 30 kg, physicians may consider diluting ALDURAZYME in a volume of 100 mL and administering at a decreased infusion rate [see Dosage and Administration (2.6)].
The initial infusion rate of ALDURAZYME is 10 mcg/kg/hr and may be increased every 15 minutes during the first hour, as tolerated, to a maximum infusion rate of 200 mcg/kg/hr. The maximum rate is then maintained for the remainder of the infusion (2 to 3 hours) [see Dosage and Administration (2.6), Warnings and Precautions (5.2, 5.3)].
If one or more doses are missed, restart ALDURAZYME treatment as soon as possible and maintain the 1-week interval between infusions thereafter. Do not double a dose to compensate for a missed dose.
2.3 Administration Modifications due to Hypersensitivity or Infusion Associated Reaction
In the event of a severe hypersensitivity reaction (e.g. anaphylaxis) or severe infusion-associated reaction (IAR), immediately discontinue ALDURAZYME administration and initiate appropriate medical treatment. For additional recommendations in the event of a severe hypersensitivity reaction, [see Warnings and Precautions (5.1)].
In the event of a mild to moderate hypersensitivity reaction or a mild to moderate IAR, consider temporarily holding the infusion for 15 to 30 minutes, or slowing the infusion rate by 25% to 50% [see Dosage and Administration (2.6)], and initiating appropriate medical treatment [see Warnings and Precautions (5.1, 5.4)].
- If symptoms persist despite holding or slowing the infusion, stop the infusion and monitor the patient. Consider re-initiating the infusion within 7 to 14 days using the incremental rate steps table [see Dosage and Administration (2.6)], up to 25% or 50% of the rate at which the reaction occurred with appropriate premedication.
- If symptoms subside after holding the infusion, resume infusion at a 25% to 50% reduced rate as tolerated. Alternatively, if symptoms subside after slowing the infusion, complete infusion at the reduced rate as tolerated.
- Starting with next infusion, increase the infusion rate by increments of 25% as tolerated until the recommended infusion rate is reached. Closely monitor the patient.
2.4 Preparation Instructions
Prepare ALDURAZYME using low-protein-binding containers. There is no information on the compatibility of diluted ALDURAZYME with glass containers. Dilute ALDURAZYME in the following manner using aseptic technique:
- Determine the infusion bag volume and number of ALDURAZYME vials to be diluted based on actual body weight in kg and the recommended dose [see Dosage and Administration (2.2)]. Round the number of vials up to the next whole number.
- Remove the appropriate number of ALDURAZYME vials from the refrigerator and allow the vials to reach room temperature 20°C to 25°C (68°F to 77°F) before use. Do not heat or microwave the vials.
- Visually inspect the solution in each vial for particulate matter and discoloration. The ALDURAZYME solution should be clear to slightly opalescent and colorless to pale yellow. Some translucency may be present in the solution. Discard if the solution is discolored or if visible particulate matter is present.
- Withdraw and discard a volume of the 0.9% Sodium Chloride Injection from an infusion bag, equal to the volume of ALDURAZYME to be added.
- Slowly withdraw the calculated volume of ALDURAZYME from the appropriate number of vials using caution to avoid excessive agitation. Do not use a filter needle, as this may cause agitation. Agitation may denature ALDURAZYME, rendering it biologically inactive. Discard any unused solution remaining in the vial.
- Slowly add the ALDURAZYME solution to the 0.9% Sodium Chloride Injection solution through the port of the infusion bag and avoid agitation. Do not use a filter needle.
- Gently rotate the infusion bag to ensure proper distribution of ALDURAZYME. Do not shake the infusion bag.
2.5. Storage Instruction for the Diluted Solution
If the diluted ALDURAZYME solution is not used immediately:
- Refrigerate the diluted solution at 2°C to 8°C (36°F to 46°F) for up to 36 hours. Discard any unused ALDURAZYME diluted solution after 36 hours. Do not store the diluted solution at room temperature.
- The solution must be infused within 8 hours after removal from the refrigerator, inclusive of the total infusion time, or discarded.
2.6 Administration Instructions
- Use an infusion set equipped with a low-protein-binding, 0.2 micron, in-line filter to administer the diluted ALDURAZYME solution.
- The total volume of infusion [see Dosage and Administration (2.2)] should be administered over approximately 3 to 4 hours as tolerated per the infusion rate steps outlined in Table 1 below.
- At the end of the infusion, flush the infusion line with 0.9% Sodium Chloride Injection, using the same infusion rate as the one used for the last part of the infusion.
- Do not infuse ALDURAZYME in the same intravenous line with other products.
| Patient Weight Range | Total Infusion Volume | Step 1 10 mcg/kg/hr | Step 2 20 mcg/kg/hr | Step 3 50 mcg/kg/hr | Step 4 100 mcg/kg/hr | Step 5 200 mcg/kg/hr |
|---|---|---|---|---|---|---|
| Infusion Rate in mL/hour | ||||||
| ≥2 to <4 kg | 50 mL | 1 | 2 | 4 | 8 | 16 |
| ≥4 to <20 kg | 100 mL | 2 | 4 | 8 | 16 | 32 |
| ≥20 kg | 250 mL | 5 | 10 | 20 | 40 | 80 |
Start infusion at rate in Step 1. In the absence of infusion-associated reactions after vital sign assessment, increase infusion rate sequentially per the steps in Table 1 every 15 minutes to reach the target rate in Step 5. Continue Step 5 until infusion is completed. The total infusion time is approximately 3 to 4 hours.
3. Dosage Forms and Strengths
Injection: 2.9 mg/5 mL (0.58 mg/mL) of laronidase as a colorless to pale yellow, clear to slightly opalescent solution in a single-dose vial.
5. Warnings and Precautions
5.1 Hypersensitivity Reactions Including Anaphylaxis
Hypersensitivity reactions including anaphylaxis have been reported in patients during or up to 3 hours after ALDURAZYME infusions. Some of these reactions were life-threatening and included respiratory failure, respiratory distress, stridor, tachypnea, bronchospasm, obstructive airways disorder, hypoxia, hypotension, bradycardia, and urticaria.
In clinical studies and postmarketing safety experience with ALDURAZYME, approximately 1% of patients experienced severe or serious hypersensitivity reactions. In patients with MPS I, pre-existing upper airway obstruction may have contributed to the severity of some reactions.
Prior to ALDURAZYME administration, consider premedicating patients with antihistamines, with or without antipyretics, 60 minutes before the start of infusion. Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available during ALDURAZYME administration. Because of the potential for recurrent reactions, some patients who experience initial severe reactions may require prolonged observation.
- If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue ALDURAZYME immediately and initiate appropriate medical treatment. Exercise caution if epinephrine is being considered for use in patients with MPS I due to the increased prevalence of coronary artery disease in these patients. Interventions have included resuscitation, mechanical ventilatory support, emergency tracheotomy, hospitalization, and treatment with inhaled beta-adrenergic agonists, epinephrine, and intravenous corticosteroids [see Adverse Reactions (6)].
- Consider the risks and benefits of re-administering ALDURAZYME following severe hypersensitivity reactions (including anaphylaxis). Patients may be rechallenged using slower infusion rates. In patients with severe hypersensitivity reaction, desensitization measures to ALDURAZYME may be considered. If the decision is made to readminister ALDURAZYME, ensure the patient tolerates the infusion. If the patient tolerates the infusion, the rate may be increased to reach the recommended rate.
- If a mild or moderate hypersensitivity reaction occurs, consider temporarily holding the infusion or slowing the infusion rate [see Dosage and Administration (2.3)].
5.2 Acute Respiratory Complications Associated with Administration
One patient with acute bronchitis and hypoxia experienced increased tachypnea during the first ALDURAZYME infusion that resolved without intervention. The patient's respiratory symptoms returned within 30 minutes of completing the infusion and responded to bronchodilator therapy. Approximately 6 hours after the infusion, the patient experienced coughing, then respiratory arrest, and died.
Patients with an acute febrile or respiratory illness at the time of ALDURAZYME infusion may be at greater risk for infusion reactions. Careful consideration should be given to the patient's clinical status prior to administration of ALDURAZYME and consider delaying ALDURAZYME infusion.
Sleep apnea is common in MPS I patients. Consider evaluating airway patency prior to initiation of treatment with ALDURAZYME. Patients using supplemental oxygen or continuous positive airway pressure (CPAP) during sleep should have these treatments readily available during infusion in the event of an infusion reaction, or extreme drowsiness/sleep induced by antihistamine use.
5.3 Acute Cardiorespiratory Failure
In postmarketing experience, reports of acute cardiorespiratory failure have been reported with ALDURAZYME treatment [see Adverse Reactions (6.2)]. Patients susceptible to fluid overload, or patients with acute underlying respiratory illness or compromised cardiac and/or respiratory function for whom fluid restriction is indicated may be at increased risk of serious exacerbation of their cardiac or respiratory status during infusions. Consider a decreased total infusion volume and infusion rate when administering ALDURAZYME to these patients [see Dosage and Administration (2.2)]. Appropriate medical monitoring and support measures should be readily available during ALDURAZYME infusion, and some patients may require prolonged observation times that should be based on the individual needs of the patient.
5.4 Infusion-Associated Reactions
ALDURAZYME may cause infusion-associated reactions (IARs). Prior to ALDURAZYME administration, consider pre-medicating with antihistamines, with or without antipyretics, 60 minutes before the start of infusion to reduce the risk of IARs. However, IARs may still occur in patients after receiving pre-medication.
- If a severe IAR occurs, discontinue ALDURAZYME immediately and initiate appropriate medical treatment. Consider the risks and benefits of re-administering ALDURAZYME following a severe IAR. Patients may be re-challenged using slower infusion rates. Once a patient tolerates the infusion, the infusion rate may be increased to reach the recommended infusion rate.
- If a mild or moderate IAR occurs, consider temporarily holding the infusion or slowing the infusion rate [see Dosage and Administration (2.3), and Adverse Reactions (6.1, 6.2)].
6. Adverse Reactions/Side Effects
Serious and or clinically significant adverse reactions described elsewhere in labeling include:
- Hypersensitivity Reactions Including Anaphylaxis [see Warnings and Precautions (5.1)]
- Acute Respiratory Complications Associated with Administration [see Warnings and Precautions (5.2)]
- Acute Cardiorespiratory Failure [see Warnings and Precautions (5.3)]
- Infusion-Associated Reactions [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Serious adverse reactions reported with ALDURAZYME treatment during clinical trials were anaphylactic and hypersensitivity reactions. The most common adverse reactions were infusion reactions. The frequency of infusion reactions decreased over time with continued use of ALDURAZYME, and the majority of reactions were classified as being mild to moderate in severity.
Clinical Trials in Patients 6 Years and Older
A 26-week, double-blind, placebo-controlled clinical study (Study 1) of ALDURAZYME was conducted in 45 patients with MPS I, ages 6 to 43 years old, gender evenly distributed (N=23 females and 22 males). Of these 45 patients, 1 was clinically assessed as having Hurler form, 37 Hurler-Scheie, and 7 Scheie. Patients were randomized to receive either 0.58 mg/kg intravenously of ALDURAZYME per week for 26 weeks or placebo. All patients were treated with antipyretics and antihistamines prior to the infusions. Infusion reactions were reported in 32% (7 of 22) of ALDURAZYME-treated patients.
The most common adverse reactions reported in patients who received ALDURAZYME were flushing, pyrexia, headache, and rash. Flushing occurred in 5 patients (23%) receiving ALDURAZYME; the other reactions were less frequent. Less common infusion reactions included angioedema (including face edema), hypotension, paresthesia, feeling hot, hyperhidrosis, tachycardia, vomiting, back pain, and cough. Other reported adverse reactions included bronchospasm, dyspnea, urticaria and pruritus.
Table 2 enumerates adverse reactions and selected laboratory abnormalities that occurred during the 26-week placebo-controlled study (Study 1) that were reported in at least 2 patients more in the ALDURAZYME group than in the placebo group.
| ALDURAZYME N=22 n (%) | Placebo N=23 n (%) |
|
|---|---|---|
| Blood and lymphatic system disorders | ||
| Thrombocytopenia | 2 (9) | 0 |
| Eye disorders | ||
| Corneal opacity | 2 (9) | 0 |
| General disorders and administration site conditions | ||
| Chest pain | 2 (9) | 0 |
| Face edema | 2 (9) | 0 |
| Gravitational edema | 2 (9) | 0 |
| Injection site pain | 2 (9) | 0 |
| Injection site reaction | 4 (18) | 2 (9) |
| Hepatobiliary disorders | ||
| Hyperbilirubinemia | 2 (9) | 0 |
| Infections and infestations | ||
| Abscess | 2 (9) | 0 |
| Upper respiratory tract infection | 7 (32) | 4 (17) |
| Nervous system disorders | ||
| Hyperreflexia | 3 (14) | 0 |
| Paresthesia | 3 (14) | 1 (4) |
| Skin and subcutaneous tissue disorders | ||
| Rash | 8 (36) | 5 (22) |
| Vascular disorders | ||
| Hypotension | 2 (9) | 0 |
| Poor venous access | 3 (14) | 0 |
All 45 patients who completed the placebo-controlled study (Study 1) continued treatment in an open-label, uncontrolled extension study (Study 2). All patients received ALDURAZYME 0.58 mg/kg of body weight once weekly for up to 182 weeks. The most serious adverse reactions reported with ALDURAZYME infusions in Study 2 were anaphylactic and hypersensitivity reactions [see Warnings and Precautions (5)]. One patient had an anaphylactic reaction consisting of urticaria and airway obstruction and tested positive for both ALDURAZYME-specific IgG and IgE binding antibodies and complement activation. The most common adverse reactions requiring intervention were infusion reactions reported in 49% (22 of 45) of patients treated with ALDURAZYME. The most common adverse reactions reported in patients who received ALDURAZYME were rash (13%), flushing (11%), pyrexia (11%), headache (9%), abdominal pain or discomfort (9%), and injection site reaction (9%). Less commonly reported infusion reactions included nausea (7%), diarrhea (7%), feeling hot or cold (7%), vomiting (4%), pruritus (4%), arthralgia (4%), and urticaria (4%). Additional common adverse reactions included back pain and musculoskeletal pain.
Clinical Trials in Patients 6 Years and Younger
Study 3 was a 52-week, open-label, uncontrolled study of 20 MPS I patients, ages 6 months to 5 years old (at enrollment). Sixteen patients were clinically assessed as having the Hurler form, and 4 had the Hurler-Scheie form. All 20 patients received ALDURAZYME at 0.58 mg/kg of body weight once weekly for 26 weeks and up to 52 weeks. All patients were treated with antipyretics and antihistamines prior to the infusions.
The nature and severity of infusion reactions were similar between the older and less severely affected patients (Studies 1 and 2) and the younger, more severely affected patients (Study 3). The most commonly reported adverse reactions in Study 3 were infusion reactions reported in 35% (7 of 20) of patients and included pyrexia (30%), chills (20%), blood pressure increased (10%), tachycardia (10%), and oxygen saturation decreased (10%). Other commonly reported infusion reactions occurring in ≥5% of patients were pallor, tremor, respiratory distress, wheezing, crepitations (pulmonary), pruritus, and rash.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of ALDURAZYME. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In postmarketing experience with ALDURAZYME, severe and serious infusion reactions have been reported, some of which were life-threatening, including anaphylactic shock [see Warnings and Precautions (5.1)] and laryngeal edema.
Adverse reactions resulting in death reported in the postmarketing setting with ALDURAZYME treatment included cardiorespiratory arrest, respiratory failure, cardiac failure, and pneumonia. These events have been reported in MPS I patients with underlying disease [see Warnings and Precautions (5.3)].
Additional adverse reactions included fatigue, peripheral edema, erythema and cyanosis.
There have been a small number of reports of extravasation in patients treated with ALDURAZYME. There have been no reports of tissue necrosis associated with extravasation.
Immunogenicity: Anti-Drug Antibody-Associated Adverse Reactions Including Anaphylaxis
In the MPS I Registry and other postmarketing setting, laronidase-specific IgE and/or IgG antibodies appeared to be associated with anaphylaxis and suspected hypersensitivity reactions in ALDURAZYME-treated patients [see Clinical Pharmacology (12.6)].
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
An MPS I Registry has been established. Pregnant women with MPS I and healthcare providers are encouraged to contact the pregnancy sub-registry by visiting www.registrynxt.com or calling 1-800-745-4447 ext. 15500.
Risk Summary
Available data from the MPS I Registry pregnancy sub-registry, published case reports, and the global pharmacovigilance database with ALDURAZYME use in more than 30 pregnant women have not identified a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. The continuation of treatment for MPS I during pregnancy should be individualized to the pregnant woman. Untreated MPS I may result in adverse pregnancy and infant outcomes (see Clinical Considerations). No evidence of fetal harm has been observed in rats when laronidase was administered during organogenesis at doses up to 6.2 times the recommended human dose (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
When laronidase was administered to pregnant female rats during organogenesis (gestation days [GD] 7-17) at doses of 0, 0.036, 0.36 or 3.6 mg/kg/day intravenously (equivalent to 7.3, 73.1, 730.8 units/kg/day) decreased maternal body weight gains and food consumption were observed with no corresponding effects on reproductive and litter parameters including number and distribution of corpora lutea, implantations and early and late resorptions at doses up to 3.6 mg/kg/day (6.2 times the recommended human dose of 0.58 mg/kg on a mg/kg basis). Laronidase has not been evaluated for effects on embryo-fetal development in any other species.
8.2 Lactation
Risk Summary
Available information from one mother:infant pair are insufficient to evaluate the presence or absence of laronidase in human milk. No adverse effects have been reported in breastfed infants in postmarketing cases of ALDURAZYME use in lactating women. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ALDURAZYME and any potential adverse effects on the breastfed child from ALDURAZYME or from the underlying maternal condition.
Lactating women with MPS I and healthcare providers are encouraged to contact the MPS I Registry by visiting www.registrynxt.com or calling 1-800-745-4447 ext. 15500.
8.4 Pediatric Use
The safety and effectiveness of ALDURAZYME have been established for the treatment of pediatric patients with Hurler and Hurler-Scheie forms of Mucopolysaccharidosis I (MPS I) and the treatment of pediatric patients with the Scheie form of MPS I who have moderate to severe symptoms. The safety and effectiveness ALDURAZYME for the treatment of mildly affected pediatric patients with the Scheie form have not been established.
Use of ALDURAZYME for these indications is supported by evidence from an adequate and well-controlled clinical study (Study1) with an open label extension (Study 2) in adult and pediatric patients with MPS I, and from an open label, uncontrolled clinical study in pediatric patients with MPS I, 6 months to 5 years of age (Study 3). The safety and effectiveness of ALDURAZYME in pediatric patients 6 months of age to 5 years of age was found to be similar to pediatric patients 6 to 18 years of age and adults for these indications [see Adverse Reactions (6.1), Clinical Studies (14)].
11. Aldurazyme Description
ALDURAZYME (laronidase) is a polymorphic variant of the human enzyme α-L-iduronidase that is produced by recombinant DNA technology in a Chinese hamster ovary cell line. α-L-iduronidase (glycosaminoglycan α-L-iduronohydrolase, EC 3.2.1.76) is a lysosomal hydrolase that catalyzes the hydrolysis of terminal α-L-iduronic acid residues of dermatan sulfate and heparan sulfate.
Laronidase is a glycoprotein with a molecular weight of approximately 83 kD. The predicted amino acid sequence of the recombinant form, as well as the nucleotide sequence that encodes it, are identical to a polymorphic form of human α-L-iduronidase. The recombinant protein is comprised of 628 amino acids after cleavage of the N-terminus and contains 6 N-linked oligosaccharide modification sites. Two oligosaccharide chains terminate in mannose-6-phosphate sugars. ALDURAZYME has a specific activity of approximately 172 U/mg.
ALDURAZYME, for intravenous infusion, is supplied as a sterile, nonpyrogenic, colorless to pale yellow, clear to slightly opalescent solution that must be diluted prior to administration in 0.9% Sodium Chloride Injection, USP. The solution in each vial contains a nominal laronidase concentration of 0.58 mg/mL and a pH of approximately 5.5. The extractable volume of 5 mL from each vial provides 2.9 mg laronidase, 43.9 mg sodium chloride, 63.5 mg sodium phosphate monobasic monohydrate, 10.7 mg sodium phosphate dibasic heptahydrate, and 0.05 mg polysorbate 80. ALDURAZYME does not contain preservatives; vials are for single dose only.
12. Aldurazyme - Clinical Pharmacology
12.1 Mechanism of Action
Mucopolysaccharide storage disorders are caused by the deficiency of specific lysosomal enzymes required for the catabolism of glycosaminoglycans (GAG). Mucopolysaccharidosis I (MPS I) is characterized by the deficiency of α-L-iduronidase, a lysosomal hydrolase which catalyzes the hydrolysis of terminal α-L-iduronic acid residues of dermatan sulfate and heparan sulfate. Reduced or absent α-L-iduronidase activity results in the accumulation of the GAG substrates, dermatan sulfate and heparan sulfate, throughout the body and leads to widespread cellular, tissue, and organ dysfunction.
The rationale of ALDURAZYME therapy in MPS I is to provide exogenous enzyme for uptake into lysosomes and increase the catabolism of GAG. ALDURAZYME uptake by cells into lysosomes is most likely mediated by the mannose-6-phosphate-terminated oligosaccharide chains of laronidase binding to specific mannose-6-phosphate receptors.
Because many proteins in the blood are restricted from entry into the central nervous system (CNS) by the blood brain barrier, effects of intravenously administered ALDURAZYME on cells within the CNS cannot be inferred from activity in sites outside the CNS. The ability of ALDURAZYME to cross the blood brain barrier has not been evaluated in animal models or in clinical studies.
12.2 Pharmacodynamics
The pharmacodynamic effect of ALDURAZYME was assessed by reductions in urinary GAG levels. The responsiveness of urinary GAG to dosage alterations of ALDURAZYME is unknown, and the relationship of urinary GAG to other measures of clinical response has also not been established [see Clinical Studies (14)].
12.3 Pharmacokinetics
The pharmacokinetics of laronidase were evaluated in 6-year-old or older patients (N=10 to 12) with MPS I who received 0.58 mg/kg of body weight once weekly of ALDURAZYME as a 4-hour infusion in the placebo-controlled clinical study (Study 1). After the 1st, 12th, and 26th weekly infusions, the mean maximum plasma concentrations (Cmax) ranged from 1.2 to 1.7 mcg/mL for the 3 time points. The mean area under the plasma concentration-time curve (AUC∞) ranged from 4.5 to 6.9 mcg ∙ hour/mL. The mean volume of distribution (Vz) ranged from 0.24 to 0.60 L/kg. Mean plasma clearance (CL) ranged from 1.7 to 2.7 mL/min/kg, and the mean elimination half-life (t1/2) ranged from 1.5 to 3.6 hours.
The pharmacokinetics of laronidase were evaluated in 6-year-old or younger patients (N=7 to 9) with MPS I disease who received 0.58 mg/kg of body weight once weekly of ALDURAZYME as a 4-hour infusion in the open label clinical study (Study 3). After the 26th infusion, the 95% confidence interval of the geometric mean values of PK parameters ranged from 0.6 to 1.6 mcg/mL for the maximum plasma concentrations (Cmax), from 1.3 to 4.4 mcg ∙ hour/mL for area under the plasma concentration-time curve (AUC∞), from 0.12 to 0.56 L/kg for volume of distribution (Vz), from 2.2 to 7.7 mL/min/kg for plasma clearance (CL), and from 0.3 to 1.9 hours for elimination half-life (t1/2).
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of ALDURAZYME or of other laronidase products.
In clinical studies 1, 2, 3 [see Clinical Studies (14.1, 14.2)] and a dose-ranging clinical study that enrolled 8 patients between 3 to 17 years old who received once weekly intravenous doses of 0.58 mg/kg of ALDURAZYME, 70 of 73 (96%) of ALDURAZYME-treated patients developed IgG anti-laronidase antibodies (referred to as ADA). The 3 patients who did not develop ADA were patients with Hurler-Scheie forms of MPS I. In the 70 patients who developed ADA, the onset of ADA positivity in most patients occurred within 2 months after starting ALDURAZYME treatment. ADA titers peaked at approximately 4 months and generally declined over time. Higher ADA titers were observed in patients with Hurler forms of MPS I. Neutralizing antibodies that inhibited cellular uptake of laronidase were detected in 38 of 70 (54%) patients who developed ADA. Neutralizing antibodies that inhibited laronidase enzyme activity were detected in 1 of 70 (1.4%) patients who developed ADA.
Anti-Drug Antibody Effects on Pharmacokinetics
In Study 1, in some ALDURAZYME-treated patients who developed ADA the plasma clearance of laronidase at Week 12 was higher than that at Week 1, and patients with higher ADA titer had higher clearance. At Week 26, plasma clearance of laronidase was comparable to that at Week 1.
Anti-Drug Antibody Effects on Pharmacodynamics
Among the 70 ALDURAZYME-treated patients who developed ADA described above, patients with lower ADA titers tended to have greater than 50% reduction in urinary GAG (uGAG) levels at Week 26, while patients with higher ADA titers tended to have less than 50% reduction in uGAG levels. This correlation was observed in patients with Hurler-Scheie or Scheie form of MPS I. There was no clear correlation in patients with Hurler form of MPS I because these patients had high ADA titers and variable urinary GAG reductions. In postmarketing studies, an inverse correlation between percent uGAG reduction and ADA titer was observed in ALDURAZYME-treated patients who developed ADA. Patients with sustained ADA titers were more likely to have high ADA titers and tended to have less reduction in uGAG.
Anti-Drug Antibody Effects on Efficacy and Safety
In clinical studies, no correlation was demonstrated between the presence of ADA and therapeutic response for FVC or 6MWT.
In Study 1 and Study 2, 9 ALDURAZYME-treated patients who experienced severe infusion-associated reactions were tested for laronidase-specific IgE antibodies. One of the 9 patients tested positive for laronidase-specific IgE antibodies.
In the MPS I Registry, laronidase-specific IgE and IgG antibodies were evaluated in 10 ALDURAZYME-treated patients who had suspected hypersensitivity reactions and had an immunogenicity sample collected within seven days of event onset [see Adverse Reactions (6.1)]. Of the 10 patients, 9 tested positive for laronidase-specific IgE and/or IgG antibodies and 1 patient tested negative for both IgE and IgG antibodies.
In the postmarketing setting, five ALDURAZYME-treated patients who experienced anaphylaxis had ADA results available within seven days of event onset. Of these 5 patients, 3 patients tested positive for laronidase-specific IgE and/or IgG antibodies and in the other 2 patients, IgE antibodies were not detected and IgG antibody results were not reported.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Animal studies to evaluate the mutagenic and carcinogenic potential of laronidase have not been conducted.
Laronidase at intravenous doses up to 3.6 mg/kg (6.2 times the recommended human dose) was found to have no effect on the fertility and reproductive performance of male and female rats.
14. Clinical Studies
14.1 Clinical Studies in Patients 6 Years and Older
Study 1 (NCT00912925) was a randomized, double-blind, placebo-controlled study in 45 patients with MPS I, including 1 patient with the Hurler form, 37 patients with Hurler-Scheie form, and 7 patients with Scheie form of MPS I. Among the 45 patients who completed Study 1, 22 (49%) were male and 23 (51%) were female ranging in age from 6 to 43 years with a mean age of 15.5 years. Thirty seven (82%) were White, 2 (4%) were Asian, 6 (13%) were Other and no patients were Black or African American. For ethnicity, 4 (9%) were Hispanic. All patients had a baseline percent predicted forced vital capacity (FVC) less than or equal to 77%. Patients received ALDURAZYME intravenously at 0.58 mg/kg of body weight once weekly or placebo once weekly for 26 weeks. All 45 randomized patients completed the study.
The primary efficacy outcome assessments were percent predicted FVC and distance walked in 6 minutes (6-minute walk test). After 26 weeks, patients treated with ALDURAZYME showed improvement in percent predicted FVC and in 6-minute walk test compared to placebo-treated patients (see Table 3).
| ALDURAZYME®
(N=22) | Placebo (N=23) |
||
|---|---|---|---|
| Forced Vital Capacity (percent of predicted normal) | |||
| Pretreatment Baseline | Mean ± s.d. | 48 ± 15 | 54 ± 16 |
| Week 26 | Mean ± s.d. | 50 ± 17 | 51 ± 13 |
| Change from Baseline to Week 26 | Mean ± s.d. | 1 ± 7 | -3 ± 7 |
| Median | 1 | -1 | |
| Difference in Change from Baseline to Week 26 Between Groups | Mean | 4 | |
| Median (95% CI) | 2 (0.4, 7), p=0.02† | ||
| 6-Minute Walk Distance (meters) | |||
| Pretreatment Baseline | Mean ± s.d. | 319 ± 131 | 367 ± 114 |
| Week 26 | Mean ± s.d. | 339 ± 127 | 348 ± 129 |
| Change from Baseline to Week 26 | Mean ± s.d. | 20 ± 69 | -18 ± 67 |
| Median | 28 | -11 | |
| Difference in Change from Baseline to Week 26 Between Groups | Mean | 38 | |
| Median (95% CI) | 39 (-2, 79), p=0.07† | ||
Evaluations of bioactivity were changes in liver size and urinary GAG levels. Liver size and urinary GAG levels decreased in patients treated with ALDURAZYME compared to patients treated with placebo. No patient in the group receiving ALDURAZYME reached the normal range for urinary GAG levels during this 6-month study.
Study 2 (NCT00146770) was a 182-week, open-label, uncontrolled extension study of all 45 patients who completed Study 1. Patients received ALDURAZYME intravenously at 0.58 mg/kg body weight once weekly. Forty (89%) patients completed the study through Week 182. Five (11%) patients, all of whom received placebo in Study 1 and subsequently received Aldurazyme in Study 2 discontinued prematurely. Of these, 2 patients discontinued due to an adverse event, 2 patients due to patient wishes, and 1 patient due to pregnancy. For patients treated with ALDURAZYME, the mean increase in 6-minute walk test distance was maintained for an additional 182 weeks through completion of Study 2.
At the end of Study 2, the decrease in mean urinary GAG was similar to the decrease in urinary GAG reported in ALDURAZYME-treated patients at the end of Study 1. The relationship of urinary GAG to other measures of clinical response has not been established.
14.2 Clinical Studies in Patients 6 Years and Younger
Study 3 (NCT00146757) was a 52-week, open-label, uncontrolled clinical study in 20 patients with MPS I, including 16 patients (80%) with the Hurler form and 4 patients (20%) with the Hurler-Scheie form. Among the 20 patients who participated in Study 3, 12 (60%) were male and 8 (40%) were female ranging in age from 6 months to 5 years old with a mean age of 2.9 years. Eighteen (90%) were White, 1 (5%) were Black or African American and 1 (5%) were Other. A total of 18 patients completed the study. All 20 patients received ALDURAZYME intravenously at 0.58 mg/kg of body weight once weekly for 26 weeks. After 26 weeks of treatment, 16 patients continued to receive 0.58 mg/kg of body weight once weekly through Week 52, and 4 patients received 1.16 mg/kg of body weight once weekly from Week 26 through Week 52.
Reduction in mean urinary GAG was demonstrated at Week 13 and was maintained through Week 52. No patient receiving ALDURAZYME reached the normal range for urinary GAG levels during this 52-week study. Changes in urinary GAG levels in children 6 years and younger were similar to changes reported in older patients in Studies 1 and 2 (6 through 43 years old). The relationship of urinary GAG to other measures of clinical response has not been established.
16. How is Aldurazyme supplied
ALDURAZYME (laronidase) injection is supplied as a colorless to pale yellow, clear to slightly opalescent solution in single-dose, clear Type I glass vial. Each vial contains 2.9 mg/5 mL (0.58 mg/mL) of laronidase. The closure consists of a siliconized butyl stopper and an aluminum seal with a plastic flip-off cap. ALDURAZYME is available as: One single-dose vial in a carton (NDC 58468-0070-1)
17. Patient Counseling Information
Hypersensitivity Reactions (Including Anaphylaxis) and Infusion-Associated Reactions (IARs)
Advise the patient or caregiver that reactions related to the infusion may occur during and up to 3 hours after ALDURAZYME treatment, including life-threatening hypersensitivity reactions, including anaphylaxis, and IARs. Inform the patient and caregiver of the signs and symptoms of hypersensitivity reactions and IARs and to seek medical care should signs and symptoms occur [see Warnings and Precaution (5.1, 5.4)].
Cardiac and Respiratory Adverse Reactions
Advise the patient and/or caregiver to report immediately to a healthcare provider if signs or symptoms of cardiac or respiratory decompensation occur during or following an infusion [see Warnings and Precautions (5.2, 5.3)]. Inform patients using supplemental oxygen or continuous positive airway pressure (CPAP) during sleep to have these treatments readily available during infusion or extreme drowsiness/sleep induced by antihistamine use.
Registry
Inform the patient and/or caregiver that a registry for MPS I patients has been established in order to better understand the MPS I disease and to track clinical outcomes of patients with MPS I over time. Additionally, the MPS I Registry also monitors the effect of ALDURAZYME on pregnant women, lactating women, and their infants. Encourage the patient and/or caregiver to contact the registry program by visiting www.registrynxt.com or by calling 1-800-745-4447 ext. 15500.
ALDURAZYME is manufactured by:
- BioMarin Pharmaceutical Inc.
Novato, CA 94949
US License Number 1649
ALDURAZYME is distributed by:
- Genzyme Corporation
Cambridge, MA 02141
A SANOFI COMPANY
ALDURAZYME® is a registered trademark of BioMarin/Genzyme LLC. All rights reserved.
PACKAGE LABEL
NDC 58468-0070-1
ALDURAZYME®
(LARONIDASE)
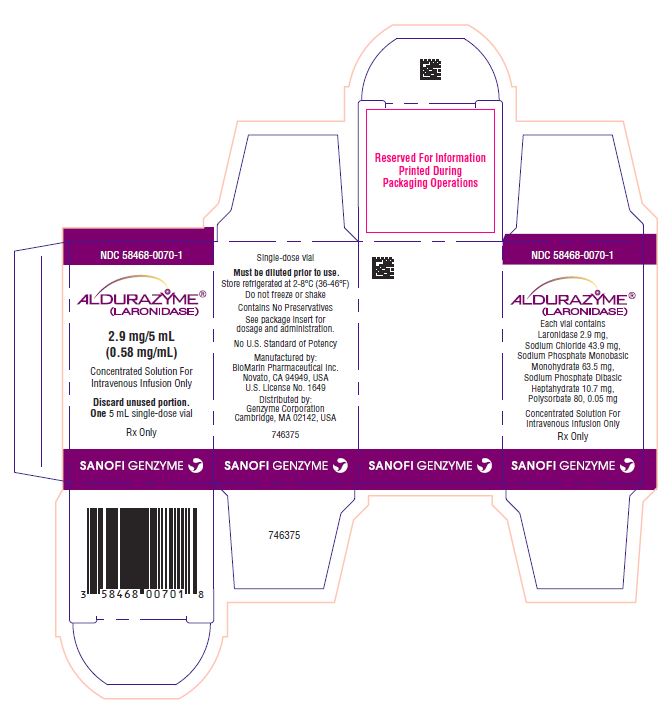
2.9 mg/5 mL
(0.58 mg/mL)
Concentrated Solution For
Intravenous Infusion Only
Discard unused portion.
One 5 mL single-dose vial
Rx Only
genzyme
Single-dose vial
Must be diluted prior to use.
Store refrigerated at 2-8°C (36-46°F)
Do not freeze or shake
Contains No Preservatives
See package insert for
dosage and administration.
No U.S. Standard of Potency
Manufactured by:
BioMarin Pharmaceutical Inc.
Novato, CA 94949, USA
U.S. License No. 1649
Distributed by:
Genzyme Corporation
Cambridge, MA 02142, USA
genzyme
NDC 58468-0070-1
ALDURAZYME®
(LARONIDASE)
Each vial contains
Laronidase 2.9 mg,
Sodium Chloride 43.9 mg,
Sodium Phosphate Monobasic Monohydrate 63.5 mg,
Sodium Phosphate Dibasic Heptahydrate 10.7 mg,
Polysorbate 80, 0.05 mg, in 5mL
Concentrated Solution For
Intravenous Infusion Only
Rx Only
genzyme
LOT
EXP

Single-dose vial
See package insert for
dosage and administration
Store at 2-8°C (36-46°F)
Manufactured by:
BioMarin Pharmaceutical Inc.
Novato, CA 94949
U.S. License No. 1649
Distributed by:
Genzyme Corporation
Cambridge, MA 02142
genzyme
NDC 58468-0070-1
ALDURAZYME®
(LARONIDASE)
2.9 mg/5 mL (0.58 mg/mL)
Concentrated Solution For
Intravenous Infusion Only
Must be diluted prior to use.
Rx Only
LOT:
EXP:
| ALDURAZYME
laronidase injection, solution, concentrate |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - Genzyme Corporation (025322157) |
More about Aldurazyme (laronidase)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- Drug class: lysosomal enzymes
- Breastfeeding
- En español