Elitek Prescribing Information
Package insert / product label
Generic name: rasburicase
Dosage form: injection
Drug class: Antihyperuricemic agents
J Code (medical billing code): J2783 (0.5 mg, injection)
Medically reviewed by Drugs.com. Last updated on Aug 22, 2023.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
ELITEK (rasburicase) for injection, for intravenous use
Initial U.S. Approval: 2002
WARNING: HYPERSENSITIVITY REACTIONS, HEMOLYSIS, METHEMOGLOBINEMIA, AND INTERFERENCE WITH URIC ACID MEASUREMENTS
See full prescribing information for complete boxed warning.
- Hypersensitivity Reactions: Elitek can cause serious and fatal hypersensitivity reactions including anaphylaxis. Immediately and permanently discontinue Elitek if a serious hypersensitivity reaction occurs (4, 5.1, 6.2).
- Hemolysis: Do not administer Elitek to patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Immediately and permanently discontinue Elitek if hemolysis occurs. Screen patients at higher risk for G6PD deficiency (e.g., patients of African or Mediterranean ancestry) prior to starting Elitek therapy (4, 5.2).
- Methemoglobinemia: Elitek can result in methemoglobinemia in some patients. Immediately and permanently discontinue Elitek if methemoglobinemia occurs (4, 5.3).
- Interference with uric acid measurements: Elitek enzymatically degrades uric acid in blood samples left at room temperature. Collect blood samples in prechilled tubes containing heparin and immediately immerse and maintain sample in an ice water bath. Assay plasma samples within 4 hours of collection (5.4).
Indications and Usage for Elitek
Elitek is a recombinant urate-oxidase indicated for initial management of plasma uric acid levels in pediatric and adult patients with leukemia, lymphoma, and solid tumor malignancies who are receiving anticancer therapy expected to result in tumor lysis and subsequent elevation of plasma uric acid. (1)
Limitations of use: Elitek is indicated only for a single course of treatment. (1)
Elitek Dosage and Administration
Dosage Forms and Strengths
Contraindications
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥20%), when used concomitantly with anticancer therapy are vomiting, nausea, fever, peripheral edema, anxiety, headache, abdominal pain, constipation, diarrhea, hypophosphatemia, pharyngolaryngeal pain, and increased alanine aminotransferase. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact sanofi-aventis U.S. LLC at 1-800-633-1610 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2022
Related/similar drugs
allopurinol, sodium bicarbonate, Zyloprim, rasburicase, Aloprim
Full Prescribing Information
WARNING: HYPERSENSITIVITY REACTIONS, HEMOLYSIS, METHEMOGLOBINEMIA, AND INTERFERENCE WITH URIC ACID MEASUREMENTS
Hypersensitivity Reactions
Elitek can cause serious and fatal hypersensitivity reactions including anaphylaxis. Immediately and permanently discontinue Elitek in patients who experience a serious hypersensitivity reaction [see Contraindications (4), Warnings and Precautions (5.1), Adverse Reactions (6.2)].
Hemolysis
Do not administer Elitek to patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Immediately and permanently discontinue Elitek in patients developing hemolysis. Screen patients at higher risk for G6PD deficiency (e.g., patients of African or Mediterranean ancestry) prior to starting Elitek [see Contraindications (4), Warnings and Precautions (5.2)].
Methemoglobinemia
Elitek can result in methemoglobinemia in some patients. Immediately and permanently discontinue Elitek in patients developing methemoglobinemia [see Contraindications (4), Warnings and Precautions (5.3)].
Interference with Uric Acid Measurements
Elitek enzymatically degrades uric acid in blood samples left at room temperature. Collect blood samples in prechilled tubes containing heparin and immediately immerse and maintain sample in an ice water bath. Assay plasma samples within 4 hours of collection [see Warnings and Precautions (5.4)].
1. Indications and Usage for Elitek
Elitek is indicated for the initial management of plasma uric acid levels in pediatric and adult patients with leukemia, lymphoma, and solid tumor malignancies who are receiving anticancer therapy expected to result in tumor lysis and subsequent elevation of plasma uric acid.
Limitations of Use
Elitek is indicated only for a single course of treatment [see Warnings and Precautions (5.1)].
2. Elitek Dosage and Administration
2.1 Dosage
The recommended dose of Elitek is 0.2 mg/kg as a 30-minute intravenous infusion daily for up to 5 days. Dosing beyond 5 days or administration of more than one course is not recommended.
2.2 Reconstitution Procedure
- Elitek must be reconstituted with the diluent provided in the carton.
- Reconstitute the 1.5 mg vial of Elitek with 1 mL of diluent. Reconstitute the 7.5 mg vial of Elitek with 5 mL of diluent. Mix by swirling gently. Do not shake or vortex.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Discard solution if particulate matter is visible or product is discolored.
2.3 Further Dilution and Administration
-
Administer Elitek as an intravenous infusion only:
- Inject the calculated dose of reconstituted Elitek solution into an infusion bag containing the appropriate volume of 0.9% sterile sodium chloride, to achieve a final total volume of 50 mL.
- Infuse over 30 minutes through a separate line or flush line with at least 15 mL of normal saline prior to and after Elitek infusion.
- Do not use filters during infusion of reconstituted Elitek drug product.
- Store reconstituted or diluted solution at 2°C–8°C.
- Discard unused product solution 24 hours following reconstitution.
3. Dosage Forms and Strengths
- For injection: 1.5 mg, lyophilized powder in single-dose vial for reconstitution
- For injection: 7.5 mg, lyophilized powder in single-dose vial for reconstitution
4. Contraindications
Elitek is contraindicated in patients with a history of anaphylaxis or severe hypersensitivity to rasburicase or in patients with development of hemolytic reactions or methemoglobinemia with rasburicase [see Boxed Warning, Warnings and Precautions (5.1, 5.2, 5.3)].
Elitek is contraindicated in individuals deficient in glucose-6-phosphate dehydrogenase (G6PD) [see Boxed Warning, Warnings and Precautions (5.2)].
5. Warnings and Precautions
5.1 Hypersensitivity Reactions
Elitek can cause serious and fatal hypersensitivity reactions including anaphylaxis. In clinical studies, anaphylaxis was reported in <1% patients receiving Elitek. This can occur at any time during treatment including the first dose. Signs and symptoms of these reactions include bronchospasm, chest pain and tightness, dyspnea, hypoxia, hypotension, shock, and urticaria. Immediately and permanently discontinue Elitek administration in any patient developing clinical evidence of a serious hypersensitivity reaction [see Boxed Warning, Contraindications (4), Adverse Reactions (6.2)].
The safety and efficacy of Elitek have been established only for a single course of treatment once daily for 5 days.
5.2 Hemolysis
Elitek is contraindicated in patients with G6PD deficiency because hydrogen peroxide is one of the major by-products of the conversion of uric acid to allantoin. In clinical studies, hemolysis occurs in <1% patients receiving Elitek; severe hemolytic reactions occurred within 2–4 days of the start of Elitek. Immediately and permanently discontinue Elitek administration in any patient developing hemolysis. Institute appropriate patient monitoring and support measures (e.g., transfusion support). Screen patients at higher risk for G6PD deficiency (e.g., patients of African or Mediterranean ancestry) prior to starting Elitek [see Boxed Warning, Contraindications (4)].
5.3 Methemoglobinemia
In clinical studies, methemoglobinemia occurred in <1% patients receiving Elitek. These included cases of serious hypoxemia requiring intervention with medical support measures. It is not known whether patients with deficiency of cytochrome b5 reductase (formerly known as methemoglobin reductase) or of other enzymes with antioxidant activity are at increased risk for methemoglobinemia or hemolytic anemia. Immediately and permanently discontinue Elitek administration in any patient identified as having developed methemoglobinemia. Institute appropriate monitoring and support measures (e.g., transfusion support, methylene-blue administration) [see Boxed Warning, Contraindications (4)].
5.4 Laboratory Test Interference
At room temperature, Elitek causes enzymatic degradation of the uric acid in blood/plasma/serum samples potentially resulting in spuriously low plasma uric acid assay readings. Special sample handling procedure must be followed to avoid ex vivo uric acid degradation [see Boxed Warning, Drug Interactions (7)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are discussed in greater detail in other sections of the prescribing information:
- Anaphylaxis [see Boxed Warning, Contraindications (4), Warnings and Precautions (5.1)]
- Hemolysis [see Boxed Warning, Contraindications (4), Warnings and Precautions (5.2)]
- Methemoglobinemia [see Boxed Warning, Contraindications (4), Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data below reflect exposure to Elitek in 265 pediatric and 82 adult patients enrolled in one active-controlled trial (Study 1), two uncontrolled trials (Studies 2 and 3), and an uncontrolled safety trial (n=82). Additional data were obtained from an expanded access program of 356 patients, for whom data collection was limited to serious adverse reactions. Among these 703 patients 63% were male, the median age was 10 years (range 10 days to 88 years), 73% were Caucasian, 9% African, 4% Asian, and 14% other/unknown.
Among the 347 patients for whom all adverse reactions regardless of severity were assessed, the most frequently observed adverse reactions (incidence ≥10%) were vomiting (50%), fever (46%), nausea (27%), headache (26%), abdominal pain (20%), constipation (20%), diarrhea (20%), mucositis (15%), and rash (13%). In Study 1, an active control study, the following adverse reactions occurred more frequently in Elitek-treated subjects than allopurinol-treated subjects: vomiting, fever, nausea, diarrhea, and headache. Although the incidence of rash was similar in the two arms, severe rash was reported only in one Elitek-treated patient.
Further studies, including one-active controlled study (Study 4) and four supportive studies, have been conducted in adult patients. In these studies, Elitek was administered to a total of 434 adult patients (58% male, 42% female; median age 56 years [range 18 years to 89 years]; 52% Caucasian, 7% African, 14% Asian, 28% other/unknown).
Of these 434 patients, 275 adult patients with leukemia, lymphoma, or solid tumor malignancies at risk for hyperuricemia and tumor lysis syndrome (TLS) were randomized in an open label trial receiving either Elitek alone, Elitek in combination with allopurinol, or allopurinol alone (Study 4).
A drug-related adverse reaction in Study 4 of any grade was experienced in 4.3% of Elitek-treated patients, 5.4% of Elitek/allopurinol-treated patients, and 1.1% of allopurinol-treated patients.
Table 1 presents the per-patient incidence of adverse reactions by study arm in Study 4.
| Adverse Reaction* | Elitek (n=92) | Elitek/Allopurinol (n=92) | Allopurinol (n=91) |
|||
|---|---|---|---|---|---|---|
| All Grades % | Grades 3,4 % | All Grades % | Grades 3,4 % | All Grades % | Grades 3,4 % |
|
| Overall incidence ≥10% in any Elitek arm and the difference between any Elitek arm versus the allopurinol arm ≥5%. | ||||||
|
||||||
| Nausea | 57.6 | 1.1 | 60.9 | 1.1 | 54.9 | 2.2 |
| Peripheral edema | 50 | 2.2 | 43.5 | 3.3 | 42.9 | 6.6 |
| Vomiting | 38 | 1.1 | 37 | 0 | 30.8 | 1.1 |
| Anxiety | 23.9 | 3.3 | 17.4 | 0 | 17.6 | 0 |
| Abdominal pain | 21.7 | 3.3 | 33.7 | 4.3 | 25.3 | 2.2 |
| Hypophosphatemia | 17.4 | 4.3 | 22.8 | 6.5 | 16.5 | 6.6 |
| Hyperbilirubinemia | 16.3 | 3.3 | 14.1 | 2.2 | 7.7 | 4.4 |
| Pharyngolaryngeal pain | 14.1 | 1.1 | 20.7 | 0 | 9.9 | 0 |
| Sepsis | 12 | 5.4 | 7.6 | 6.5 | 4.4 | 4.4 |
| Fluid overload | 12 | 0 | 6.5 | 0 | 3.3 | 1.1 |
| Increased alanine aminotransferase | 10.9 | 3.3 | 27.2 | 4.3 | 17.6 | 2.2 |
| Hyperphosphatemia | 9.8 | 0 | 15.2 | 0 | 8.8 | 1.1 |
Hypersensitivity reactions occurred in 4.3% of Elitek-treated patients and 1.1% of Elitek/allopurinol-treated patients in Study 4. Clinical manifestations of hypersensitivity included arthralgia, injection site irritation, peripheral edema, and rash.
The following serious adverse reactions occurred at a difference in incidence of ≥2% in patients receiving Elitek compared to patients receiving allopurinol in randomized studies (Study 1 and Study 4): pulmonary hemorrhage, respiratory failure, supraventricular arrhythmias, ischemic coronary artery disorders, and abdominal and gastrointestinal infections.
The incidence of anaphylaxis, hemolysis, and methemoglobinemia was less than 1% of the 887 Elitek-treated patients entered on these clinical trials.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. Elitek can elicit antiproduct antibodies that bind to rasburicase and in some instances inhibit the activity of rasburicase in vitro [see Boxed Warning, Warnings and Precautions (5.1)].
In clinical trials of pediatric patients with hematologic malignancies, 24/218 patients tested (11%) developed antibodies by day 28 following Elitek administration as assessed by qualitative ELISA.
Using quasi-quantitative immunoassays in rasburicase-naive adult patients with hematological malignancies, 47/260 (18%) patients were positive for anti-rasburicase immunoglobulin G (IgG), 21/260 (8%) patients were positive for anti-rasburicase neutralizing IgG, and 16/260 (6%) patients were positive for anti-rasburicase immunoglobulin E (IgE) from day 14 to 24 months after 5 daily doses of Elitek.
The incidence of antibody responses detected is highly dependent on the sensitivity and specificity of the assay, which have not been fully evaluated. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors, including serum sampling, timing and methodology, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Elitek with the incidence of antibodies to other products may be misleading.
6.3 Postmarketing Experience
The following adverse reactions have been identified from clinical trials and/or postmarketing surveillance. Because they are reported from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Central nervous system disorders: convulsion, muscle contractions involuntary.
Immune system disorders: cases of anaphylaxis with fatal outcome have been reported.
7. Drug Interactions
Laboratory Test Interference
At room temperature, Elitek causes enzymatic degradation of the uric acid in blood/plasma/serum samples potentially resulting in spuriously low plasma uric acid assay readings. The following special sample handling procedure must be followed to avoid ex vivo uric acid degradation.
Uric acid must be analyzed in plasma. Blood must be collected into prechilled tubes containing heparin anticoagulant. Immediately immerse plasma samples for uric acid measurement in an ice water bath. Plasma samples must be prepared by centrifugation in a precooled centrifuge (4°C). Finally, the plasma must be maintained in an ice water bath and analyzed for uric acid within four hours of collection [see Boxed Warning].
Rasburicase does not metabolize allopurinol, cytarabine, methylprednisolone, methotrexate, 6-mercaptopurine, thioguanine, etoposide, daunorubicin, cyclophosphamide or vincristine in vitro. No metabolic-based drug interactions are therefore anticipated with these agents in patients.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings in animals, Elitek may cause fetal harm when administered to pregnant women. In animal reproduction studies, intravenous administration of rasburicase to pregnant rabbits during organogenesis at 5-times the human exposure (based on AUC) at the recommended human dose of 0.2 mg/kg resulted in adverse developmental outcomes, including structural abnormalities, embryo-fetal mortality, and alterations to growth (see Data). The limited available data with Elitek use in pregnant women are insufficient to inform a drug-associated risk of major birth defects, miscarriage, or adverse maternal fetal outcomes. Consider the benefits and risks of Elitek and possible risks to the fetus when prescribing Elitek to a pregnant woman.
The estimated background risk of major birth defects and miscarriage in the indicated population is unknown. All pregnancies have a background risk of birth defect, miscarriage, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal data
Intravenous administration of rasburicase at doses of 10, 20, or 50 mg/kg/day (approximately 14, 34, and 100 times the exposure at the recommended human dose of 0.2 mg/kg) to pregnant rats from gestation days (GD) 6 to 17 produced multiple heart and great vessel malformations at 50 mg/kg/day (approximately 100 times the exposure at the recommended human dose). Intravenous administration of rasburicase from GD 6 to 19 at doses of 2, 10, or 20 mg/kg/day (approximately 5, 26, and 54 times the exposure at the recommended human dose) to pregnant rabbits produced increased pre and postimplantation loss, abortion, decreased uterine weight, decreased fetal body weights, and heart and great vessel malformations at all dose levels.
8.2 Lactation
Risk Summary
There are no available data on the presence of rasburicase in human breast milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise patients that breastfeeding is not recommended during treatment with Elitek, and for 2 weeks after the last dose.
8.4 Pediatric Use
The safety and effectiveness of Elitek have been established in pediatric patients ages 1 month to 17 years for initial management of plasma uric acid levels in patients with leukemia, lymphoma, and solid tumor malignancies who are receiving anticancer therapy expected to result in tumor lysis and subsequent elevation of plasma uric acid. Elitek was studied in 246 pediatric patients. There were insufficient numbers of patients between 0 and 6 months (n=7) to determine whether they respond differently than older children. Mean uric acid AUC0–96 hr was higher in children <2 years of age (n=24; 150 ± s.e. 16 mg hr/dL) than those age 2 to 17 years (n=222; 108 ± s.e. 4 mg hr/dL). Children <2 years of age had a lower rate of achieving normal uric acid concentration by 48 hours (83% [95% CI: 62, 95]) than those 2 to 17 years (93% [95% CI: 89, 95]).
10. Overdosage
Of the six reported cases of overdosage, five cases had no adverse events reported; nonserious adverse events in the sixth case (a single dose of 1.3 mg/kg) included decreased levels of blood potassium and blood albumin, and increased levels of carbon dioxide, blood lactate dehydrogenase, blood urea, blood phosphorus, blood sodium, and blood alkaline phosphatase. Monitor patients who receive an overdose and initiate supportive measures if required.
11. Elitek Description
Elitek (rasburicase) is a recombinant urate-oxidase produced by a genetically modified Saccharomyces cerevisiae strain. The cDNA coding for rasburicase was cloned from a strain of Aspergillus flavus.
Rasburicase is a tetrameric protein with identical subunits. Each subunit is made up of a single 301 amino acid polypeptide chain with a molecular mass of about 34 kDa. The drug product is a sterile, white to off-white, lyophilized powder intended for intravenous administration following reconstitution with a diluent. Elitek is supplied in 2 mL or 3 mL and 10 mL colorless, glass vials containing rasburicase at a concentration of 1.5 mg/mL after reconstitution.
Elitek 1.5 mg presentation contains 1.5 mg rasburicase, 10.6 mg mannitol, 15.9 mg L-alanine, between 12.6 and 14.3 mg of dibasic sodium phosphate (lyophilized powder), and a diluent (1 mL Water for Injection, USP, and 1 mg Poloxamer 188).
Elitek 7.5 mg presentation contains 7.5 mg of rasburicase, 53 mg mannitol, 79.5 mg L-alanine, and between 63 and 71.5 mg dibasic sodium phosphate (lyophilized powder) and a diluent (5 mL Water for Injection, USP, and 5 mg Poloxamer 188).
12. Elitek - Clinical Pharmacology
12.1 Mechanism of Action
In humans, uric acid is the final step in the catabolic pathway of purines. Rasburicase catalyzes enzymatic oxidation of poorly soluble uric acid into an inactive and more soluble metabolite (allantoin).
12.2 Pharmacodynamics
The measurement of plasma uric acid was used to evaluate the effectiveness of rasburicase in clinical studies. Following administration of either 0.15 or 0.20 mg/kg rasburicase daily for up to 5 days, plasma uric acid levels decreased within 4 hours and were maintained below 7.5 mg/dL in 98% of adult and 90% of pediatric patients for at least 7 days. There was no evidence of a dose response effect on uric acid control for doses between 0.15 and 0.20 mg/kg rasburicase.
In preclinical in vivo studies, rasburicase did not affect the activity of isoenzymes CYP1A, CYP2A, CYP2B, CYP2C, CYP2E, and CYP3A, suggesting no induction or inhibition potential. Clinically relevant P450-mediated drug-drug interactions are therefore not anticipated in patients treated with the recommended Elitek dose and dosing schedule.
12.3 Pharmacokinetics
The pharmacokinetics of rasburicase was evaluated in both pediatric and adult patients with leukemia, lymphoma or other hematological malignancies. Rasburicase exposure, as measured by AUC0–24 hr and Cmax, tended to increase with a dose range from 0.15 to 0.2 mg/kg. The mean terminal half-life was similar between pediatric and adult patients and ranged from 15.7 to 22.5 hours. The mean volume of distribution of rasburicase ranged from 110 to 127 mL/kg in pediatric patients and from 75.8 to 138 mL/kg in adult patients, respectively. Minimal accumulation of rasburicase (<1.3-fold) was observed between days 1 and 5 of dosing. In adults, age, gender, baseline liver enzymes and creatinine clearance did not impact the pharmacokinetics of rasburicase. A cross-study comparison revealed that after administration of rasburicase at 0.15 or 0.20 mg/kg, the geometric mean values of body-weight normalized clearance were approximately 40% lower in Japanese (n=20) than that in Caucasians (n=22).
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies in animals to evaluate tumorigenic potential of rasburicase have not been performed. Rasburicase was not mutagenic in the Ames, unscheduled DNA synthesis, chromosome analysis, mouse lymphoma, and micronucleus tests.
Rasburicase did not affect reproductive performance or fertility in male or female rats with an exposure 11-times greater at a dose of 10 mg/kg than the recommended human dose of 0.2 mg/kg.
14. Clinical Studies
14.1 Pediatrics
Elitek was administered in three studies to 265 patients with acute leukemia or non-Hodgkin's lymphoma. These clinical studies were largely limited to pediatric patients (246 of 265). Elitek was administered as a 30-minute infusion once (n=251) or twice (n=14) daily at a dose of 0.15 or 0.2 mg/kg/dose (total daily dose 0.2–0.4 mg/kg/day). Elitek was administered prior to and concurrent with antitumor therapy, which consisted of either systemic chemotherapy (n=196) or steroids (n=69).
Study 1
Study 1 was a randomized, open-label, controlled study conducted at six institutions, in which 52 pediatric patients were randomized to receive either Elitek (n=27) or allopurinol (n=25). The dose of allopurinol varied according to local institutional practice. Elitek was administered as an intravenous infusion over 30 minutes once (n=26) or twice (n=1) daily at a dose of 0.2 mg/kg/dose (total daily dose 0.2–0.4 mg/kg/day). Initiation of dosing was permitted at any time between 4 to 48 hours before the start of antitumor therapy and could be continued for 5 to 7 days after initiation of antitumor therapy. Patients were stratified at randomization on the basis of underlying malignant disease (leukemia or lymphoma) and baseline serum or plasma uric acid levels (<8 mg/dL and ≥8 mg/dL). The primary study objective was to demonstrate a greater reduction in uric acid concentration over 96 hours (AUC0–96 hr) in the Elitek group as compared to the allopurinol group. Uric acid AUC0–96 hr was defined as the area under the curve for plasma uric acid levels (mg hr/dL), measured from the last value prior to the first dose of Elitek until 96 hours after that first dose. Plasma uric acid levels were used for all uric acid AUC0–96 hr calculations [see Warnings and Precautions (5.4)].
The demographics of the two study arms (Elitek vs allopurinol) were as follows: age <13 years (82% vs 76%), males (59% vs 72%), Caucasian (59% vs 72%), ECOG performance status 0 (89% vs 84%), and leukemia (74% vs 76%). The median interval, in hours, between initiation of Elitek and of antitumor treatment was 20 hours, with a range of 70 hours before to 10 hours after the initiation of antitumor treatment (n=24, data not reported for 3 patients).
The uric acid AUC0–96 hr was significantly lower in the Elitek group (128 ± s.e. 14 mg hr/dL) as compared to the allopurinol group (328 ± s.e. 26 mg hr/dL). All but one patient in the Elitek arm had reduction and maintenance of uric acid levels to within or below the normal range during the treatment. The incidence of renal dysfunction was similar in the two study arms; one patient in the allopurinol arm developed acute renal failure.
Study 2
Study 2 was a multi-institutional, single-arm study conducted in 89 pediatric and 18 adult patients with hematologic malignancies. Patients received Elitek at a dose of 0.15 mg/kg/day. The primary efficacy objective was determination of the proportion of patients with maintained plasma uric acid concentration at 48 hours where maintenance of uric acid concentration was defined as: 1) achievement of uric acid concentration ≤6.5 mg/dL (patients <13 years) or ≤7.5 mg/dL (patients ≥13 years) within a designated time point (48 hours) from initiation of Elitek and maintained until 24 hours after the last administration of study drug; and 2) control of uric acid level without the need for allopurinol or other agents.
The study population demographics were: age <13 years (76%), males (61%), Caucasian (91%), ECOG performance status=0 (92%), and leukemia (89%).
The proportion of patients with maintenance of uric acid concentration at 48 hours in Study 2 was 99% (106/107).
Study 3
Study 3 was a multi-institutional, single-arm study conducted in 130 pediatric patients and 1 adult patient with hematologic malignancies. Patients received Elitek at either a dose of 0.15 mg/kg/day (n=12) or 0.2 mg/kg/day (n=119). The primary efficacy objective was determination of the proportion of patients with maintained plasma uric acid concentration at 48 hours as defined for Study 2 above.
The study population demographics were: age <13 years (76%), Caucasian (83%), males (67%), ECOG=0 (67%), and leukemia (88%).
The proportion of patients with maintenance of uric acid concentration at 48 hours in Study 3 was 92% in the 0.15 mg/kg group (n=12) and 95% in the 0.2 mg/kg group (n=119).
Pooled Analyses of Studies 1, 2, and 3
Data from the 3 studies (n=265) were pooled and analyzed according to the plasma uric acid levels over time. The pretreatment plasma uric acid concentration was ≥8 mg/dL in 61 patients and was <8 mg/dL in 200 patients. The median uric acid concentration at baseline, at 4 hours following the first dose of Elitek, and the per-patient fall in plasma uric acid concentration from baseline to 4 hours, were calculated in those patients with both pretreatment and 4-hour post-treatment values. Among patients with pretreatment uric acid ≥8 mg/dL (baseline median 10.6 mg/dL [range 8.1-36.4]), the median per-patient change in plasma uric acid concentration by 4 hours after the first dose was a decrease of 9.1 mg/dL (0.3-19.3 mg/dL). Among the patients with a pretreatment plasma uric acid level <8 mg/dL (baseline median 4.6 mg/dL [range 0.2-7.9 mg/dL]), the median per-patient change in plasma uric acid concentration by 4 hours after the first dose was a decrease of 4.1 mg/dL (0.1-7.6 mg/dL).
Figure 1: Box and Whisker Plot of Uric Acid Concentration at Designated Time Blocks – Elitek Administration Began Immediately after Baseline
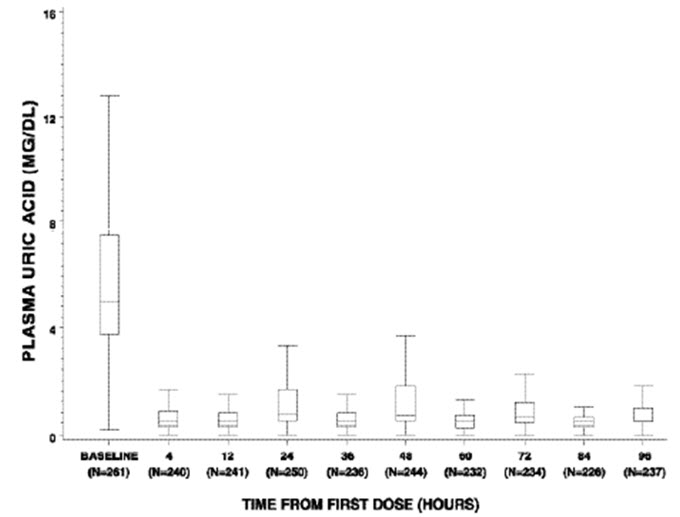
Figure 1 is a box and whisker plot of plasma uric acid levels inclusive of 261 of the 265 Elitek-treated patients from Studies 1, 2, and 3. Of the 261 evaluable patients, plasma uric acid concentration was maintained (see Study 2 for the definition of uric acid concentration maintenance) by 4 hours for 92% of patients (240/261), by 24 hours for 93% of patients (245/261), by 48 hours for 97% of patients (254/261), by 72 hours for 99% of patients (260/261), and by 96 hours for 100% of patients (261/261). Of the subset of 61 patients whose plasma uric acid level was elevated at baseline (≥8 mg/dL), plasma uric acid concentration was maintained by 4 hours for 72% of patients (44/61), by 24 hours for 80% of patients (49/61), by 48 hours for 92% of patients (56/61), by 72 hours for 98% of patients (60/61), and by 96 hours for 100% (61/61).
14.2 Studies in Adults
A total of 342 adults with either leukemia, lymphoma, or other hematologic malignancy received Elitek in five studies (one randomized study, Study 4, and four uncontrolled studies). Across the five studies, Elitek was administered at a dose of 0.15 mg/kg/day (n=38) or 0.2 mg/kg/day (n=304).
Study 4 was a randomized (1:1:1), multi-center, open-label study conducted in patients with leukemia, lymphoma, and solid tumor malignancies at risk for hyperuricemia and TLS. A total of 275 adult patients received at least one dose of study drug. The median age was 56 years, 62% were males, 80% were Caucasian, 66% had leukemia, 29% had lymphoma; 18% were hyperuricemic (uric acid >7.5 mg/dL) at study entry. Patients in Arm A received Elitek for 5 days (n=92). Patients in Arm B received Elitek from day 1 through day 3 followed by oral allopurinol from day 3 through day 5 (overlap on day 3: Elitek and allopurinol administered approximately 12 hours apart) (n=92). Patients in Arm C received oral allopurinol for 5 days (n=91). Elitek was administered at the dose of 0.2 mg/kg/day as a 30-minute infusion once daily. Allopurinol was administered orally at the dose of 300 mg once a day. Patients were eligible for the study if they were either at high risk or potential risk for TLS. The major endpoint of this study was the uric acid response rate defined as the proportion of patients with plasma uric acid levels ≤7.5 mg/dL from day 3 to day 7, after initiation of antihyperuricemic treatment.
Table 2 presents the response rates in the three treatment arms. The response rate in arm A was significantly greater than in arm C (p=0.0009). The response rate was higher for arm B compared to arm C; this difference was not statistically significant.
| Arm A Elitek n=92 | Arm B Elitek/Allopurinol n=92 | Arm C Allopurinol n=91 |
|
|---|---|---|---|
| Response Rate % (95% CI) | 87% (80%, 94%) | 78% (70%, 87%) | 66% (56%, 76%) |
| Nonresponse Rate % | 13% | 22% | 34% |
| Failed to control uric acid | 0 | 0 | 11% |
| Hyperuricemic treatment extended beyond 5 days | 0 | 6.5% | 4.4% |
| Missing uric acid samples | 13% | 15% | 19% |
There were no patients with documented failure to control uric acid in arms A or B. In arm C, 34% of patients did not have a uric acid response; 11% due to failure to control uric acid and 4.4% due to the need for extended antihyperuricemic treatment.
Box and whisker plots of uric acid over time for the patient population (Figure 2) show that in the two arms containing Elitek, uric acid levels were ≤2 mg/dL in 96% of patients at 4 hours of the day 1 dose.
| Figure 2: Uric Acid Concentration Over Time – Patient Population Box and Whisker Plot |
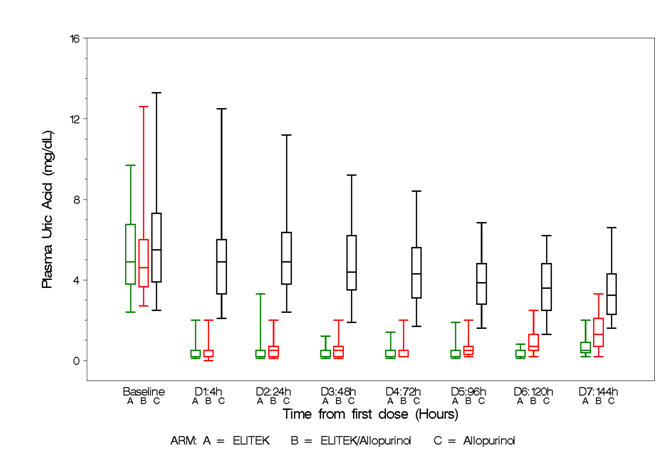 |
Tumor Lysis Syndrome (TLS)
Clinical TLS was defined by changes in at least two or more laboratory parameters for hyperuricemia, hyperkalemia, hyperphosphatemia and hypocalcemia and at least one of the following events occurring within 7 days of treatment: renal failure/injury, need for renal dialysis, and/or serum creatinine increase >1.5 ULN, arrhythmia or seizure. Clinical TLS occurred in 3% of Elitek-treated patients, 3% of Elitek/allopurinol-treated patients, and 4% of allopurinol-treated patients.
16. How is Elitek supplied
17. Patient Counseling Information
Hypersensitivity Reactions
Instruct patients to notify their physician immediately if any of the following occur: allergic reaction, bronchospasm, chest pain or tightness, dyspnea, hypoxia, hypotension, shock or urticaria [see Boxed Warning, Warnings and Precautions (5.1)].
Lactation
Advise females not to breastfeed during treatment with Elitek and for at least 2 weeks after the last dose [see Use in Specific Populations (8.2)].
Manufactured by:
sanofi-aventis U.S. LLC
Bridgewater, NJ 08807
U.S. License No. 1752
©2022 sanofi-aventis U.S. LLC
PRINCIPAL DISPLAY PANEL - 1.5 mg Kit Carton
NDC 0024-5150-10
Rx only
ELITEK®
rasburicase
1.5 mg
FOR INTRAVENOUS INFUSION
Reconstitute with Accompanying Diluent Before Use
Must be Reconstituted and Diluted Before Use
Sterile – Contains No Preservatives
Contents:
Three single-dose vials
Three diluent ampules
sanofi

PRINCIPAL DISPLAY PANEL - 7.5 mg Kit Carton
NDC 0024-5151-75
Rx only
ELITEK®
rasburicase
7.5 mg
FOR INTRAVENOUS INFUSION
Reconstitute with Accompanying Diluent Before Use
Must be Reconstituted and Diluted Before Use
Sterile – Contains No Preservatives
Contents:
One single-dose vial
One diluent ampule
sanofi

| ELITEK
rasburicase kit |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ELITEK
rasburicase kit |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - Sanofi-Aventis U.S. LLC (824676584) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi Chimie | 262699775 | ANALYSIS(0024-5150, 0024-5151) , API MANUFACTURE(0024-5150, 0024-5151) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi S.r.l. | 338454274 | ANALYSIS(0024-5150, 0024-5151) , LABEL(0024-5150, 0024-5151) , MANUFACTURE(0024-5150, 0024-5151) , PACK(0024-5150, 0024-5151) | |
More about Elitek (rasburicase)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- Drug class: antihyperuricemic agents
- Breastfeeding
- En español