Banzel Prescribing Information
Package insert / product label
Generic name: rufinamide
Dosage form: tablet, film coated
Drug class: Dibenzazepine anticonvulsants
Medically reviewed by Drugs.com. Last updated on Nov 23, 2023.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
BANZEL® (rufinamide) film-coated tablet, for oral use
BANZEL® (rufinamide) oral suspension
Initial U.S. Approval: 2008
Indications and Usage for Banzel
BANZEL is indicated for adjunctive treatment of seizures associated with Lennox-Gastaut Syndrome (LGS) in pediatric patients 1 year of age and older, and in adults (1)
Banzel Dosage and Administration
- BANZEL should be given with food. Tablets can be administered whole, as half tablets, or crushed (2.2)
- Measure oral suspension using provided adapter and dosing syringe (2.2)
Pediatric patients 1 year and older:
- Starting daily dose: 10 mg/kg per day in two equally divided doses (2.1)
- Increase by 10 mg/kg increments every other day to maximum dose of 45 mg/kg per day, not to exceed 3200 mg per day, in two divided doses (2.1)
Adults:
Dosage Forms and Strengths
Contraindications
BANZEL is contraindicated in patients with Familial Short QT syndrome (4)
Warnings and Precautions
- Monitor patients for new or worsening depression, suicidal thoughts/behavior, and unusual changes in mood or behavior (5.1)
- Central nervous system reactions can occur (5.2)
- Use caution when administering BANZEL with other drugs that shorten the QT interval (5.3)
- Discontinue BANZEL if multi-organ hypersensitivity reaction occurs (5.4)
- Withdraw BANZEL gradually to minimize the risk of precipitating seizures, seizure exacerbation, or status epilepticus (5.5)
Adverse Reactions/Side Effects
Most common adverse reactions (≥ 10% and greater than placebo) were headache, dizziness, fatigue, somnolence, and nausea (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eisai Inc. at 1-888-274-2378 or www.banzel.com or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2022
Full Prescribing Information
1. Indications and Usage for Banzel
BANZEL is indicated for adjunctive treatment of seizures associated with Lennox-Gastaut Syndrome in pediatric patients 1 year of age and older and in adults.
2. Banzel Dosage and Administration
2.1 Dosage Information
Pediatric patients (1 year to less than 17 years)
The recommended starting daily dose of BANZEL in pediatric patients with Lennox-Gastaut Syndrome is approximately 10 mg/kg administered in two equally divided doses. The dose should be increased by approximately 10 mg/kg increments every other day until a maximum daily dose of 45 mg/kg, not to exceed 3200 mg, administered in two equally divided doses, is reached. It is not known whether doses lower than the target doses are effective.
Adults (17 years and older)
The recommended starting daily dose of BANZEL in adults with Lennox-Gastaut Syndrome is 400 to 800 mg per day administered in two equally divided doses. The dose should be increased by 400-800 mg every other day until a maximum daily dose of 3200 mg, administered in two equally divided doses, is reached. It is not known whether doses lower than 3200 mg are effective.
2.2 Administration Information
Administer BANZEL with food. BANZEL film-coated tablets can be administered whole, as half tablets or crushed.
BANZEL oral suspension should be shaken well before every administration. The provided adapter and calibrated oral dosing syringe should be used to administer the oral suspension. The adapter which is supplied in the product carton should be inserted firmly into the neck of the bottle before use and remain in place for the duration of the usage of the bottle. The dosing syringe should be inserted into the adapter and the dose withdrawn from the inverted bottle. The cap should be replaced after each use. The cap fits properly when the adapter is in place [see Patient Counseling Information (17)].
2.3 Dosing in Patients Undergoing Hemodialysis
Hemodialysis may reduce exposure to a limited (about 30%) extent. Accordingly, adjusting the BANZEL dose during the dialysis process should be considered [see Clinical Pharmacology (12.3)].
2.4 Dosing in Patients with Hepatic Disease
Use of BANZEL in patients with hepatic impairment has not been studied. Therefore, use in patients with severe hepatic impairment is not recommended. Caution should be exercised in treating patients with mild to moderate hepatic impairment [see Use in Specific Populations (8.7)].
2.5 Dosing in Patients Taking Valproate
Patients taking valproate should begin BANZEL at a dose lower than 10 mg/kg per day in pediatric patients or 400 mg per day in adults [see Drug Interactions (7.2)].
3. Dosage Forms and Strengths
Film-coated Tablets: 200 mg (pink) and 400 mg (pink). Tablets are scored on both sides.
Oral Suspension: 40 mg/mL. White to off-white opaque liquid.
4. Contraindications
BANZEL is contraindicated in patients with Familial Short QT syndrome [see Warnings and Precautions (5.3)].
5. Warnings and Precautions
5.1 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including BANZEL, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as 1 week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5-100 years) in the clinical trials analyzed. Table 1 shows absolute and relative risk by indication for all evaluated AEDs.
| Indication | Placebo Patients with Events Per 1000 Patients | Drug Patients with Events Per 1000 Patients | Relative Risk: Incidence of Events in Drug Patients/Incidence in Placebo Patients | Risk Difference: Additional Drug Patients with Events Per 1000 Patients |
| Epilepsy | 1.0 | 3.4 | 3.5 | 2.4 |
| Psychiatric | 5.7 | 8.5 | 1.5 | 2.9 |
| Other | 1.0 | 1.8 | 1.9 | 0.9 |
| Total | 2.4 | 4.3 | 1.8 | 1.9 |
The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing BANZEL or any other AED must balance the risk of suicidal thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
5.2 Central Nervous System Reactions
Use of BANZEL has been associated with central nervous system-related adverse reactions in the controlled clinical trial of patients 4 years or older with Lennox-Gastaut Syndrome. The most significant of these can be classified into two general categories: 1) somnolence or fatigue, and 2) coordination abnormalities, dizziness, gait disturbances, and ataxia.
Somnolence was reported in 24% of BANZEL-treated patients compared to 13% of patients on placebo, and led to study discontinuation in 3% of BANZEL-treated patients compared to 0% of patients on placebo. Fatigue was reported in 10% of BANZEL-treated patients compared to 8% of patients on placebo. It led to study discontinuation in 1% of BANZEL-treated patients and 0% of patients on placebo.
Dizziness was reported in 2.7% of BANZEL-treated patients compared to 0% of patients on placebo, and did not lead to study discontinuation.
Ataxia and gait disturbance were reported in 5.4% and 1.4% of BANZEL-treated patients, respectively, compared to no patient on placebo. None of these reactions led to study discontinuation.
Accordingly, patients should be advised not to drive or operate machinery until they have gained sufficient experience on BANZEL to gauge whether it adversely affects their ability to drive or operate machinery.
5.3 QT Shortening
Formal cardiac ECG studies demonstrated shortening of the QT interval (mean = 20 msec, for doses >2400 mg twice daily) with BANZEL. In a placebo-controlled study of the QT interval, a higher percentage of BANZEL-treated subjects (46% at 2400 mg, 46% at 3200 mg, and 65% at 4800 mg) had a QT shortening of greater than 20 msec at Tmax compared to placebo (5-10%).
Reductions of the QT interval below 300 msec were not observed in the formal QT studies with doses up to 7200 mg per day. Moreover, there was no signal for drug-induced sudden death or ventricular arrhythmias.
The degree of QT shortening induced by BANZEL is without any known clinical risk. Familial Short QT syndrome is associated with an increased risk of sudden death and ventricular arrhythmias, particularly ventricular fibrillation. Such events in this syndrome are believed to occur primarily when the corrected QT interval falls below 300 msec. Non-clinical data also indicate that QT shortening is associated with ventricular fibrillation.
Patients with Familial Short QT syndrome should not be treated with BANZEL. Caution should be used when administering BANZEL with other drugs that shorten the QT interval [see Contraindications (4)].
5.4 Multi-organ Hypersensitivity/Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as multi-organ hypersensitivity, has been reported in patients taking antiepileptic drugs, including BANZEL. DRESS may be fatal or life-threatening. DRESS typically, although not exclusively, presents with fever, rash, and/or lymphadenopathy, and/or facial swelling, in association with other organ system involvement, such as hepatitis, nephritis, hematological abnormalities, myocarditis, or myositis, sometimes resembling an acute viral infection. Eosinophilia is often present. It is important to note that early manifestations of hypersensitivity, such as fever or lymphadenopathy, may be present even though rash is not evident. Because this disorder is variable in its expression, other organ systems not noted here may be involved.
All cases of DRESS identified in clinical trials with BANZEL occurred in pediatric patients less than 12 years of age, occurred within 4 weeks of treatment initiation, and resolved or improved with BANZEL discontinuation. DRESS has also been reported in adult and pediatric patients taking BANZEL in the postmarketing setting.
If DRESS is suspected, the patient should be evaluated immediately, BANZEL should be discontinued, and alternative treatment should be started.
5.5 Withdrawal of AEDs
As with all antiepileptic drugs, BANZEL should be withdrawn gradually to minimize the risk of precipitating seizures, seizure exacerbation, or status epilepticus. If abrupt discontinuation of the drug is medically necessary, the transition to another AED should be made under close medical supervision. In clinical trials, BANZEL discontinuation was achieved by reducing the dose by approximately 25% every 2 days.
5.6 Status Epilepticus
Estimates of the incidence of treatment emergent status epilepticus among patients treated with BANZEL are difficult because standard definitions were not employed. In a controlled Lennox-Gastaut Syndrome trial, 3 of 74 (4.1%) BANZEL-treated patients had episodes that could be described as status epilepticus in the BANZEL-treated patients compared with none of the 64 patients in the placebo-treated patients. In all controlled trials that included patients with different epilepsies, 11 of 1240 (0.9%) BANZEL-treated patients had episodes that could be described as status epilepticus compared with none of 635 patients in the placebo-treated patients.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described below and elsewhere in the labeling:
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.1)]
- Central Nervous System Reactions [see Warnings and Precautions (5.2)]
- QT Shortening [see Warnings and Precautions (5.3)]
- Multi-Organ Hypersensitivity/Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) [see Warnings and Precautions (5.4)]
- Leukopenia [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Adult and Pediatric Patients ages 3 to 17 years of age
In the pooled, double-blind, adjunctive therapy studies in adult and pediatric patients ages 3 to 17 years of age, the most common (≥10%) adverse reactions in BANZEL-treated patients, in all doses studied (200 to 3200 mg per day) with a higher frequency than in patients on placebo were: headache, dizziness, fatigue, somnolence, and nausea.
Table 2 lists adverse reactions that occurred in at least 3% of pediatric patients (ages 3 to less than 17 years) with epilepsy treated with BANZEL in controlled adjunctive studies and were numerically more common in patients treated with BANZEL than in patients on placebo.
At the target dose of 45 mg/kg per day for adjunctive therapy in pediatric patients (ages 3 to less than 17 years), the most common (≥3%) adverse reactions with an incidence greater than in placebo for BANZEL were somnolence, vomiting, and headache.
| Adverse Reaction | BANZEL
(N=187) % | Placebo
(N=182) % |
| Somnolence | 17 | 9 |
| Vomiting | 17 | 7 |
| Headache | 16 | 8 |
| Fatigue | 9 | 8 |
| Dizziness | 8 | 6 |
| Nausea | 7 | 3 |
| Influenza | 5 | 4 |
| Nasopharyngitis | 5 | 3 |
| Decreased Appetite | 5 | 2 |
| Rash | 4 | 2 |
| Ataxia | 4 | 1 |
| Diplopia | 4 | 1 |
| Bronchitis | 3 | 2 |
| Sinusitis | 3 | 2 |
| Psychomotor Hyperactivity | 3 | 1 |
| Upper Abdominal Pain | 3 | 2 |
| Aggression | 3 | 2 |
| Ear Infection | 3 | 1 |
| Disturbance in Attention | 3 | 1 |
| Pruritis | 3 | 0 |
Table 3 lists adverse reactions that occurred in at least 3% of adult patients with epilepsy treated with BANZEL (up to 3200 mg per day) in adjunctive controlled studies and were numerically more common in patients treated with BANZEL than in patients on placebo. In these studies, either BANZEL or placebo was added to the current AED therapy.
At all doses studied of up to 3200 mg per day given as adjunctive therapy in adults, the most common (≥ 3%) adverse reactions, and with the greatest increase in incidence compared to placebo, for BANZEL were dizziness, fatigue, nausea, diplopia, vision blurred, and ataxia.
| Adverse Reaction | BANZEL
(N=823) % | Placebo
(N=376) % |
| Headache | 27 | 26 |
| Dizziness | 19 | 12 |
| Fatigue | 16 | 10 |
| Nausea | 12 | 9 |
| Somnolence | 11 | 9 |
| Diplopia | 9 | 3 |
| Tremor | 6 | 5 |
| Nystagmus | 6 | 5 |
| Blurred Vision | 6 | 2 |
| Vomiting | 5 | 4 |
| Ataxia | 4 | 0 |
| Upper Abdominal Pain | 3 | 2 |
| Anxiety | 3 | 2 |
| Constipation | 3 | 2 |
| Dyspepsia | 3 | 2 |
| Back Pain | 3 | 1 |
| Gait Disturbance | 3 | 1 |
| Vertigo | 3 | 1 |
Discontinuation in Controlled Clinical Studies
In controlled, double-blind, adjunctive clinical studies, 9% of pediatric and adult patients receiving BANZEL as adjunctive therapy and 4% receiving placebo discontinued as a result of an adverse reaction. The adverse reactions most commonly leading to discontinuation of BANZEL (>1%) used as adjunctive therapy were generally similar in adults and pediatric patients.
In pediatric patients (ages 4 to less than 17 years) double-blind adjunctive clinical studies, 8% of patients receiving BANZEL as adjunctive therapy (at the recommended dose of 45 mg/kg per day) and 2% receiving placebo discontinued as a result of an adverse reaction. The adverse reactions most commonly leading to discontinuation of BANZEL (>1%) used as adjunctive therapy are presented in Table 4.
| Adverse Reaction | BANZEL
(N=187) % | Placebo
(N=182) % |
| Convulsion | 2 | 1 |
| Rash | 2 | 1 |
| Fatigue | 2 | 0 |
| Vomiting | 1 | 0 |
In adult double-blind, adjunctive clinical studies, 10% of patients receiving BANZEL as adjunctive therapy (at doses up to 3200 mg per day) and 6% receiving placebo discontinued as a result of an adverse reaction. The adverse reactions most commonly leading to discontinuation of BANZEL (>1%) used as adjunctive therapy are presented in Table 5.
| Adverse Reaction | BANZEL
(N=823) % | Placebo
(N=376) % |
| Dizziness | 3 | 1 |
| Fatigue | 2 | 1 |
| Headache | 2 | 1 |
| Nausea | 1 | 0 |
| Ataxia | 1 | 0 |
Pediatric Patients ages 1 to less than 4 years
In a multicenter, parallel group, open-label study comparing BANZEL (45 mg/kg per day) adjunctive treatment (n=25) to the adjunctive treatment with an AED of the investigator’s choice (n=11) in pediatric patients (1 year to less than 4 years of age) with inadequately controlled Lennox-Gastaut Syndrome, the adverse reaction profile was generally similar to that observed in adults and pediatric patients 4 years of age and older treated with BANZEL. Adverse reactions that occurred in at least 2 (8%) BANZEL-treated patients and with a higher frequency than in the AED comparator group were: vomiting (24%), somnolence (16%), bronchitis (12%), constipation (12%), cough (12%), decreased appetite (12%), rash (12%), otitis media (8%), pneumonia (8%), decreased weight (8%), gastroenteritis (8%), nasal congestion (8%), and pneumonia aspiration (8%).
Other Adverse Reactions Observed During Clinical Trials
BANZEL has been administered to 1978 individuals during all epilepsy clinical trials (placebo-controlled and open-label). Adverse reactions occurring during these studies were recorded by the investigators using terminology of their own choosing. To provide a meaningful estimate of the proportion of patients having adverse reactions, these events were grouped into standardized categories using the MedDRA dictionary. Adverse events occurring at least three times and considered possibly related to treatment are included in the System Organ Class listings below. Terms not included in the listings are those already included in the tables above, those too general to be informative, those related to procedures, and terms describing events common in the population. Some events occurring fewer than 3 times are also included based on their medical significance. Because the reports include events observed in open-label, uncontrolled observations, the role of BANZEL in their causation cannot be reliably determined.
Events are classified by body system and listed in order of decreasing frequency as follows: frequent adverse events—those occurring in at least 1/100 patients; infrequent adverse events—those occurring in 1/100 to 1/1000 patients; rare—those occurring in fewer than 1/1000 patients.
Blood and Lymphatic System Disorders: Frequent: anemia. Infrequent: lymphadenopathy, leukopenia, neutropenia, iron deficiency anemia, thrombocytopenia.
Cardiac Disorders: Infrequent: bundle branch block right, atrioventricular block first degree.
Metabolic and Nutritional Disorders: Frequent: decreased appetite, increased appetite.
Renal and Urinary Disorders: Frequent: pollakiuria. Infrequent: urinary incontinence, dysuria, hematuria, nephrolithiasis, polyuria, enuresis, nocturia, incontinence.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of BANZEL. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Dermatologic: Stevens-Johnson syndrome and other serious skin rashes with mucosal involvement.
7. Drug Interactions
7.1 Effects of BANZEL on other AEDs
Population pharmacokinetic analysis of average concentration at steady state of carbamazepine, lamotrigine, phenobarbital, phenytoin, topiramate, and valproate showed that typical rufinamide Cavss levels had little effect on the pharmacokinetics of other AEDs. Any effects, when they occur, have been more marked in the pediatric population.
Table 6 summarizes the drug-drug interactions of BANZEL with other AEDs.
| AED
Co-administered | Influence of Rufinamide on AED concentrationa) | Influence of AED on Rufinamide concentration |
| Carbamazepine | Decrease by 7 to 13%b) | Decrease by 19 to 26% Dependent on dose of carbamazepine |
| Lamotrigine | Decrease by 7 to 13%b) | No Effect |
| Phenobarbital | Increase by 8 to 13%b) | Decrease by 25 to 46%c)’ d)
Independent of dose or concentration of phenobarbital |
| Phenytoin | Increase by 7 to 21%b) | Decrease by 25 to 46%c)’ d)
Independent of dose or concentration of phenytoin |
| Topiramate | No Effect | No Effect |
| Valproate | No Effect | Increase by <16 to 70%c)
Dependent on concentration of valproate |
| Primidone | Not Investigated | Decrease by 25 to 46%c)’ d)
Independent of dose or concentration of primidone |
| Benzodiazepines e) | Not Investigated | No Effect |
| a) Predictions are based on BANZEL concentrations at the maximum recommended dose of BANZEL. b) Maximum changes predicted to be in pediatric patients and in adult patients who achieve significantly higher levels of BANZEL, as the effect of rufinamide on these AEDs is concentration-dependent. c) Larger effects in pediatric patients at high doses/concentrations of AEDs. d) Phenobarbital, primidone and phenytoin were treated as a single covariate (phenobarbital-type inducers) to examine the effect of these agents on BANZEL clearance. e) All compounds of the benzodiazepine class were pooled to examine for ‘class effect’ on BANZEL clearance. |
||
Phenytoin: The decrease in clearance of phenytoin estimated at typical levels of rufinamide (Cavss 15 μg/mL) is predicted to increase plasma levels of phenytoin by 7 to 21%. As phenytoin is known to have non-linear pharmacokinetics (clearance becomes saturated at higher doses), it is possible that exposure will be greater than the model prediction.
7.2 Effects of Other AEDs on BANZEL
Potent cytochrome P450 enzyme inducers, such as carbamazepine, phenytoin, primidone, and phenobarbital, appear to increase the clearance of BANZEL (see Table 6). Given that the majority of clearance of BANZEL is via a non-CYP-dependent route, the observed decreases in blood levels seen with carbamazepine, phenytoin, phenobarbital, and primidone are unlikely to be entirely attributable to induction of a P450 enzyme. Other factors explaining this interaction are not understood. Any effects, where they occurred, were likely to be more marked in the pediatric population.
Valproate
Patients stabilized on BANZEL before being prescribed valproate should begin valproate therapy at a low dose, and titrate to a clinically effective dose. Similarly, patients on valproate should begin at a BANZEL dose lower than 10 mg/kg per day (pediatric patients) or 400 mg per day (adults) [see Dosage and Administration (2.5), Clinical Pharmacology (12.3)].
7.3 Effects of BANZEL on Hormonal Contraceptives
Female patients of childbearing age should be warned that the concurrent use of BANZEL with hormonal contraceptives may render this method of contraception less effective. Additional non-hormonal forms of contraception are recommended when using BANZEL [see Use in Specific Populations (8.3), Clinical Pharmacology (12.3) and Patient Counseling Information (17)].
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to AEDs, such as BANZEL, during pregnancy. Encourage women who are taking BANZEL during pregnancy to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry by calling 1-888-233-2334 or visiting http://www.aedpregnancyregistry.org
Risk Summary
There are no adequate data on the developmental risks associated with use of BANZEL in pregnant women. In animal reproduction studies, oral administration of rufinamide resulted in developmental toxicity in pregnant rats and rabbits at clinically relevant doses [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal data
Oral administration of rufinamide (0, 20, 100, or 300 mg/kg/day) to pregnant rats throughout organogenesis resulted in decreased fetal weight and increased incidence of fetal skeletal abnormalities at 100 and 300 mg/kg/day, which were associated with maternal toxicity. The maternal plasma exposure (AUC) at the no-adverse effect dose (20 mg/kg/day) for developmental toxicity was less than that in humans at the maximum recommended human dose (MRHD) of 3200 mg/day.
Oral administration of rufinamide (0, 30, 200, or 1000 mg/kg/day) to pregnant rabbits throughout organogenesis resulted in embryofetal death, decreased fetal body weight, and increased incidence of fetal visceral and skeletal abnormalities at doses of 200 and 1000 mg/kg/day. The high dose (1000 mg/kg/day) was associated with abortion. Plasma exposure (AUC) at the no-adverse effect dose (30 mg/kg/day) was less than that in humans at the MRHD.
When rufinamide was orally administered (0, 5, 30, or 150 mg/kg/day) to pregnant rats throughout pregnancy and lactation, decreased offspring growth and survival were observed at all doses tested. A no-effect dose for adverse effects on pre- and postnatal development was not established. At the lowest dose tested (5 mg/kg/day), plasma exposure (AUC) was less than that in humans at the MRHD.
8.2 Lactation
Risk Summary
There are no data on the presence of rufinamide in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for BANZEL and any potential adverse effects on the breastfed infant from BANZEL or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Use of BANZEL may reduce the effectiveness of hormonal contraceptives containing ethinyl estradiol or norethindrone. Advise women of reproductive potential taking BANZEL who are using a contraceptive containing ethinyl estradiol and norethindrone to use an additional non-hormonal form of contraception [see Drug Interactions (7.3) and Clinical Pharmacology (12.3)].
Infertility
The effect of rufinamide on fertility in humans has not been established. Oral administration of rufinamide (20, 60, 200, and 600 mg/kg/day) to male and female rats prior to mating, during mating, and during early gestation (females only) resulted in the impairment of fertility at all dose levels tested. The no-effect dose was not established. The plasma exposure level at 20 mg/kg was approximately 0.2 times the human plasma AUC at the MRHD [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness have been established in pediatric patients 1 to 17 years of age. The effectiveness of BANZEL in pediatric patients 4 years of age and older was based upon an adequate and well-controlled trial of BANZEL that included both adults and pediatric patients, 4 years of age and older, with Lennox-Gastaut Syndrome. The effectiveness in patients 1 to less than 4 years was based upon a bridging pharmacokinetic and safety study [see Dosage and Administration (2.1), Adverse Reactions (6.1), and Clinical Studies (14)]. The pharmacokinetics of rufinamide in the pediatric patients, ages 1 to less than 4 years of age is similar to children older than 4 years of age and adults [see Clinical Pharmacology (12.3)].
Safety and effectiveness in pediatric patients below the age of 1 year has not been established.
Oral administration of rufinamide (0, 15, 50, or 150 mg/kg) to young rats for 10 weeks starting on postnatal day 7 resulted in decreased brain weights at the mid and high doses and neurobehavioral impairment (learning and memory deficit, altered startle response, decreased locomotor activity) and decreased growth (decreased body weight) at the highest dose tested. The no-effect dose for adverse effects on postnatal development in rats (15 mg/kg) was associated with a plasma exposure (AUC) lower than that in humans at the maximum recommended human dose (MRHD) of 3200 mg/day.
8.5 Geriatric Use
Clinical studies of BANZEL did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Pharmacokinetics of rufinamide in the elderly are similar to that in the young subjects [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Rufinamide pharmacokinetics in patients with severe renal impairment (creatinine clearance < 30 mL/min) was similar to that of healthy subjects. Dose adjustment in patients undergoing dialysis should be considered [see Clinical Pharmacology (12.3)].
10. Overdosage
Because strategies for the management of overdose are continually evolving, it is advisable to contact a Certified Poison Control Center to determine the latest recommendations for the management of an overdose of any drug.
One overdose of 7200 mg per day BANZEL was reported in an adult during the clinical trials. The overdose was associated with no major signs or symptoms, no medical intervention was required, and the patient continued in the study at the target dose.
Treatment or Management of Overdose: There is no specific antidote for overdose with BANZEL. If clinically indicated, elimination of unabsorbed drug should be attempted by induction of emesis or gastric lavage. Usual precautions should be observed to maintain the airway. General supportive care of the patient is indicated including monitoring of vital signs and observation of the clinical status of the patient.
Hemodialysis: Standard hemodialysis procedures may result in limited clearance of rufinamide. Although there is no experience to date in treating overdose with hemodialysis, the procedure may be considered when indicated by the patient’s clinical state.
11. Banzel Description
BANZEL (rufinamide) is a triazole derivative structurally unrelated to currently marketed antiepileptic drugs (AEDs). Rufinamide has the chemical name 1-[(2,6-difluorophenyl)methyl]-1H-1,2,3-triazole-4 carboxamide. It has an empirical formula of C10H8F2N4O and a molecular weight of 238.2. The drug substance is a white, crystalline, odorless, and slightly bitter tasting neutral powder. Rufinamide is practically insoluble in water, slightly soluble in tetrahydrofuran and in methanol, and very slightly soluble in ethanol and in acetonitrile.
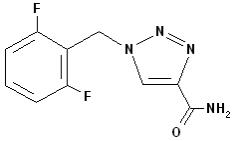
BANZEL is available for oral administration in film-coated tablets, scored on both sides, containing 200 and 400 mg of rufinamide. Inactive ingredients are colloidal silicon dioxide, corn starch crosscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulphate. The film coating contains hypromellose, iron oxide red, polyethylene glycol, talc, and titanium dioxide.
BANZEL is also available for oral administration as a liquid containing rufinamide at a concentration of 40 mg/mL. Inactive ingredients include microcrystalline cellulose and carboxymethylcellulose sodium, hydroxyethylcellulose, anhydrous citric acid, simethicone emulsion 30%, poloxamer 188, methylparaben, propylparaben, propylene glycol, potassium sorbate, noncrystallizing sorbitol solution 70%, and an orange flavor.
12. Banzel - Clinical Pharmacology
12.1 Mechanism of Action
The precise mechanism(s) by which rufinamide exerts its antiepileptic effect is unknown.
The results of in vitro studies suggest that the principal mechanism of action of rufinamide is modulation of the activity of sodium channels and, in particular, prolongation of the inactive state of the channel. Rufinamide (≥ 1 μM) significantly slowed sodium channel recovery from inactivation after a prolonged prepulse in cultured cortical neurons, and limited sustained repetitive firing of sodium-dependent action potentials (EC50 of 3.8 μM).
12.3 Pharmacokinetics
Overview
BANZEL oral suspension is bioequivalent on a mg per mg basis to BANZEL tablets. BANZEL is well absorbed after oral administration. However, the rate of absorption is relatively slow and the extent of absorption is decreased as dose is increased. The pharmacokinetics does not change with multiple dosing. Most elimination of rufinamide is via metabolism, with the primary metabolite resulting from enzymatic hydrolysis of the carboxamide moiety to form the carboxylic acid. This metabolic route is not cytochrome P450 dependent. There are no known active metabolites. Plasma half-life of rufinamide is approximately 6-10 hours.
Absorption and Distribution
Following oral administration of BANZEL, peak plasma concentrations occur between 4 and 6 hours (Tmax) both under fed and fasted conditions. BANZEL tablets display decreasing bioavailability with increasing dose after single and multiple dose administration. Based on urinary excretion, the extent of absorption was at least 85% following oral administration of a single dose of 600 mg rufinamide tablet under fed conditions.
Multiple dose pharmacokinetics can be predicted from single dose data for both rufinamide and its metabolite. Given the dosing frequency of every 12 hours and the half-life of 6 to 10 hours, the observed steady-state peak concentration of about two to three times the peak concentration after a single dose is expected.
Food increased the extent of absorption of rufinamide in healthy volunteers by 34% and increased peak exposure by 56% after a single dose of 400 mg tablet, although the Tmax was not elevated [see Dosage and Administration (2.2)].
Only a small fraction of rufinamide (34%) is bound to human serum proteins, predominantly to albumin (27%), giving little risk of displacement drug-drug interactions. Rufinamide was evenly distributed between erythrocytes and plasma. The apparent volume of distribution is dependent upon dose and varies with body surface area. The apparent volume of distribution was about 50 L at 3200 mg per day.
Metabolism
Rufinamide is extensively metabolized but has no active metabolites. Following a radiolabeled dose of rufinamide, less than 2% of the dose was recovered unchanged in urine. The primary biotransformation pathway is carboxylesterase(s) mediated hydrolysis of the carboxamide group to the acid derivative CGP 47292. A few minor additional metabolites were detected in urine, which appeared to be acyl-glucuronides of CGP 47292. There is no involvement of oxidizing cytochrome P450 enzymes or glutathione in the biotransformation process.
Rufinamide is a weak inhibitor of CYP 2E1. It did not show significant inhibition of other CYP enzymes. Rufinamide is a weak inducer of CYP 3A4 enzymes.
Rufinamide did not show any significant inhibition of P-glycoprotein in an in vitro study.
Elimination/Excretion
Renal excretion is the predominant route of elimination for drug related material, accounting for 85% of the dose based on a radiolabeled study. Of the metabolites identified in urine, at least 66% of the rufinamide dose was excreted as the acid metabolite CGP 47292, with 2% of the dose excreted as rufinamide.
The plasma elimination half-life is approximately 6-10 hours in healthy subjects and patients with epilepsy.
Special Populations
Age
- Pediatrics
Based on a population analysis which included a total of 115 patients, including 85 pediatric patients (24 patients ages 1 to 3 years, 40 patients ages 4 to 11 years, and 21 patients ages 12 to 17 years), the pharmacokinetics of rufinamide was similar across all age groups.
- Elderly
The results of a study evaluating single-dose (400 mg) and multiple dose (800 mg per day for 6 days) pharmacokinetics of rufinamide in 8 healthy elderly subjects (65-80 years old) and 7 younger healthy subjects (18-45 years old) found no significant age-related differences in the pharmacokinetics of rufinamide.
Sex
Population pharmacokinetic analyses of females show a 6-14% lower apparent clearance of rufinamide compared to males. This effect is not clinically important.
Race
In a population pharmacokinetic analysis of clinical studies, no difference in clearance or volume of distribution of rufinamide was observed between the black and Caucasian subjects, after controlling for body size. Information on other races could not be obtained because of smaller numbers of these subjects.
Renal Impairment
Rufinamide pharmacokinetics in 9 patients with severe renal impairment (creatinine clearance < 30 mL per min) was similar to that of healthy subjects. Patients undergoing dialysis 3 hours post rufinamide dosing showed a reduction in AUC and Cmax by 29% and 16%, respectively.
Drug Interactions
Based on in vitro studies, rufinamide shows little or no inhibition of most cytochrome P450 enzymes at clinically relevant concentrations, with weak inhibition of CYP 2E1. Drugs that are substrates of CYP 2E1 (e.g., chlorzoxazone) may have increased plasma levels in the presence of rufinamide, but this has not been studied.
Based on a population pharmacokinetic analysis, rufinamide clearance was decreased by valproate. In pediatric patients, valproate administration may lead to elevated levels of rufinamide by up to 70% [see Drug Interactions (7.2)].
Based on in vivo drug interaction studies with triazolam and oral contraceptives, rufinamide is a weak inducer of the CYP 3A4 enzyme and can decrease exposure of drugs that are substrates of CYP 3A4.
- Co-administration and pre-treatment of BANZEL (400 mg twice daily) and triazolam resulted in a 37% decrease in AUC and a 23% decrease in Cmax of triazolam, a CYP 3A4 substrate.
- Co-administration of BANZEL (800 mg twice daily for 14 days) and Ortho-Novum 1/35® resulted in a mean decrease in the ethinyl estradiol AUC0-24 of 22% and Cmax by 31% and norethindrone AUC0-24 by 14% and Cmax by 18%, respectively. The clinical significance of this decrease is unknown [see Drug Interactions (7.3) and Use in Specific Populations (8.3)].
Rufinamide is metabolized by carboxylesterases. Drugs that may induce the activity of carboxylesterases may increase the clearance of rufinamide. Broad-spectrum inducers such as carbamazepine and phenobarbital may have minor effects on rufinamide metabolism via this mechanism. Drugs that are inhibitors of carboxylesterases may decrease metabolism of rufinamide.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Rufinamide was given in the diet to mice at 40, 120, and 400 mg/kg per day and to rats at 20, 60, and 200 mg/kg per day for 2 years. The doses in mice were associated with plasma AUCs 0.1 to 1 times the human plasma AUC at the maximum recommended human dose (MRHD, 3200 mg/day). Increased incidences of tumors (benign bone tumors (osteomas) and/or hepatocellular adenomas and carcinomas) were observed in mice at all doses. Increased incidences of thyroid follicular adenomas were observed in rats at all but the low dose; the low dose is < 0.1 times the MRHD on a mg/m2 basis.
Mutagenesis
Rufinamide was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay or the in vitro mammalian cell point mutation assay. Rufinamide was not clastogenic in the in vitro mammalian cell chromosomal aberration assay or the in vivo rat bone marrow micronucleus assay.
Impairment of Fertility
Oral administration of rufinamide (doses of 20, 60, 200, and 600 mg/kg per day) to male and female rats prior to mating and throughout mating, and continuing in females up to day 6 of gestation resulted in impairment of fertility (decreased conception rates and mating and fertility indices; decreased numbers of corpora lutea, implantations, and live embryos; increased preimplantation loss; decreased sperm count and motility) at all doses tested. Therefore, a no-effect dose was not established. The lowest dose tested was associated with a plasma AUC ≈ 0.2 times the human plasma AUC at the MRHD.
14. Clinical Studies
Adult and Pediatric Patients ages 4 years and older
The effectiveness of BANZEL as adjunctive treatment for the seizures associated with Lennox-Gastaut Syndrome (LGS) in adult and pediatric patients ages 4 years and older was established in a single multicenter, double-blind, placebo-controlled, randomized, parallel-group study (N=138). Male and female patients (between 4 and 30 years of age) were included if they had a diagnosis of inadequately controlled seizures associated with LGS (including both atypical absence seizures and drop attacks) and were being treated with 1 to 3 concomitant stable dose AEDs. Each patient must have had at least 90 seizures in the month prior to study entry. After completing a 4-week Baseline Phase on stable therapy, patients were randomized to have BANZEL or placebo added to their ongoing therapy during the 12 -week Double-blind Phase. The Double-blind Phase consisted of 2 periods: the Titration Period (1 to 2 weeks) and the Maintenance Period (10 weeks). During the Titration Period, the dose was increased to a target dosage of approximately 45 mg/kg per day (3200 mg in adults of > 70 kg), given on a twice daily schedule. Dosage reductions were permitted during titration if problems in tolerability were encountered. Final doses at titration were to remain stable during the maintenance period. Target dosage was achieved in 88% of the BANZEL-treated patients. The majority of these patients reached the target dose within 7 days, with the remaining patients achieving the target dose within 14 days.
The primary efficacy variables were:
- The percent change in total seizure frequency per 28 days;
- The percent change in tonic-atonic (drop attacks) seizure frequency per 28 days;
- Seizure severity from the Parent/Guardian Global Evaluation of the patient’s condition. This was a 7-point assessment performed at the end of the Double-blind Phase. A score of +3 indicated that the patient’s seizure severity was very much improved, a score of 0 that the seizure severity was unchanged, and a score of -3 that the seizure severity was very much worse.
The results of the three primary endpoints are shown in Table 7 below.
| Variable | Placebo | Rufinamide |
| Median percent change in total seizure frequency per 28 days | -11.7 | -32.7 (p=0.0015) |
| Median percent change in tonic-atonic seizure frequency per 28 days | 1.4 | -42.5 (p<0.0001) |
| Improvement in Seizure Severity Rating from Global Evaluation | 30.6 | 53.4 (p=0.0041) |
Pediatric Patients ages 1 to less than 4 years
The effectiveness of BANZEL as adjunctive treatment for the seizures associated with Lennox-Gastaut Syndrome in pediatric patients ages 1 year to less than 4 years was established based on a single multi-center, open-label, active-controlled, randomized, pharmacokinetic bridging study. The pharmacokinetic profile of BANZEL is not significantly affected by age either as a continuous covariate (1 to 35 years) or as a categorical covariate (age categories: 1 to less than 4 years and 4 years of age and older), after body weight is taken into consideration.
16. How is Banzel supplied
16.1 How Supplied
BANZEL 200 mg tablets (containing 200 mg rufinamide) are pink in color, film-coated, oblong-shape tablets, with a score on both sides, imprinted with “ 262” on one side. They are available in bottles of 120 (NDC 62856-582-52).
262” on one side. They are available in bottles of 120 (NDC 62856-582-52).
BANZEL 400 mg tablets (containing 400 mg rufinamide) are pink in color, film-coated, oblong-shape tablets, with a score on both sides, imprinted with “ 263” on one side. They are available in bottles of 120 (NDC 62856-583-52).
BANZEL oral suspension is an orange flavored liquid supplied in a polyethylene terephthalate (PET) bottle with child-resistant closure. The oral suspension is packaged with a dispenser set which contains a calibrated oral dosing syringe and an adapter. Store the oral suspension in an upright position. Use within 90 days of first opening the bottle, then discard any remainder. The oral suspension is available in bottles of 460 mL (NDC 62856-584-46).
16.2 Storage and Handling
Store the tablets at 25ºC (77ºF); excursions permitted to 15º- 30ºC (59ºF - 86ºF). Protect from moisture. Replace cap securely after opening.
Store the oral suspension at 25ºC (77ºF); excursions permitted to 15º- 30ºC (59ºF - 86ºF). Replace cap securely after opening. The cap fits properly in place when the adapter is in place.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Administration Information
- Advise patients to take BANZEL with food [see Dosage and Administration (2.2)].
- Advise patients who are prescribed the oral suspension to shake the bottle vigorously before every administration and to use the adaptor and oral dosing syringe [see Dosage and Administration (2.2)].
Suicidal Thinking and Behavior
Inform patients, their caregivers, and families that antiepileptic drugs increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers [see Warnings and Precautions (5.1)].
Central Nervous System Reactions
Inform patients about the potential for somnolence or dizziness and advise them not to drive or operate machinery until they have gained sufficient experience on BANZEL to gauge whether it adversely affects their mental and/or motor performance [see Warnings and Precautions (5.2)].
Multi-Organ Hypersensitivity Reactions
Advise patients to notify their physician if they experience a rash associated with fever [see Warnings and Precautions (5.4)].
Drug Interactions
- Inform female patients of childbearing age that the concurrent use of BANZEL with hormonal contraceptives may render this method of contraception less effective. Recommend patients use additional non-hormonal forms of contraception when using BANZEL [see Drug Interactions (7.3) and Use in Specific Populations (8.3)].
- Inform patients that alcohol in combination with BANZEL may cause additive central nervous system effects.
Pregnancy
Advise patients to notify their physician if they become pregnant or intend to become pregnant during therapy. Encourage patients to enroll in the North American Antiepileptic Drug Pregnancy Registry if they become pregnant. To enroll, patients can call the toll free number 1-888-233-2334 [see Use in Specific Populations (8.1)].
Breast-feeding
Advise patients to notify their physician if they are breast-feeding or intend to breast-feed [see Use in Specific Populations (8.2)].
Medication Guide
Medication Guide
BANZEL (ban-‘zel)
[rufinamide]
Tablets and Oral Suspension
Read this Medication Guide before you start taking BANZEL and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment.
What is the most important information I should know about BANZEL?
Do not stop taking BANZEL without first talking to your healthcare provider.
Stopping BANZEL suddenly can cause serious problems.
BANZEL can cause serious side effects, including:
1. Like other antiepileptic drugs, BANZEL may cause suicidal thoughts or actions in a very small number of people, about 1 in 500.
Call a healthcare provider right away if you have any of these symptoms, especially if they are new, worse, or worry you:
- thoughts about suicide or dying
- attempt to commit suicide
- new or worse depression
- new or worse anxiety
- feeling agitated or restless
- panic attacks
- trouble sleeping (insomnia)
- new or worse irritability
- acting aggressive, being angry, or violent
- acting on dangerous impulses
- an extreme increase in activity and talking (mania)
- other unusual changes in behavior or mood
- Suicidal thoughts or actions can be caused by things other than medicines. If you have suicidal thoughts or actions, your healthcare provider may check for other causes.
How can I watch for early symptoms of suicidal thoughts and actions?
- Pay attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings.
- Keep all follow-up visits with your healthcare provider as scheduled.
Call your healthcare provider between visits as needed, especially if you are worried about symptoms.
Do not stop BANZEL without first talking to a healthcare provider.
- Stopping BANZEL suddenly can cause serious problems. Stopping a seizure medicine suddenly in a patient who has epilepsy can cause seizures that will not stop (status epilepticus).
2. BANZEL may cause you to feel sleepy, tired, weak, dizzy, or have problems with coordination and walking.
What is BANZEL?
BANZEL is a prescription medicine used with other medicines to treat seizures associated with Lennox-Gastaut Syndrome (LGS) in adults and pediatric patients 1 year of age and older.
It is not known if BANZEL is safe and effective in the treatment of Lennox-Gastaut Syndrome in pediatric patients under 1 year of age.
Who should not take BANZEL?
Do not take BANZEL if you have a genetic condition called familial short QT syndrome, a problem that affects the electrical system of the heart.
What should I tell my healthcare provider before taking BANZEL?
Before you take BANZEL, tell your healthcare provider if you:
- have heart problems
- have liver problems
- have any other medical problems
- have or have had suicidal thoughts or actions, depression or mood problems
- are pregnant or plan to become pregnant. It is not known if BANZEL can harm your unborn baby. Tell your healthcare provider right away if you become pregnant while taking BANZEL. You and your healthcare provider will decide if you should take BANZEL while you are pregnant.
- BANZEL may make certain types of birth control less effective. Talk to your healthcare provider about the best birth control methods for you while you take BANZEL.
○ If you become pregnant while taking BANZEL, talk to your healthcare provider about registering with the North American Antiepileptic Drug Pregnancy Registry. You can enroll in this registry by calling 1-888-233-2334. The purpose of this registry is to collect information about the safety of antiepileptic medicines during pregnancy.
- are breastfeeding or plan to breastfeed. It is not known if BANZEL will pass into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you take BANZEL.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
Taking BANZEL with certain other medicines can cause side effects or affect how well they work. Do not start or stop other medicines without talking to your healthcare provider.
Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist each time you get a new medicine.
How should I take BANZEL?
- Take BANZEL exactly as your healthcare provider tells you. Your healthcare provider will tell you how much BANZEL to take.
- Your healthcare provider may change your dose. Do not change your dose of BANZEL without talking to your healthcare provider.
- Take BANZEL with food.
- BANZEL tablets can be swallowed whole, cut in half or crushed.
- If you take BANZEL Oral Suspension instead of BANZEL tablets, shake the bottle well before you take each dose. Measure your dose of BANZEL Oral Suspension using the bottle adapter and dosing syringes provided.
See the complete Instructions for Use below for information on how to use the dosing syringes and measure your dose of BANZEL Oral Suspension.
- If you take too much BANZEL, call your local Poison Control Center or get emergency medical help right away.
What should I avoid while taking BANZEL?
- Do not drink alcohol or take other medicines that make you sleepy or dizzy while taking BANZEL until you talk to your healthcare provider. BANZEL taken with alcohol or medicines that cause sleepiness or dizziness may make your sleepiness or dizziness worse.
- Do not drive, operate heavy machinery, or do other dangerous activities until you know how BANZEL affects you. BANZEL can slow your thinking and motor skills.
What are the possible side effects of BANZEL?
See “What is the most important information I should know about BANZEL?”
BANZEL may cause serious side effects including:
- BANZEL can also cause allergic reactions or serious problems which may affect organs and other parts of your body like the liver or blood cells. You may or may not have a rash with these types of reactions.
Call your healthcare provider right away if you have any of the following. Symptoms may include:
- swelling of your face, eyes, lips, or tongue
- trouble swallowing or breathing
- a skin rash
- hives
- fever, swollen glands, or sore throat that do not go away or come and go
- swollen glands
- yellowing of your skin or eyes
- dark urine
- unusual bruising or bleeding
- severe fatigue or weakness
- severe muscle pain
- your seizures happen more often or become worse
Call your healthcare provider right away if you have any of the symptoms listed above.
The most common side effects of BANZEL include:
- headache
- dizziness
- tiredness
- sleepiness
- nausea
- vomiting
Tell your healthcare provider about any side effect that bothers you or that does not go away. These are not all of the possible side effects of BANZEL. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store BANZEL?
- Store BANZEL tablets and oral suspension at 59ºF to 86ºF (15ºC to 30ºC).
Tablets
- Keep BANZEL tablets in a dry place.
Oral Suspension
- Replace the cap securely after opening.
- Keep BANZEL Oral Suspension in an upright position.
- Use BANZEL Oral Suspension within 90 days of first opening the bottle.
Keep BANZEL and all medicines out of the reach of children.
General Information about the safe and effective use of BANZEL
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use BANZEL for a condition for which it was not prescribed. Do not give BANZEL to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about BANZEL. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about BANZEL that is written for health professionals.
For more information, go to www.banzel.com or call 1-888-274-2378.
What are the ingredients in BANZEL?
Tablets
Active ingredient: rufinamide
Inactive ingredients: colloidal silicon dioxide, corn starch crosscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulphate, iron oxide red, polyethylene glycol, talc, and titanium dioxide.
Oral Suspension
Active ingredient: rufinamide
Inactive ingredients: microcrystalline cellulose and carboxymethylcellulose sodium, hydroxyethylcellulose, anhydrous citric acid, simethicone emulsion 30%, poloxamer 188, methylparaben, propylparaben, propylene glycol, potassium sorbate, noncrystallizing sorbitol solution 70%, orange flavor.
The oral suspension does not contain lactose or gluten and is dye-free. The oral suspension does contain carbohydrates.
Distributed by Eisai Inc., Nutley, NJ 07110
Revised: 12/2021
This Medication Guide has been approved by the U.S. Food and Drug Administration.
© 2008-2021 Eisai Inc.
Instructions for Use
BANZEL (ban-‘zel)
[rufinamide]
Oral Suspension
Read the Instructions for Use before using BANZEL Oral Suspension and each time you get a refill. There may be new information. This leaflet does not take the place of talking with the doctor about your medical condition or treatment.
Prepare the BANZEL Oral Suspension dose
You will need the following supplies: See Figure A
- BANZEL Oral Suspension bottle
- Bottle adapter
- Dosing syringe (2 dosing syringes are included in the BANZEL Oral Suspension box)
Figure A
| Your total daily dose of BANZEL Oral Suspension is______mL.
Take BANZEL in 2 equally divided doses: Morning dose = _____mL Evening dose =______mL Note: The doctor may change your dose, especially when you are first starting BANZEL Oral Suspension. If your morning and evening doses are more than 20 mL each, measure each dose using either:
|
Step 1. Remove the BANZEL Oral Suspension bottle, bottle adapter, and 2 syringes from the box. See Figure A
Step 2. Shake the bottle well before each use. See Figure B
Figure B
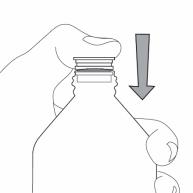
Step 3. Uncap the bottle and insert the bottle adapter into the bottle. See Figure C
Figure C
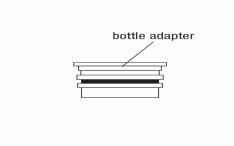
Once the bottle adapter is installed, it cannot be removed. See Figure D
Figure D
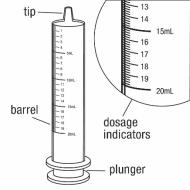
Step 4. Check the morning or evening dose in milliliters (mL) as prescribed by your doctor. Locate this number on the syringe. See Figure E
Figure E
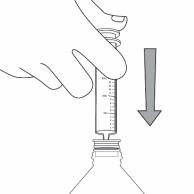
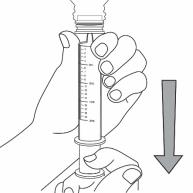
Step 5. Insert the syringe into the upright bottle and push the plunger all the way down. See Figure F
Figure F
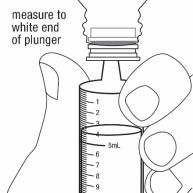
Step 6. With the syringe in place, turn the bottle upside down. Pull the plunger to the number of mL needed (the amount of liquid medicine in Step 4). See Figure G
Figure G
Measure the mLs of medicine from the end of the plunger. See Figure H
Figure H
Step 7. If the dose is more than 20 mL, you can either use:
• 2 syringes, or
• 1 syringe, taking two steps to draw up the medicine in that same syringe
For example:
If your dose is 30 mL, draw up 20 mL in the first syringe and the remaining 10 mL in the second syringe.
or
If your dose is 30 mL, draw up 20 mL in the single syringe and squirt the medicine into your mouth, then draw up the remaining 10 mL in that same syringe.
Repeat Steps 4 through 6 when drawing up the remaining dose of medicine, if your dose is more than 20 mL.
Step 8. Remove the syringe from the bottle adapter.
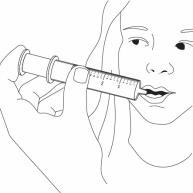
Step 9. Slowly squirt BANZEL directly into the corner of your mouth. If you need 2 syringes for your dose, slowly squirt the medicine from the first syringe into your mouth, then slowly squirt the medicine from the second syringe into your mouth. See Figure I
Figure I
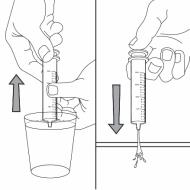
Step 10. Rinse the syringe (or syringes) with tap water after each use. See Figure J
- Fill a cup with water
- Pull back on the plunger and draw the water from the cup into the syringe
- Push on the plunger to release the water into the sink
Figure J
Step 11. Cap the bottle tightly. The cap will fit over the bottle adapter. Store the bottle upright at 59°F to 86°F (15°C to 30°C). See Figure K
Figure K
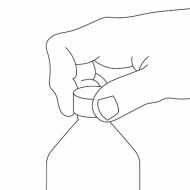
This Instructions for Use has been approved by the U. S. Food and Drug Administration. Revised: 12/2022




| BANZEL
rufinamide tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| BANZEL
rufinamide tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| BANZEL
rufinamide suspension |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Eisai Inc. (189246791) |
| Registrant - Eisai Co., Ltd. (695153262) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Ajinomoto Omnichem | 400344442 | api manufacture(62856-582, 62856-583, 62856-584) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bushu Pharmaceuticals, Ltd. | 692386648 | manufacture(62856-582, 62856-583) , pack(62856-582, 62856-583) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Catalent Pharma Solutions, LLC | 014167995 | analysis(62856-582, 62856-583) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Delpharm Huningue | 262639390 | analysis(62856-584) , manufacture(62856-584) , pack(62856-584) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pharma Packaging Solutions, LLC dba Tjoapack, LLC | 928861723 | label(62856-582, 62856-583, 62856-584) , pack(62856-582, 62856-583, 62856-584) | |
Frequently asked questions
More about Banzel (rufinamide)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (7)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- FDA approval history
- Drug class: dibenzazepine anticonvulsants
- Breastfeeding
- En español
